Synthesis of Pyrazolo-Fused 4-Azafluorenones in an Ionic Liquid. Mechanistic Insights by Joint Studies Using DFT Analysis and Mass Spectrometry

 ,
,  and
and
Abstract
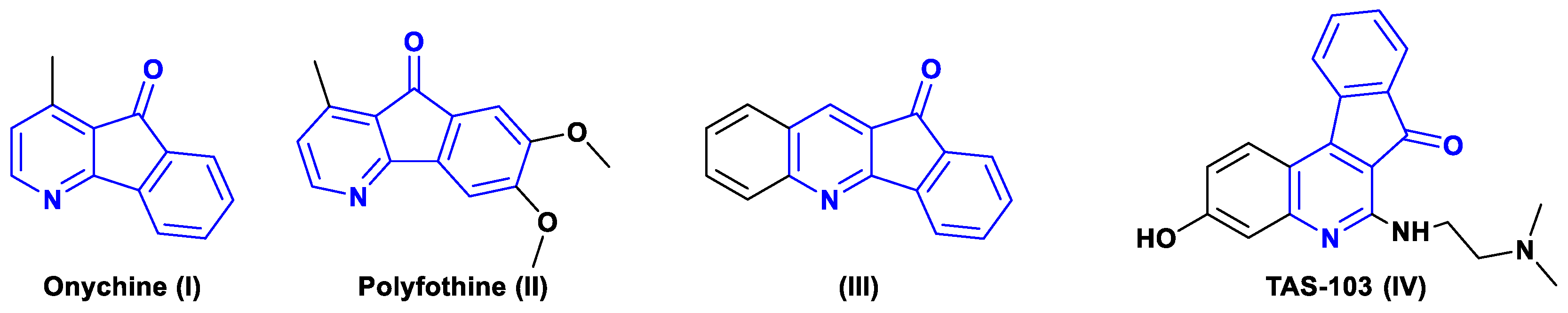
1. Introduction
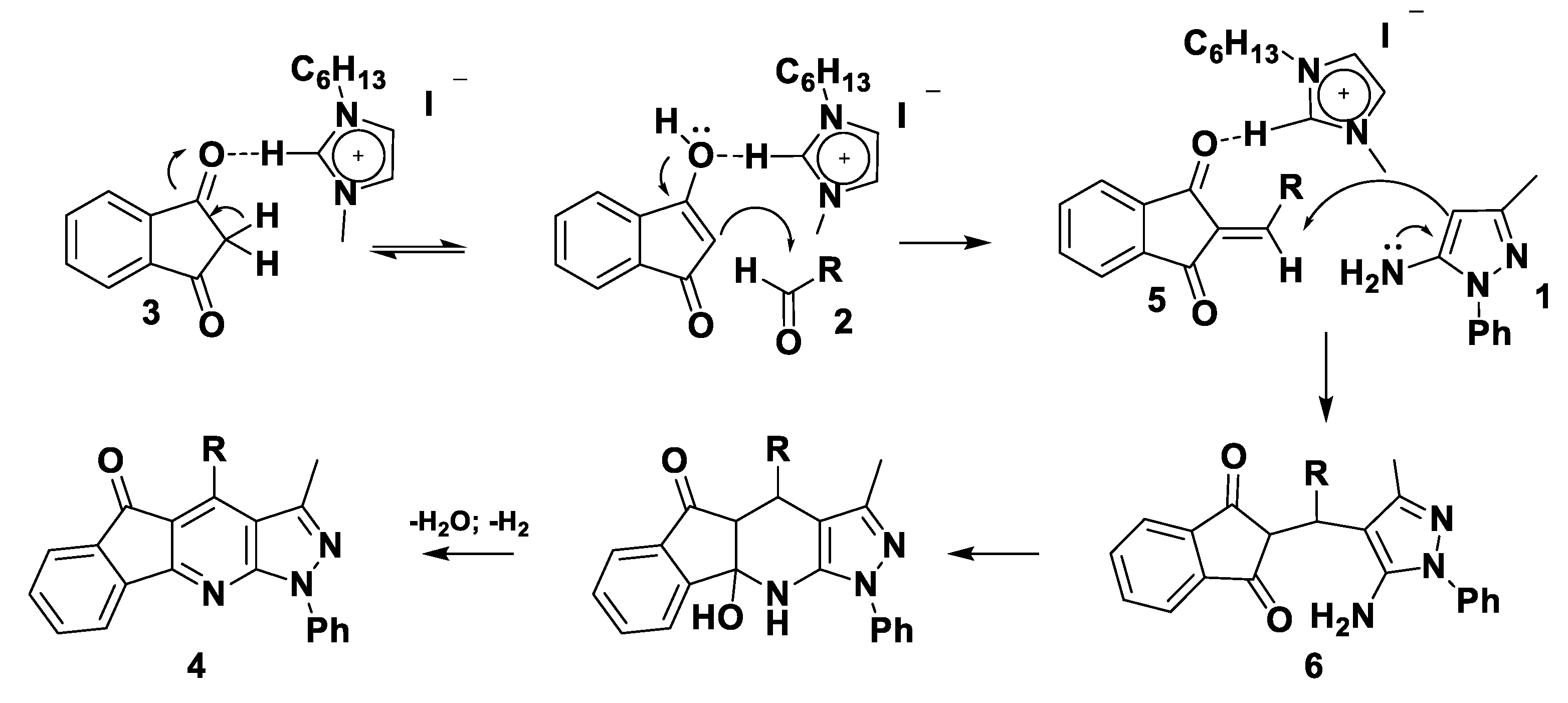
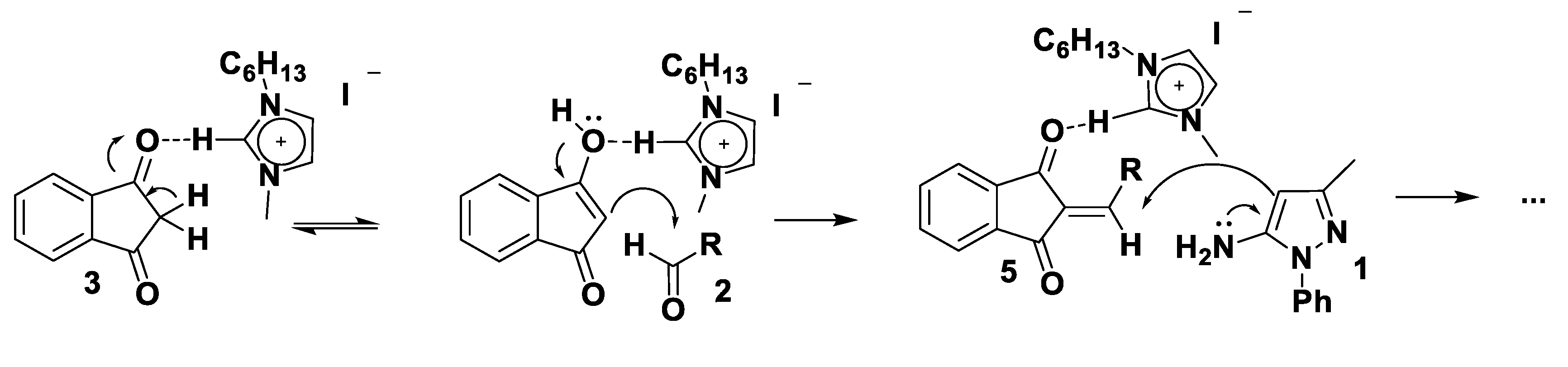
2. Results
3. Theoretical Analysis
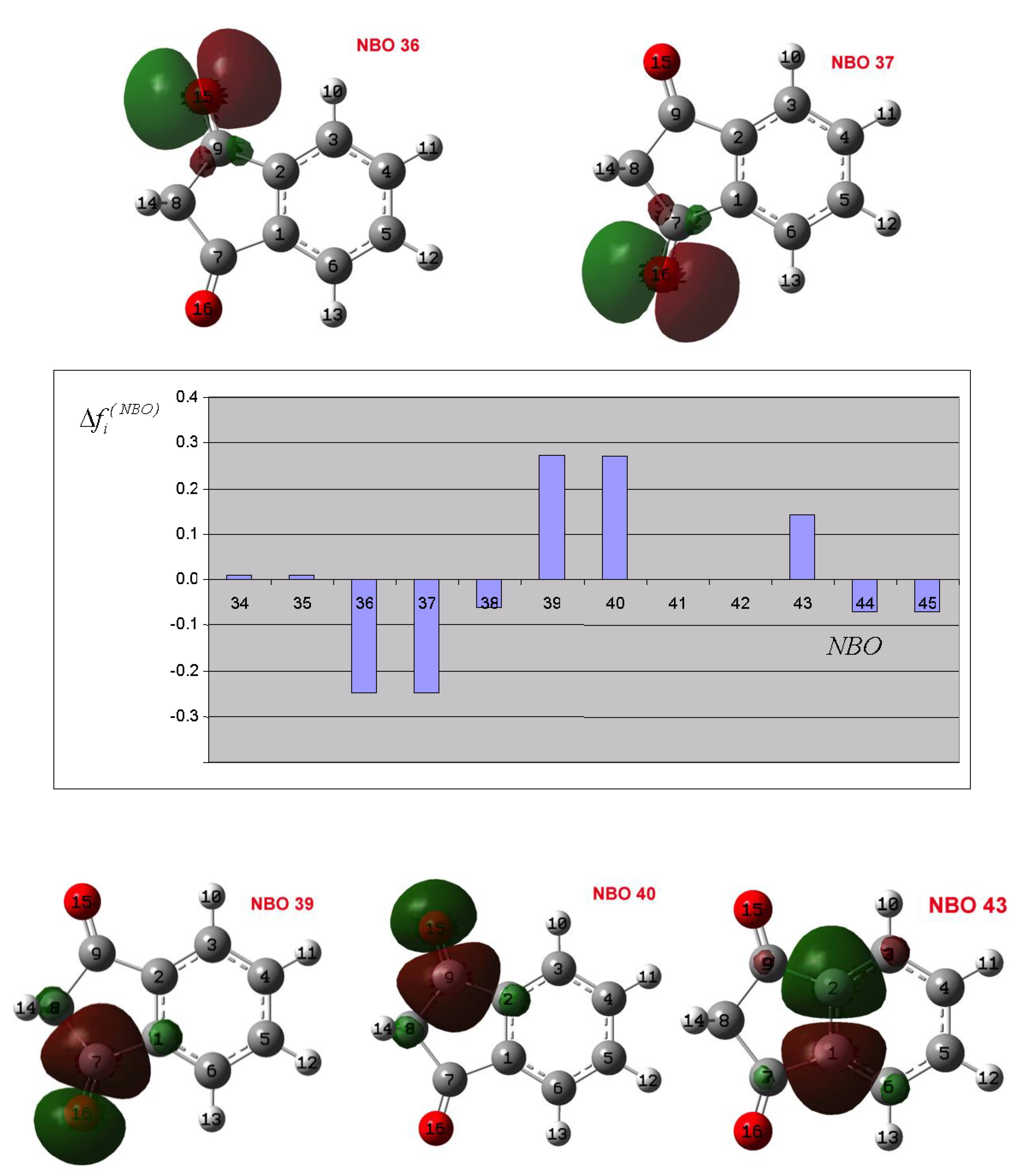
3.1. Effect of the Field Generated by the Catalyst
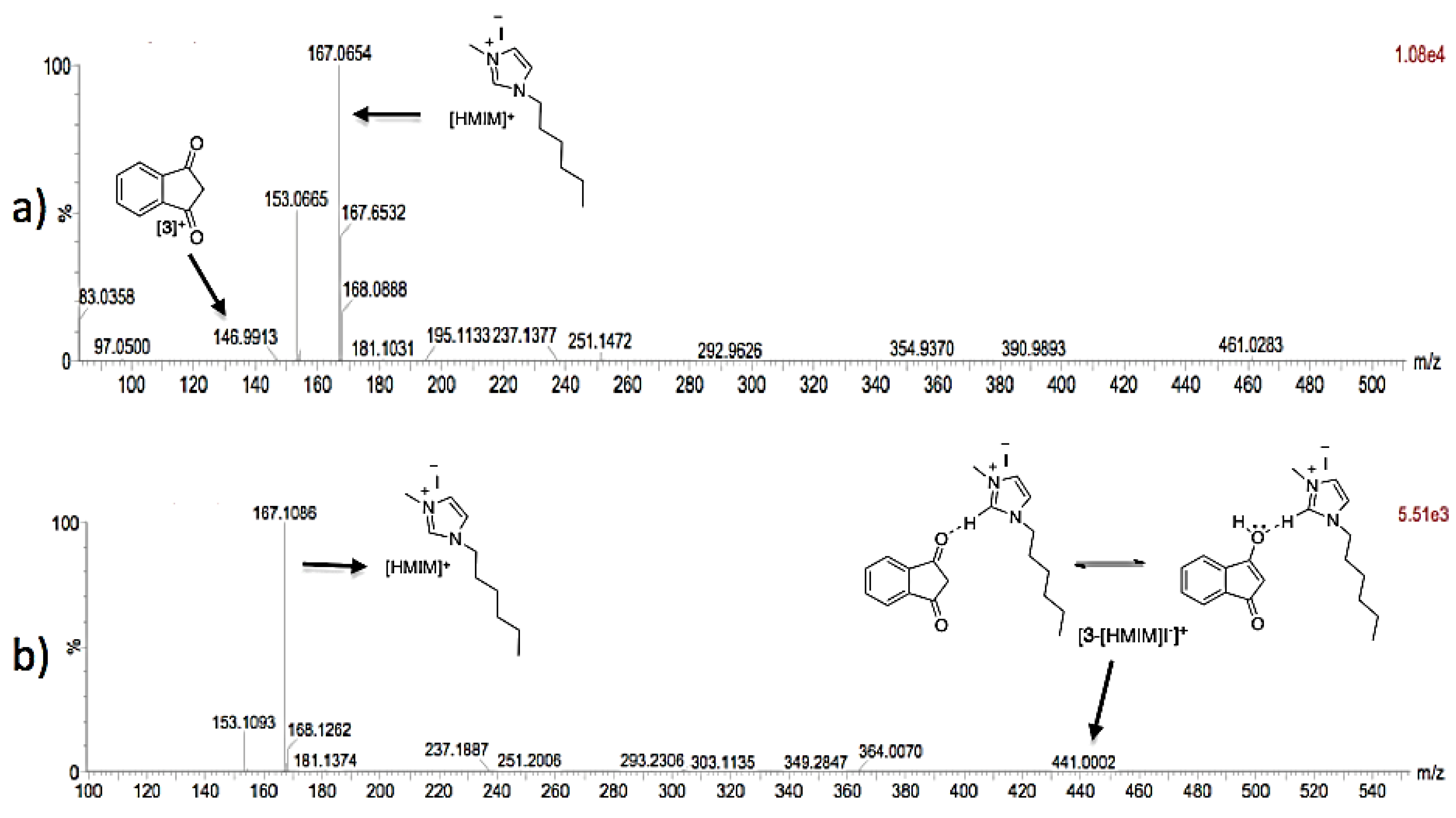
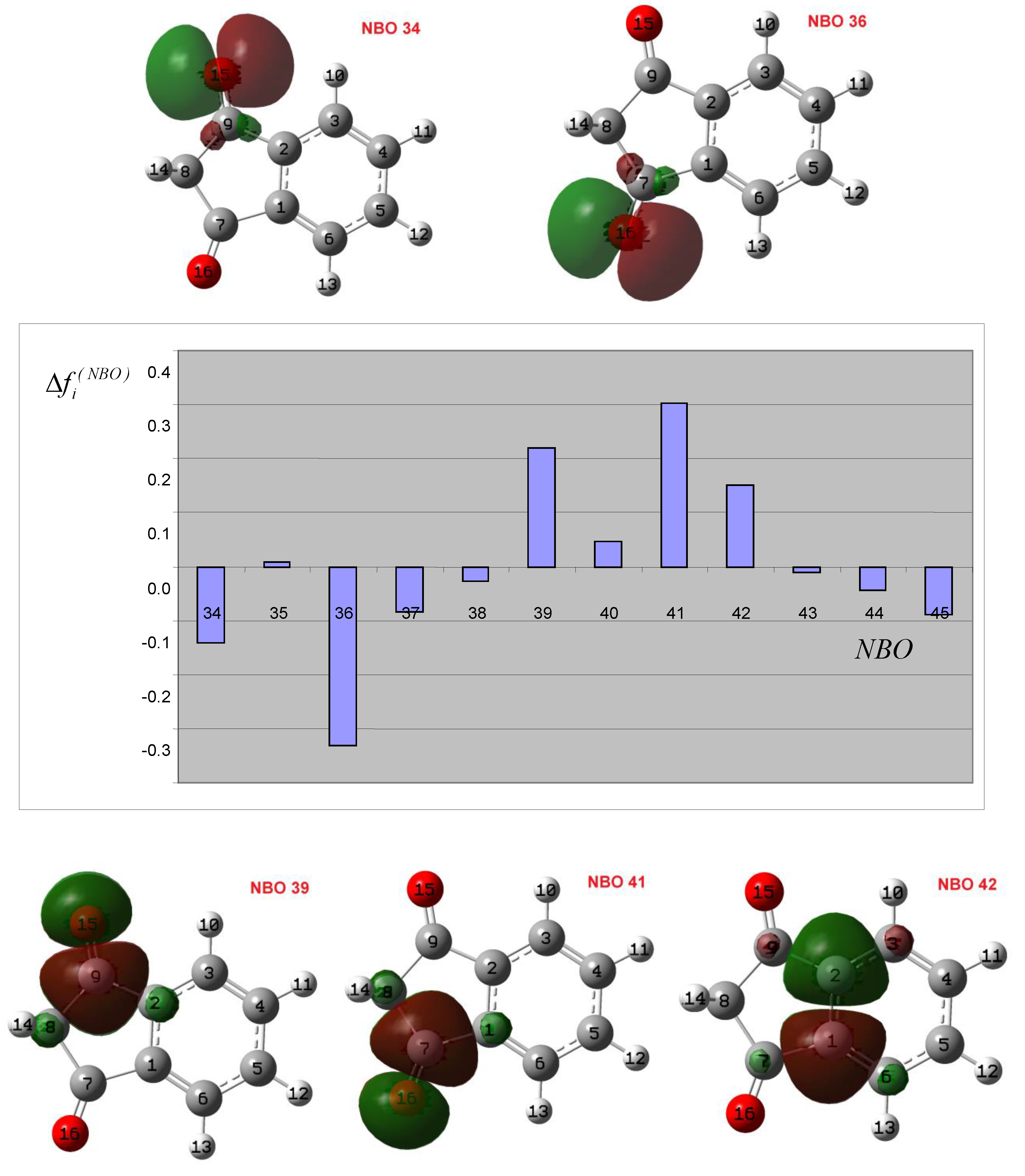
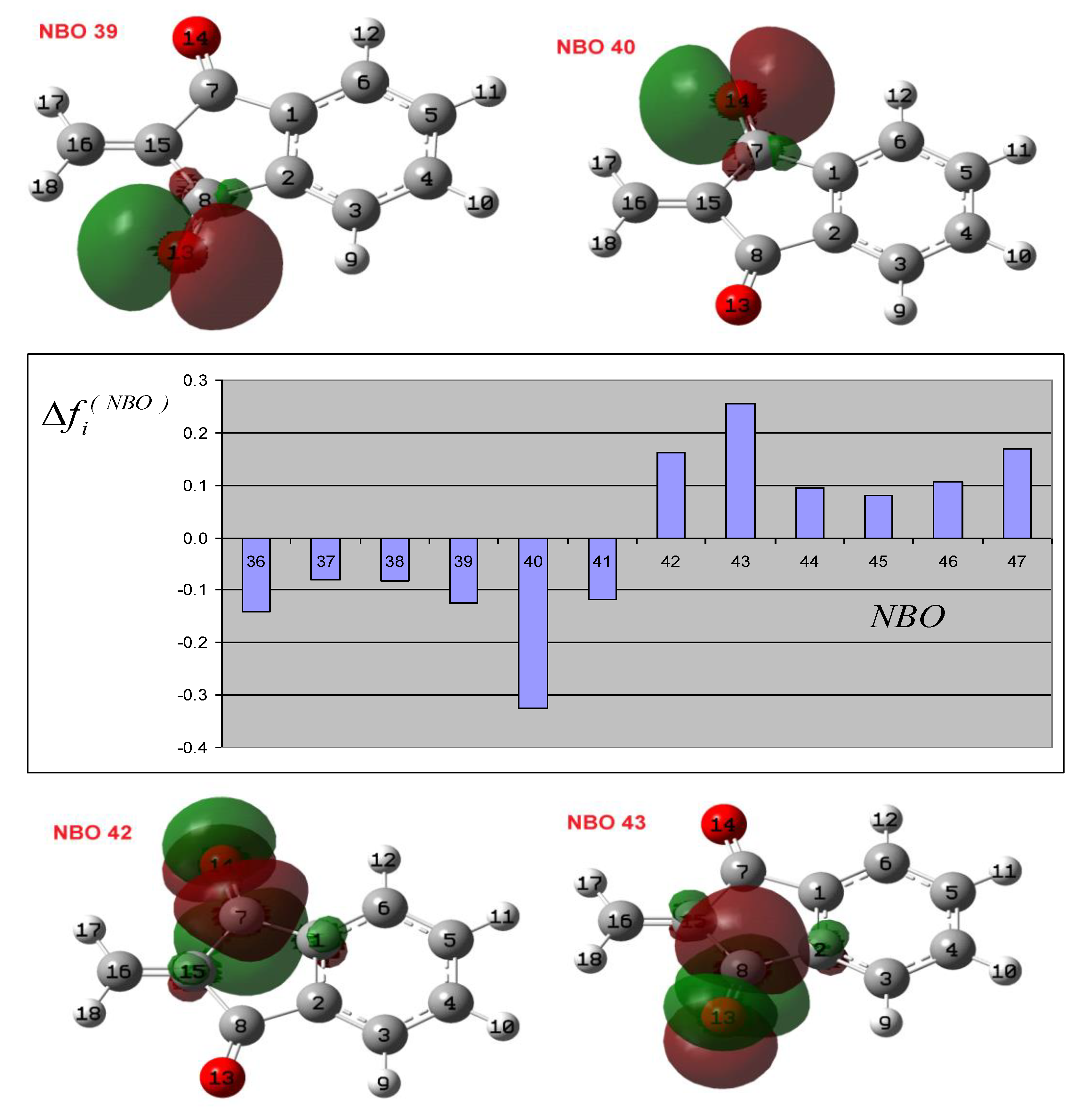
3.2. Theoretical Analysis of the First System
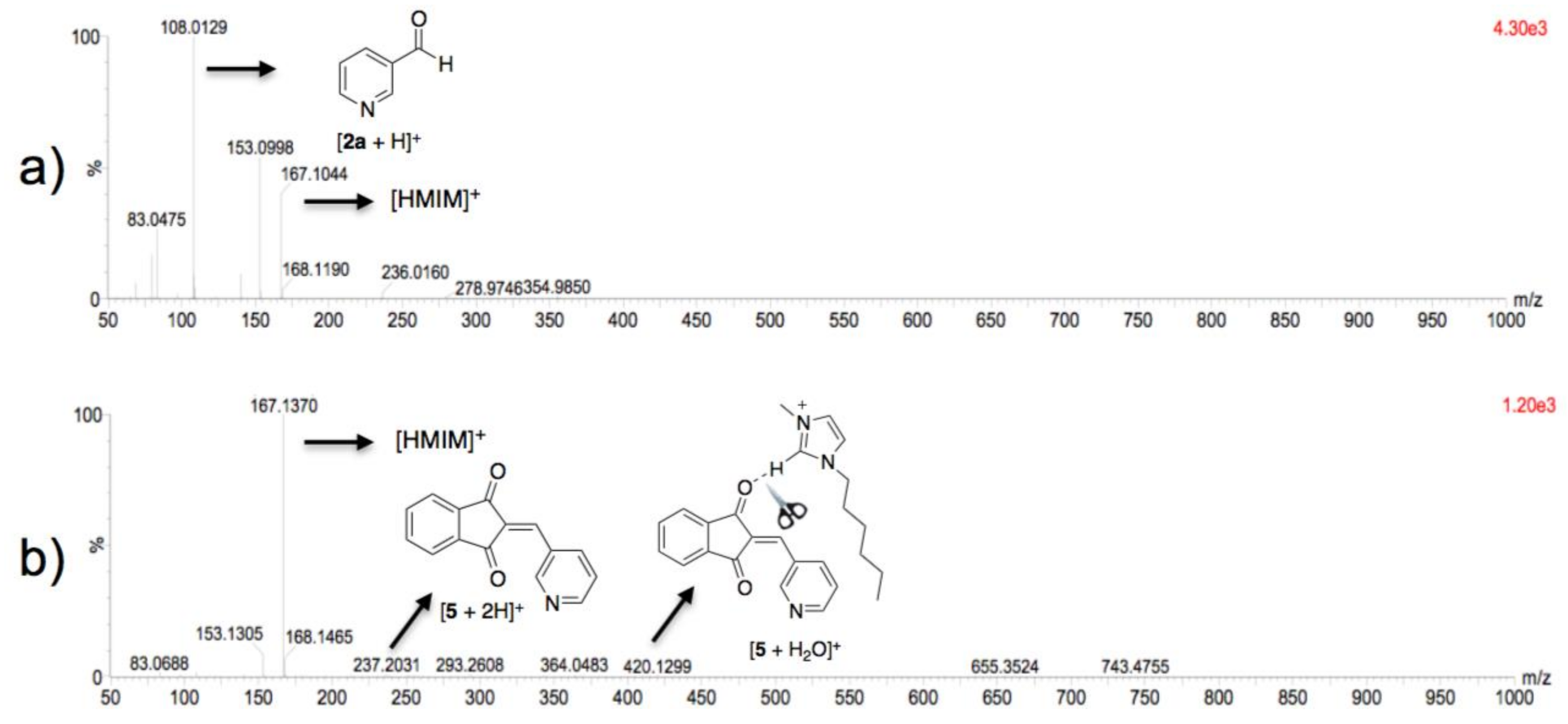
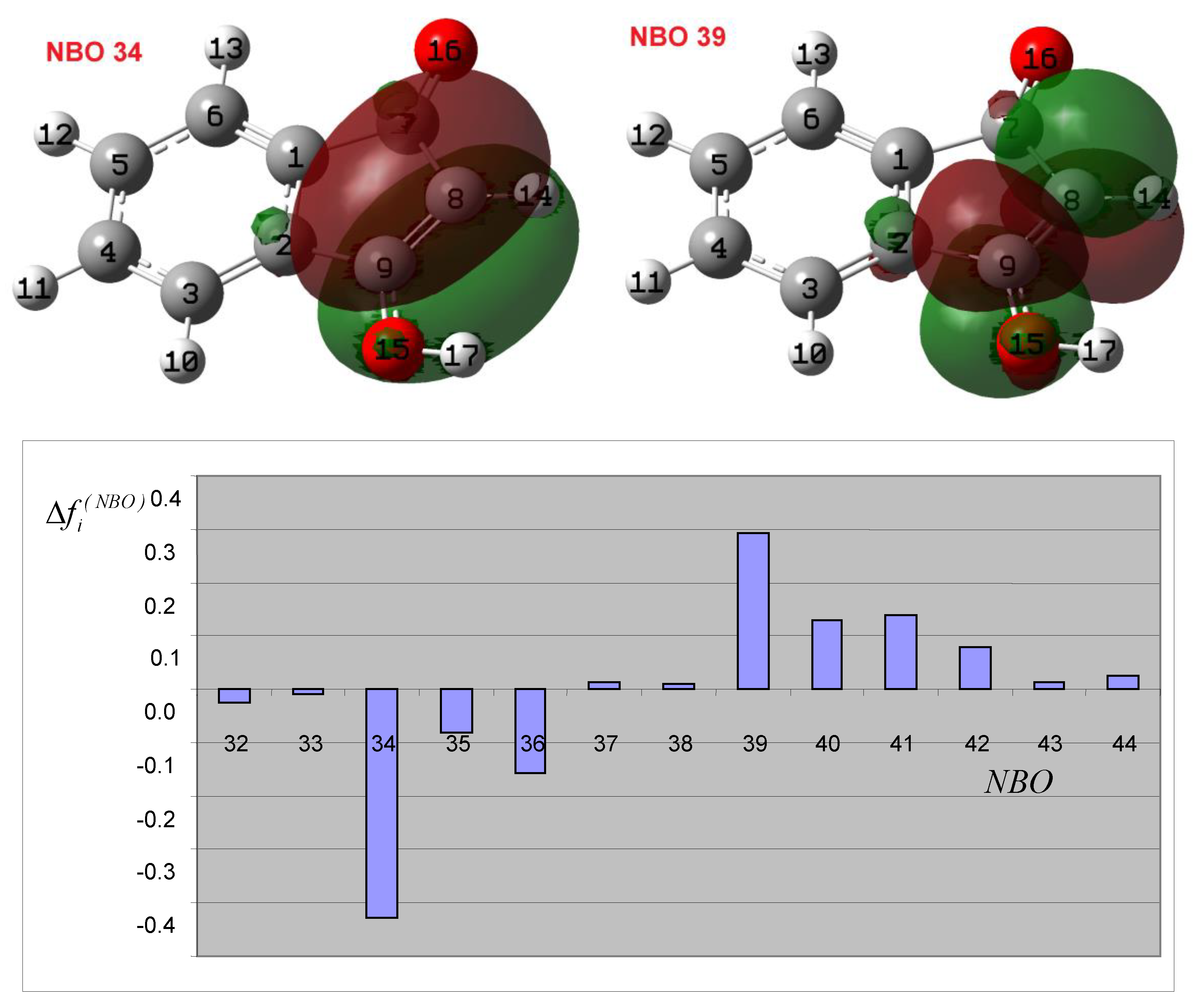
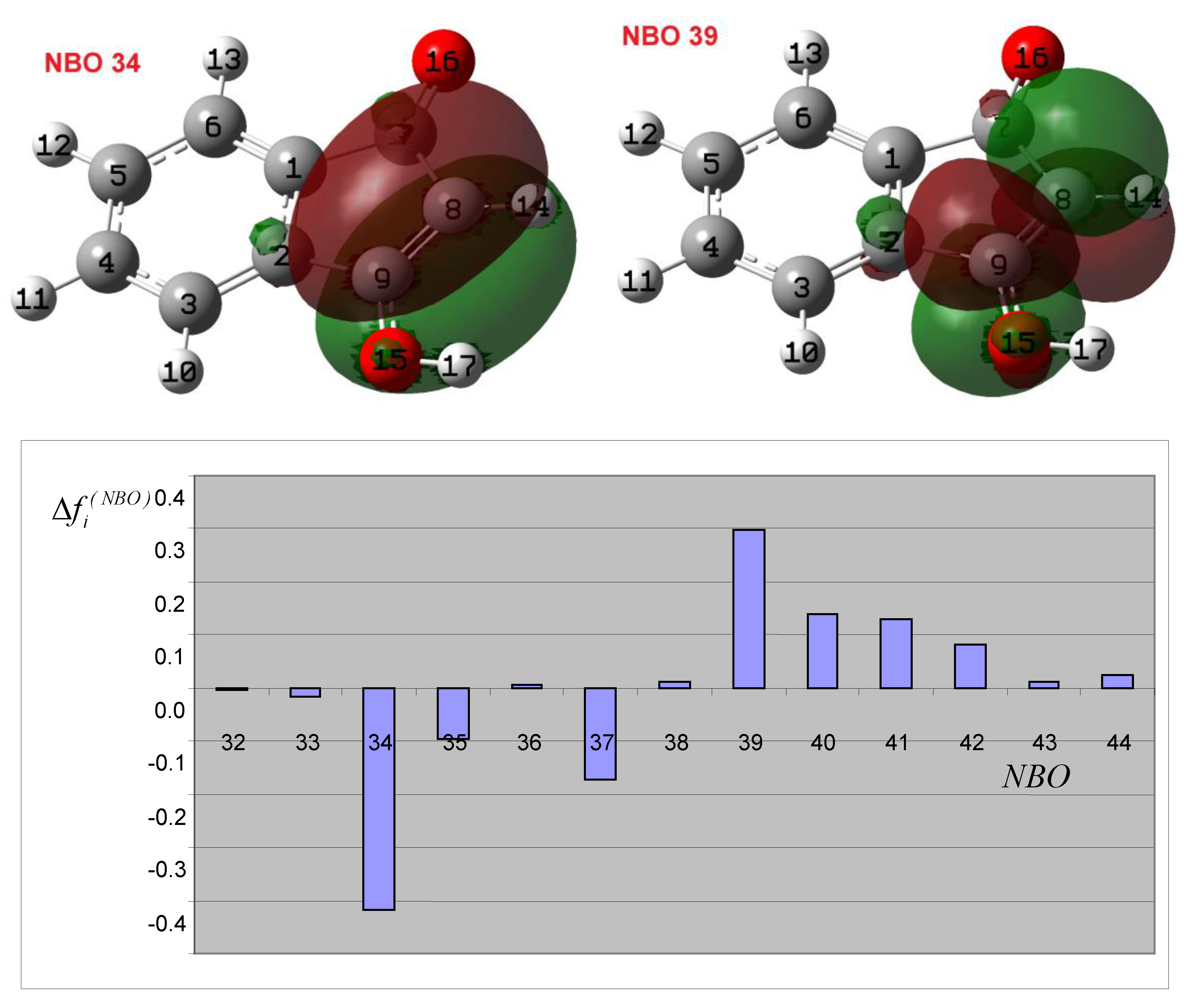
3.3. Theoretical Analysis of the Second System
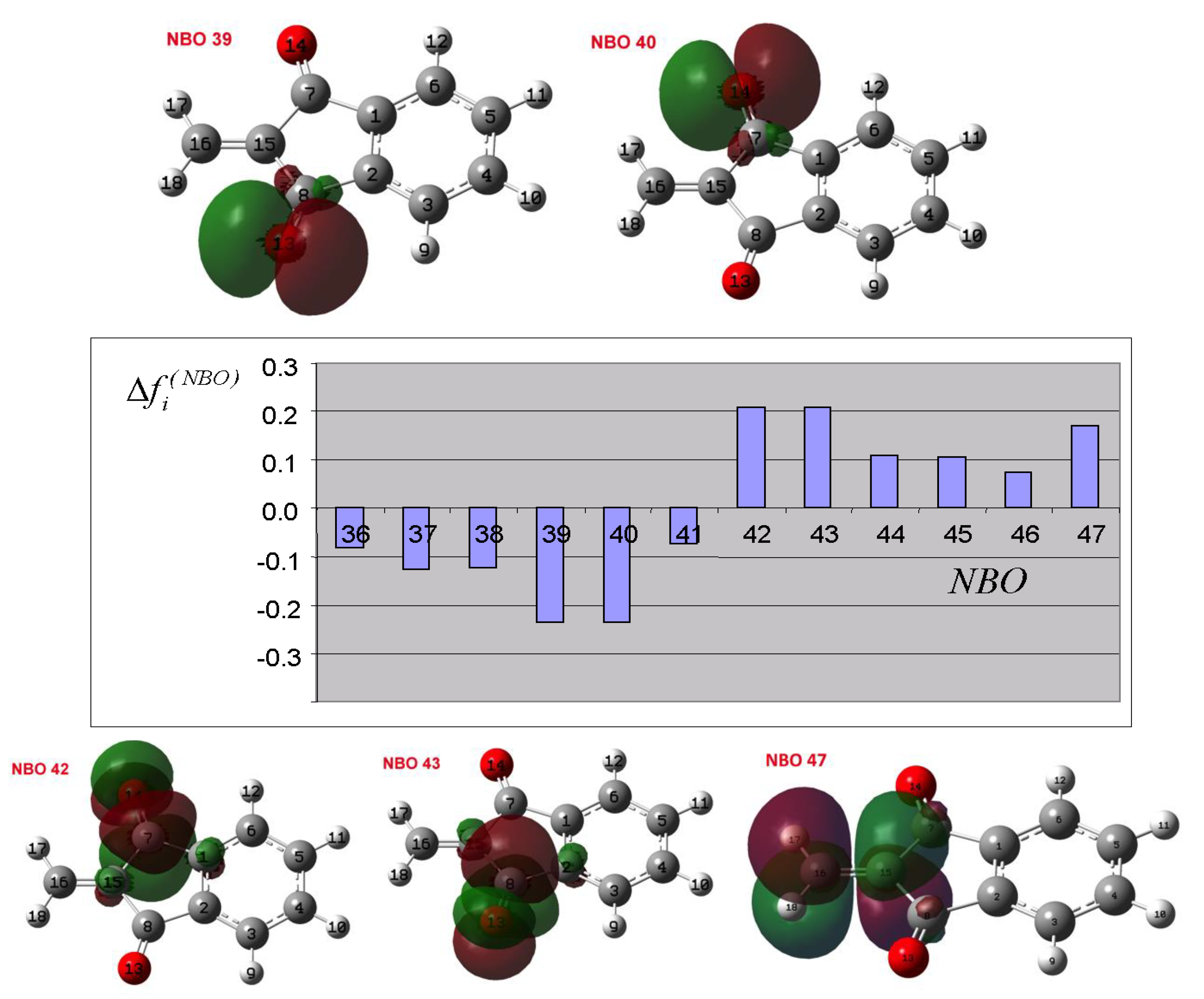
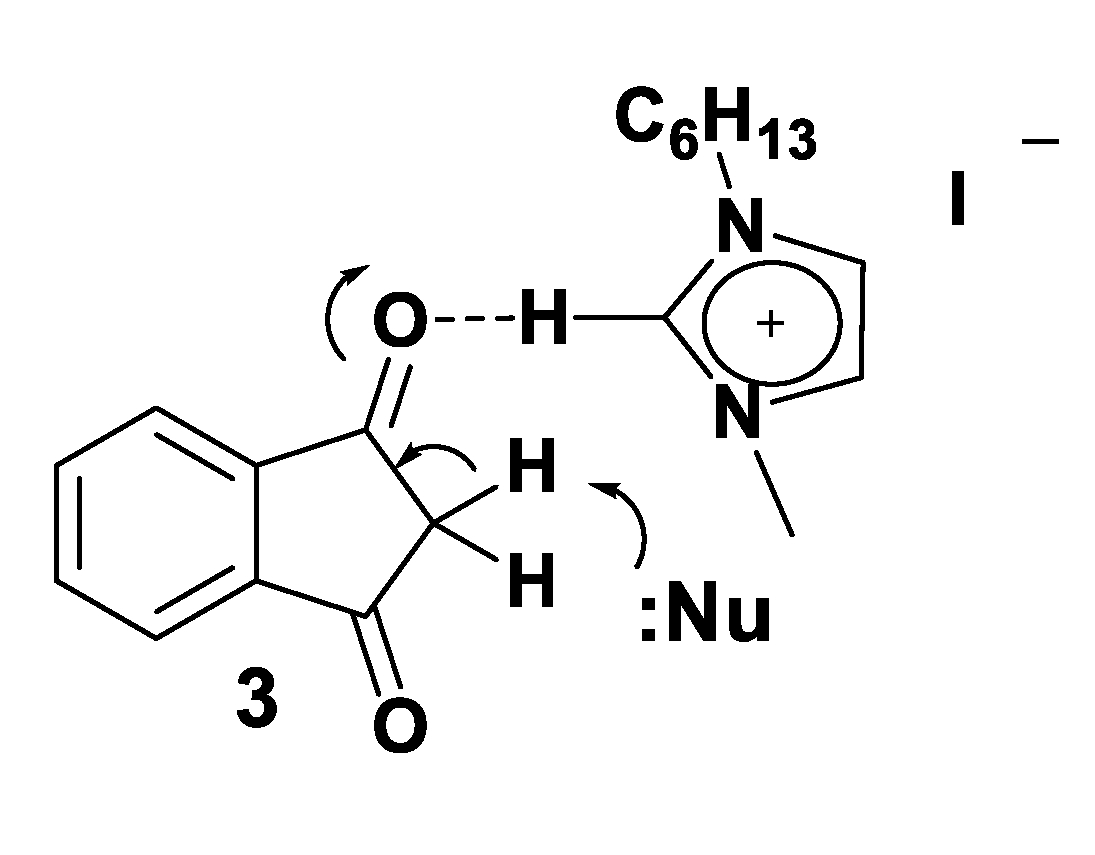
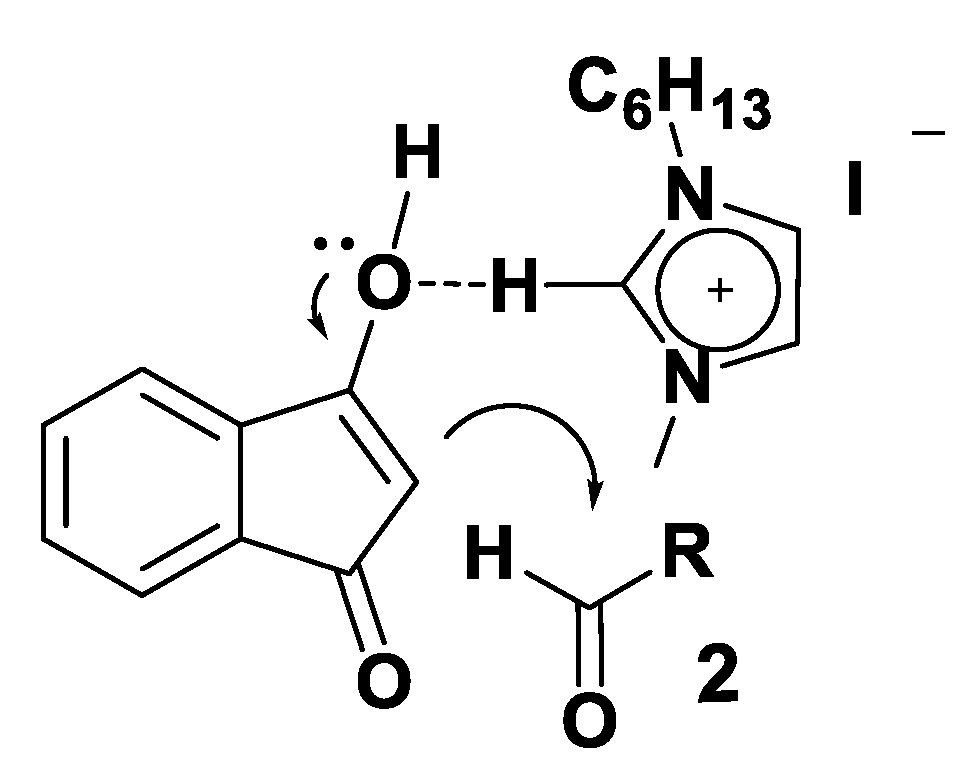
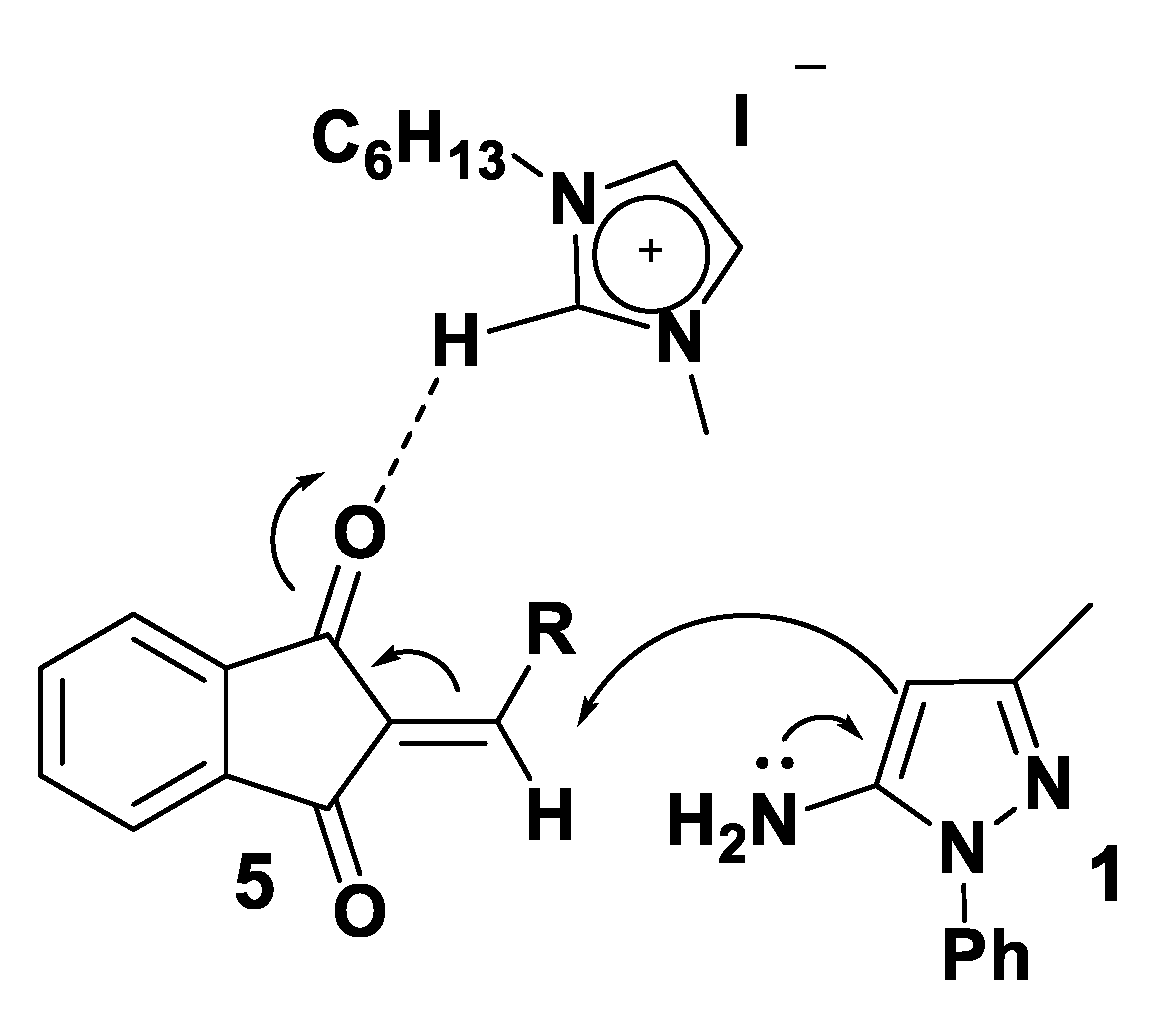
3.4. Theoretical Analysis of the Third System
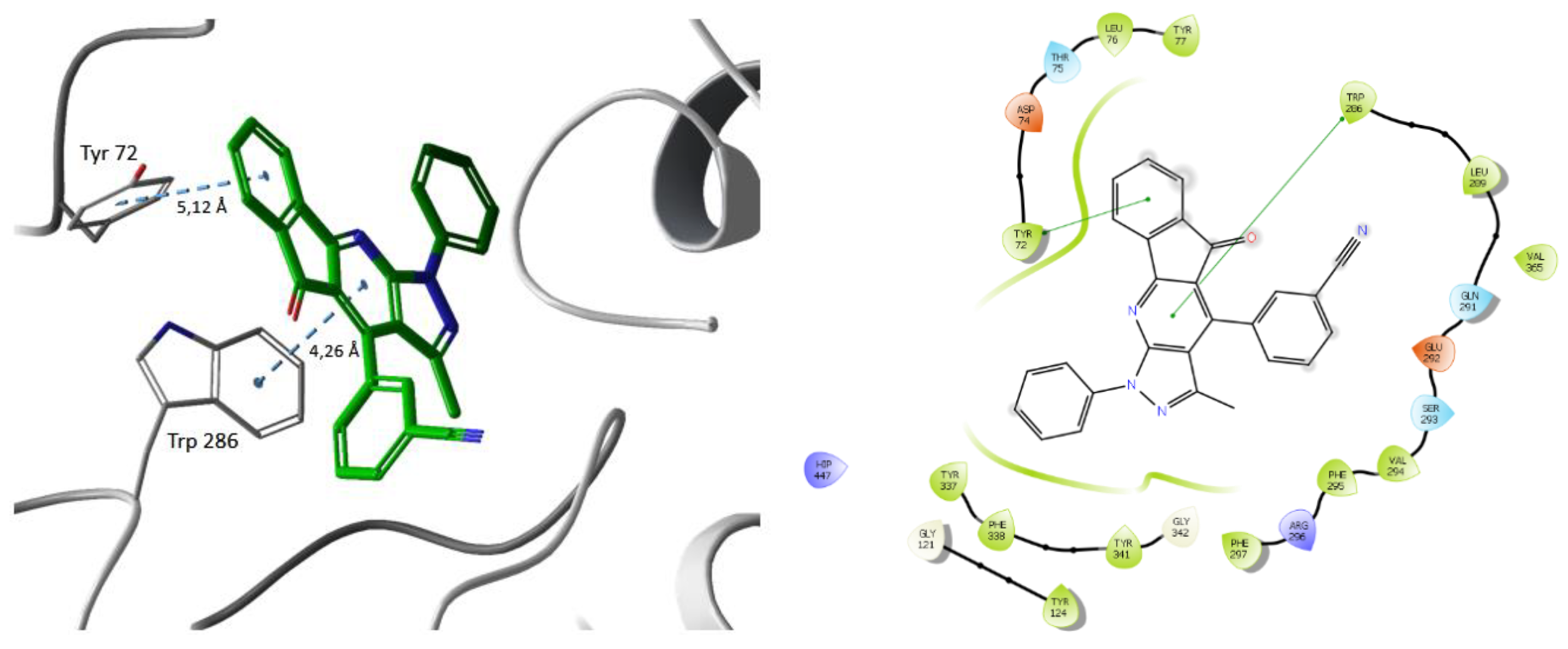
4. Biological Activities
5. Materials and Methods
5.1. Chemicals and Apparatus
5.2. Procedure for the Synthesis of 1-Hexyl-3-Methyl-Imidazolium Iodide [HMIM]I
5.3. General Procedure for the Synthesis of Indeno[2,1-e]Pyrazolo[5,4-b]Pyridines
6. Computational Details
7. Biological Evaluation
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Addla, D.; Sridhar, B.; Devi, A.; Kantevari, S. Design, synthesis and antimicrobial evaluation of novel 1-benzyl 2-butyl-4-chloroimidazole embodied 4-azafluorenones via molecular hybridization approach. Bioorg. Med. Chem. Lett. 2012, 22, 7475–7480. [Google Scholar] [CrossRef] [PubMed]
- Anand, D.; Yadav, P.K.; Patel, O.P.S.; Parmar, N.; Maurya, R.K.; Vishwakarma, P.; Raju, K.S.R.; Taneja, I.; Wahajuddin, M.; Kar, S.; et al. Antileishmanial Activity of Pyrazolopyridine Derivatives and Their Potential as an Adjunct Therapy with Miltefosine. J. Med. Chem. 2017, 60, 1041–1059. [Google Scholar] [CrossRef] [PubMed]
- Heintzelman, G.; Averill, K.; Dodd, J.; Demarest, K.; Tang, Y.; Jackson, P. Arylindenopyridines and Related Therapeutic and Prophylactic Methods. U.S. Patent No. 6,903,109, 7 July 2005. [Google Scholar]
- Heintzelman, G.R.; Averill, K.M.; Dodd, J.H.; Demarest, K.T.; Tang, Y.; Jackson, P.F. 5-oxo and 5-thio Derivatives of 5h-Indeno’1,2-Bipyridine with Adenosine a2a Receptor Binding and Phosphodiesterase Inhibiting Activity for the Treatment of Neurodegenerative Disorders and Inflammation Related Diseases. Int. Patent WO 03/088963, 30 October 2003. [Google Scholar]
- Safak, C.; Simsek, R.; Altas, Y.; Boydag, S.; Erol, K. 2-methyl-3-acetyl-4-aryl-5-oxo-1,4-dihydro-5H indeno(1,2-b) pyridine derivatives studies and their calcium antagonistic activities. Boll. Chim. Farm. 1997, 136, 665–669. [Google Scholar] [PubMed]
- Ashton, M.J.; Cook, D.C.; Fenton, G.; Karlsson, J.-A.; Palfreyman, M.N.; Raeburn, D.; Ratcliffe, A.J.; Souness, J.E.; Thurairatnam, S.; Vicker, N. Selective Type IV Phosphodiesterase Inhibitors as Antiasthmatic Agents. The Syntheses and Biological Activities of 3-(Cyclopentyloxy)-4-methoxybenzamides and Analogs. J. Med. Chem. 1994, 37, 1696–1703. [Google Scholar] [CrossRef]
- Manpadi, M.; Uglinskii, P.Y.; Rastogi, S.K.; Cotter, K.M.; Wong, Y.-S.C.; Anderson, L.A.; Ortega, A.J.; Van slambrouck, S.; Steelant, W.F.A.; Rogelj, S.; et al. Three-component synthesis and anticancer evaluation of polycyclic indenopyridines lead to the discovery of a novel indenoheterocycle with potent apoptosis inducing properties. Org. Biomol. Chem. 2007, 5, 3865. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, S.; Manam, P.; Chinworrungsee, M.; Isarankura-Na-Ayudhya, C.; Ruchirawat, S.; Prachayasittikul, V. Bioactive Azafluorenone Alkaloids from Polyalthia debilis (Pierre) Finet & Gagnep. Molecules 2009, 14, 4414–4424. [Google Scholar] [PubMed]
- Wu, Y.-C.; Duh, C.-Y.; Wang, S.-K.; Chen, K.-S.; Yang, T.-H. Two New Natural Azafluorene Alkaloids and a Cytotoxic Aporphine Alkaloid from Polyalthia longifolia. J. Nat. Prod. 1990, 53, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Naaz, H.; Singh, S.; Pandey, V.P.; Singh, P.; Dwivedi, U.N. Anti-cholinergic alkaloids as potential therapeutic agents for Alzheimer’s disease: An in silico approach. Indian J. Biochem. Biophys. 2013, 50, 120–125. [Google Scholar] [PubMed]
- Marquise, N.; Harford, P.J.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Gagez, A.-L.; Sablé, S.; Picot, L.; Thiéry, V.; Wheatley, A.E.H.; et al. Synthesis of azafluorenones and related compounds using deprotocupration–aroylation followed by intramolecular direct arylation. Tetrahedron 2013, 69, 10123–10133. [Google Scholar] [CrossRef]
- Utsugi, T.; Aoyagi, K.; Asao, T.; Okazaki, S.; Aoyagi, Y.; Sano, M.; Wierzba, K.; Yamada, Y. Antitumor activity of a novel quinoline derivative, TAS-103, with inhibitory effects on topoisomerases I and II. Jpn. J. Cancer Res. Gann 1997, 88, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Petrova, O.N.; Lipson, V.V.; Zamigailo, L.L.; Shirobokova, M.G.; Musatov, V.I.; Baumer, V.N.; Sofronov, D.S. Synthesis and chemical properties of 4-aroyl-3-methyl-4,10-dihydroindeno[1,2-b]pyrazolo-[4,3-e]pyridin-5-ones. Russ. J. Org. Chem. 2015, 51, 1597–1605. [Google Scholar] [CrossRef]
- Nikpassand, M.; Mamaghani, M.; Shirini, F.; Tabatabaeian, K. A convenient ultrasound-promoted regioselective synthesis of fused polycyclic 4-aryl-3-methyl-4,7-dihydro-1H-pyrazolo[3,4-b]pyridines. Ultrason. Sonochem. 2010, 17, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Zhang, Y.; Tu, S.-J.; Zhou, D.-X.; Li, C.-M.; Shao, Q.-Q.; Cao, L.-J. A Green Approach to the Synthesis of Biologically Important Indeno[2,1- e ]pyrazolo[5,4- b ]pyridines via Microwave-assisted Multi-component Reactions in Water. Chin. J. Chem. 2008, 26, 1262–1266. [Google Scholar] [CrossRef]
- Khurana, J.M.; Chaudhary, A.; Nand, B.; Lumb, A. Aqua mediated indium(III) chloride catalyzed synthesis of fused pyrimidines and pyrazoles. Tetrahedron Lett. 2012, 53, 3018–3022. [Google Scholar] [CrossRef]
- Quiroga, J.; Cobo, D.; Insuasty, B.; Abonía, R.; Cruz, S.; Nogueras, M.; Cobo, J. Regioselective three-component synthesis of novel indeno[1,2- b ]-pyrazolo[4,3- e ]pyridines-fused derivatives of 4-azafluorenone alkaloid. J. Heterocycl. Chem. 2008, 45, 155–159. [Google Scholar] [CrossRef]
- Shi, C.-L.; Shi, D.-Q.; Kim, S.H.; Huang, Z.-B.; Ji, S.-J.; Ji, M. A novel and efficient one-pot synthesis of furo[3′,4′:5,6]pyrido[2,3-c]pyrazole derivatives using organocatalysts. Tetrahedron 2008, 64, 2425–2432. [Google Scholar] [CrossRef]
- Shi, D.-Q.; Shi, J.-W.; Yao, H. Three-Component One-Pot Synthesis of Indeno[2′,1′:5,6]Pyrido[2,3- d ]Pyrazole Derivatives in Aqueous Media. J. Chin. Chem. Soc. 2009, 56, 504–509. [Google Scholar] [CrossRef]
- Shi, D.-Q.; Yang, F.; Ni, S.-N. A facile synthesis of furo[3,4- e ]pyrazolo[3,4- b ]pyridine-5(7 H)-one derivatives via three-component reaction in ionic liquid without any catalyst. J. Heterocycl. Chem. 2009, 46, 469–476. [Google Scholar] [CrossRef]
- Simon, M.-O.; Li, C.-J. Green chemistry oriented organic synthesis in water. Chem. Soc. Rev. 2012, 41, 1415–1427. [Google Scholar] [CrossRef]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and Biology of Multicomponent Reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef]
- Rotstein, B.H.; Zaretsky, S.; Rai, V.; Yudin, A.K. Small Heterocycles in Multicomponent Reactions. Chem. Rev. 2014, 114, 8323–8359. [Google Scholar] [CrossRef] [PubMed]
- Brauch, S.; van Berkel, S.S.; Westermann, B. Higher-order multicomponent reactions: Beyond four reactants. Chem. Soc. Rev. 2013, 42, 4948. [Google Scholar] [CrossRef] [PubMed]
- de Graaff, C.; Ruijter, E.; Orru, R.V.A. Recent developments in asymmetric multicomponent reactions. Chem. Soc. Rev. 2012, 41, 3969. [Google Scholar] [CrossRef] [PubMed]
- Cintas, P.; Luche, J.-L. Green chemistry. Green Chem. 1999, 1, 115–125. [Google Scholar] [CrossRef]
- Constable, D.J.C.; Curzons, A.D.; Cunningham, V.L. Metrics to ‘green’ chemistry—Which are the best? Green Chem. 2002, 4, 521–527. [Google Scholar] [CrossRef]
- Poliakoff, M. Green Chemistry: Science and Politics of Change. Science 2002, 297, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Egorova, K.S.; Gordeev, E.G.; Ananikov, V.P. Biological Activity of Ionic Liquids and Their Application in Pharmaceutics and Medicine. Chem. Rev. 2017, 117, 7132–7189. [Google Scholar] [CrossRef]
- Xu, L.-W.; Li, J.-W.; Zhou, S.-L.; Xia, C.-G. A green, ionic liquid and quaternary ammonium salt-catalyzed aza-Michael reaction of α,β-ethylenic compounds with amines in water. New. J. Chem. 2004, 28, 183–184. [Google Scholar] [CrossRef]
- Howarth, J. An exploration of the catalytic Sakurai reaction in the moisture stable ionic liquids [bmim]PF6 and [bmim]BF4. J. Mol. Catal. Chem. 2004, 214, 143–146. [Google Scholar] [CrossRef]
- Palimkar, S.S.; Siddiqui, S.A.; Daniel, T.; Lahoti, R.J.; Srinivasan, K.V. Ionic Liquid-Promoted Regiospecific Friedlander Annulation: Novel Synthesis of Quinolines and Fused Polycyclic Quinolines. J. Org. Chem. 2003, 68, 9371–9378. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Venkataiah, B.; Dong, G. The use of potassium alkynyltrifluoroborates in Mannich reactions. Tetrahedron Lett. 2004, 45, 729–731. [Google Scholar] [CrossRef]
- Carmichael, A.J.; Earle, M.J.; Holbrey, J.D.; McCormac, P.B.; Seddon, K.R. The Heck Reaction in Ionic Liquids: A Multiphasic Catalyst System. Org. Lett. 1999, 1, 997–1000. [Google Scholar] [CrossRef]
- Mathews, C.J.; Smith, P.J.; Welton, T. Palladium catalysed Suzuki cross-coupling reactions in ambient temperature ionic liquids. Chem. Commun. 2000, 14, 1249–1250. [Google Scholar] [CrossRef]
- Schreiner, P.R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289. [Google Scholar] [CrossRef] [PubMed]
- Rosa, J.N.; Afonso, C.A.M.; Santos, A.G. Ionic liquids as a recyclable reaction medium for the Baylis–Hillman reaction. Tetrahedron 2001, 57, 4189–4193. [Google Scholar] [CrossRef]
- Gholap, A.R.; Venkatesan, K.; Daniel, T.; Lahoti, R.J.; Srinivasan, K.V. Ionic liquid promoted novel and efficient one pot synthesis of 3,4-dihydropyrimidin-2-(1H)-ones at ambient temperature under ultrasound irradiation. Green Chem. 2004, 6, 147. [Google Scholar] [CrossRef]
- Shelke, K.F.; Madje, B.R.; Sapkal, S.B.; Shingate, B.B.; Shingare, M.S. An efficient ionic liquid promoted Knoevenagel condensation of 4-oxo-4 H -benzopyran-3-carbaldehyde with Meldrum’s acid. Green Chem. Lett. Rev. 2009, 2, 3–7. [Google Scholar] [CrossRef][Green Version]
- Karmee, S.K.; Hanefeld, U. Ionic Liquid Catalysed Synthesis of β-Hydroxy Ketones. ChemSusChem 2011, 4, 1118–1123. [Google Scholar] [CrossRef]
- Peng, J.; Deng, Y. Cycloaddition of carbon dioxide to propylene oxide catalyzed by ionic liquids. New J. Chem. 2001, 25, 639–641. [Google Scholar] [CrossRef]
- Carmichael, A.J.; Haddleton, D.M.; Bon, S.A.F.; Seddon, K.R. Copper(i) mediated living radical polymerisation in an ionic liquid. Chem. Commun. 2000, 1, 1237–1238. [Google Scholar] [CrossRef]
- Khan, F.A.; Dash, J.; Satapathy, R.; Upadhyay, S.K. Hydrotalcite catalysis in ionic liquid medium: A recyclable reaction system for heterogeneous Knoevenagel and nitroaldol condensation. Tetrahedron Lett. 2004, 45, 3055–3058. [Google Scholar] [CrossRef]
- Wong, H.; Pink, C.J.; Ferreira, F.C.; Livingston, A.G. Recovery and reuse of ionic liquids and palladium catalyst for Suzuki reactions using organic solvent nanofiltration. Green Chem. 2006, 8, 373. [Google Scholar] [CrossRef]
- Hunt, P.A.; Kirchner, B.; Welton, T. Characterising the Electronic Structure of Ionic Liquids: An Examination of the 1-Butyl-3-Methylimidazolium Chloride Ion Pair. Chem. Eur. J. 2006, 12, 6762–6775. [Google Scholar] [CrossRef] [PubMed]
- Weingärtner, H. Understanding Ionic Liquids at the Molecular Level: Facts, Problems, and Controversies. Angew. Chem. Int. Ed. 2008, 47, 654–670. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Márquez, J. Introducing new reactivity descriptors: “Bond reactivity indices.” Comparison of the new definitions and atomic reactivity indices. J. Chem. Phys. 2016, 145, 194105. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Márquez, J.; Zorrilla, D.; García, V.; Fernández, M. Introducing a new bond reactivity index: Philicities for natural bond orbitals. J. Mol. Model. 2018, 24, 25. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods: Natural bond orbital methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Ritchie, J.P.; Bachrach, S.M. Some methods and applications of electron density distribution analysis. J. Comput. Chem. 1987, 8, 499–509. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept®): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef]
- Gutiérrez, M.; Theoduloz, C.; Rodríguez, J.; Lolas, M.; Schmeda-Hirschmann, G. Bioactive Metabolites from the Fungus Nectria galligena, the Main Apple Canker Agent in Chile. J. Agric. Food Chem. 2005, 53, 7701–7708. [Google Scholar] [CrossRef] [PubMed]
- Polo, E.; Ferrer-Pertuz, K.; Trilleras, J.; Quiroga, J.; Gutiérrez, M. Microwave-assisted one-pot synthesis in water of carbonylpyrazolo[3,4-b]pyridine derivatives catalyzed by InCl 3 and sonochemical assisted condensation with aldehydes to obtain new chalcone derivatives containing the pyrazolopyridinic moiety. RSC Adv. 2017, 7, 50044–50055. [Google Scholar] [CrossRef]
- Gupta, A.K.; Vaishanv, N.K.; Kant, R.; Mohanan, K. Rapid and selective synthesis of spiropyrazolines and pyrazolylphthalides employing Seyferth–Gilbert reagent. Org. Biomol. Chem. 2017, 15, 6411–6415. [Google Scholar] [CrossRef]
- Sigalov, M.; Shainyan, B.; Chipanina, N.; Ushakov, I.; Shulunova, A. Intra- and intermolecular N—H⋯O hydrogen bonds in pyrrolyl derivatives of indane-1,3-dione—Experimental and theoretical study: HYDROGEN BONDS IN PYRROLYL DERIVATIVES OF 1,3-INDANDIONE. J. Phys. Org. Chem. 2009, 22, 1178–1187. [Google Scholar] [CrossRef]
- Ramachary, D.B.; Anebouselvy, K.; Chowdari, N.S.; Barbas, C.F. Direct Organocatalytic Asymmetric Heterodomino Reactions: The Knoevenagel/Diels−Alder/Epimerization Sequence for the Highly Diastereoselective Synthesis of Symmetrical and Nonsymmetrical Synthons of Benzoannelated Centropolyquinanes. J. Org. Chem. 2004, 69, 5838–5849. [Google Scholar] [CrossRef]
- El-Gaby, M.S.A. Syntheses of Hitherto Unknown Thiazole, Ylidene and Pyridinethione Derivatives Having a Piperidin-1-yl Moiety and Their Use as Antimicrobial Agents. J. Chin. Chem. Soc. 2004, 51, 125–134. [Google Scholar] [CrossRef]
- Qi, J.; Zheng, J.; Cui, S. Facile synthesis of carbo- and heterocycles via Fe(iii)-catalyzed alkene hydrofunctionalization. Org. Chem. Front. 2018, 5, 222–225. [Google Scholar] [CrossRef]
- Zhao, B.; Liang, H.-W.; Yang, J.; Yang, Z.; Wei, Y. Copper-Catalyzed Intermolecular Cyclization between Oximes and Alkenes: A Facile Access to Spiropyrrolines. ACS Catal. 2017, 7, 5612–5617. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Montgomery, J.; Vreven, T.; Kudin, K.; Burant, J.; et al. Gaussian 03, Revision C.02; Gaussian. Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Sánchez-Márquez, J.; Zorrilla, D.; Sánchez-Coronilla, A.; de los Santos, D.M.; Navas, J.; Fernández-Lorenzo, C.; Alcántara, R.; Martín-Calleja, J. Introducing “UCA-FUKUI” software: Reactivity-index calculations. J. Mol. Model. 2014, 20, 2492. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J.; Eppinnett, K.; Hovell, W.L.; Gilliland, R. GaussView, Version 3.09; Semichem, Inc.: Shawnee Mission, KS, USA, 2003. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
|---|
| Previous Work |
| Water, MW, 100 °C, 5–9 min (10 examples; 95%–97%) [15] |
| Water, InCl3 (20% mol), reflux, 45–60 min (3 examples; 87–93%) [16] |
| DMF, Et3N (cat) reflux 6-8 h (9 examples; 47%–69%) [17] |
| L-proline (10% mol), EtOH 80 °C, 0.5–2 h (17 examples; 85%–97%) [18] |
| SDS, Water, 90 °C, 4–13 h (14 examples; 86%–98%) [19] |
| [bmim]Br, 95 °C, 2–5 h (17 examples; 79%–96%) [20] |
| This work; [HMIM]I, H2O), 10–20 min (8 examples; 27%–95%) |
| Entry | Conditions | Yield (%) |
|---|---|---|
| 1 | EtOH, Sonication, 20 min | 60 |
| 2 | H2O, Sonication, 20 min | - |
| 3 | AcOH, , Sonication, 1,5 h | 65 |
| 4 | [HMIM]I (10%), Sonication, 10 min | 95 |
| 5 | [HMIM]I (15%), Sonication, 10 min | 92 |
| 6 | [HMIM]I (20%), Sonication, 10 min | 92 |
| 7 | InCl3, (10%), Water, Sonication 30min | 54 |
| 8 | InCl3, (20%), Water, Sonication 30min | 68 |
 |
 |
| Cycle | Time (min) | Product | Conv (%) |
|---|---|---|---|
| 1 | 10 | 4a | 95 |
| 2 | 10 | 4a | 72 |
| 3 | 10 | 4a | 41 |
| Property | Optimal Range | 4a | 4b | 4c | 4d | 4e | 4f | 4g | 4h | 4i | 4j | 4k |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MW | <500 | 388 | 412 | 412 | 438 | 391 | 431 | 311 | 377 | 416 | 414 | 266 |
| Log P | <5 | 4.88 | 5.37 | 5.35 | 5.77 | 4.00 | 5.53 | 3.89 | 4.76 | 6.08 | 4.69 | 1.87 |
| H Bond donors | <5 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| H Bon Acceptors | <10 | 5 | 5 | 5 | 5 | 6 | 6 | 4 | 5 | 4 | 6 | 5 |
| Rotable Bonds | <5 | 2 | 2 | 2 | 2 | 2 | 3 | 1 | 2 | 3 | 6 | 2 |
| TPSA | <89 | 60.68 | 71.58 | 71.58 | 60.68 | 65.61 | 85.09 | 47.79 | 60.93 | 33.96 | 60.96 | 71.54 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polo, E.; Arce-Parada, V.; López-Cortés, X.A.; Sánchez-Márquez, J.; Morales-Bayuelo, A.; Forero-Doria, O.; Gutiérrez, M. Synthesis of Pyrazolo-Fused 4-Azafluorenones in an Ionic Liquid. Mechanistic Insights by Joint Studies Using DFT Analysis and Mass Spectrometry. Catalysts 2019, 9, 820. https://doi.org/10.3390/catal9100820
Polo E, Arce-Parada V, López-Cortés XA, Sánchez-Márquez J, Morales-Bayuelo A, Forero-Doria O, Gutiérrez M. Synthesis of Pyrazolo-Fused 4-Azafluorenones in an Ionic Liquid. Mechanistic Insights by Joint Studies Using DFT Analysis and Mass Spectrometry. Catalysts. 2019; 9(10):820. https://doi.org/10.3390/catal9100820
Chicago/Turabian StylePolo, Efraín, Valentina Arce-Parada, Xaviera A. López-Cortés, Jesús Sánchez-Márquez, Alejandro Morales-Bayuelo, Oscar Forero-Doria, and Margarita Gutiérrez. 2019. "Synthesis of Pyrazolo-Fused 4-Azafluorenones in an Ionic Liquid. Mechanistic Insights by Joint Studies Using DFT Analysis and Mass Spectrometry" Catalysts 9, no. 10: 820. https://doi.org/10.3390/catal9100820
APA StylePolo, E., Arce-Parada, V., López-Cortés, X. A., Sánchez-Márquez, J., Morales-Bayuelo, A., Forero-Doria, O., & Gutiérrez, M. (2019). Synthesis of Pyrazolo-Fused 4-Azafluorenones in an Ionic Liquid. Mechanistic Insights by Joint Studies Using DFT Analysis and Mass Spectrometry. Catalysts, 9(10), 820. https://doi.org/10.3390/catal9100820