Decoding Essential Amino Acid Residues in the Substrate Groove of a Non-Specific Nuclease from Pseudomonas syringae



Abstract

1. Introduction
2. Results
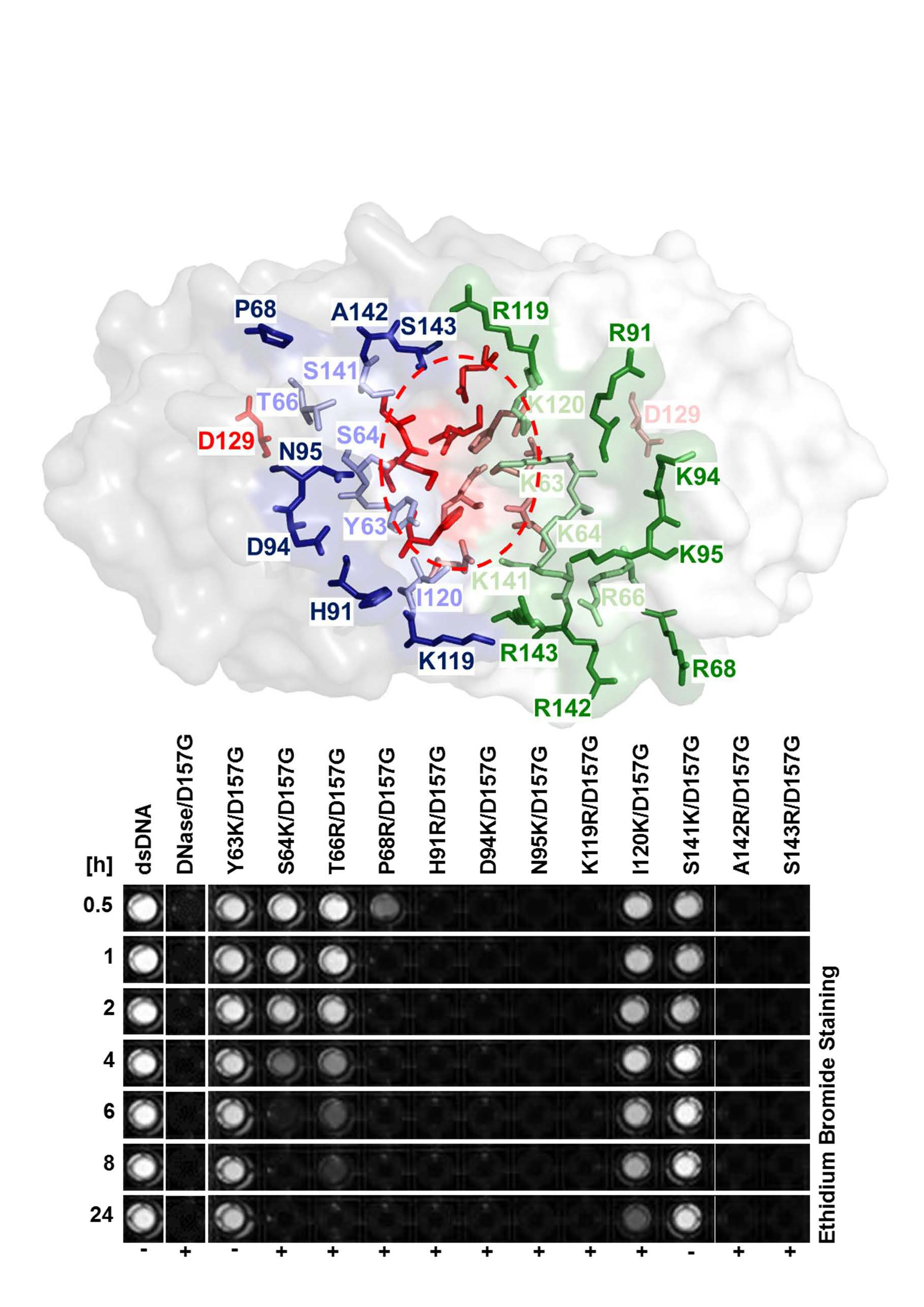
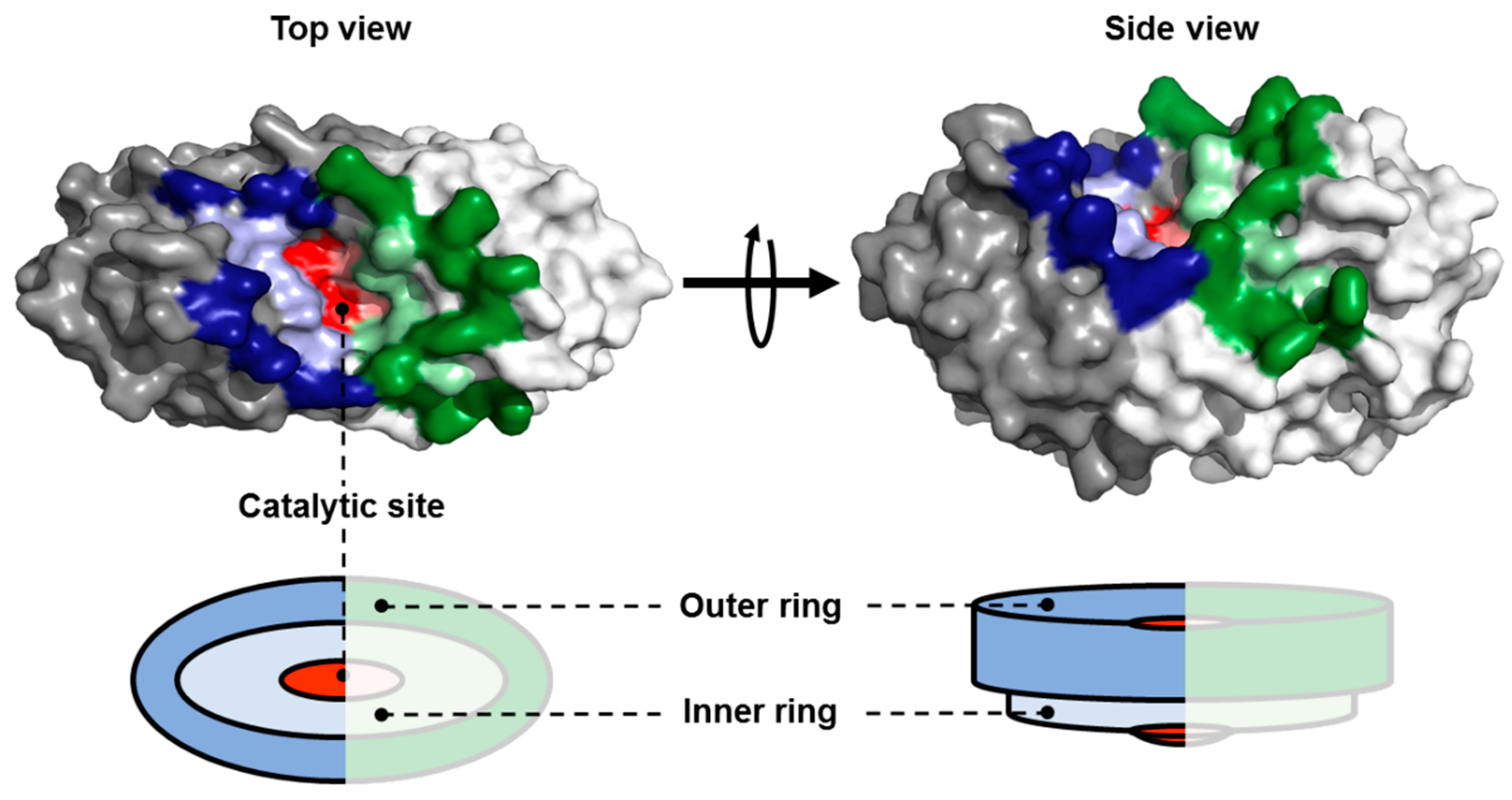
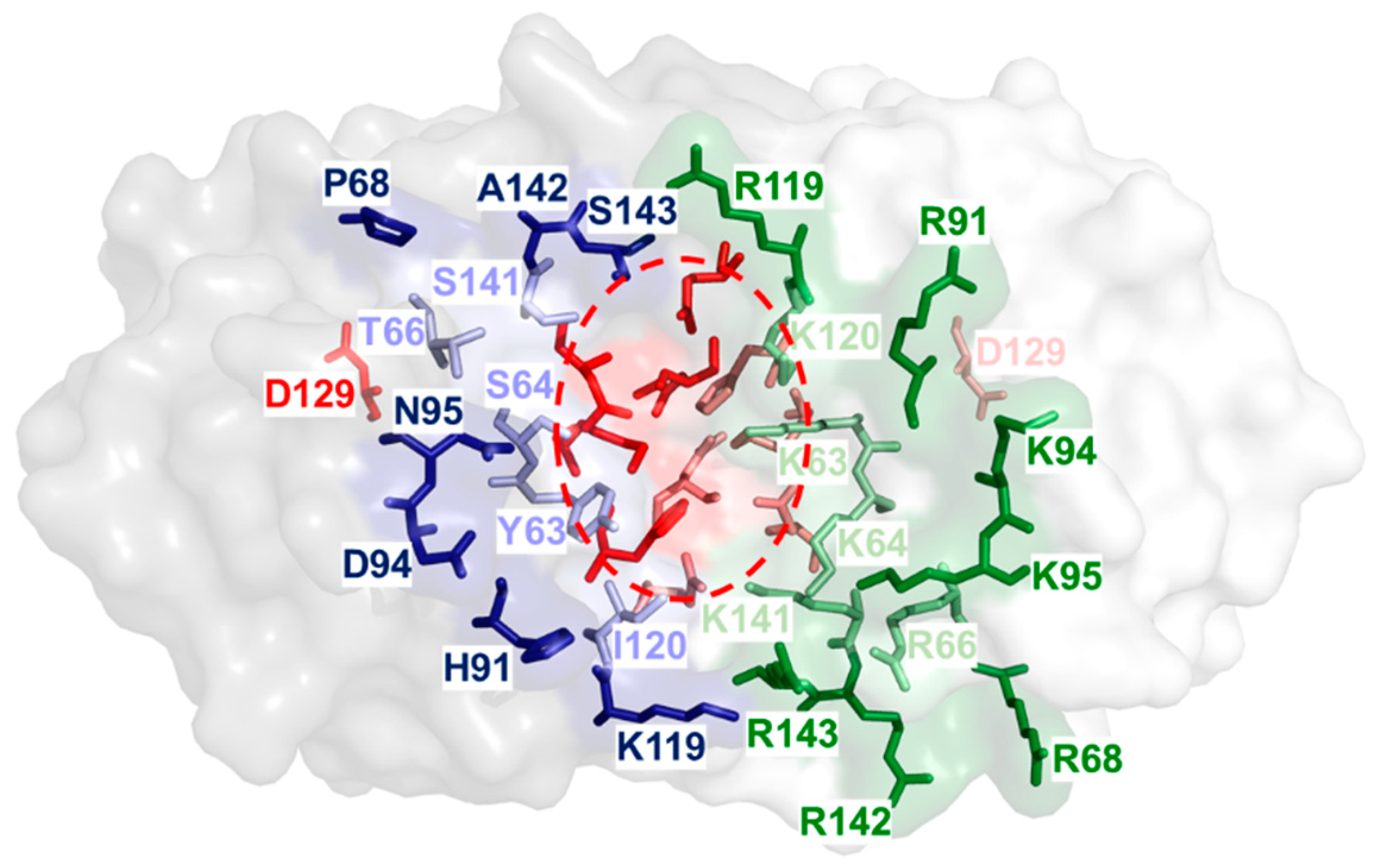
2.1. Identification of Amino Acid Residues within the Substrate Groove of a NSN from Pseudomonas sp.
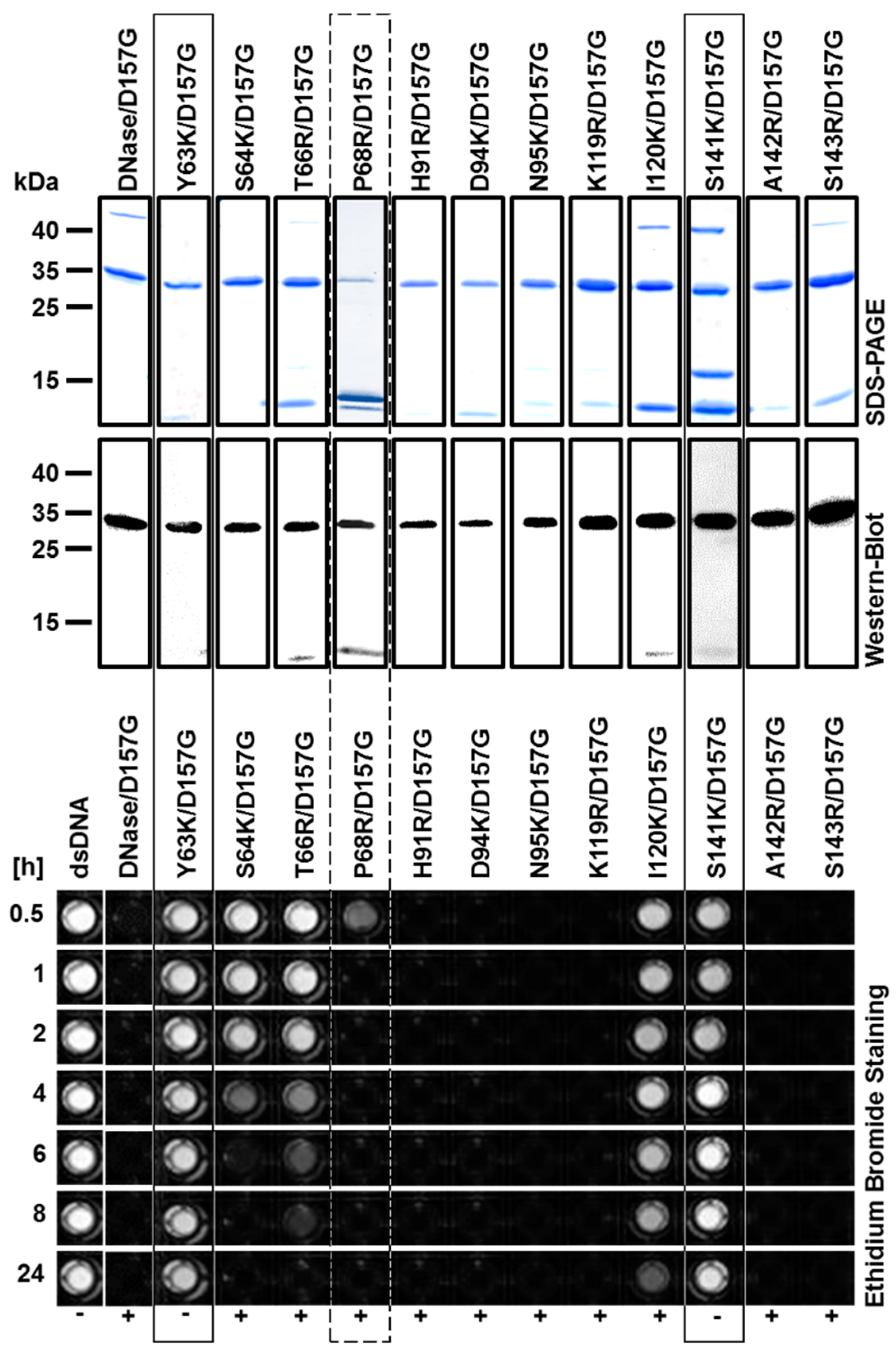
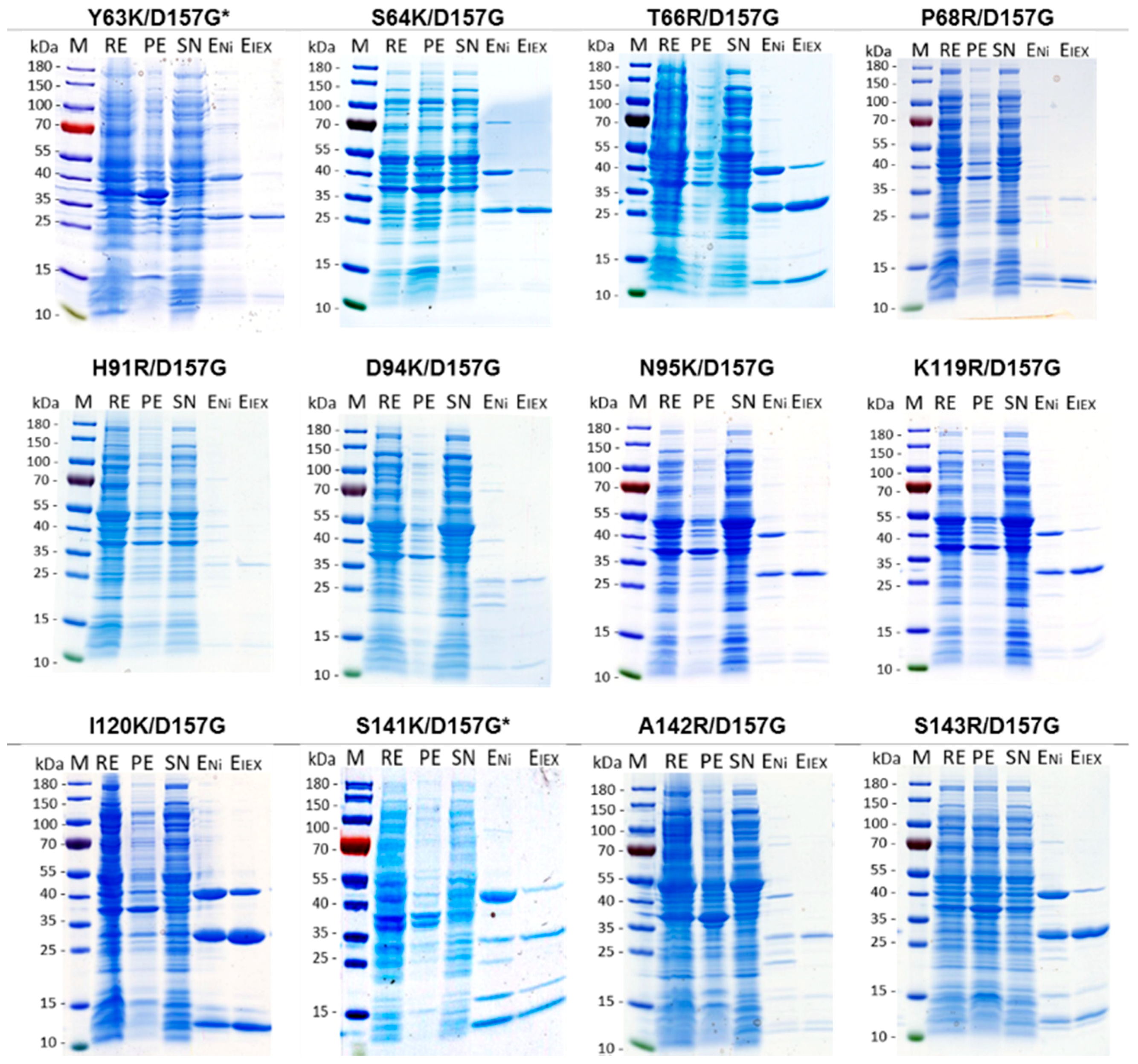
2.2. Production and Purification of Recombinant Nuclease Mutants
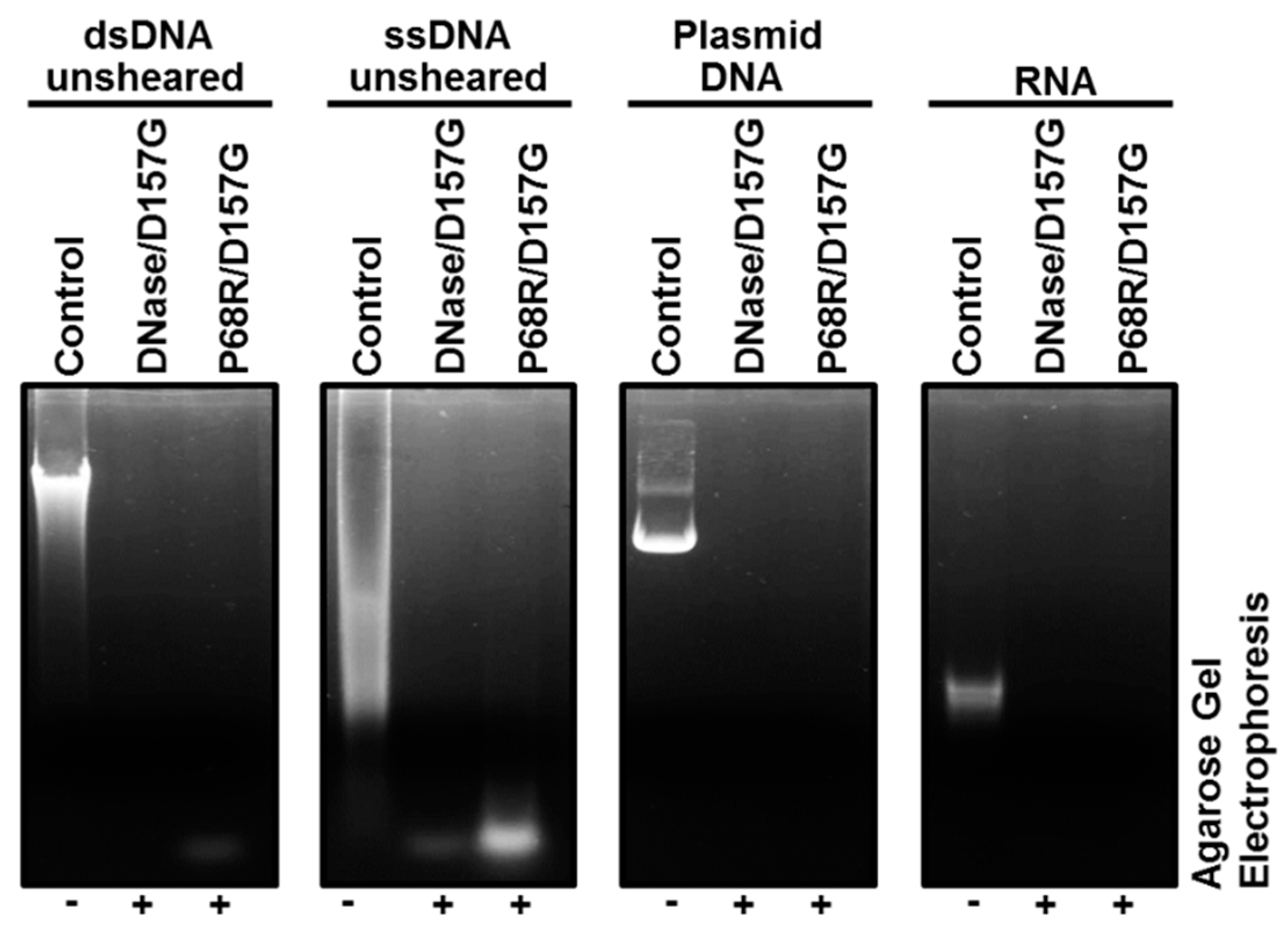
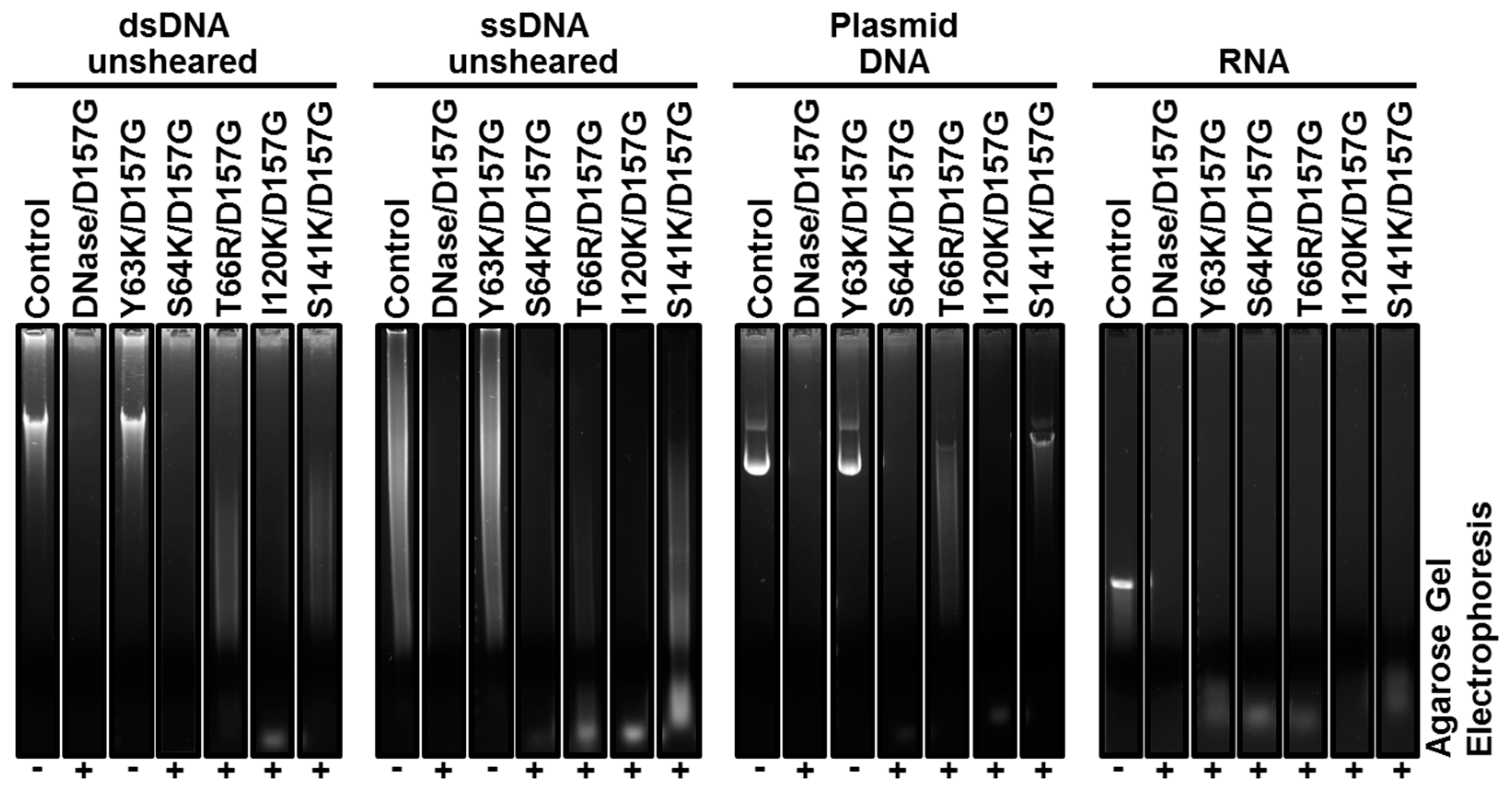
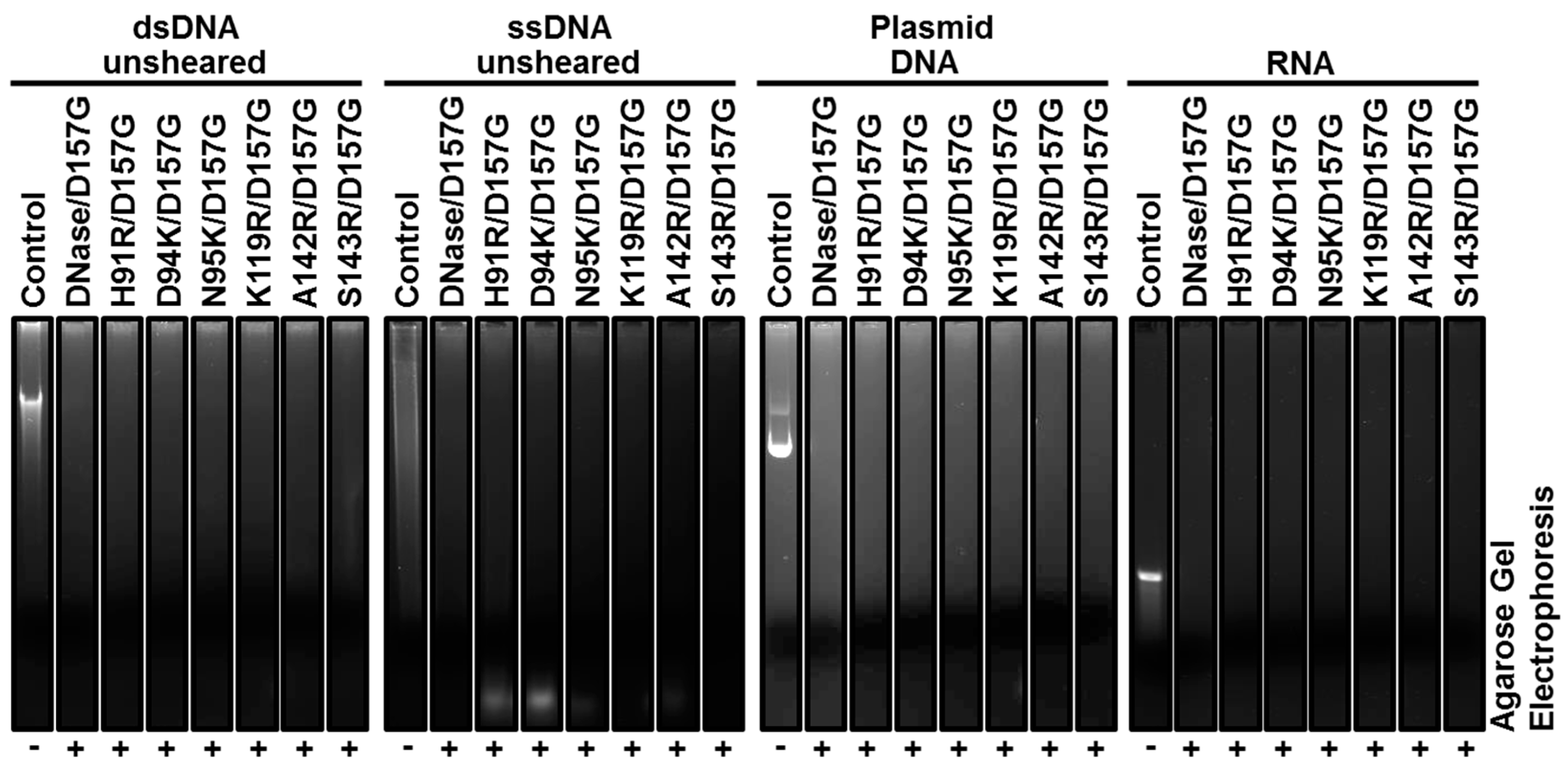
2.3. Biochemical Properties of Nucleases
2.4. Enzyme Kinetics
2.5. EDTA Tolerance of Nuclease Mutants
3. Discussion
4. Materials and Methods
4.1. Strain and Culture Conditions
4.2. Computational Sequence Analysis and Structure Modelling
4.3. Cloning of Nuclease Variants
4.4. Gene Expression and Protein Purification
4.5. Enzyme Activity Assays
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



References
- Rangarajan, E.S.; Shankar, V. Sugar non-specific endonucleases. FEMS Microbiol. Rev. 2001, 25, 583–613. [Google Scholar] [CrossRef]
- Benedik, M.J.; Strych, U. Serratia marcescens and its extracellular nuclease. FEMS Microbiol. Lett. 1998, 165, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dang, G.; Cui, Y.; Wang, L.; Li, T.; Cui, Z.; Song, N.; Chen, L.; Pang, H.; Liu, S. Extracellular sphingomyelinase Rv0888 of Mycobacterium tuberculosis contributes to pathological lung injury of Mycobacterium smegmatis in mice via inducing formation of neutrophil extracellular traps. Front. Immunol. 2018, 9, 677. [Google Scholar] [CrossRef] [PubMed]
- Vafina, G.; Zainutdinova, E.; Bulatov, E.; Filimonova, M.N. Endonuclease from gram-negative Bacteria Serratia marcescens is as effective as Pulmozyme in the hydrolysis of DNA in sputum. Front. Pharmacol. 2018, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wang, J.; Sun, Y.; Zhu, J.; Wu, S. Facilitating the evolution of esterase activity from a promiscuous enzyme (Mhg) with catalytic functions of amide hydrolysis and carboxylic acid perhydrolysis by engineering the substrate entrance tunnel. Appl. Environ. Microbiol. 2016, 82, 6748–6756. [Google Scholar] [CrossRef]
- Rao, S.J.; Shukla, E.; Bhatia, V.; Lohiya, B.; Gaikwad, S.M.; Kar, A.; Pal, J.K. The Leishmania donovani IMPACT-like protein possesses non-specific nuclease activity. Int. J. Biol. Macromol. 2018, 119, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Kerr, I.D. A novel family of phospholipase D homologues that includes phospholipid synthases and putative endonucleases: Identification of duplicated repeats and potential active site residues. Protein Sci. A Publ. Protein Soc. 1996, 5, 914–922. [Google Scholar] [CrossRef]
- Rudolph, A.E.; Stuckey, J.A.; Zhao, Y.; Matthews, H.R.; Patton, W.A.; Moss, J.; Dixon, J.E. Expression, characterization, and mutagenesis of the Yersinia pestis murine toxin, a phospholipase D superfamily member. J. Biol. Chem. 1999, 274, 11824–11831. [Google Scholar] [CrossRef]
- Nelson, R.K.; Frohman, M.A. Physiological and pathophysiological roles for phospholipase D. J. Lipid Res. 2015, 56, 2229–2237. [Google Scholar] [CrossRef]
- Liscovitch, M.; Czarny, M.; Fiucci, G.; Tang, X. Phospholipase D: Molecular and cell biology of a novel gene family. Biochem. J. 2000, 345, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, J.A.; Dixon, J.E. Crystal structure of a phospholipase D family member. Nat. Struct. Biol. 1999, 6, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Stuckey, J.A.; Lohse, D.L.; Dixon, J.E. Expression, characterization, and crystallization of a member of the novel phospholipase D family of phosphodiesterases. Protein Sci. 1997, 6, 2655–2658. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Higgins, L.; Zhang, P.; Chan, S.H.; Laget, S.; Sweeney, S.; Lunnen, K.; Xu, S.Y. Expression and purification of BmrI restriction endonuclease and its N-terminal cleavage domain variants. Protein Expr. Purif. 2008, 58, 42–52. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grazulis, S.; Manakova, E.; Roessle, M.; Bochtler, M.; Tamulaitiene, G.; Huber, R.; Siksnys, V. Structure of the metal-independent restriction enzyme BfiI reveals fusion of a specific DNA-binding domain with a nonspecific nuclease. Proc. Natl. Acad. Sci. USA 2005, 102, 15797–15802. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Zhang, X. Characterization of a novel non-specific nuclease from thermophilic bacteriophage GBSV1. BMC Biotechnol. 2008, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Miyazono, K.I.; Tanokura, M. Tetrameric structure of the restriction DNA glycosylase R.PabI in complex with nonspecific double-stranded DNA. Sci. Rep. 2016, 6, 35197. [Google Scholar] [CrossRef]
- Li, L.; Lin, S.; Yanga, F. Functional identification of the non-specific nuclease from white spot syndrome virus. Virology 2005, 337, 399–406. [Google Scholar] [CrossRef]
- Schmitz, S.; Börner, P.; Nölle, V.; Elleuche, S. Comparative analysis of two non-specific nucleases of the phospholipase D family from the plant pathogen competitor bacterium Pantoea Agglomerans. Appl. Microbiol. Biotechnol. 2019, 103, 2635–2648. [Google Scholar] [CrossRef]
- Schmitz, S.; Nölle, V.; Elleuche, S. A non-specific nucleolytic enzyme and its application potential in EDTA-containing buffer solutions. Biotechnol. Lett. 2019, 41, 129–136. [Google Scholar] [CrossRef]
- Pan, C.Q.; Lazarus, R.A. Hyperactivity of human DNase I variants. Dependence on the number of positively charged residues and concentration, length, and environment of DNA. J. Biol. Chem. 1998, 273, 11701–11708. [Google Scholar] [CrossRef] [PubMed]
- Miltenyi, S.; Hübel, T.; Nölle, V. Process for Sorting Cells by Microfabricated Components Using a Nuclease. US Patant 10,018,541 B2, 10 July 2018. [Google Scholar]
- Belkebir, A.; Azeddoug, H. Characterization of LlaKI, a new metal ion-independent restriction endonuclease from Lactococcus lactis KLDS4. ISRN Biochem. 2012, 2012, 287230. [Google Scholar] [CrossRef] [PubMed]
- Friedhoff, P.; Kolmes, B.; Gimadutdinow, O.; Wende, W.; Krause, K.L.; Pingoud, A. Analysis of the mechanism of the Serratia nuclease using site-directed mutagenesis. Nucleic Acids Res. 1996, 24, 2632–2639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Z.H.; Zheng, W.; Tang, Z.X.; Shi, L.E. Enzyme activity and thermostability of a non-specific nuclease from Yersinia enterocolitica supsp. palearctica by site-directed mutagenesis. Electron. J. Biotechnol. 2016, 24, 32–37. [Google Scholar] [CrossRef]
- Franke, I.; Meiss, G.; Pingoud, A. On the advantage of being a dimer, a case study using the dimeric Serratia nuclease and the monomeric nuclease from Anabaena sp. strain PCC 7120. J. Biol. Chem. 1999, 274, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Bommarius, A.S.; Paye, M.F. Stabilizing biocatalysts. Chem. Soc. Rev. 2013, 42, 6534–6565. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.; Rost, B. Predict impact of single amino acid change upon protein structure. BMC Genom. 2012, 13, S4. [Google Scholar] [CrossRef]
- Elleuche, S.; Fodor, K.; Klippel, B.; von der Heyde, A.; Wilmanns, M.; Antranikian, G. Structural and biochemical characterisation of a NAD(+)-dependent alcohol dehydrogenase from Oenococcus oeni as a new model molecule for industrial biotechnology applications. Appl. Microbiol. Biotechnol. 2013, 97, 8963–8975. [Google Scholar] [CrossRef]
- Marcal, D.; Rego, A.T.; Fogg, M.J.; Wilson, K.S.; Carrondo, M.A.; Enguita, F.J. Crystallization and preliminary X-ray characterization of 1,3-propanediol dehydrogenase from the human pathogen Klebsiella pneumoniae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63, 249–251. [Google Scholar] [CrossRef]
- Xin, X.F.; Kvitko, B.; He, S.Y. Pseudomonas syringae: What it takes to be a pathogen. Nat. Rev. Microbiol. 2018, 16, 316–328. [Google Scholar] [CrossRef]
- Daniel, R.M.; Danson, M.J.; Eisenthal, R.; Lee, C.K.; Peterson, M.E. The effect of temperature on enzyme activity: New insights and their implications. Extremophiles 2008, 12, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Feller, G.; Narinx, E.; Arpigny, J.L.; Zekhnini, Z.; Swings, J.; Gerday, C. Temperature dependence of growth, enzyme secretion and activity of psychrophilic Antarctic bacteria. Appl. Microbiol. Biotechnol. 1994, 41, 477–479. [Google Scholar] [CrossRef]
- Erb, T.J. Back to the future: Why we need enzymology to build a synthetic metabolism of the future. Beilstein J. Org. Chem. 2019, 15, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.J.; Tang, Z.X.; Li, Z.H.; Zhang, Z.L.; Shi, L.E. Production of a new non-specific nuclease from Yersinia enterocolitica subsp. palearctica: Optimization of induction conditions using response surface methodology. Biotechnol. Biotechnol. Equip. 2014, 28, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska, N.; Walkusz, R.; Olszewski, M.; Szymanska, A. New nuclease from extremely psychrophilic microorganism Psychromonas ingrahamii 37: Identification and characterization. Mol. Biotechnol. 2019, 61, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, V.E.; Shcheglov, A.S.; Bogdanova, E.A.; Rebrikov, D.V.; Nekrasov, A.N.; Barsova, E.V.; Shagin, D.A.; Lukyanov, S.A. Is crab duplex-specific nuclease a member of the Serratia family of non-specific nucleases? Gene 2008, 418, 41–48. [Google Scholar] [CrossRef] [PubMed]
- MacLellan, S.R.; Forsberg, C.W. Properties of the major non-specific endonuclease from the strict anaerobe Fibrobacter succinogenes and evidence for disulfide bond formation in vivo. Microbiology 2001, 147, 315–323. [Google Scholar] [CrossRef][Green Version]
- Friedhoff, P.; Meiss, G.; Kolmes, B.; Pieper, U.; Gimadutdinow, O.; Urbanke, C.; Pingoud, A. Kinetic analysis of the cleavage of natural and synthetic substrates by the Serratia nuclease. Eur. J. Biochem. 1996, 241, 572–580. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Theoretical MW (Da) 1 | Measured MW (Da) |
|---|---|---|
| DNase/D157G | 28,900.30 | 28,900.61 |
| Y63K/D157G | 28,780.19 | 28,780.30 |
| S64K/D157G | 28,856.29 | 28,856.19 |
| T66R/D157G | 28,870.28 | 28,871.01 |
| P68R/D157G | 28,874.27 | n.d. 2 |
| H91R/D157G | 28,834.24 | 28,834.07 |
| D94K/D157G | 28,828.28 | 28,828.56 |
| N95K/D157G | 28,829.26 | 28,829.40 |
| K119R/D157G | 28,843.21 | 28,842.25 |
| I120K/D157G | 28,830.21 | 28,829.26 |
| S141K/D157G | 28,856.29 | 28,856.10 |
| A142R/D157G | 28,900.30 | 28,900.58 |
| S143R/D157G | 28,884.30 | 28,884.27 |
| Variant | KM (µM) | kcat (s−1) | kcat/KM (s−1 µM−1) | Relative Turnover Rate (%) |
|---|---|---|---|---|
| DNase/D157G | 357 | 924 | 2.58 | 100 |
| Y63K/D157G | n.d. 2 | n.d. | n.d. | - |
| S64K/D157G | 115 | 8.0 | 0.07 | 0.9 |
| T66R/D157G | 335 | 62 | 0.19 | 6.7 |
| P68R/D157G | 358 | 106 | 0.3 | 11 |
| H91R/D157G | 284 | 290 | 1.02 | 31 |
| D94K/D157G | 224 | 170 | 0.76 | 18 |
| N95K/D157G | 273 | 455 | 1.66 | 49 |
| K119R/D157G | 428 | 1034 | 2.42 | 112 |
| I120K/D157G | 115 | 3.0 | 0.03 | 0.3 |
| S141K/D157G | n.d. | n.d. | n.d. | - |
| A142R/D157G | 352 | 707 | 2.01 | 77 |
| S143R/D157G | 83 | 153 | 1.84 | 17 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwardmann, L.S.; Schmitz, S.; Nölle, V.; Elleuche, S. Decoding Essential Amino Acid Residues in the Substrate Groove of a Non-Specific Nuclease from Pseudomonas syringae. Catalysts 2019, 9, 941. https://doi.org/10.3390/catal9110941
Schwardmann LS, Schmitz S, Nölle V, Elleuche S. Decoding Essential Amino Acid Residues in the Substrate Groove of a Non-Specific Nuclease from Pseudomonas syringae. Catalysts. 2019; 9(11):941. https://doi.org/10.3390/catal9110941
Chicago/Turabian StyleSchwardmann, Lynn Sophie, Sarah Schmitz, Volker Nölle, and Skander Elleuche. 2019. "Decoding Essential Amino Acid Residues in the Substrate Groove of a Non-Specific Nuclease from Pseudomonas syringae" Catalysts 9, no. 11: 941. https://doi.org/10.3390/catal9110941
APA StyleSchwardmann, L. S., Schmitz, S., Nölle, V., & Elleuche, S. (2019). Decoding Essential Amino Acid Residues in the Substrate Groove of a Non-Specific Nuclease from Pseudomonas syringae. Catalysts, 9(11), 941. https://doi.org/10.3390/catal9110941