Synthesis of Tri- and Disubstituted Fluorenols and Derivatives Thereof Using Catalytic [2+2+2] Cyclotrimerization

 , , and
, , and 
Abstract

1. Introduction
2. Results
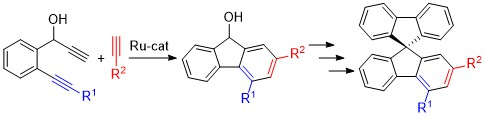
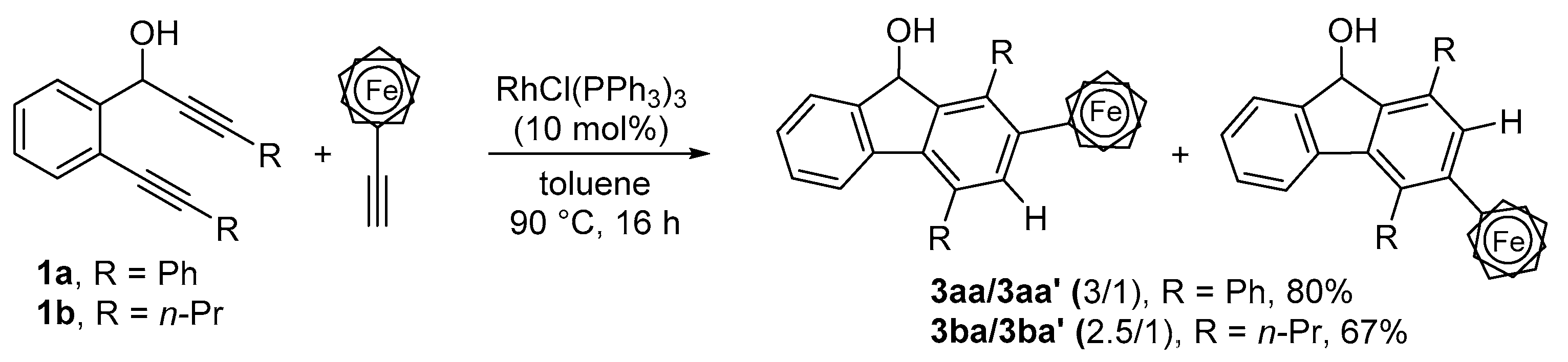
2.1. Synthesis of 1,2,4- and 1,3,4-Trisubstituted Fluorenols
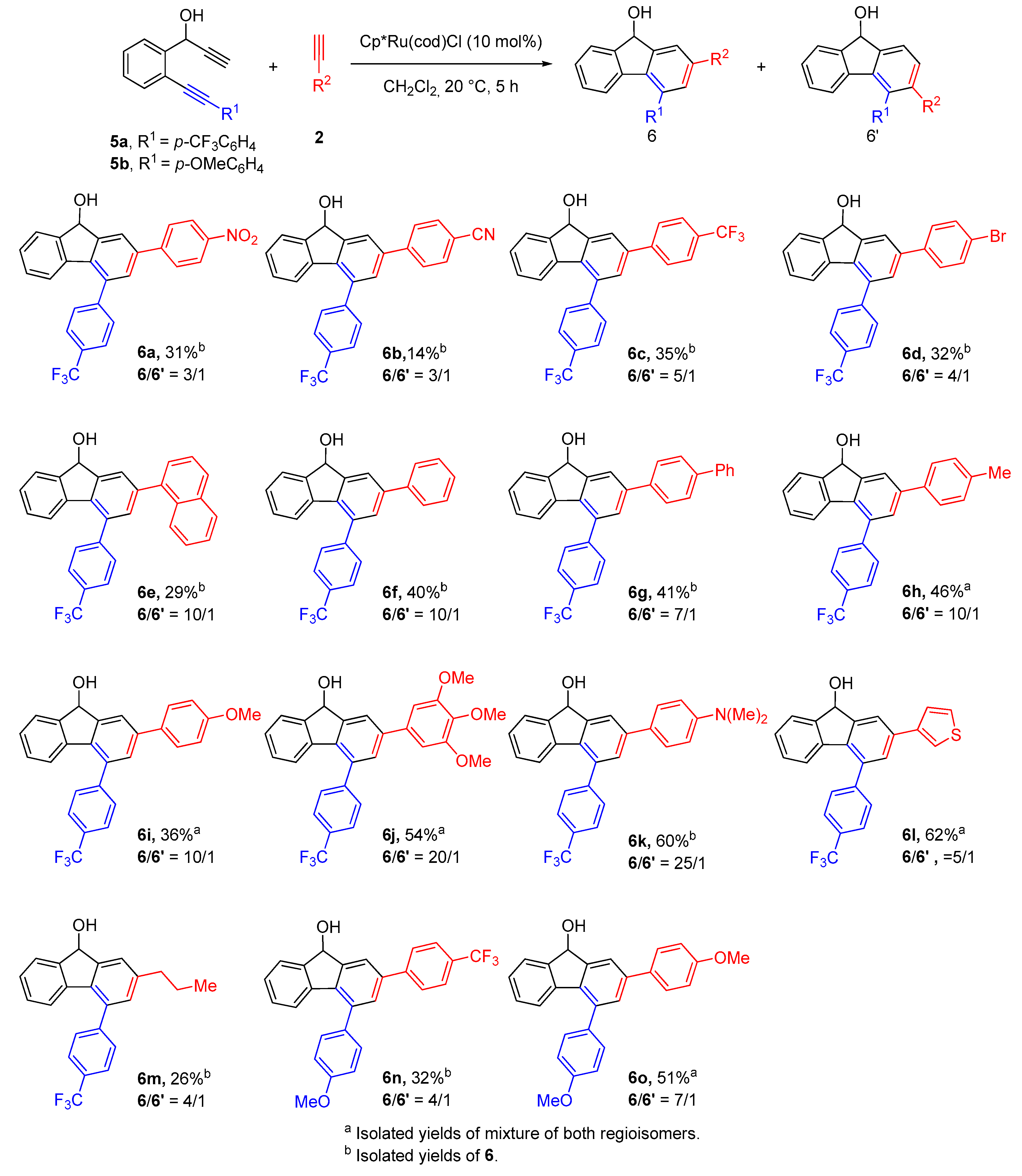
2.2. Synthesis of 2,4-Disubstituted Fluorenols
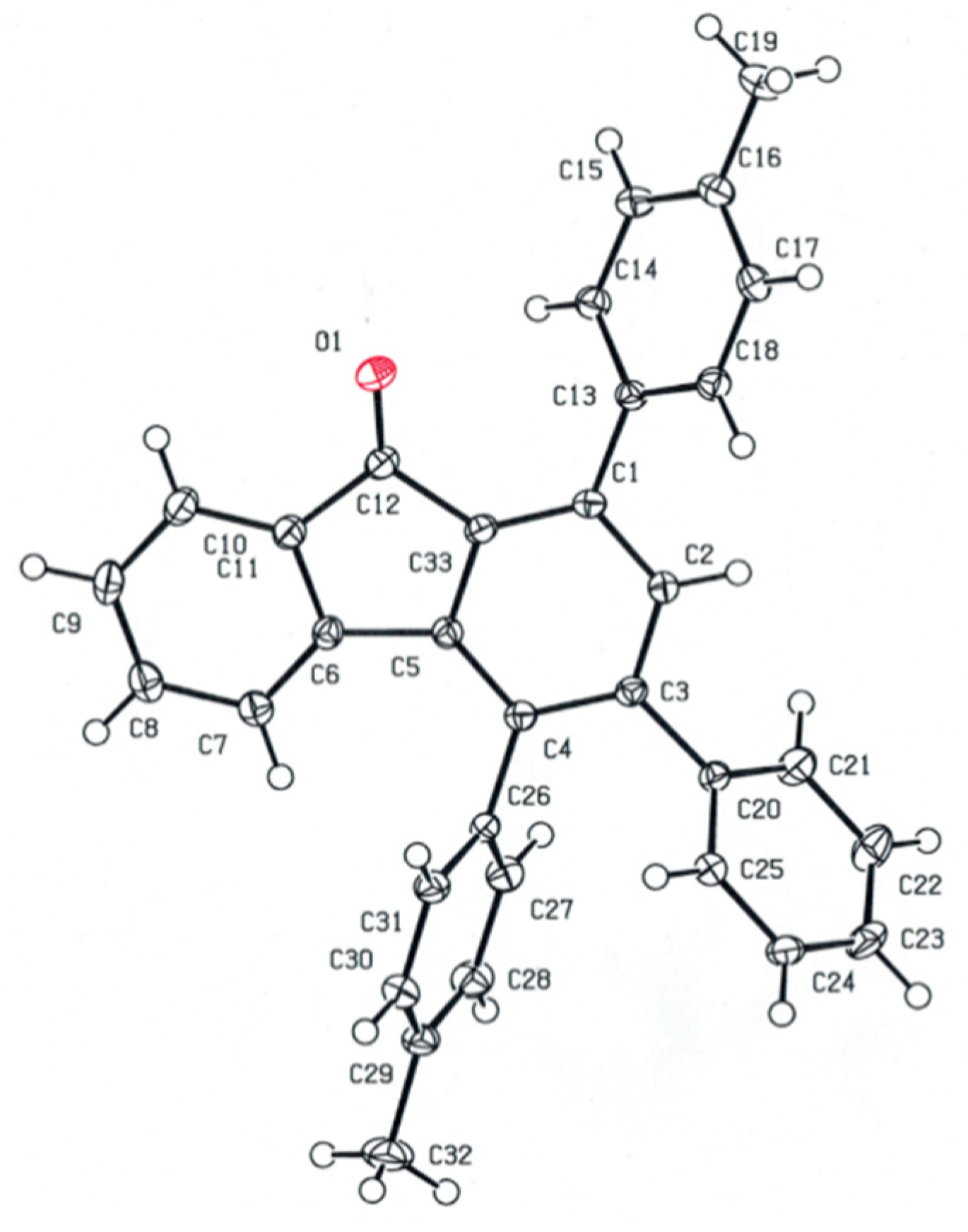
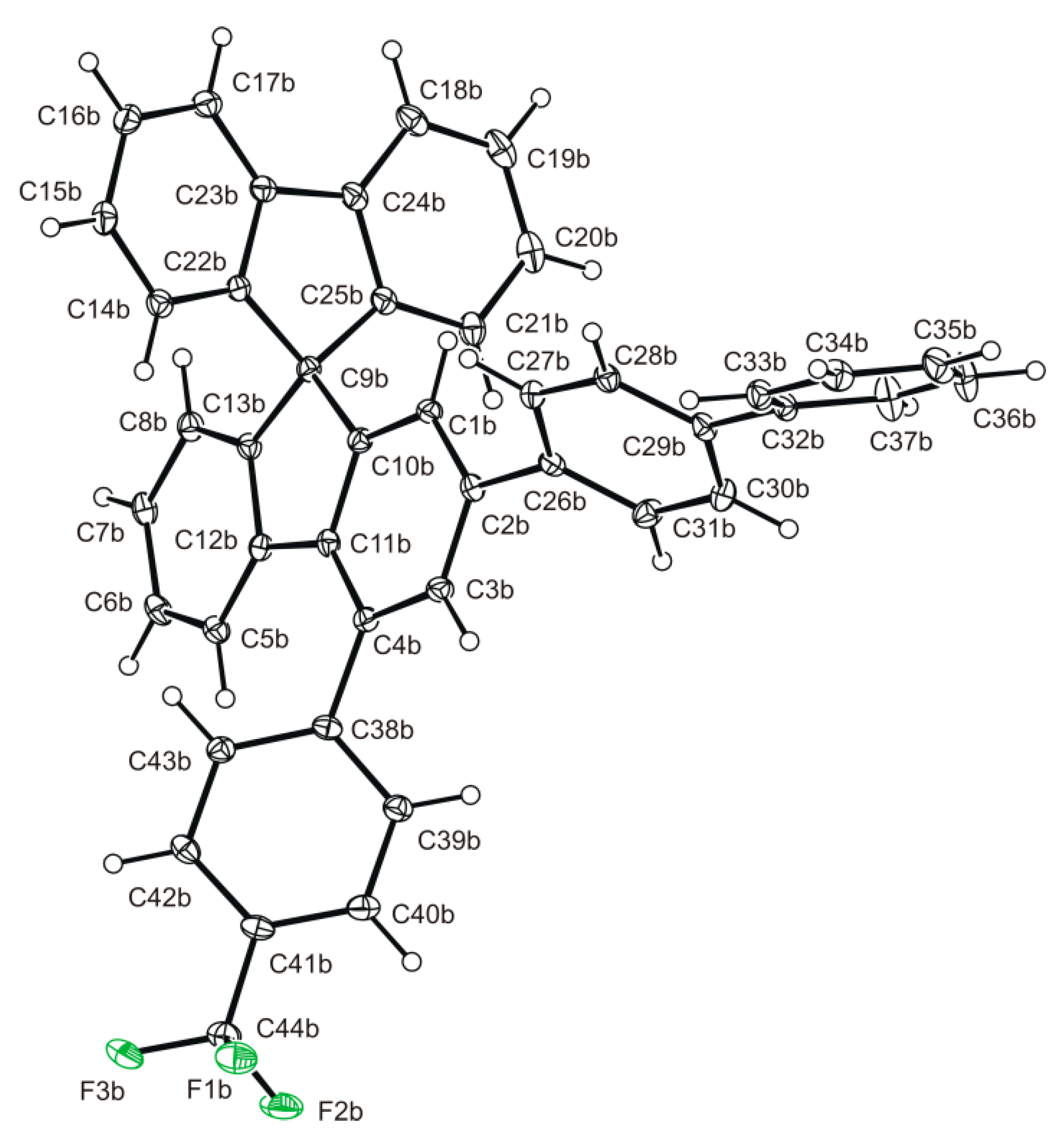
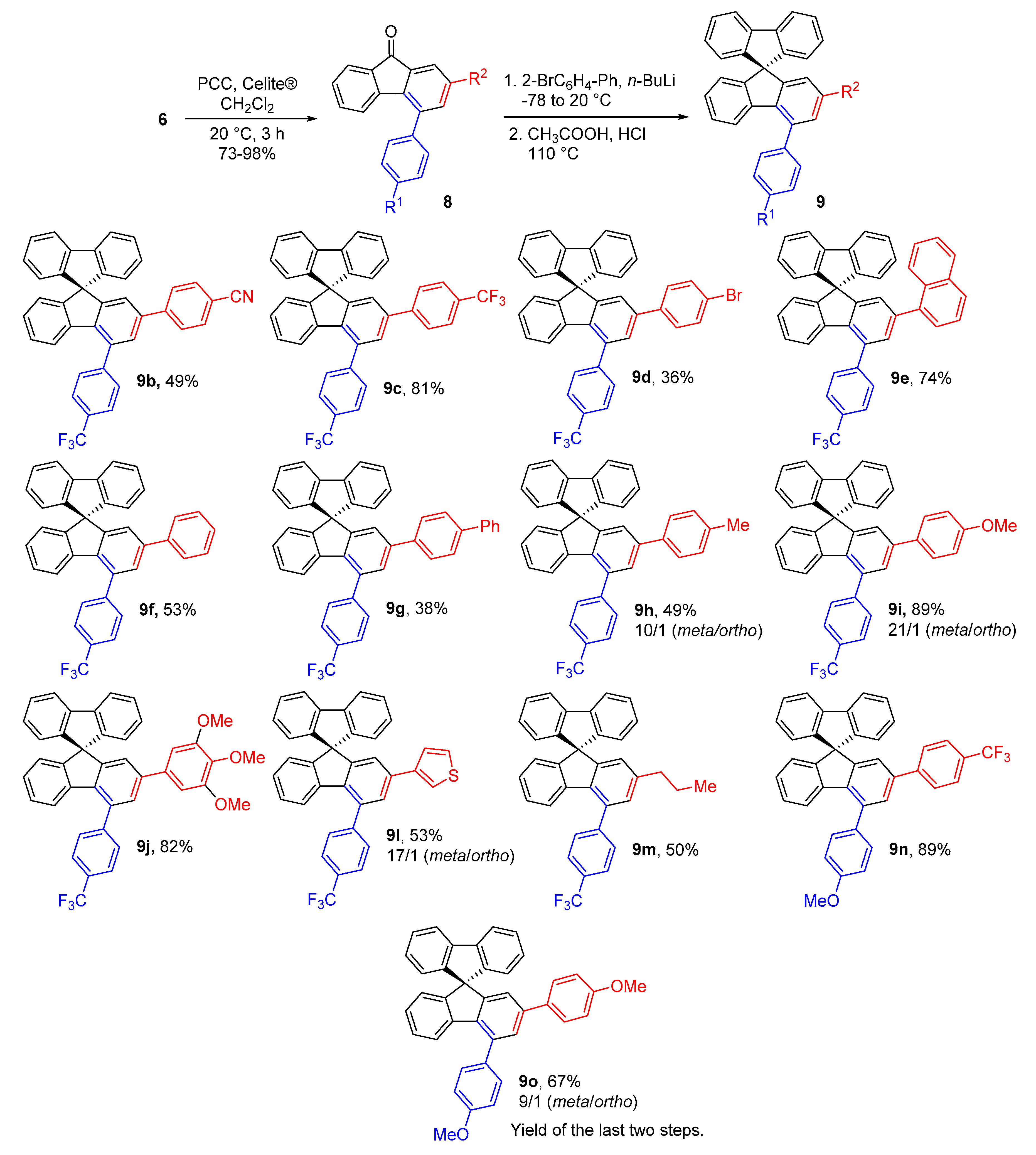
2.3. Structural Analysis
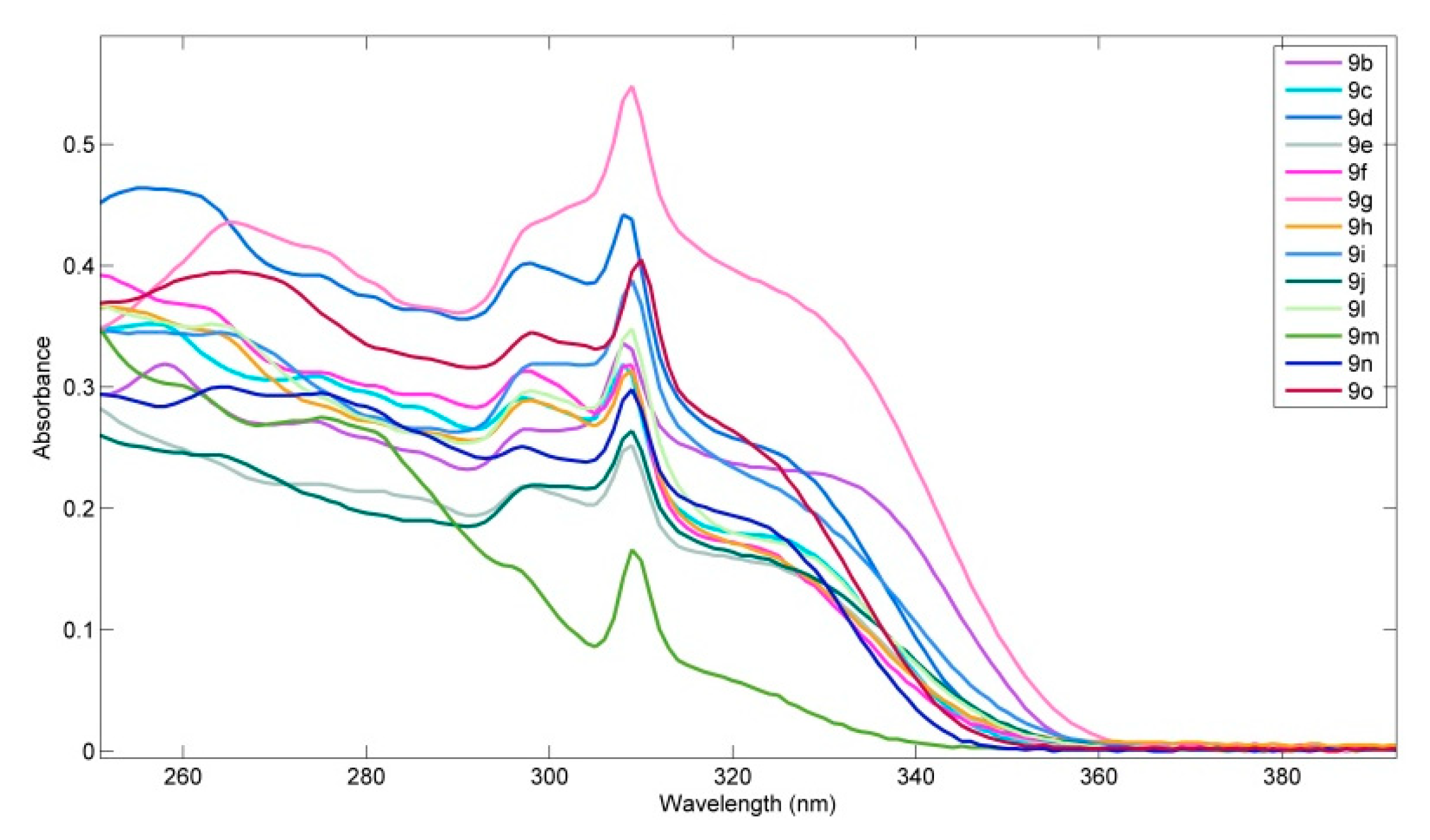
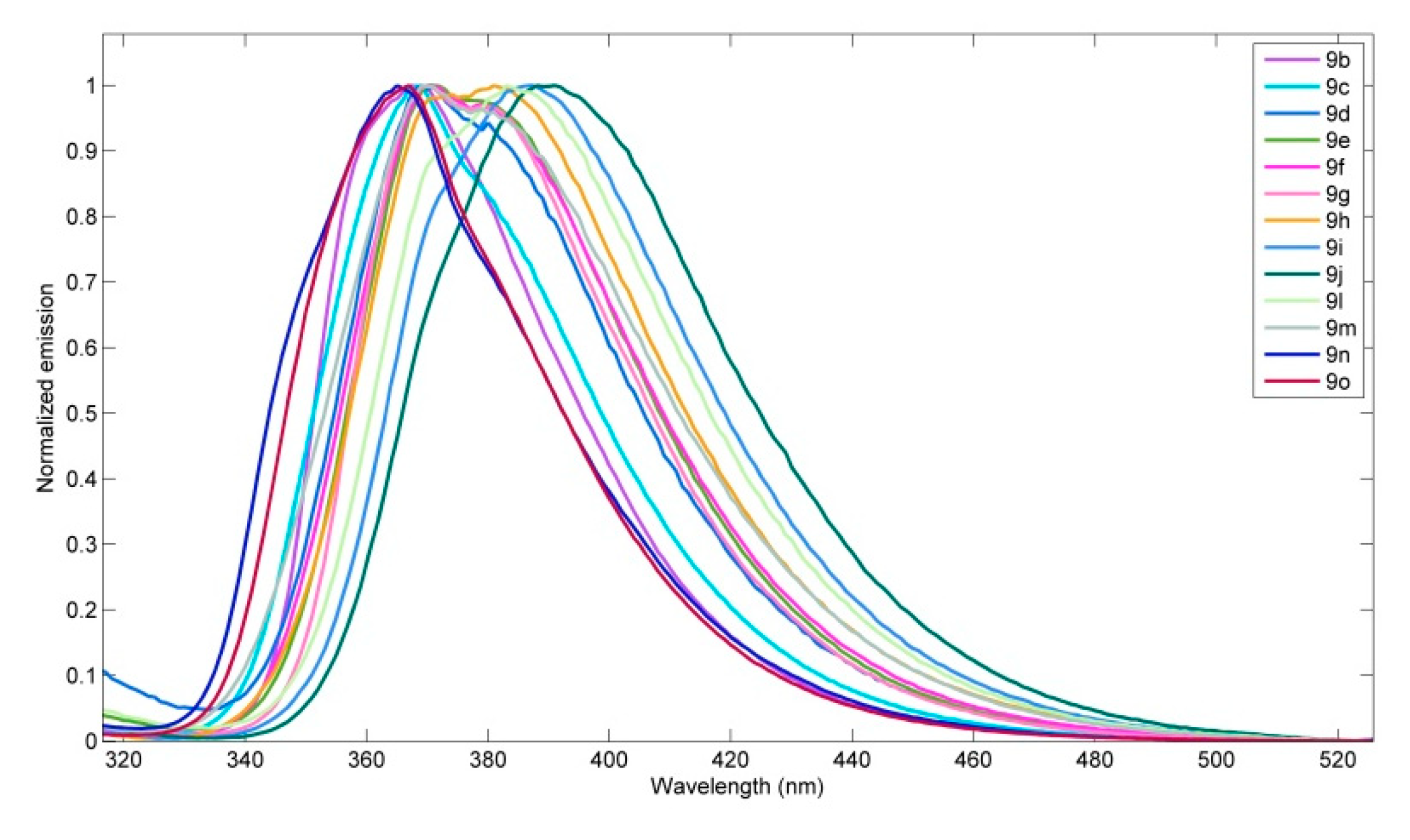
2.4. Photophysical Properties of Spirobifluorenes 9
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shi, Y.; Gao, S. Recent Advances of Synthesis of Fluorenone and Fluorene Containing Natural Products. Tetrahedron 2016, 14, 1717–1735. [Google Scholar] [CrossRef]
- Fleckenstein, C.A.; Plenio, H. 9-Fluorenylphosphines for the Pd-Catalyzed Sonogashira, Suzuki, and Buchwald–Hartwig Coupling Reactions in Organic Solvents and Water. Chem. Eur. J. 2007, 13, 2701–2716. [Google Scholar] [CrossRef] [PubMed]
- King, D.S.; Fields, C.G.; Fields, G.B. A Cleavage Method Which Minimizes Side Reactions Following Fmoc Solid Phase Peptide Synthesis. Int. J. Pept. Protein Res. 1990, 36, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.; Bryce, M.R. Electron-transporting Materials for Organic Electroluminescent and Electrophosphorescent Devices. J. Mater. Chem. 2005, 15, 94–107. [Google Scholar] [CrossRef]
- Saragi, T.P.I.; Spehr, T.; Siebert, A.; Fuhrmann-Lieker, T.; Salbeck, J. Spiro Compounds for Organic Optoelectronics. Chem. Rev. 2007, 107, 1011–1065. [Google Scholar] [CrossRef]
- Wu, C.C.; Lin, Y.T.; Chiang, H.H.; Cho, T.Y.; Chen, C.W.; Wong, K.T.; Liao, Y.L.; Lee, G.H.; Peng, S.M. Highly bright blue organic light-emitting devices using spirobifluorene-cored conjugated compounds. Appl. Phys. Lett. 2002, 81, 577–581. [Google Scholar] [CrossRef]
- Atul, G.; Sumit, C.; Manish, D.; Kumar, V.; Prakash, S.; Jena, B.; Verma, J.K.; Jain, M.; Anand, R.S.; Manoharan, S.S. Donor−Acceptor 9-Uncapped Fluorenes and Fluorenones as Stable Blue Light Emitters. Org. Lett. 2009, 6, 1289–1292. [Google Scholar]
- Ingans, O.; Zhang, F.; Andersson, M.R. Alternating Polyfluorenes Collect Solar Light in Polymer Photovoltaics. Acc. Chem. Res. 2009, 42, 1731–1739. [Google Scholar] [CrossRef]
- Etoria, H.; Jin, X.L.; Yasuda, T.; Mataka, S.; Tsutsui, T. Spirobifluorene Derivatives for Ultraviolet Organic Light-Emitting Diodes. Syn. Met. 2006, 156, 1090–1096. [Google Scholar] [CrossRef]
- Lyu, Y.-Y.; Kwak, J.; Jeon, W.-S.; Byun, Y.; Lee, H.S.; Kim, D.; Lee, C.; Char, K. Highly Efficient Red Phosphorescent OLEDs based on Non-Conjugated Silicon-Cored Spirobifluorene Derivative Doped with Ir-Complexes. Adv. Funct. Mater. 2009, 19, 420–427. [Google Scholar] [CrossRef]
- Moreau, F.; Audebrabd, N.; Poriel, C.; Moizan-Baslé, V.; Ouvry, J. A 9,9′-spirobifluorene Based Metal–Organic Framework: Synthesis, Structure Analysis and Gas Sorption Properties. J. Mater. Chem. 2011, 21, 18715–18722. [Google Scholar] [CrossRef]
- Thiery, S.; Declairieux, C.; Tondelier, D.; Seo, G.; Geoffry, B.; Jeannin, O.; Metivier, R.; Rault-Berthelot, J.; Poriel, C. 2-Substituted vs. 4-substituted-9,9′-spirobifluorene host materials for green and blue phosphorescent OLEDs: A structure—Property for green and blue phosphorescent OLEDs: A structure—Property. Tetrahedron 2014, 70, 6337–6351. [Google Scholar]
- Thiery, S.; Tondelier, D.; Declairieux, S.; Seo, G.; Geoffry, B.; Jeannin, O.; Metivier, R.; Rault-Berthelot, J.; Poriel, C. 9,9′-Spirobifluorene and 4-phenyl-9,9′-spirobifluorene: Pure hydrocarbon small molecules as hosts for efficient green and blue PhOLEDs. J. Mater. Chem. C 2014, 2, 4156–4166. [Google Scholar] [CrossRef]
- Thiery, S.; Tondelier, D.; Declairieux, S.; Geoffry, B.; Jeannin, O.; Metivier, R.; Rault-Berthelot, J.; Poriel, C. 4-Pyridyl-9,9′-spirobifluorenes as Host Materials for Green and SkyBlue Phosphorescent OLEDs. J. Phys. Chem. 2015, 119, 5790–5805. [Google Scholar]
- Poriel, C.; Rault-Berthelot, J. Structure–property relationship of 4-substitutedspirobifluorenes as hosts for phosphorescent organic light emitting diodes: An overview. J. Mater. Chem. C 2017, 5, 3869–3897. [Google Scholar] [CrossRef]
- Sicard, L.; Quinton, C.; Peltier, J.-D.; Tondelier, D.; Geffroy, B.; Biapo, U.; Métivier, R.; Jeannin, O.; Rault-Berthelot, J.; Poriel, C. Spirobifluorene Regioisomerism: A Structure–Property Relationship Study. Chem. Eur. J. 2017, 23, 7719–7727. [Google Scholar] [CrossRef]
- Clarkson, R.G.; Gomberg, M. Spirans with FourAromatic Radicals on the Spiro Carbon Atom. J. Am. Chem. Soc. 1930, 52, 2881–2891. [Google Scholar] [CrossRef]
- Kaiser, R.P.; Caivano, I.; Kotora, M. Transition-metal-catalyzed methods for synthesis of fluorenes. Tetrahedron report 1186. Tetrahedron 2019, 75, 2981–2992. [Google Scholar] [CrossRef]
- Reppe, W.; Schlichting, O.; Klager, K.; Toepel, T. Cyclisierende Polymerisation von Acetylen I Über Cyclooctatetraen. Liebigs Ann. Chem. 1948, 560, 1–92. [Google Scholar] [CrossRef]
- Reppe, W.; Schweckendiek, W. Cyclisierende Polymerisation von Acetylen. III Benzol, Benzolderivate und hydroaromatische Verbindungen. Liebigs Ann. Chem. 1948, 560, 104–116. [Google Scholar]
- Kaiser, R.P.; Hessler, F.; Mosinger, J.; Císařová, I.; Kotora, M. A [2+2+2]Cyclotrimerization Approach to Selectively Substituted Fluorenes and Fluorenols, and Their Conversion to 9,9′-Spirobifluorenes. Chem. Eur. J. 2015, 21, 13577–13582. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.P.; Mosinger, J.; Císařová, I.; Kotora, M. Synthesis of selectively 4-substituted 9,9′-spirobifluorenes and modulation of their photophysical properties. Org. Biomol. Chem. 2017, 15, 6913–6920. [Google Scholar] [CrossRef] [PubMed]
- Broere, D.L.J.; Ruijter, E. Transition-Metal-Catalyzed [2+2+2]Cyclo(co)trimerizations. Synthesis 2012, 44, 2639–2672. [Google Scholar]
- Hilt, G.; Hess, W.; Vogler, T.; Hengst, C. Ligand and Solvent Effects on Cobalt(I)-Catalysed Reactions: Alkyne Dimerisation Versus [2+2+2]Cyclotrimerisation Versus Diels–Alder Reaction Versus [4+2+2]cycloaddition. J. Organomet. Chem. 2005, 690, 5170–5181. [Google Scholar] [CrossRef]
- Hilt, G.; Hengst, C.; Hess, W. Solvent-Dependent Regiochemical Cyclotrimerisation of Phenylacetylene with Cobalt Catalysts Containing Disulfide Ligands: A Case Study. Eur. J. Org. Chem. 2008, 13, 2293–2297. [Google Scholar] [CrossRef]
- Komine, Y.; Miyauchi, Y.; Kobayashi, M.; Tanaka, K. Synthesis of Tri- and Diaryloxybenzenes by Rhodium-Catalyzed Complete Intermolecular [2+2+2]Cycloaddition of Aryl Ethynyl Ethers. Synlett 2010, 20, 3092–3098. [Google Scholar] [CrossRef]
- Komine, Y.; Kamisawa, A.; Tanaka, K. Flexible Synthesis of Fused Benzofuran Derivatives by Rhodium-Catalyzed [2+2+2]Cycloaddition with Phenol-Linked 1,6-Diynes. Org. Lett. 2009, 11, 2361–2364. [Google Scholar] [CrossRef]
- Tanaka, K.; Sawada, Y.; Aida, Y.; Thammathevo, M.; Tanaka, R.; Sagae, H.; Otake, Y. Rhodium-catalyzed Convenient Synthesis of Functionalized Tetrahydronaphthalenes. Tetrahedron 2010, 66, 1563–1569. [Google Scholar] [CrossRef]
- Nishigaki, S.; Shivata, Y.; Tanaka, K. Rhodium-Catalyzed Chemo- and Regioselective Intermolecular Cross-Cyclotrimerization of Nonactivated Terminal and Internal Alkynes. J. Org. Chem. 2017, 82, 11117–11125. [Google Scholar] [CrossRef]
- Fabbian, M.; Marsich, N.; Farnetti, E. Organoiridium compounds with bidentate phosphines as highly regioselective catalysts for alkynes cyclotrimerization. Inorg. Chim. Acta 2004, 357, 2881–2888. [Google Scholar] [CrossRef]
- Takeuchi, R.; Tanaka, S.; Nakaya, Y. Iridium Complex-catalyzed [2+2+2]Cycloaddition of α,ω-Diynes with Monoalkynes: A New and Efficient Catalyst for Cyclotrimerization of Alkynes. Tetrahedron Lett. 2001, 42, 2991–2994. [Google Scholar] [CrossRef]
- Kezuka, S.; Tanaka, S.; Ohe, T.; Nakaya, Y.; Takeuchi, R. Iridium Complex-Catalyzed [2+2+2]Cycloaddition of α,ω-Diynes with Monoynes and Monoene. J. Org. Chem. 2006, 71, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Okabe, A.; Mizuno, T.; Izawa, M.; Takeuchi, R. Iridium-catalyzed [2+2+2] cycloaddition of α,ω-diynes with alkynyl ketones and alkynyl esters. Tetrahedron 2014, 70, 8681–8689. [Google Scholar] [CrossRef]
- Matoušová, E.; Gyepes, R.; Císařová, I.; Kotora, M. [2+2+2]Cyclotrimerization of 1-Cyclopropyl-1,6-diynes with Alkynes: Formation of Cyclopropylarenes. Adv. Synth. Catal. 2016, 358, 254–267. [Google Scholar] [CrossRef]
- Van Leeuwen, P.W.N.M.; Kamer, P.C.J.; Reek, J.N.H.; Dierkes, P. Ligand Bite Angle Effects in Metal-catalyzed C−C Bond Formation. Chem. Rev. 2000, 100, 2741–2769. [Google Scholar] [CrossRef]
- Birkholz (née Gensow), M.-N.; Freixa, Z.; van Leeuwen, P.W.N.M. Bite angle effects of diphosphines in C–C and C–X bond forming cross coupling reactions. Chem. Soc. Rev. 2009, 38, 1099–1118. [Google Scholar] [CrossRef]
- Lee, T.-J.; Lee, D.H.; Kim, B.; Oh, H.S.; Kang, H.-R. Organic Electroluminiscent Device. WO 2018/182221 Al, 4 October 2018. [Google Scholar]
- Yamamoto, Y.; Kinpara, K.; Saigoku, T.; Nishiyama, H.; Itoh, K. Synthesis of Benzo-fused Lactams and Lactonesvia Ru(ii)-Catalyzed Cycloaddition of Amide- and Ester-tethered α,ω-Diynes with Terminal Alkynes: Electronic Directing Effect of Internal Conjugated Carbonyl Group. Org. Biomol. Chem. 2004, 2, 1287–1294. [Google Scholar] [CrossRef]
- Jiang, Z.; Yao, H.; Zhang, Z.; Yang, C.; Liu, Z.; Tao, Y.; Qin, J.; Ma, D. Novel Oligo-9,9′-spirobifluorenes through ortho-Linkage as Full Hydrocarbon Host for Highly Efficient Phosphorescent OLEDs. Org. Lett. 2009, 11, 2607–2610. [Google Scholar] [CrossRef]
- Fan, C.; Chen, Y.; Gan, P.; Yang, C.; Zhong, C.; Qin, J.; Ma, D. Tri-, Tetra- and Pentamers of 9,9′-Spirobifluorenes through Full ortho-Linkage: High Triplet-Energy Pure Hydrocarbon Host for Blue Phosphorescent Emitter. Org. Lett. 2010, 12, 5648–5651. [Google Scholar] [CrossRef]
- Cui, L.-S.; Xie, Y.-M.; Wang, Y.-K.; Zhong, C.; Deng, Y.-L.; Liu, X.-Y.; Jiang, Z.-Q.; Liao, L.-S. Pure Hydrocarbon Hosts for ≈100% Exciton Harvesting in Both Phosphorescent and Fluorescent Light-Emitting Devices. Adv. Mater. 2015, 27, 4213–4217. [Google Scholar] [CrossRef]
- Jang, S.E.; Joo, C.W.; Jeon, S.O.; Yook, K.S.; Lee, J.Y. Pure Hydrocarbon Hosts for ≈100% Exciton Harvesting in Both Phosphorescent and Fluorescent. The Relationship between the Substitution Position of the Diphenylphosphine oxide on the Spirobifluorene and Device Performances of Blue Phosphorescent Organic Light-emitting Diodes. Org. Electron. 2010, 11, 1059–1065. [Google Scholar]
- Dong, S.-C.; Gao, C.-H.; Yuan, X.D.; Cui, L.-S.; Jiang, Z.-Q.; Lee, S.-T.; Liao, L.S. Novel Dibenzothiophene Based Host Materials Incorporating Spirobifluorene for High-efficiency White Phosphorescent Organic Light-emitting Diodes. Org. Electron. 2013, 14, 902–908. [Google Scholar] [CrossRef]
- Dong, S.-C.; Gao, C.-H.; Zhang, Z.-H.; Jiang, Z.-Q.; Lee, S.-T.; Liao, L.-S. New Dibenzofuran/spirobifluorene Hybrids as Thermally Stable Host Materials for Efficient Phosphorescent Organic Light-emitting Diodes with Low Efficiency Roll-off. Phys. Chem. Chem. Phys. 2012, 14, 14224–14228. [Google Scholar] [CrossRef] [PubMed]
- Quinton, C.; Thiery, S.; Jeannin, O.; Tondelier, D.; Geffroy, B.; Jacques, E.; Rault-Berthelot, J.; Poriel, C. Electron-Rich 4-Substituted Spirobifluorenes: Toward a New Family of High Triplet Energy Host Materials for High-Efficiency Green and Sky Blue Phosphorescent OLEDs. ACS Appl. Mater. Interfaces 2017, 9, 6194–6206. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R | 2 | Yield (%) 1 | 4:4‘ 2 |
|---|---|---|---|---|
| 1 | 4-MeOC6H4- | 2a | 43 | 1:3.2 |
| 2 | 4-MeC6H4- | 2b | 31 | 1:1.6 |
| 3 | 4-PhC6H4- | 2c | 42 | 1:0.7 |
| 4 | Ph- | 2d | 23 | 1:0.8 |
| 5 | 4-CF3C6H4- | 2e | 26 | 1:1 |
| 6 | 3-thienyl | 2f | 18 | 1:1.3 |
| 7 | n-Pr- | 2g | 25 | 1:0.7 |
| 8 | Me3Si- | 2h | 15 | 1:2.4 |

| Entry | Ligand | β (°) 1 | 4d:4d‘ 2 |
|---|---|---|---|
| 1 | dppp | 91 | 1:3.2 |
| 2 | dppb | 94 | 1:1.6 |
| 3 | (R)-BINAP | 93 | 1:0.7 |
| 4 | (S)-BINAP | 93 | 1:0.8 |
| 5 | dppf | 99 | 1:2.4 |

| Entry | Catalytic system | Conditions | 6h:6h‘ 1 | Yield (%) 2 |
|---|---|---|---|---|
| 1 | [RhCl(PPh3)3] (3 mol%), Ag2CO3 (6 mol%) | 180 °C, MW, 1h | 1:2 | 62 |
| 2 | [Rh(cod)2]BF4 (10 mol%), dppp | THF, 60 °C, 16 h | 1:1.25 | 40 |
| 3 | Cp*Ru(cod)Cl (10 mol%), | CH2Cl2, 20 °C, 5 h | 10:1 | 46 |

| Entry | 2b (eq) | 6h + 6h′, Yield (%) 1 | 7, Yield (%) 1 |
|---|---|---|---|
| 1 | 2 | 46 | 40 |
| 2 | 3 | 47 | 40 |
| 3 | 4 | 66 | 19 |
| 4 | 5 | 66 | 18 |
| 5 | 7 | 75 | 14 |
| 6 | 9 | 76 | 14 |
| 7 | 10 | 77 | 15 2 |
| 9 | λabs/nm (ε/104 mol−1∙dm3∙cm−1) | λem/nm | Φs 1 |
|---|---|---|---|
| 9b | 258 (3.1), 274 (sh), 297 (2.6), 308 (3.3), 331 (sh) | 367 | 0.92 |
| 9c | 256 (3.5), 274 (sh), 297 (2.9), 308 (3.2), 326 (sh) | 368, 378 | 0.88 |
| 9d | 258 (4.6), 275 (sh), 298 (4.0), 308 (4.4), 326 (sh) | 369, 380 | 0.14 |
| 9e | 255 (sh), 275 (sh), 297 (2.1), 309 (2.4), 323 (sh) | 371, 379 | 0.77 |
| 9f | 250 (sh), 262 (sh), 297 (3.1), 309 (3.1), 323 (sh) | 370. 379 | 0.86 |
| 9g | 265(4.4), 275 (sh), 298 (sh), 309 (5.5), 326 (sh) | 370, 380 | 1.00 |
| 9h | 252 (sh), 263 (sh), 298 (2.9), 309 (3.1), 325 (sh) | 373, 380 | 0.69 |
| 9i | 264 (sh), 299 (3.2), 309 (3.9), 323 (sh) | 386 | 0.78 |
| 9j | 264 (sh), 298 (2.2), 309 (2.7), 324 (sh) | 391 | 0.37 |
| 9l | 264 (3.5), 298 (3.0), 309 (3.5), 326 (sh) | 372, 385 | 0.44 |
| 9m | 260 (sh), 278 (2.5), 297 (sh), 309 (1.6) | 370, 380 | 0.64 |
| 9n | 264 (3.0), 275 (sh), 297 (2.5), 309 (3.0), 323 (sh) | 365 | 0.86 |
| 9o | 265 (3.9), 298 (3.4), 310 (4.0), 323 (sh) | 367 | 0.91 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caivano, I.; Kaiser, R.P.; Schnurrer, F.; Mosinger, J.; Císařová, I.; Nečas, D.; Kotora, M. Synthesis of Tri- and Disubstituted Fluorenols and Derivatives Thereof Using Catalytic [2+2+2] Cyclotrimerization. Catalysts 2019, 9, 942. https://doi.org/10.3390/catal9110942
Caivano I, Kaiser RP, Schnurrer F, Mosinger J, Císařová I, Nečas D, Kotora M. Synthesis of Tri- and Disubstituted Fluorenols and Derivatives Thereof Using Catalytic [2+2+2] Cyclotrimerization. Catalysts. 2019; 9(11):942. https://doi.org/10.3390/catal9110942
Chicago/Turabian StyleCaivano, Ilaria, Reinhard P. Kaiser, Florian Schnurrer, Jiří Mosinger, Ivana Císařová, David Nečas, and Martin Kotora. 2019. "Synthesis of Tri- and Disubstituted Fluorenols and Derivatives Thereof Using Catalytic [2+2+2] Cyclotrimerization" Catalysts 9, no. 11: 942. https://doi.org/10.3390/catal9110942
APA StyleCaivano, I., Kaiser, R. P., Schnurrer, F., Mosinger, J., Císařová, I., Nečas, D., & Kotora, M. (2019). Synthesis of Tri- and Disubstituted Fluorenols and Derivatives Thereof Using Catalytic [2+2+2] Cyclotrimerization. Catalysts, 9(11), 942. https://doi.org/10.3390/catal9110942