Proteases Immobilization for In Situ Time-Limited Proteolysis on MALDI Chips



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
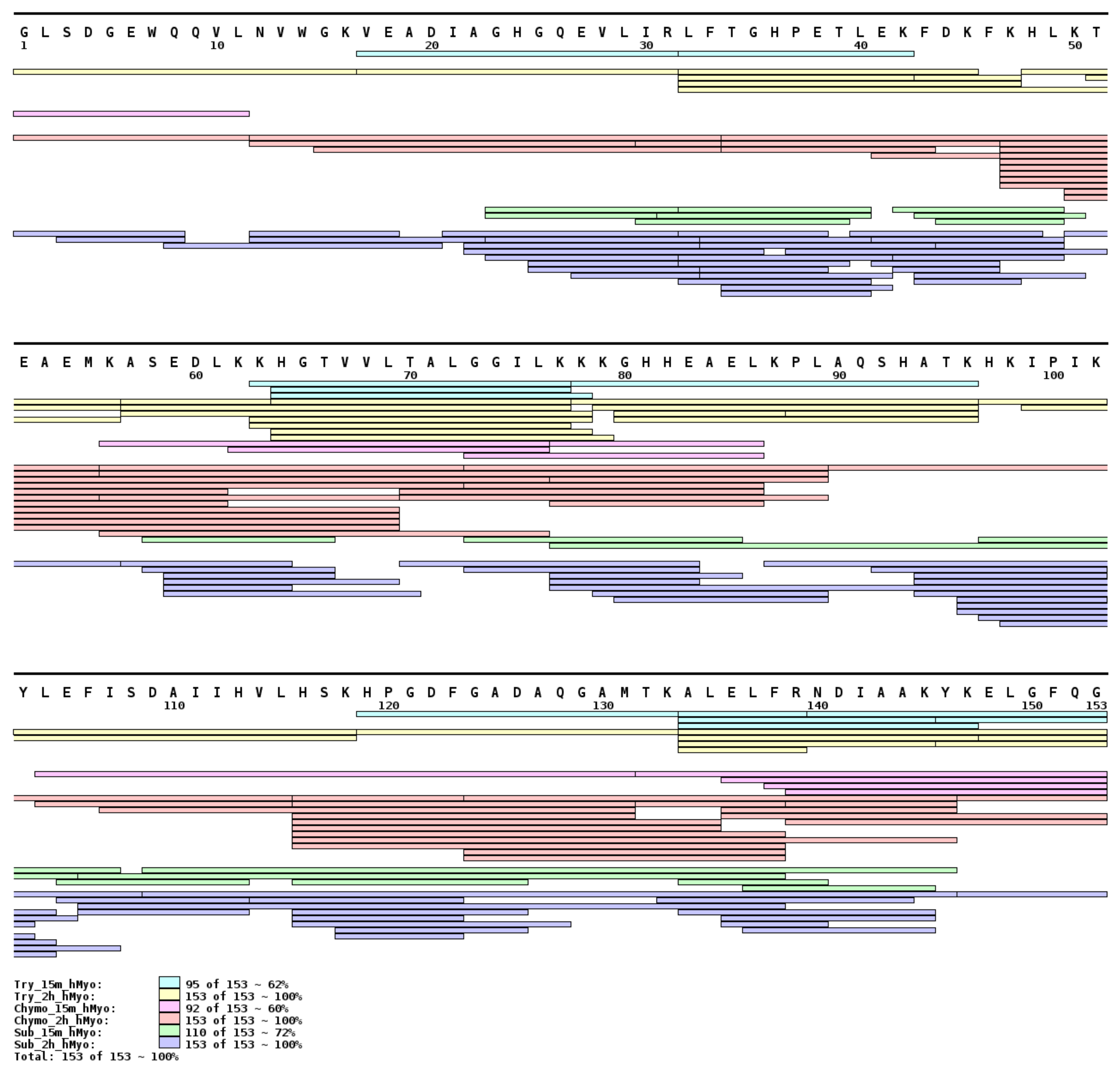
2.1. Protease Immobilization
2.2. Data Interpretation Procedure
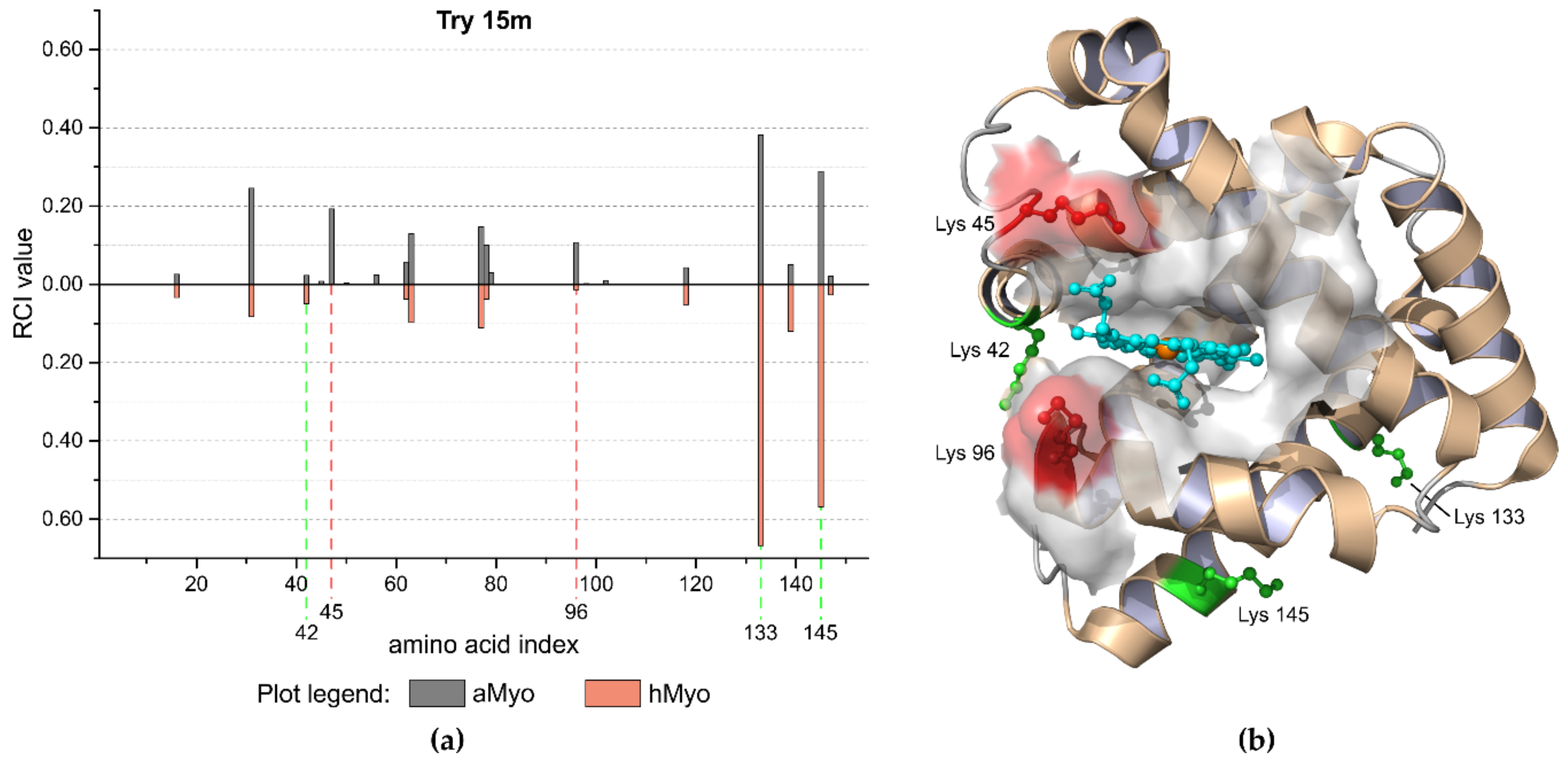
2.3. Method Feasibility
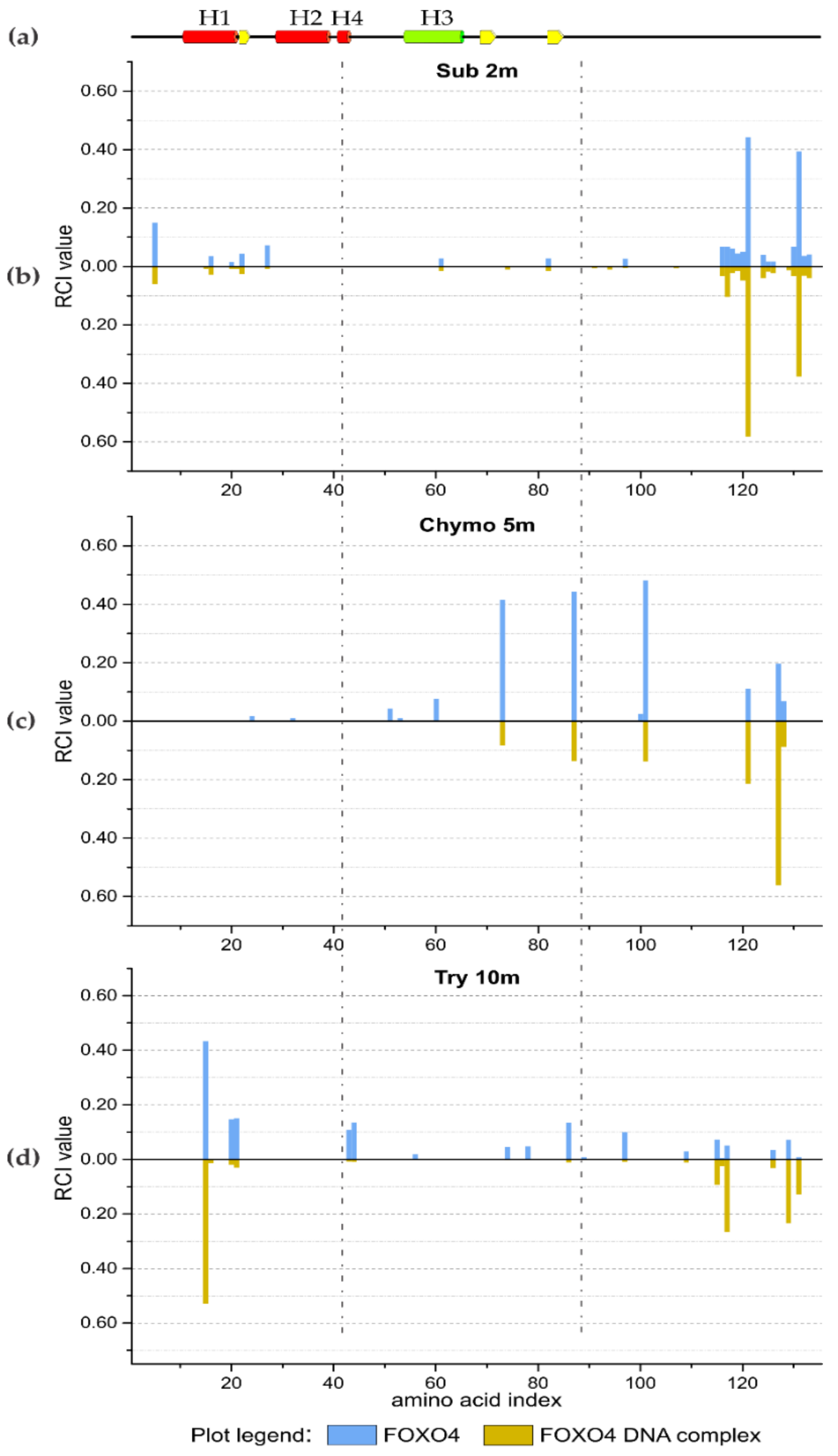
2.4. Limited Proteolysis of FOXO4 DNA Complex
3. Materials and Methods
3.1. Proteolytic Enzymes
3.2. Protein Systems
3.2.1. Apomyoglobin and Holomyoglobin Model System
3.2.2. FOXO4 Transcription Factor and Oligonucleotide Duplex
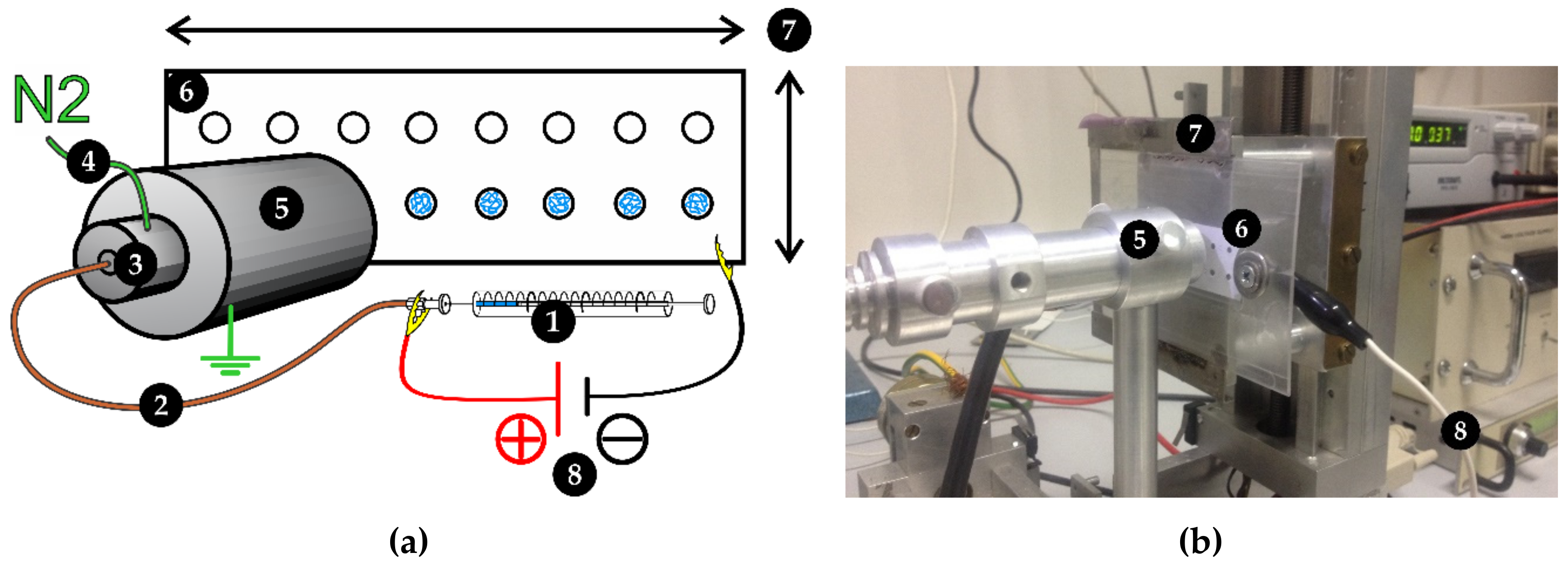
3.3. Ambient Ion-Landing Immobilization
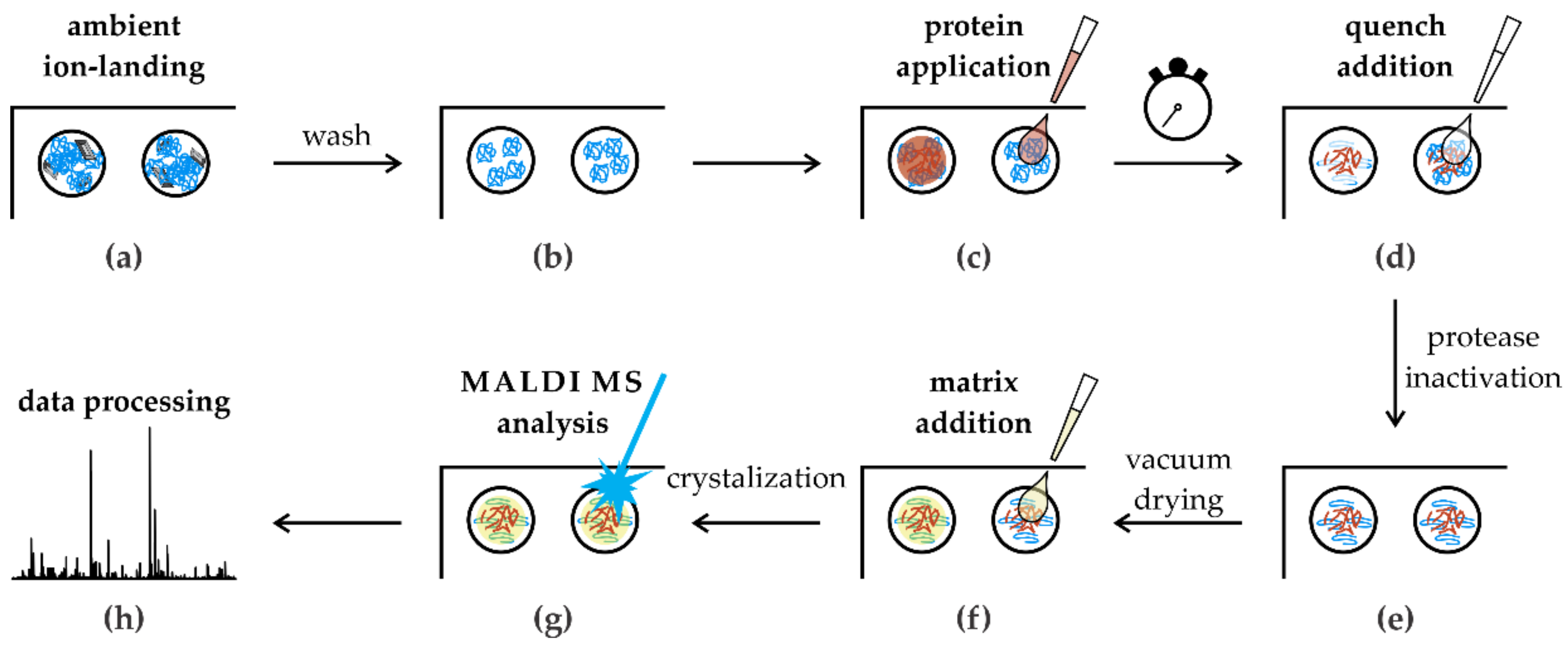
3.4. In Situ Limited Proteolysis Workflow
3.5. Control Spots
3.6. Mass Spectrometry
3.7. Data Interpretation
3.8. Data Visualization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rusmini, F.; Zhong, Z.; Feijen, J. Protein Immobilization Strategies for Protein Biochips. Biomacromolecules 2007, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Christena, L.R.; Rajaram, Y.R. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pompach, P.; Benada, O.; Rosulek, M.; Darebna, P.; Hausner, J.; Ruzicka, V.; Volny, M.; Novak, P. Protein Chips Compatible with MALDI Mass Spectrometry Prepared by Ambient Ion Landing. Anal. Chem. 2016, 88, 8526–8534. [Google Scholar] [CrossRef] [PubMed]
- Grill, V.; Shen, J.; Evans, C.; Cooks, R.G. Collisions of ions with surfaces at chemically relevant energies: Instrumentation and phenomena. Rev. Sci. Instrum. 2001, 72, 3149. [Google Scholar] [CrossRef]
- Franchetti, V.; Solka, B.H.; Baitinger, W.E.; Amy, J.W.; Cooks, R.G. Soft landing of ions as a means of surface modification. Int. J. Mass Spectrom. Ion Phys. 1977, 23, 29–35. [Google Scholar] [CrossRef]
- Miller, S.A.; Luo, H.; Pachuta, S.J.; Cooks, R.G. Soft-Landing of Polyatomic Ions at Fluorinated Self-Assembled Monolayer Surfaces. Science 1997, 275, 1447–1450. [Google Scholar] [CrossRef]
- Feng, B.; Wunschel, D.S.; Masselon, C.D.; Pasa-Tolic, L.; Smith, R.D. Retrieval of DNA Using Soft-Landing after Mass Analysis by ESI-FTICR for Enzymatic Manipulation. J. Am. Chem. Soc. 1999, 121, 8961–8962. [Google Scholar] [CrossRef]
- Geiger, R.; Melnyk, M.; Busch, K.; Bartlett, M. Modifications to an analytical mass spectrometer for the soft-landing experiment. Int. J. Mass Spectrom. 1999, 182–183, 415–422. [Google Scholar] [CrossRef]
- Ouyang, Z.; Takats, Z.; Blake, T.A.; Gologan, B.; Guymon, A.J.; Wiseman, J.M.; Oliver, J.C.; Davisson, V.J.; Cooks, R.G. Preparing protein microarrays by soft-landing of mass-selected ions. Science 2003, 301, 1351–1354. [Google Scholar] [CrossRef]
- Volny, M.; Elam, W.T.; Branca, A.; Ratner, B.D.; Turecek, F. Preparative soft and reactive landing of multiply charged protein ions on a plasma-treated metal surface. Anal. Chem. 2005, 77, 4890–4896. [Google Scholar] [CrossRef]
- Volny, M.; Elam, W.T.; Ratner, B.D.; Turecek, F. Preparative soft and reactive landing of gas-phase ions on plasma-treated metal surfaces. Anal. Chem. 2005, 77, 4846–4853. [Google Scholar] [CrossRef] [PubMed]
- Badu-Tawiah, A.K.; Wu, C.; Cooks, R.G. Ambient ion soft landing. Anal. Chem. 2011, 83, 2648–2654. [Google Scholar] [CrossRef] [PubMed]
- Krasny, L.; Pompach, P.; Strohalm, M.; Obsilova, V.; Strnadova, M.; Novak, P.; Volny, M. In-situ enrichment of phosphopeptides on MALDI plates modified by ambient ion landing. J. Mass Spectrom. 2012, 47, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zheng, Q.; Badu-Tawiah, A.K.; Xiong, C.; Guan, C.; Chen, S.; Nie, Z.; Wang, D.; Wan, L. Electrospray soft-landing for the construction of non-covalent molecular nanostructures using charged droplets under ambient conditions. Chem. Commun. (Camb) 2016, 52, 13660–13663. [Google Scholar] [CrossRef] [PubMed]
- Pompach, P.; Novakova, J.; Kavan, D.; Benada, O.; Ruzicka, V.; Volny, M.; Novak, P. Planar Functionalized Surfaces for Direct Immunoaffinity Desorption/Ionization Mass Spectrometry. Clin. Chem. 2015, 68, 270–278. [Google Scholar] [CrossRef]
- Darebna, P.; Spicka, J.; Kucera, R.; Topolcan, O.; Navratilova, E.; Ruzicka, V.; Volny, M.; Novak, P.; Pompach, P. Detection and Quantification of Carbohydrate-Deficient Transferrin by MALDI-Compatible Protein Chips Prepared by Ambient Ion Soft Landing. Clin. Chem. 2018, 64, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Thontasen, N.; Malinowski, N.; Rinke, G.; Harnau, L.; Rauschenbach, S.; Kern, K. A close look at proteins: Submolecular resolution of two- and three-dimensionally folded cytochrome c at surfaces. Nano Lett. 2012, 12, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, M. Induction of biological activity by limited proteolysis. Annu. Rev. Biochem. 1967, 36, 55–76. [Google Scholar] [CrossRef]
- Schejter, A.; Goldkorn, T.; Sokolovsky, M. Limited Proteolysis of Horse Heart Cytochrome c. Eur. J. Biochem. 1971, 20, 414–419. [Google Scholar] [CrossRef]
- Foster, J.F.; Wilson, W.D. Conformation-dependent limited proteolysis of bovine plasma albumin by an enzyme present in commercial albumin preparations. Biochemistry 1971, 10, 1772–1780. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Fischer, S.G.; Kirschner, M.W.; Laemmli, U.K. Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by gel electrophoresis. J. Biol. Chem. 1977, 252, 1102–1106. [Google Scholar] [PubMed]
- Brockerhoff, S.E.; Edmonds, C.G.; Davis, T.N. Structural analysis of wild-type and mutant yeast calmodulins by limited proteolysis and electrospray ionization mass spectrometry. Protein Sci. Publ. Protein Soc. 1992, 1, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Schopper, S.; Kahraman, A.; Leuenberger, P.; Feng, Y.; Piazza, I.; Muller, O.; Boersema, P.J.; Picotti, P. Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nat. Protoc. 2017, 12, 2391–2410. [Google Scholar] [CrossRef]
- Suckau, D.; Kohl, J.; Karwath, G.; Schneider, K.; Casaretto, M.; Bitter-Suermann, D.; Przybylski, M. Molecular epitope identification by limited proteolysis of an immobilized antigen-antibody complex and mass spectrometric peptide mapping. Proc. Natl. Acad. Sci. USA 1990, 87, 9848–9852. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chait, B.T. Protein Epitope Mapping By Mass Spectrometry. Anal. Chem. 1994, 66, 3723–3726. [Google Scholar] [CrossRef]
- Cohen, S.L.; Ferre-D’Amare, A.R.; Burley, S.K.; Chait, B.T. Probing the solution structure of the DNA-binding protein Max by a combination of proteolysis and mass spectrometry. Protein Sci. Publi. Protein Soc. 1995, 4, 1088–1099. [Google Scholar] [CrossRef]
- Feng, Y.; De Franceschi, G.; Kahraman, A.; Soste, M.; Melnik, A.; Boersema, P.J.; de Laureto, P.P.; Nikolaev, Y.; Oliveira, A.P.; Picotti, P. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 2014, 32, 1036–1044. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Lin, L.; Pinker, R.J.; Forde, K.; Rose, G.D.; Kallenbach, N.R. Molten globular characteristics of the native state of apomyoglobin. Nat. Struct. Mol. Biol. 1994, 1, 447–452. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, Y.A.; Park, N.; Son, H.S.; Kim, K.S.; Hahn, J.H. Structural characterization of the molten globule state of apomyoglobin by limited proteolysis and HPLC-mass spectrometry. Biochemistry 2005, 44, 7490–7496. [Google Scholar] [CrossRef] [PubMed]
- Boura, E.; Rezabkova, L.; Brynda, J.; Obsilova, V.; Obsil, T. Structure of the human FOXO4-DBD-DNA complex at 1.9 A resolution reveals new details of FOXO binding to the DNA. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Laboratory of Structural Biology and Cell Signaling, Institute of Microbiology of the CAS. Linx: Software for Interpretation of High-Resolution MS Data Obtained after Protein Chemical Cross-Linking. 2019. Available online: http://peterslab.org/downloads.php (accessed on 9 August 2019).
- Mrazova, B.; Martinkova, M.; Martinek, V.; Frei, E.; Stiborova, M. Optimalization of preparation of apo-cytochrome b(5) utilizing apo-myoglobin. Interdiscip. Toxicol. 2008, 1, 190–192. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kavan, D.; Man, P. MSTools—Web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom. 2011, 302, 53–58. [Google Scholar] [CrossRef]
- Maurus, R.; Overall, C.M.; Bogumil, R.; Luo, Y.; Mauk, A.G.; Smith, M.; Brayer, G.D. A myoglobin variant with a polar substitution in a conserved hydrophobic cluster in the heme binding pocket. Biochim. Biophys. Acta 1997, 1341, 1–13. [Google Scholar] [CrossRef]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosulek, M.; Darebna, P.; Pompach, P.; Slavata, L.; Novak, P. Proteases Immobilization for In Situ Time-Limited Proteolysis on MALDI Chips. Catalysts 2019, 9, 833. https://doi.org/10.3390/catal9100833
Rosulek M, Darebna P, Pompach P, Slavata L, Novak P. Proteases Immobilization for In Situ Time-Limited Proteolysis on MALDI Chips. Catalysts. 2019; 9(10):833. https://doi.org/10.3390/catal9100833
Chicago/Turabian StyleRosulek, Michal, Petra Darebna, Petr Pompach, Lukas Slavata, and Petr Novak. 2019. "Proteases Immobilization for In Situ Time-Limited Proteolysis on MALDI Chips" Catalysts 9, no. 10: 833. https://doi.org/10.3390/catal9100833
APA StyleRosulek, M., Darebna, P., Pompach, P., Slavata, L., & Novak, P. (2019). Proteases Immobilization for In Situ Time-Limited Proteolysis on MALDI Chips. Catalysts, 9(10), 833. https://doi.org/10.3390/catal9100833