Conversion of 5-Methyl-3-Heptanone to C8 Alkenes and Alkane over Bifunctional Catalysts



Abstract
:1. Introduction
2. Results and Discussions
2.1. Characterization of Catalysts
2.1.1. X-Ray Diffraction (XRD)
2.1.2. Transmission Electron Microscopy (TEM)
2.1.3. N2 Adsorption
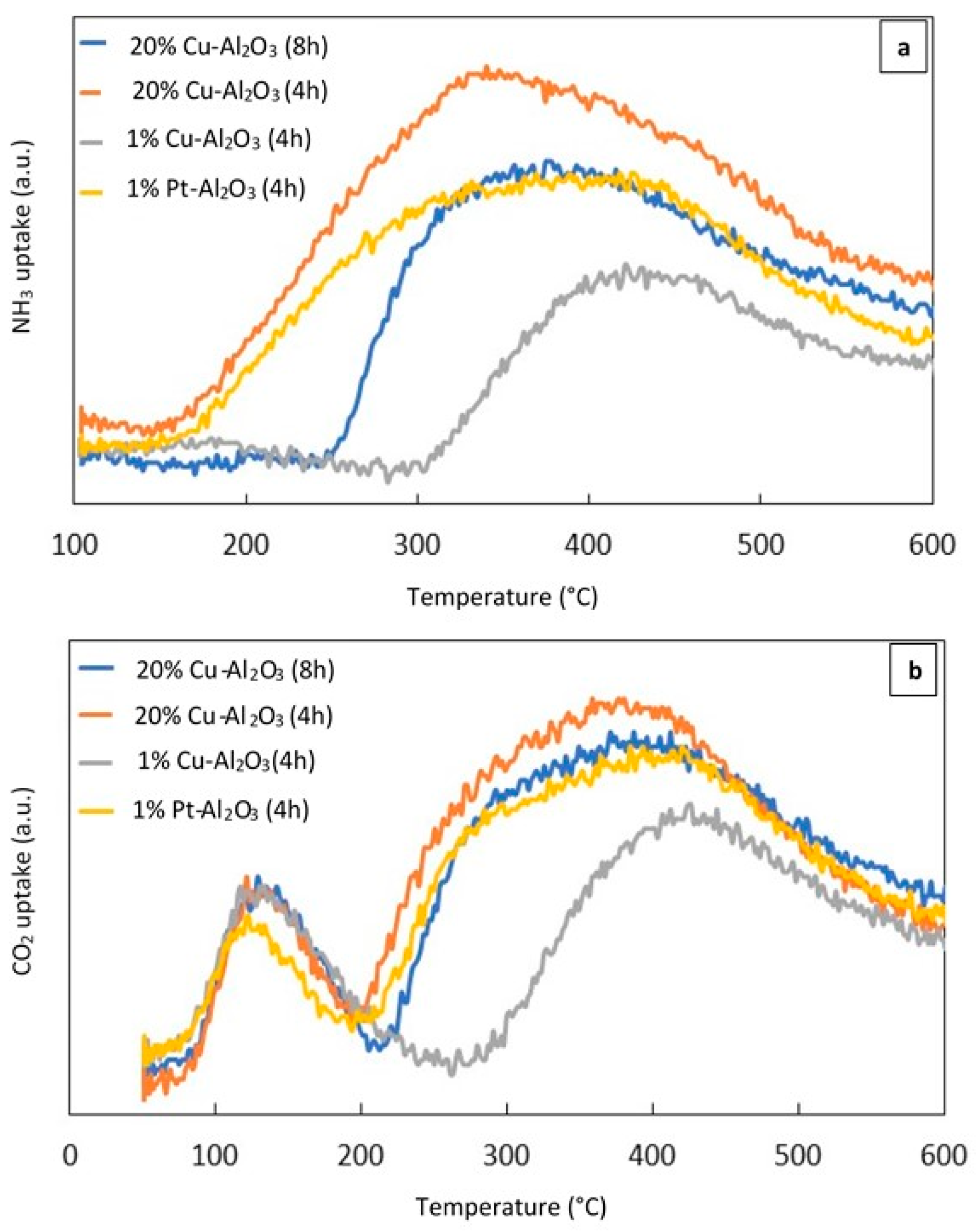
2.1.4. Temperature Programmed Desorption (NH3–TPD) and (CO2–TPD)
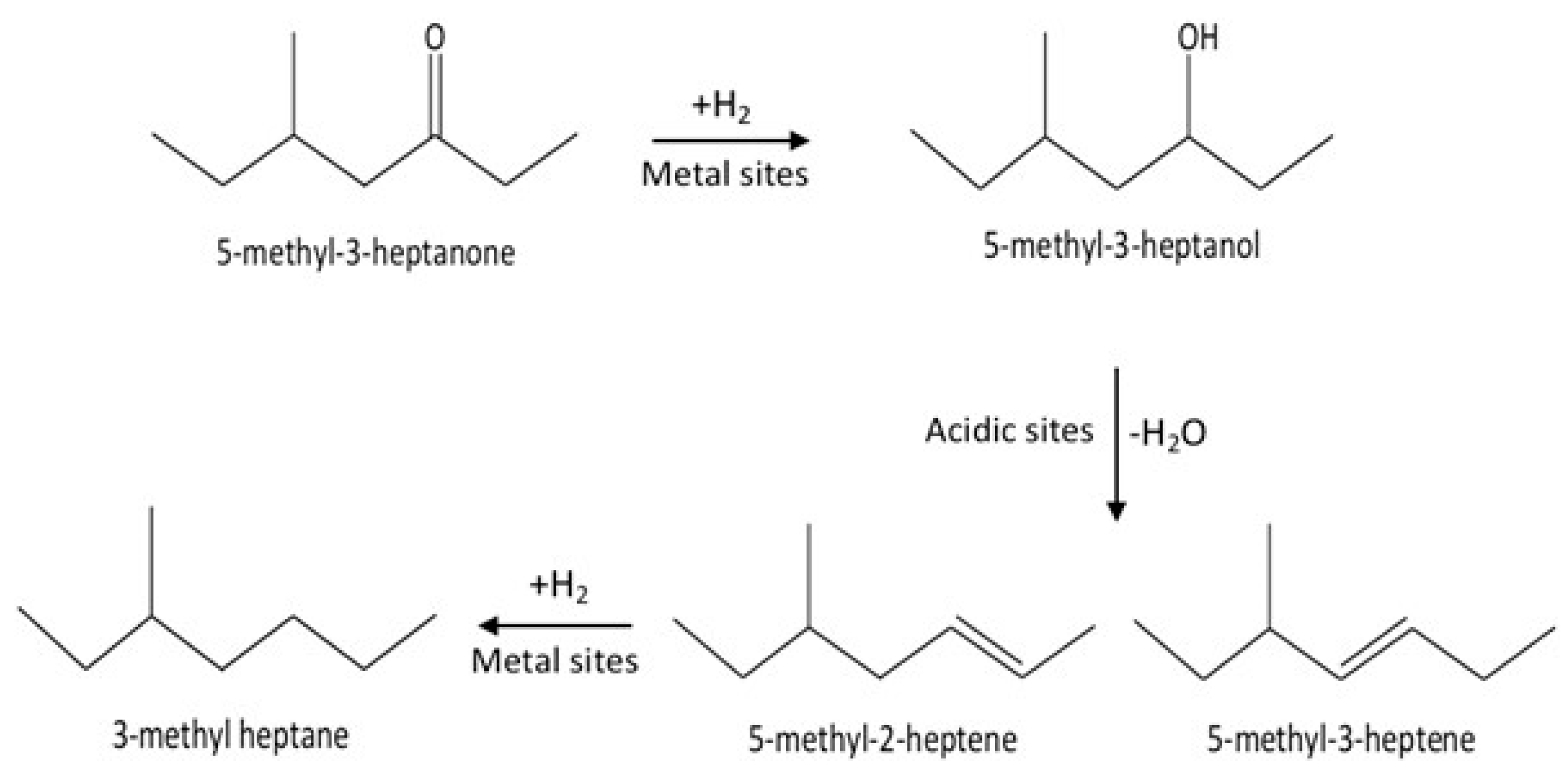
2.2. Catalytic Conversion of C8 Ketone to C8 Alkenes and C8 Alkane in a Fixed Bed Reactor
2.2.1. Effect of Different Supported Catalysts on the Reaction of C8 Ketone
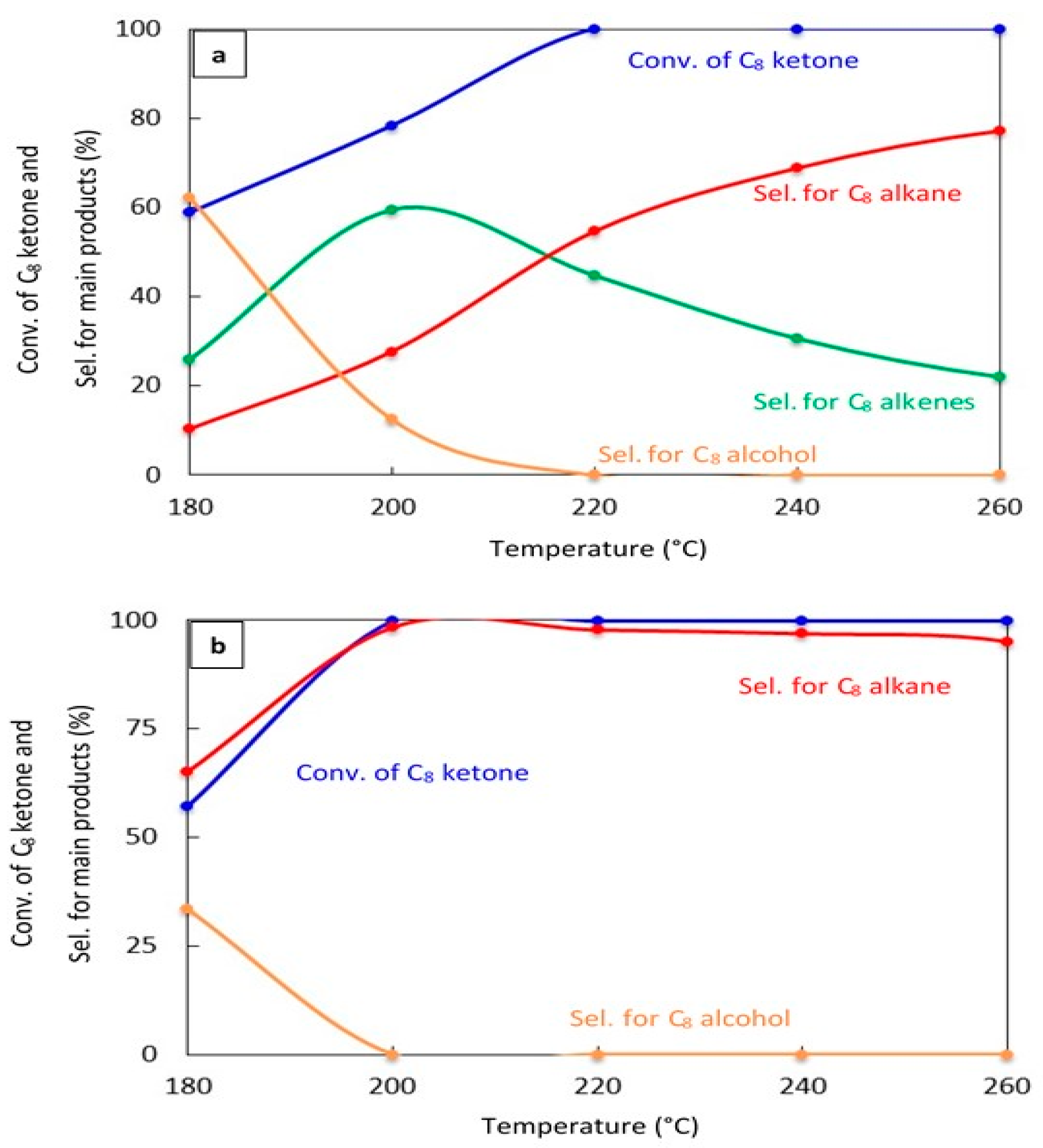
2.2.2. Effect of Reaction Temperature
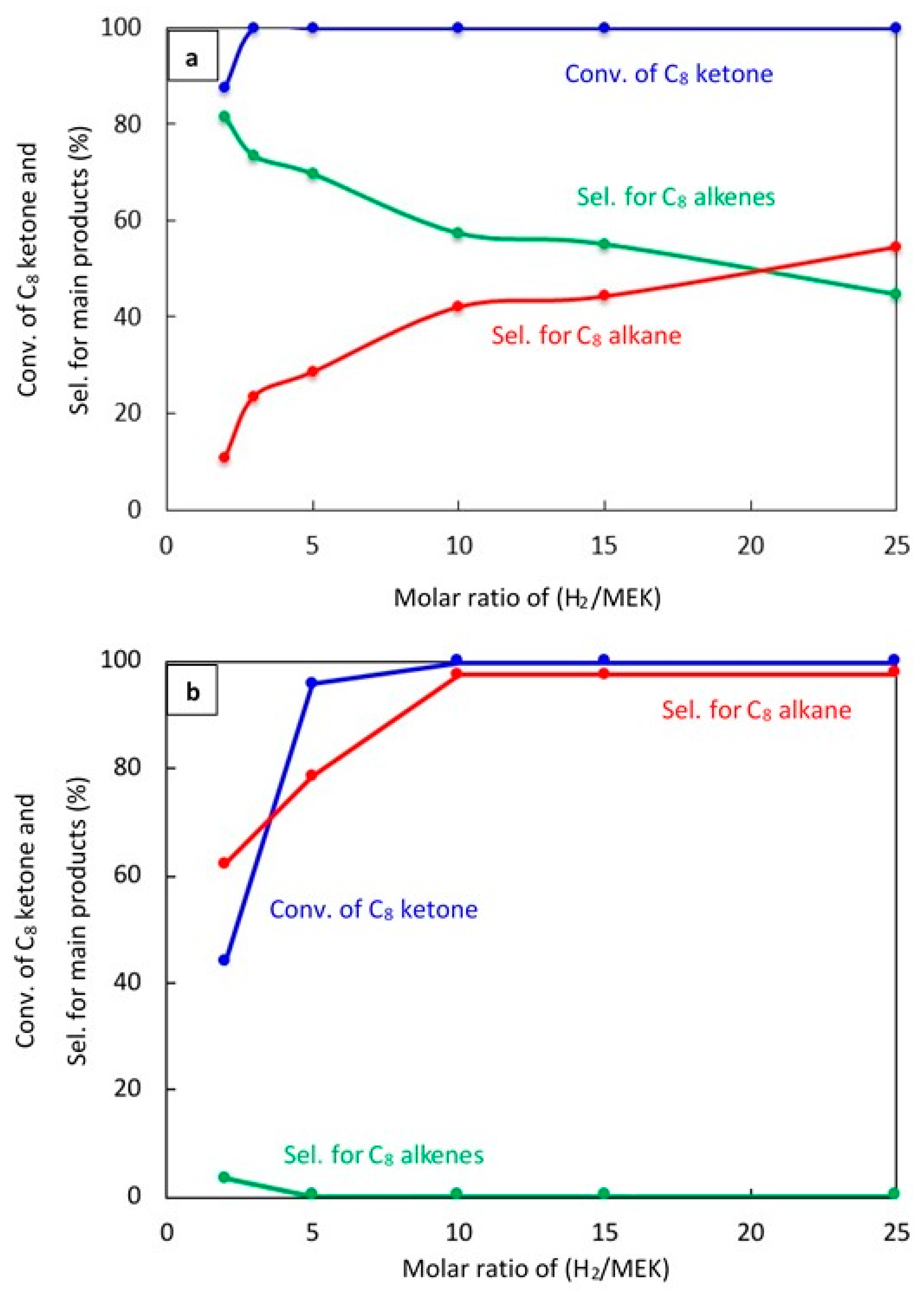
2.2.3. Effect of H2/C8 Ketone Molar Ratio
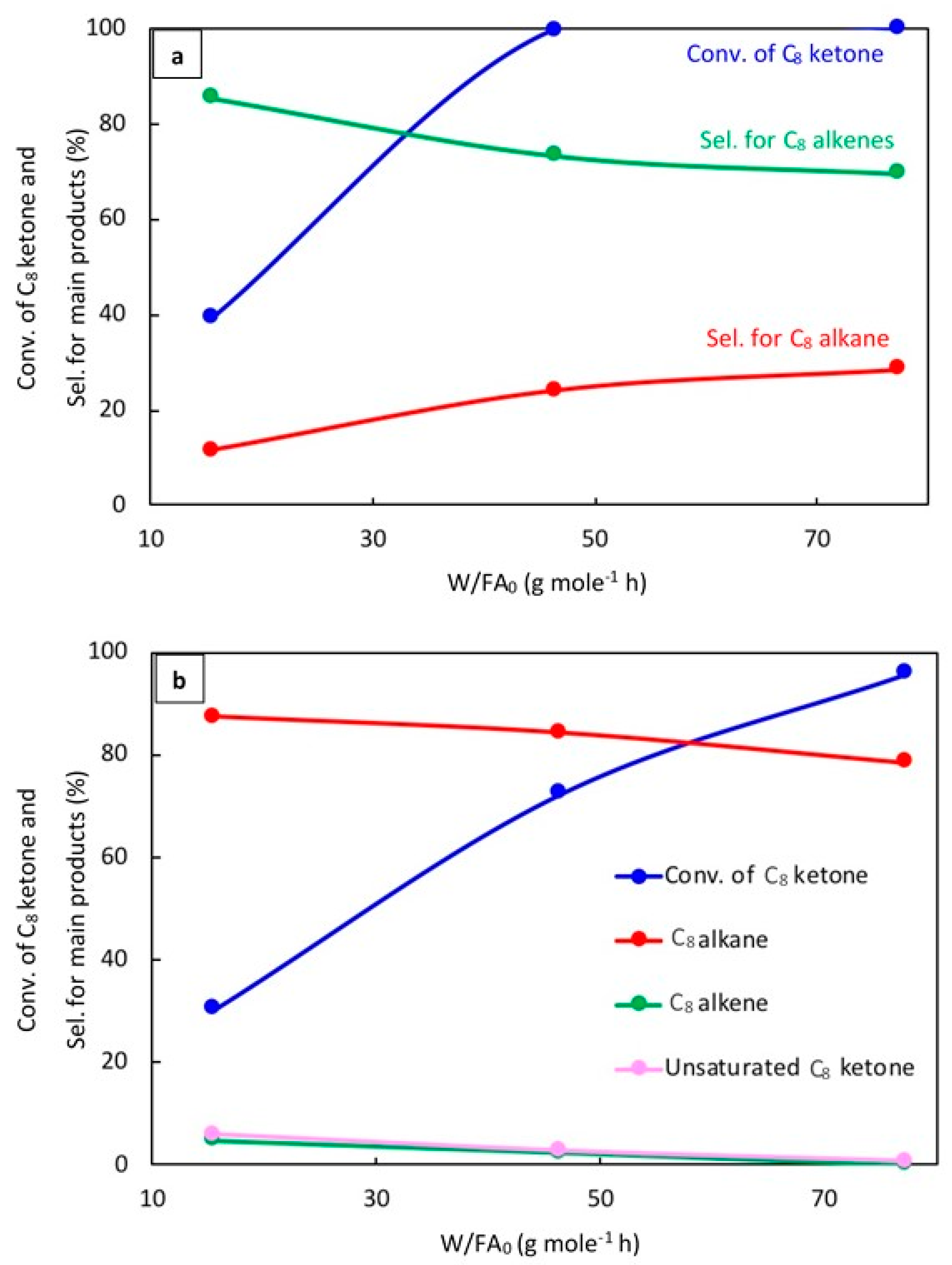
2.2.4. Effect of Space Time
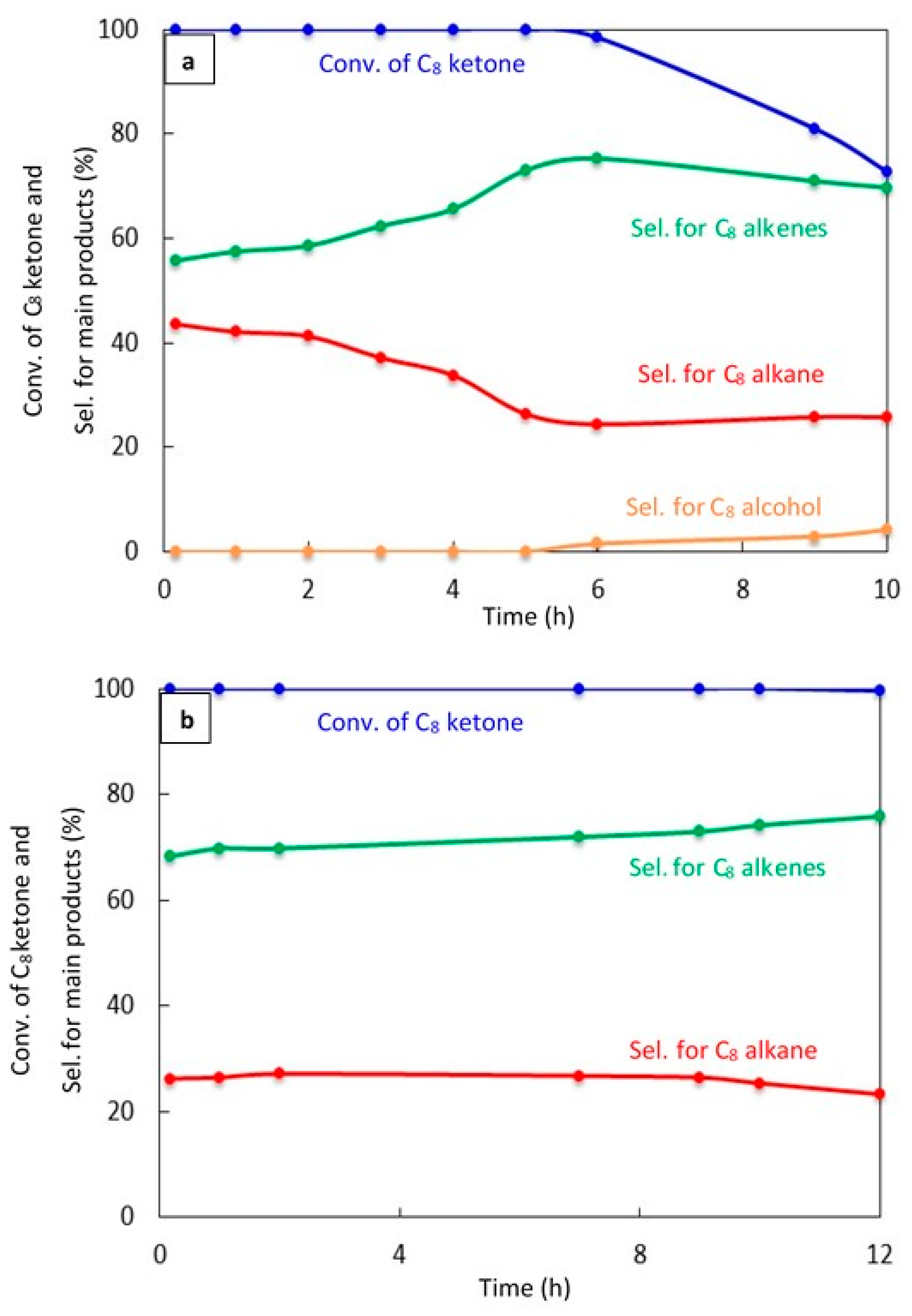
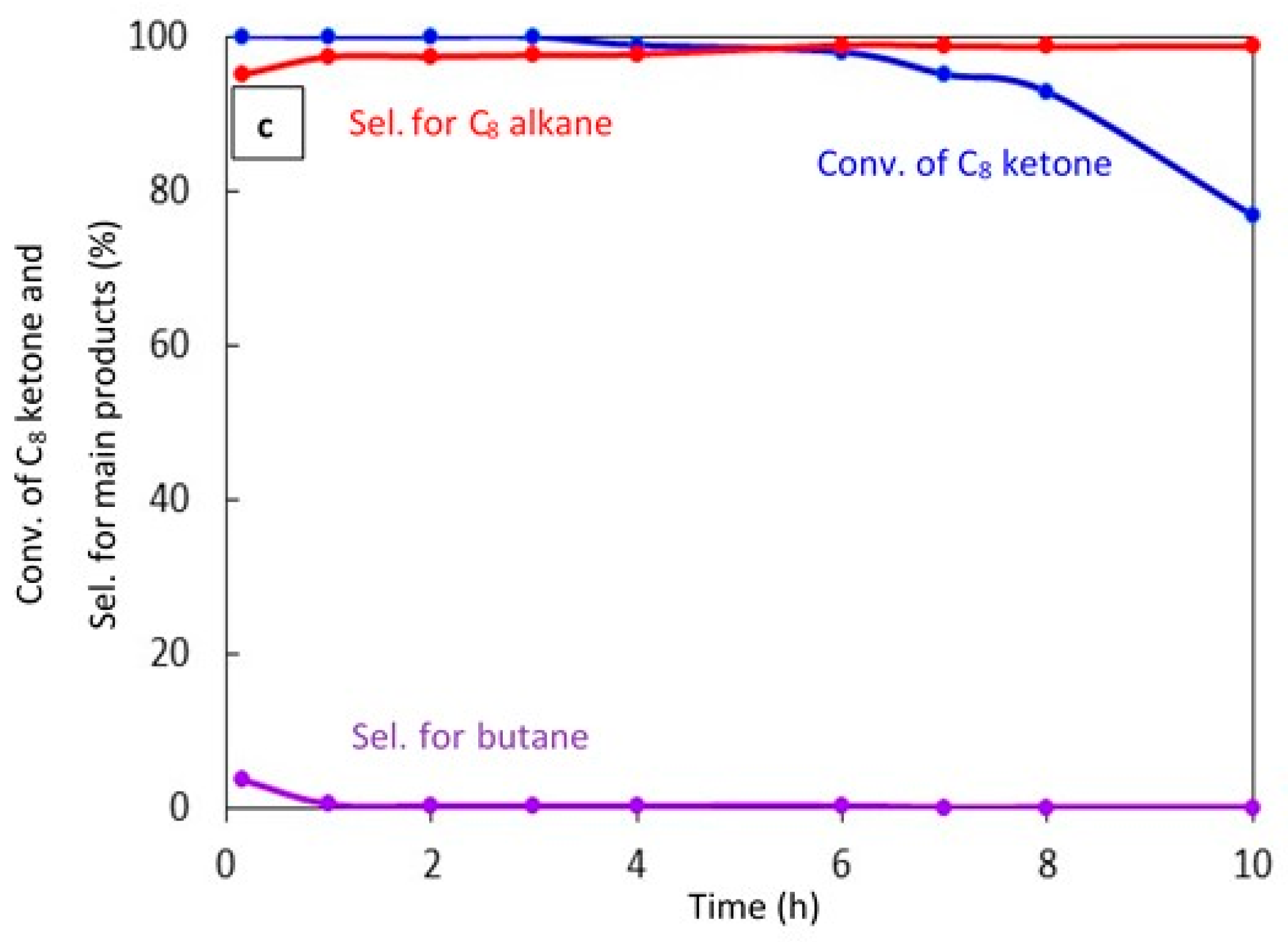
2.2.5. Catalyst Stability
3. Materials and Methods
3.1. Materials
3.2. Preparation of Supported Catalysts
3.3. Catalytic Reaction
3.4. Catalyst Characterization
3.4.1. X-Ray Diffraction (XRD)
3.4.2. Brunauer–Emmett–Teller (BET)
3.4.3. Transmission Electron Microscopy (TEM)
3.4.4. Temperature Programmed Desorption (NH3–TPD and CO2–TPD)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yu, E.K.; Saddler, J.N. Fed-batch approach to production of 2,3-butanediol by Klebsiella pneumoniae grown on high substrate concentrations. Appl. Environ. Microbiol. 1983, 46, 630–635. [Google Scholar] [PubMed]
- Zeng, A.P.; Biebl, H.; Deckwer, W.D. Effect of pH and acetic acid on growth and 2,3-butanediol production of Enterobacter aerogenes in continuous culture. Appl. Microbiol. Biotechnol. 1990, 33, 485–489. [Google Scholar] [CrossRef]
- De Mas, C.; Jansen, N.B.; Tsao, G.T. Production of optically active 2,3-butanediol by Bacillus polymyxa. Biotechnol. Bioeng. 1988, 31, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Ledingham, G.A.; Neish, A.C. Fermentative Production of 2,3-Butanediol. In Industrial Fermentations; Underkofler, L.A., Hickey, R.J., Eds.; Chemical Publishing Co: New York, NY, USA, 1954; Volume 2, pp. 27–93. [Google Scholar]
- Jansen, N.B.; Flickinger, M.C.; Tsao, G.T. Production of 2,3-butanediol from D-xylose by Klebsiella oxytoca ATCC 8724. Biotechnol. Bioeng. 1984, 26, 362–369. [Google Scholar] [CrossRef]
- Qureshi, N.; Cheryan, M. Effects of aeration on 2,3-butanediol production from glucose by Klebsiella oxytoca. J. Ferment. Bioeng. 1989, 67, 415–418. [Google Scholar] [CrossRef]
- Ji, X.J.; Huang, H.; Du, J.; Zhu, J.G.; Ren, L.J.; Hu, N.; Li, S. Enhanced 2,3-butanediol production by Klebsiella oxytoca using a two-stage agitation speed control strategy. Bioresour. Technol. 2009, 100, 3410–3414. [Google Scholar] [CrossRef]
- Saha, B.C.; Bothast, R.J. Production of 2,3-butanediol by newly isolated Enterobacter cloacae. Appl. Microbiol. Biotechnol. 1999, 52, 321–326. [Google Scholar] [CrossRef]
- Molnár, Á.; Bucsi, I.; Bartók, M. Pinacol Rearrangement on Zeolites. Stud. Surf. Sci. Catal. 1988, 41, 203–210. [Google Scholar]
- Multer, A.; McGraw, N.; Hohn, K.; Vadlani, P. Production of methyl ethyl ketone from biomass using a hybrid biochemical/catalytic approach. Ind. Eng. Chem. Res. 2012, 52, 56–60. [Google Scholar] [CrossRef]
- Emerson, R.R.; Flickinger, M.C.; Tsao, G.T. Kinetics of dehydration of aqueous 2,3-butanediol to methyl ethyl ketone. Ind. Eng. Chem. Prod. Res. Dev. 1982, 21, 473–477. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, D.; Ji, X.; Huang, H. Efficient dehydration of bio-based 2,3-butanediol to butanone over boric acid modified HZSM-5 zeolites. Green Chem. 2012, 14, 3441–3450. [Google Scholar] [CrossRef]
- Bucsi, I.; Molnár, Á.; Bartók, M.; Olah, G.A. Transformation of 1,2-diols over perfluorinated resinsulfonic acids (Nafion-H). Tetrahedron 1994, 50, 8195–8202. [Google Scholar] [CrossRef]
- Török, B.; Bucsi, I.; Beregszászi, T.; Kapocsi, I.; Molnár, Á. Transformation of diols in the presence of heteropoly acids under homogeneous and heterogeneous conditions. J. Mol. Catal. A Chem. 1996, 107, 305–311. [Google Scholar] [CrossRef]
- Nikitina, M.A.; Ivanova, I.I. Conversion of 2,3-Butanediol over Phosphate Catalysts. ChemCatChem 2016, 8, 1346–1353. [Google Scholar] [CrossRef]
- Lee, J.; Grutzner, J.B.; Walters, W.E.; Delgass, W.N. The conversion of 2,3-butanediol to methyl ethyl ketone over zeolites. Stud. Surf. Sci. Catal. 2000, 130, 2603–2608. [Google Scholar]
- Kelly, G.J. Aldol Condensation Reaction and Catalyst Therefor. U.S. Patent 6,706,928, 16 March 2004. [Google Scholar]
- Zheng, Q.; Wales, M.D.; Heidlage, M.G.; Rezac, M.; Wang, H.; Bossmann, S.H.; Hohn, K.L. Conversion of 2,3-butanediol to butenes over bifunctional catalysts in a single reactor. J. Catal. 2015, 330, 222–237. [Google Scholar] [CrossRef]
- Zheng, Q.; Grossardt, J.; Almkhelfe, H.; Xu, J.; Grady, B.P.; Douglas, J.T.; Amama, P.B.; Hohn, K.L. Study of mesoporous catalysts for conversion of 2,3-butanediol to butenes. J. Catal. 2017, 354, 182–196. [Google Scholar] [CrossRef]
- Al-Auda, Z.; Al-Atabi, H.; Li, X.; Zheng, Q.; Hohn, K.L. Conversion of methyl ethyl ketone to butenes over bifunctional catalysts. Appl. Catal. A Gen. 2019, 570, 173–182. [Google Scholar] [CrossRef]
- Al-Auda, Z.; Al-Atabi, H.; Hohn, K. Metals on ZrO2: Catalysts for the Aldol Condensation of Methyl Ethyl Ketone (MEK) to C8 Ketones. Catalysts 2018, 8, 622. [Google Scholar] [CrossRef]
- Brands, D.S.; Poels, E.K.; Bliek, A. Ester hydrogenolysis over promoted Cu/SiO2 catalysts. Appl. Catal. A Gen. 1999, 184, 279–289. [Google Scholar] [CrossRef]
- Fan, X.; Liu, S.; Yan, X.; Du, X.; Chen, L. A continuous process for the production of 2,2,6,6-tetramethylpiperidin-4-ol catalyzed by Cu–Cr/γ-Al2O3. Catal. Commun. 2010, 11, 960–963. [Google Scholar] [CrossRef]
- Saadi, A.; Rassoul, Z.; Bettahar, M.M. Gas phase hydrogenation of benzaldehyde over supported copper catalysts. J. Mol. Catal. A Chem. 2000, 164, 205–216. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Kumar, V.S.; Shasikala, V.; Padmasri, A.H.; Sreedhar, B.; Raju, B.D.; Rao, K.S.R. A highly efficient Cu/MgO catalyst for vapour phase hydrogenation of furfural to furfuryl alcohol. Catal. Commun. 2003, 4, 287–293. [Google Scholar] [CrossRef]
- Chambers, A.; Jackson, S.D.; Stirling, D.; Webb, G. Selective hydrogenation of cinnamaldehyde over supported copper catalysts. J. Catal. 1997, 168, 301–314. [Google Scholar] [CrossRef]
- Poondi, D.; Vannice, M.A. The influence of MSI (metal-support interactions) on phenylacetaldehyde hydrogenation over Pt catalysts. J. Mol. Catal. A Chem. 1997, 124, 79–89. [Google Scholar] [CrossRef]
- Vannice, M.A.; Sen, B. Metal-support effects on the intramolecular selectivity of crotonaldehyde hydrogenation over platinum. J. Catal. 1989, 115, 65–78. [Google Scholar] [CrossRef]
- Vannice, M.A.; Poondi, D. The effect of metal-support interactions on the hydrogenation of benzaldehyde and benzyl alcohol. J. Catal. 1997, 169, 166–175. [Google Scholar] [CrossRef]
- Kijeński, J.; Winiarek, P.; Paryjczak, T.; Lewicki, A.; Mikołajska, A. Platinum deposited on monolayer supports in selective hydrogenation of furfural to furfuryl alcohol. Appl. Catal. A Gen. 2002, 233, 171–182. [Google Scholar] [CrossRef]
- Shi, B.C.; Davis, B.H. Alcohol dehydration: Mechanism of ether formation using an alumina catalyst. J. Catal. 1995, 157, 359–367. [Google Scholar] [CrossRef]
- Kostestkyy, P.; Yu, J.; Gorte, R.J.; Mpourmpakis, G. Structure–activity relationships on metal-oxides: Alcohol dehydration. Catal. Sci. Technol. 2014, 4, 3861–3869. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Liu, S.; Tamura, M.; Tomishige, K. Catalytic total hydrodeoxygenation of biomass-derived polyfunctionalized substrates to alkanes. ChemSusChem 2015, 8, 1114–1132. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, S.; Kong, X.; Fan, X.; Yan, X.; Chen, L. A continuous process for the reductive deoxygenation of aromatic ketones over Cu30Cr10/γ-Al2O3. Res. Chem. Intermed. 2012, 38, 1341–1349. [Google Scholar] [CrossRef]
- Alotaibi, M.A.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Efficient hydrodeoxygenation of biomass-derived ketones over bifunctional Pt-polyoxometalate catalyst. Chem. Commun. 2012, 48, 7194–7196. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, K.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Hydrogenation of ketones over bifunctional Pt-heteropoly acid catalyst in the gas phase. Appl. Catal. A Gen. 2015, 504, 457–462. [Google Scholar] [CrossRef]
- Yang, J.; Li, N.; Li, G.; Wang, W.; Wang, A.; Wang, X.; Cong, Y.; Zhang, T. Synthesis of renewable high-density fuels using cyclopentanone derived from lignocellulose. Chem. Commun. 2014, 50, 2572–2574. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhu, H.; Niu, L.; Xiao, G.; Xiao, R. Catalytic hydroprocessing of furfural to cyclopentanol over Ni/CNTs catalysts: Model reaction for upgrading of bio-oil. Catal. Lett. 2014, 144, 235–241. [Google Scholar] [CrossRef]
- Li, X.L.; Deng, J.; Shi, J.; Pan, T.; Yu, C.G.; Xu, H.J.; Fu, Y. Selective conversion of furfural to cyclopentanone or cyclopentanol using different preparation methods of Cu–Co catalysts. Green Chem. 2015, 17, 1038–1046. [Google Scholar] [CrossRef]
- Guo, J.; Xu, G.; Han, Z.; Zhang, Y.; Fu, Y.; Guo, Q. Selective conversion of furfural to cyclopentanone with CuZnAl catalysts. ACS Sustain. Chem. Eng. 2014, 2, 2259–2266. [Google Scholar] [CrossRef]
- Chary, K.V.R.; Sagar, G.V.; Srikanth, C.S.; Rao, V.V. Characterization and catalytic functionalities of copper oxide catalysts supported on zirconia. J. Phys. Chem. B. 2007, 111, 543–550. [Google Scholar] [CrossRef]
- Sagar, G.V.; Rao, P.V.R.; Srikanth, C.S.; Chary, K.V.R. Dispersion and Reactivity of Copper Catalysts Supported on Al2O3−ZrO2. J. Phys. Chem. B 2006, 110, 13881–13888. [Google Scholar] [CrossRef]
- Marella, R.K.; Koppadi, K.S.; Jyothi, Y.; Rao, K.S.R.; Burri, D.R. Selective gas-phase hydrogenation of benzonitrile into benzylamine over Cu–MgO catalysts without using any additives. New J. Chem. 2013, 37, 3229–3235. [Google Scholar] [CrossRef]
- Zheng, Q.; Xu, J.; Liu, B.; Hohn, K.L. Mechanistic study of the catalytic conversion of 2,3-butanediol to butenes. J. Catal. 2018, 360, 221–239. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Augustine, R.L. Heterogeneous Catalysis for the Synthetic Chemist; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Particle Size of Metal Species (nm) a | Particle Size of Metal Species (nm) b | Surface Area (m2/g) c | Total Pore Volume (cm3/g) c | Average Pore Dimeter (Å) c | NH3 Desorbed (mmol/g) d | CO2 Desorbed (mmol/g) d |
|---|---|---|---|---|---|---|---|
| Al2O3 | _ | _ | 225 | 0.698 | 124 | 0.16 | 0.039 |
| 1% Pt–Al2O3 | _ | ~7 | 195 | 0.633 | 130 | 0.44 | 0.114 |
| 1% Cu–Al2O3 | _ | ~8 | 193 | 0.64 | 133 | 0.21 | 0.066 |
| 20% Cu–Al2O3 (4 h) | 20 | ~18 | 168 | 0.513 | 122 | 0.54 | 0.155 |
| 20% Cu–Al2O3 (8 h) | 20.5 | ~18 | 168 | 0.532 | 126 | 0.4 | 0.115 |
| Catalyst | Conv. of C8 Ketone (%) | Sel. for C8 Alkenes (%) | Sel. for C8 Alkane (%) | Others (%) a |
|---|---|---|---|---|
| 20% Cu–Al2O3 | 100 | 45 | 54 | 1 |
| 1% Cu–Al2O3 | 51 | 66 | 10 | 24 |
| 1% Pt–Al2O3 | 100 | 0 | 98 | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Auda, Z.; Al-Atabi, H.; Li, X.; Thapa, P.; Hohn, K. Conversion of 5-Methyl-3-Heptanone to C8 Alkenes and Alkane over Bifunctional Catalysts. Catalysts 2019, 9, 845. https://doi.org/10.3390/catal9100845
Al-Auda Z, Al-Atabi H, Li X, Thapa P, Hohn K. Conversion of 5-Methyl-3-Heptanone to C8 Alkenes and Alkane over Bifunctional Catalysts. Catalysts. 2019; 9(10):845. https://doi.org/10.3390/catal9100845
Chicago/Turabian StyleAl-Auda, Zahraa, Hayder Al-Atabi, Xu Li, Prem Thapa, and Keith Hohn. 2019. "Conversion of 5-Methyl-3-Heptanone to C8 Alkenes and Alkane over Bifunctional Catalysts" Catalysts 9, no. 10: 845. https://doi.org/10.3390/catal9100845
APA StyleAl-Auda, Z., Al-Atabi, H., Li, X., Thapa, P., & Hohn, K. (2019). Conversion of 5-Methyl-3-Heptanone to C8 Alkenes and Alkane over Bifunctional Catalysts. Catalysts, 9(10), 845. https://doi.org/10.3390/catal9100845