Effect of a Substituent in Cyclopentadienyl Ligand on Iridium-Catalyzed Acceptorless Dehydrogenation of Alcohols and 2-Methyl-1,2,3,4-tetrahydroquinoline



Abstract
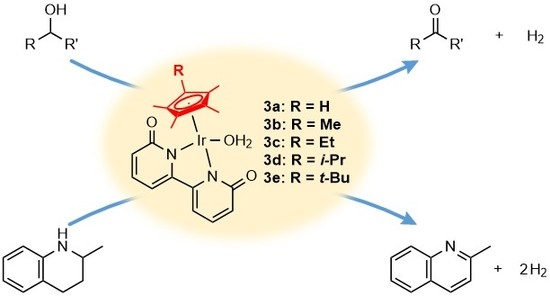
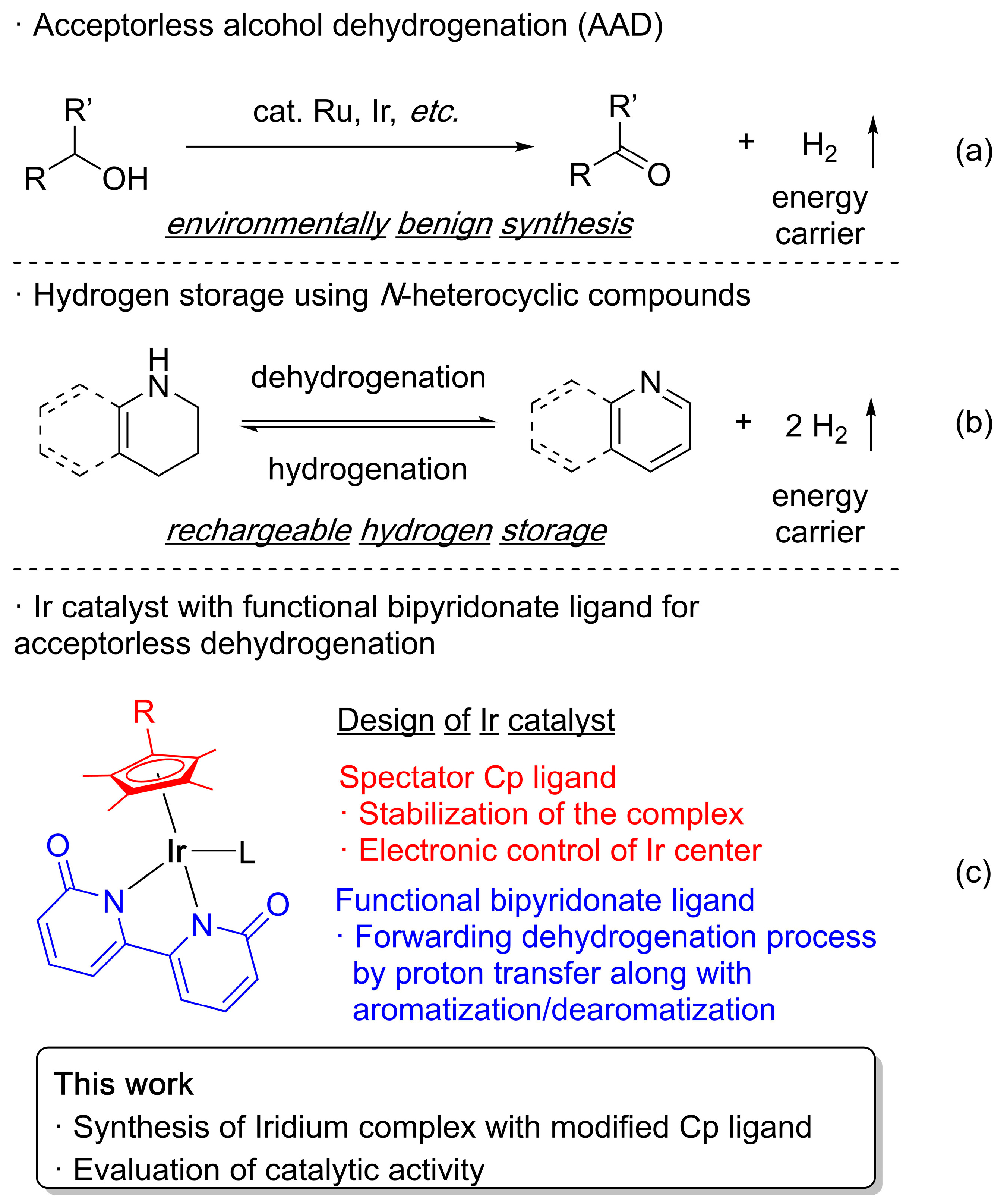
1. Introduction
2. Results
3. Materials and Methods
3.1. General
3.2. Procedures for the Synthesis of (CpRIrCl2)2
3.2.1. (η5-C5Me4H)IrCl2)2 (CAS: 835614-43-2) (1a)
3.2.2. (Cp*EthylIrCl2)2 (CAS: 2050480-26-5) (1c)
3.2.3. (Cp*iPrIrCl2)2 (CAS: 1621315-48-7) (1d)
3.2.4. (Cp*tBuIrCl2)2 (1e)
3.3. Procedures for the Synthesis of (CpRIr(6,6′-dihydroxy-2,2′-bipyridine)Cl)Cl
3.3.1. ((η5-C5Me4H)Ir(6,6′-dihydroxy-2,2′-bipyridine)Cl)Cl (2a)
3.3.2. (Cp*EthylIr(6,6′-dihydroxy-2,2′-bipyridine)Cl)Cl (2c)
3.3.3. (Cp*iPrIr(6,6′-dihydroxy-2,2′-bipyridine)Cl)Cl (2d)
3.3.4. (Cp*tBuIr(6,6′-dihydroxy-2,2′-bipyridine)Cl)Cl (2e)
3.4. Procedures for the Synthesis of CpRIr(2.2′-bipyridine-6,6′-dionato)H2O
3.4.1. (η5-C5Me4H)Ir(2,2′-bipyridine-6,6′-dionato)H2O (3a)
3.4.2. Cp*EthylIr(2,2′-bipyridine-6,6′-dionato)H2O (3c)
3.4.3. Cp*iPrIr(2,2′-bipyridine-6,6′-dionato)H2O (3d)
3.4.4. Cp*tBuIr(2,2′-bipyridine-6,6′-dionato)H2O (3e)
3.5. Investigation of Catalytic Activity in Dehydrogenation of 1-Phenylethanol (4)
3.6. Investigation of Catalytic Activity in Dehydrogenation of Benzyl alcohol (6)
3.7. Investigation of Catalytic Activity in Dehydrogenation of 2-Octanol (8)
3.8. Investigation of Catalytic Activity in Dehydrogenation of 2-MeTHQ (10)
3.9. X-ray Crystallographic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedrich, A.; Schneider, S. Acceptorless Dehydrogenation of Alcohols: Perspectives for Synthesis and H2 Storage. ChemCatChem 2009, 1, 72–73. [Google Scholar] [CrossRef]
- Gunanathan, C.; Milstein, D. Applications of Acceptorless Dehydrogenation and Related Transformations in Chemical Synthesis. Science 2013, 341, 1229712. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H. Homogeneous Transition Metal Catalysis of Acceptorless Dehydrogenative Alcohol Oxidation: Applications in Hydrogen Storage and to Heterocycle Synthesis. Chem. Rev. 2017, 117, 9228–9246. [Google Scholar] [CrossRef] [PubMed]
- Siddiki, S.M.A.H.; Toyao, T.; Shimizu, K. Acceptorless dehydrogenative coupling reactions with alcohols over heterogeneous catalysts. Green Chem. 2018, 20, 2933–2952. [Google Scholar] [CrossRef]
- Trincado, M.; Banerjee, D.; Grützmacher, H. Molecular catalysts for hydrogen production from alcohols. Energy Environ. Sci. 2014, 7, 2464–2503. [Google Scholar] [CrossRef]
- Jiang, Z.; Pan, Q.; Xu, J.; Fang, T. Current situation and prospect of hydrogen storage technology with new organic liquid. Int. J. Hydrogen Energy 2014, 39, 17442–17451. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef]
- Aakko-Saksa, P.T.; Cook, C.; Kiviaho, J.; Repo, T. Liquid organic hydrogen carriers for transportation and storing of renewable energy—Review and discussion. J. Power Sources 2018, 396, 803–823. [Google Scholar] [CrossRef]
- Modisha, P.M.; Ouma, C.N.M.; Garidzirai, R.; Wasserscheid, P.; Bessarabov, D. The Prospect of Hydrogen Storage Using Liquid Organic Hydrogen Carriers. Energy Fuels 2019, 33, 2778–2796. [Google Scholar] [CrossRef]
- Dobson, A.; Robinson, S.D. Catalytic dehydrogenation of primary and secondary alcohols by Ru(OCOCF3)2(CO)(PPh3)2. J. Organomet. Chem. 1975, 87, C52–C53. [Google Scholar] [CrossRef]
- Dobson, A.; Robinson, S.D. Complexes of the platinum metals. 7. Homogeneous ruthenium and osmium catalysts for the dehydrogenation of primary and secondary alcohols. Inorg. Chem. 1977, 16, 137–142. [Google Scholar] [CrossRef]
- Ligthart, G.B.W.L.; Meijer, R.H.; Donners, M.P.J.; Meuldijk, J.; Vekemans, J.A.J.M.; Hulshof, L.A. Highly sustainable catalytic dehydrogenation of alcohols with evolution of hydrogen gas. Tetrahedron Lett. 2003, 44, 1507–1509. [Google Scholar] [CrossRef]
- Zhang, J.; Gandelman, M.; Shimon, L.J.W.; Rozenberg, H.; Milstein, D. Electron-Rich, Bulky Ruthenium PNP-Type Complexes. Acceptorless Catalytic Alcohol Dehydrogenation. Organometallics 2004, 23, 4026–4033. [Google Scholar] [CrossRef]
- Adair, G.R.A.; Williams, J.M.J. Oxidant-free oxidation: Ruthenium catalyzed dehydrogenation of alcohols. Tetrahedron Lett. 2005, 46, 8233–8235. [Google Scholar] [CrossRef]
- van Buijtenen, J.; Meuldijk, J.; Vekemans, J.A.J.M.; Hulshof, L.A.; Kooijman, H.; Spek, A.L. Dinuclear Ruthenium Complexes Bearing Dicarboxylate and Phosphine Ligands. Acceptorless Catalytic Dehydrogenation of 1-Phenylethanol. Organometallics 2006, 25, 873–881. [Google Scholar] [CrossRef]
- Royer, A.M.; Rauchfuss, T.B.; Wilson, S.R. Coordination Chemistry of a Model for the GP Cofactor in the Hmd Hydrogenase: Hydrogen-Bonding and Hydrogen-Transfer Catalysis. Inorg. Chem. 2008, 47, 395–397. [Google Scholar] [CrossRef]
- Royer, A.M.; Rauchfuss, T.B.; Gray, D.L. Organoiridium Pyridonates and Their Role in the Dehydrogenation of Alcohols. Organometallics 2010, 29, 6763–6768. [Google Scholar] [CrossRef]
- Baratta, W.; Bossi, G.; Putignano, E.; Rigo, P. Pincer and Diamine Ru and Os Diphosphane Complexes as Efficient Catalysts for the Dehydrogenation of Alcohols to Ketones. Chem. Eur. J. 2011, 17, 3474–3481. [Google Scholar] [CrossRef]
- Prades, A.; Peris, E.; Albrecht, M. Oxidations and Oxidative Couplings Catalyzed by Triazolylidene Ruthenium Complexes. Organometallics 2011, 30, 1162–1167. [Google Scholar] [CrossRef]
- Zhang, J.; Balaraman, E.; Leitus, G.; Milstein, D. Electron-Rich PNP- and PNN-Type Ruthenium(II) Hydrido Borohydride Pincer Complexes. Synthesis, Structure, and Catalytic Dehydrogenation of Alcohols and Hydrogenation of Esters. Organometallics 2011, 30, 5716–5724. [Google Scholar] [CrossRef]
- Musa, S.; Shaposhnikov, I.; Cohen, S.; Gelman, D. Ligand–Metal Cooperation in PCP Pincer Complexes: Rational Design and Catalytic Activity in Acceptorless Dehydrogenation of Alcohols. Angew. Chem. Int. Ed. 2011, 50, 3533–3537. [Google Scholar] [CrossRef] [PubMed]
- Gülcemal, S.; Gülcemal, D.; Whitehead, G.F.S.; Xiao, J. Acceptorless Dehydrogenative Oxidation of Secondary Alcohols Catalysed by Cp*IrIII-NHC Complexes. Chem. Eur. J. 2016, 22, 10513–10522. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H. Hydrogen storage in liquid organic heterocycles. Energy Environ. Sci. 2008, 1, 134–138. [Google Scholar] [CrossRef]
- Moores, A.; Poyatos, M.; Luo, Y.; Crabtree, R.H. Catalysed low temperature H2 release from nitrogen heterocycles. New J. Chem. 2006, 30, 1675–1678. [Google Scholar] [CrossRef]
- Wang, Z.; Tonks, I.; Belli, J.; Jensen, C.M. Dehydrogenation of N-ethyl perhydrocarbazole catalyzed by PCP pincer iridium complexes: Evaluation of a homogenous hydrogen storage system. J. Organomet. Chem. 2009, 694, 2854–2857. [Google Scholar] [CrossRef]
- Wang, Z.; Belli, J.; Jensen, C.M. Homogeneous dehydrogenation of liquid organic hydrogen carriers catalyzed by an iridium PCP complex. Faraday Discuss. 2011, 151, 297–305. [Google Scholar] [CrossRef]
- Chakraborty, S.; Brennessel, W.W.; Jones, W.D. A Molecular Iron Catalyst for the Acceptorless Dehydrogenation and Hydrogenation of N-Heterocycles. J. Am. Chem. Soc. 2014, 136, 8564–8567. [Google Scholar] [CrossRef]
- Xu, R.; Chakraborty, S.; Yuan, H.; Jones, W.D. Acceptorless, Reversible Dehydrogenation and Hydrogenation of N-Heterocycles with a Cobalt Pincer Catalyst. ACS Catal. 2015, 5, 6350–6354. [Google Scholar] [CrossRef]
- Manas, M.G.; Sharninghausen, L.S.; Crabtree, R.H. Iridium catalyzed reversible dehydrogenation e Hydrogenation of quinoline derivatives under mild conditions. J. Organomet. Chem. 2015, 792, 184–189. [Google Scholar] [CrossRef]
- Vivancos, Á.; Beller, M.; Albrecht, M. NHC-Based Iridium Catalysts for Hydrogenation and Dehydrogenation of N-Heteroarenes in Water under Mild Conditions. ACS Catal. 2018, 8, 17–21. [Google Scholar] [CrossRef]
- Fujita, K.; Yamaguchi, R. Cp*Ir Complex-Catalyzed Hydrogen Transfer Reactions Directed toward Environmentally Benign Organic Synthesis. Synlett 2005, 560–571. [Google Scholar] [CrossRef]
- Fujita, K.; Enoki, Y.; Yamaguchi, R. Cp*Ir-catalyzed N-alkylation of amines with alcohols. A versatile and atom economical method for the synthesis of amines. Tetrahedron 2008, 64, 1943–1954. [Google Scholar] [CrossRef]
- Kawahara, R.; Fujita, K.; Yamaguchi, R. Multialkylation of Aqueous Ammonia with Alcohols Catalyzed by Water-Soluble Cp*Ir–Ammine Complexes. J. Am. Chem. Soc. 2010, 132, 15108–15111. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, R.; Fujita, K.; Yamaguchi, R. N-Alkylation of Amines with Alcohols Catalyzed by a Water-Soluble Cp*Iridium Complex: An Efficient Method for the Synthesis of Amines in Aqueous Media. Adv. Synth. Catal. 2011, 353, 1161–1168. [Google Scholar] [CrossRef]
- Fujita, K.; Furukawa, S.; Morishima, N.; Shimizu, M.; Yamaguchi, R. N-Alkylation of Aqueous Ammonia with Alcohols Leading to Primary Amines Catalyzed by Water-Soluble N-Heterocyclic Carbene Complexes of Iridium. ChemCatChem 2018, 10, 1993–1997. [Google Scholar] [CrossRef]
- Fujita, K. Development and Application of New Iridium Catalysts for Efficient Dehydrogenative Reactions of Organic Molecules. Bull. Chem. Soc. Jpn. 2019, 92, 344–351. [Google Scholar] [CrossRef]
- Fujita, K.; Tanino, N.; Yamaguchi, R. Ligand-Promoted Dehydrogenation of Alcohols Catalyzed by Cp*Ir Complexes. A New Catalytic System for Oxidant-Free Oxidation of Alcohols. Org. Lett. 2007, 9, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Yoshida, T.; Imori, Y.; Yamaguchi, R. Dehydrogenative Oxidation of Primary and Secondary Alcohols Catalyzed by a Cp*Ir Complex Having a Functional C,N-Chelate Ligand. Org. Lett. 2011, 13, 2278–2281. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, R.; Fujita, K.; Yamaguchi, R. Dehydrogenative Oxidation of Alcohols in Aqueous Media Using Water-Soluble and Reusable Cp*Ir Catalysts Bearing a Functional Bipyridine Ligand. J. Am. Chem. Soc. 2012, 134, 3643–3646. [Google Scholar] [CrossRef]
- Kawahara, R.; Fujita, K.; Yamaguchi, R. Cooperative Catalysis by Iridium Complexes with a Bipyridonate Ligand: Versatile Dehydrogenative Oxidation of Alcohols and Reversible Dehydrogenation–Hydrogenation between 2-Propanol and Acetone. Angew. Chem. Int. Ed. 2012, 51, 12790–12794. [Google Scholar] [CrossRef]
- Fujita, K.; Tamura, R.; Tanaka, Y.; Yoshida, M.; Onoda, M.; Yamaguchi, R. Dehydrogenative Oxidation of Alcohols in Aqueous Media Catalyzed by a Water-Soluble Dicationic Iridium Complex Bearing a Functional N-Heterocyclic Carbene Ligand without Using Base. ACS Catal. 2017, 7, 7226–7230. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Kobayashi, D.; Shimizu, M.; Fujita, K. Synthesis of a series of iridium complexes bearing substituted 2-pyridonates and their catalytic performance for acceptorless dehydrogenation of alcohols under neutral conditions. J. Organomet. Chem. 2017, 843, 14–19. [Google Scholar] [CrossRef]
- Yoshida, M.; Wang, H.; Shimbayashi, T.; Fujita, K. Dehydrogenative Transformation of Alcoholic Substrates in Aqueous Media Catalyzed by an Iridium Complex Having a Functional Ligand with α-Hydroxypyridine and 4,5-Dihydro-1H-imidazol-2-yl Moieties. Catalysis 2018, 8, 312. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Ikeda, C.; Takahashi, Y.; Fujita, K. Homogeneous Catalytic System for Reversible Dehydrogenation−Hydrogenation Reactions of Nitrogen Heterocycles with Reversible Interconversion of Catalytic Species. J. Am. Chem. Soc. 2009, 131, 8410–8412. [Google Scholar] [CrossRef]
- Fujita, K.; Tanaka, Y.; Kobayashi, M.; Yamaguchi, R. Homogeneous Perdehydrogenation and Perhydrogenation of Fused Bicyclic N-Heterocycles Catalyzed by Iridium Complexes Bearing a Functional Bipyridonate Ligand. J. Am. Chem. Soc. 2014, 136, 4829–4832. [Google Scholar] [CrossRef]
- Fujita, K.; Wada, T.; Shiraishi, T. Reversible Interconversion between 2,5-Dimethylpyrazine and 2,5-Dimethylpiperazine by Iridium-Catalyzed Hydrogenation/Dehydrogenation for Efficient Hydrogen Storage. Angew. Chem. Int. Ed. 2017, 56, 10886–10889. [Google Scholar] [CrossRef]
- Onoda, M.; Nagano, Y.; Fujita, K. Iridium-catalyzed dehydrogenative lactonization of 1,4-butanediol and reversal hydrogenation: New hydrogen storage system using cheap organic resources. Int. J. Hydrogen Energy 2019. [Google Scholar] [CrossRef]
- Zeng, G.; Sakaki, S.; Fujita, K.; Sano, H.; Yamaguchi, R. Efficient Catalyst for Acceptorless Alcohol Dehydrogenation: Interplay of Theoretical and Experimental Studies. ACS Catal. 2014, 4, 1010–1020. [Google Scholar] [CrossRef]
- Newton, C.G.; Kossler, D.; Cramer, N. Asymmetric Catalysis Powered by Chiral Cyclopentadienyl Ligands. J. Am. Chem. Soc. 2016, 138, 3935–3941. [Google Scholar] [CrossRef]
- Piou, T.; Rovis, T. Electronic and Steric Tuning of a Prototypical Piano Stool Complex: Rh(III) Catalysis for C–H Functionalization. Acc. Chem. Res. 2018, 51, 170–180. [Google Scholar] [CrossRef]
- Field, L.D.; Lindall, C.M.; Masters, A.F.; Clentsmith, G.K.B. Penta-arylcyclopentadienyl complexes. Coord. Chem. Rev. 2011, 255, 1733–1790. [Google Scholar] [CrossRef]
- Piou, T.; Romanov-Michailidis, F.; Momanova-Michaelides, M.; Jackson, K.E.; Semakul, N.; Taggart, T.D.; Newell, B.S.; Rithner, C.D.; Paton, R.S.; Rovis, T. Correlating Reactivity and Selectivity to Cyclopentadienyl Ligand Properties in Rh(III)-Catalyzed C–H Activation Reactions: An Experimental and Computational Study. J. Am. Chem. Soc. 2017, 139, 1296–1310. [Google Scholar] [CrossRef]
- Hong, S.Y.; Jeong, J.; Chang, S. [4+2] or [4+1] Annulation: Changing the Reaction Pathway of a Rhodium-Catalyzed Process by Tuning the Cp Ligand. Angew. Chem. Int. Ed. 2017, 56, 2408–2412. [Google Scholar] [CrossRef]
- Yoshizaki, S.; Shibata, Y.; Tanaka, K. Fulvene Synthesis by Rhodium(I)-Catalyzed [2+2+1] Cycloaddition: Synthesis and Catalytic Activity of Tunable Cyclopentadienyl Rhodium(III) Complexes with Pendant Amides. Angew. Chem. Int. Ed. 2017, 56, 3590–3593. [Google Scholar] [CrossRef]
- Hyster, T.K.; Rovis, T. An improved catalyst architecture for rhodium(III) catalyzed C–H activation and its application to pyridine synthesis. Chem. Sci. 2011, 2, 1606–1610. [Google Scholar] [CrossRef]
- Hyster, T.K.; Rovis, T. Pyridine synthesis from oximes and alkynes via rhodium(III) catalysis: Cp* and Cpt provide complementary selectivity. Chem. Commun. 2011, 47, 11846–11848. [Google Scholar] [CrossRef]
- Yoshimura, R.; Shibata, Y.; Yamada, T.; Tanaka, K. Aerobic Oxidative Olefination of Benzamides with Styrenes Catalyzed by a Moderately Electron-Deficient CpRh(III) Complex with a Pendant Amide. J. Org. Chem. 2019, 84, 2501–2511. [Google Scholar] [CrossRef]
- Yamada, T.; Shibata, Y.; Kawauchi, S.; Yoshizaki, S.; Tanaka, K. Formal Lossen Rearrangement/[3+2] Annulation Cascade Catalyzed by a Modified Cyclopentadienyl RhIII Complex. Chem. Eur. J. 2018, 24, 5723–5727. [Google Scholar] [CrossRef]
- Yamada, T.; Shibata, Y.; Tanaka, K. Functionalized Cyclopentadienyl Ligands and Their Substituent Effects on a Rhodium(III)-Catalyzed Oxidative [4+2] Annulation of Indole- and Pyrrole-1-Carboxamides with Alkynes. Asian. J. Org. Chem. 2018, 7, 1396–1402. [Google Scholar] [CrossRef]
- Terasawa, J.; Shibata, Y.; Kimura, Y.; Tanaka, K. Synthesis of Functionalized (η5-Indenyl)rhodium(III) Complexes and Their Application to C–H Bond Functionalization. Chem. Asian. J. 2018, 13, 505–509. [Google Scholar] [CrossRef]
- Piou, T.; Rovis, T. Rh(III)-Catalyzed Cyclopropanation Initiated by C–H Activation: Ligand Development Enables a Diastereoselective [2+1] Annulation of N-Enoxyphthalimides and Alkenes. J. Am. Chem. Soc. 2014, 136, 11292–11295. [Google Scholar] [CrossRef]
- Mahr, A.; Nürnberg, O.; Werner, H. Halfsandwich-Type Complexes of Iridium with Tetramethylcyclopentadienyl as Ligand. Z. Anorg. Allg. Chem. 2003, 629, 91–98. [Google Scholar] [CrossRef]
- Dooley, T.; Fairhurst, G.; Chalk, C.D.; Tabatabaian, K.; White, C. Ethyltetramethylcyclopentadienyl Complexes of Cobalt, Rhodium, Iridium and Ruthenium. Transit. Met. Chem. 1978, 3, 299–302. [Google Scholar] [CrossRef]
- Morris, D.M.; McGeagh, M.; Peña, D.D.; Merola, J.S. Extending the range of pentasubstituted cyclopentadienyl compounds: The synthesis of a series of tetramethyl(alkyl or aryl)cyclopentadienes (Cp*R), their iridium complexes and their catalytic activity for asymmetric transfer hydrogenation. Polyhedron 2014, 84, 120–135. [Google Scholar] [CrossRef]
- Brown, L.C.; Ressegue, E.; Merola, J.S. Rapid Access to Derivatized, Dimeric, Ring-Substituted Dichloro(cyclopentadienyl)rhodium(III) and Iridium(III) Complexes. Organometallics 2016, 35, 4014–4022. [Google Scholar] [CrossRef]
- Du Plooy, K.E.; du Toit, J.; Levendis, D.C.; Coville, N.J. Multiply substituted cyclopentadienyl metal complexes: I. Solid-state and solution conformational studies on (η5-C5Me4R)Fe( CO)(L) I(R = H, tBu). J. Organomet. Chem. 1996, 508, 231–242. [Google Scholar] [CrossRef]
- DePasquale, J.; Nieto, I.; Reuther, L.E.; Herbst-Gervasoni, C.J.; Paul, J.J.; Mochalin, V.; Zeller, M.; Thomas, C.M.; Addison, A.W.; Papish, E.T. Iridium Dihydroxybipyridine Complexes Show That Ligand Deprotonation Dramatically Speeds Rates of Catalytic Water Oxidation. Inorg. Chem. 2013, 52, 9175–9183. [Google Scholar] [CrossRef]
- Umemoto, T.; Nagayoshi, M.; Adachi, K.; Tomizawa, G. Synthesis, Properties, and Reactivity of N,N′-Difluorobipyridinium and Related Salts and Their Applications as Reactive and Easy-To-Handle Electrophilic Fluorinating Agents with High Effective Fluorine Content. J. Org. Chem. 1998, 63, 3379–3385. [Google Scholar] [CrossRef]
- Randles, M.D.; Simpson, P.V.; Gupta, V.; Fu, J.; Moxey, G.J.; Schwich, T.; Criddle, A.L.; Petrie, S.; MacLellan, J.G.; Batten, S.R.; et al. Syntheses of Pentanuclear Group 6 Iridium Clusters by Core Expansion of Tetranuclear Clusters with Ir(CO)2(η5-C5Me4R) (R = H, Me). Inorg. Chem. 2013, 52, 11256–11268. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| entry | catalyst | conv. (%) [a, b] | yield (%) [a, b] |
|---|---|---|---|
| 1 | 3a | 35 | 35 |
| 2 | 3b | 57 | 57 |
| 3 | 3c | 53 | 53 |
| 4 | 3d | 55 | 55 |
| 5 | 3e | 64 | 64 |

| entry | catalyst | conv. (%) [a] | yield (%) [a] |
|---|---|---|---|
| 1 | 3a | 19 | 18 |
| 2 | 3b | 49 | 49 |
| 3 | 3e | 57 | 57 |

| entry | catalyst | yield (%) [a] |
|---|---|---|
| 1 | 3a | 41 |
| 2 | 3b | 46 |
| 3 | 3e | 48 |

| entry | catalyst | conv. (%) [a] | yield (%) [a] |
|---|---|---|---|
| 1 | 3a | 55 | 55 |
| 2 | 3b | 91 | 91 |
| 3 | 3c | 83 | 83 |
| 4 | 3d | 62 | 62 |
| 5 | 3e | 99 | 99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, J.; Shimbayashi, T.; Fujita, K.-i. Effect of a Substituent in Cyclopentadienyl Ligand on Iridium-Catalyzed Acceptorless Dehydrogenation of Alcohols and 2-Methyl-1,2,3,4-tetrahydroquinoline. Catalysts 2019, 9, 846. https://doi.org/10.3390/catal9100846
Jeong J, Shimbayashi T, Fujita K-i. Effect of a Substituent in Cyclopentadienyl Ligand on Iridium-Catalyzed Acceptorless Dehydrogenation of Alcohols and 2-Methyl-1,2,3,4-tetrahydroquinoline. Catalysts. 2019; 9(10):846. https://doi.org/10.3390/catal9100846
Chicago/Turabian StyleJeong, Jaeyoung, Takuya Shimbayashi, and Ken-ichi Fujita. 2019. "Effect of a Substituent in Cyclopentadienyl Ligand on Iridium-Catalyzed Acceptorless Dehydrogenation of Alcohols and 2-Methyl-1,2,3,4-tetrahydroquinoline" Catalysts 9, no. 10: 846. https://doi.org/10.3390/catal9100846
APA StyleJeong, J., Shimbayashi, T., & Fujita, K.-i. (2019). Effect of a Substituent in Cyclopentadienyl Ligand on Iridium-Catalyzed Acceptorless Dehydrogenation of Alcohols and 2-Methyl-1,2,3,4-tetrahydroquinoline. Catalysts, 9(10), 846. https://doi.org/10.3390/catal9100846