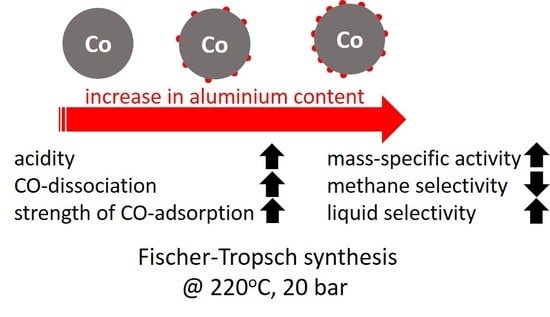
2. Results
Cobalt oxide (Co
3O
4) with an average crystallite size of 14 nm was prepared by the calcination of a cobalt carbonate precursor at 300 °C [
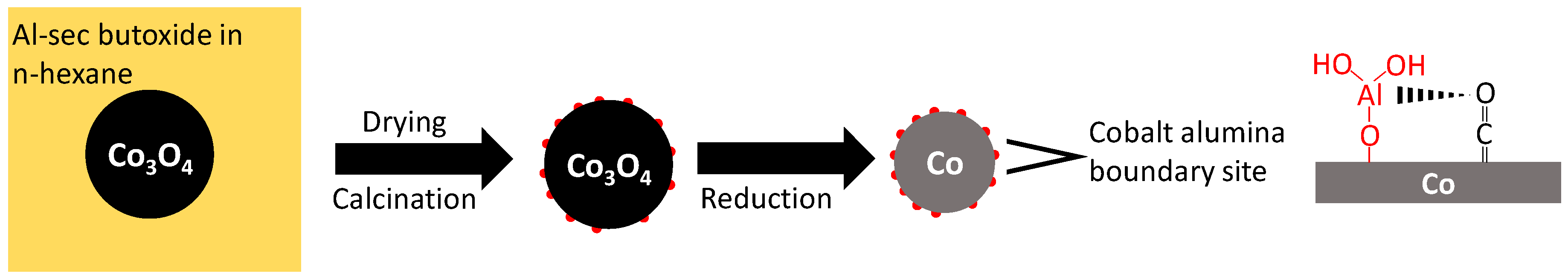
18], which was impregnated with aluminum sec-butoxide in dry n-hexane to achieve a weight loading of 0.1, 0.5 or 2.5 wt.% Al [
18]. The formation of a separate, alumina phase could not be observed using X-ray diffraction (XRD), although analysis of transmission electron microscopy (TEM) images showed the presence of small alumina islands (<1 nm) on the surface of Co
3O
4 [
18].
Metallic cobalt is the catalytic active phase in the Fischer–Tropsch synthesis [
4], and the precursor needs to be reduced prior to the FT synthesis. Temperature programmed reduction profiles of the calcined material showed a significant broadening of the reduction peak upon modification with aluminum, which was accompanied by a shift of the reduction peaks to higher reduction temperatures [
18]. This was ascribed to a blockage of defect sites by alumina, thereby hindering hydrogen activation and the nucleation of reduced cobalt phases. The appearance of reduction processes taking place at temperatures in excess of 400 °C were ascribed to the reduction of cobalt ions in proximity to alumina in the catalyst. These materials were reduced prior to the Fischer–Tropsch synthesis isothermally at 350 °C for 10 h. The degree of reduction (DoR) decreased gradually from 96% to 86% upon increasing alumina loading from 0 to 2.5 wt.% (see
Table 1). This further implies that the modification of alumina impedes the reduction of Co
3O
4 as observed in the profiles obtained from temperature-programmed reduction (TPR) [
18].
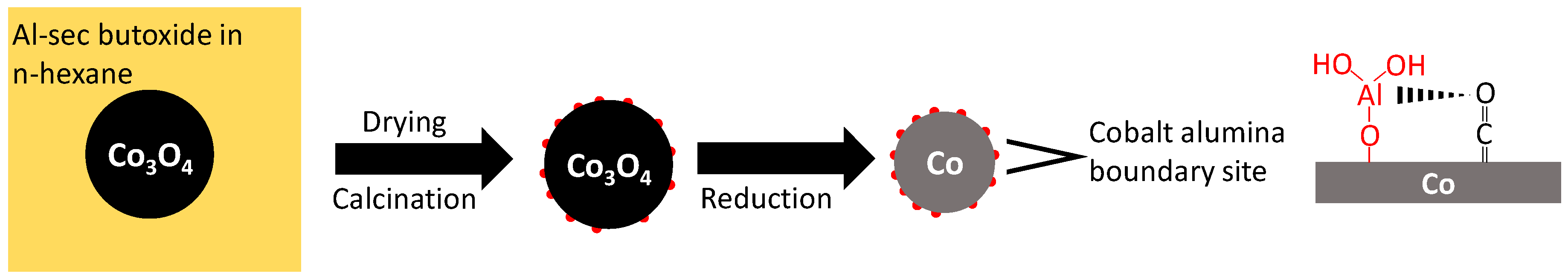
The catalysts studied in this work were essentially bulk cobalt catalysts, and these materials were expected to sinter extensively during catalyst activation, despite the low reduction temperature (the chosen reduction temperature (350 °C) is above the Hüttig temperature of metallic cobalt (253 °C), implying the significant mobility of cobalt atoms at defect sites [
20], which may lead to sintering if cobalt particles are in contact with each other). The metal dispersion after reduction as determined using H
2 chemisorption varied between 0.23% (particle size approximately 412 nm) for Al0 and 3.30% (particle size approximately 29 nm) for Al2.5, showing severe sintering for specifically the sample containing no aluminum (Al0). Interestingly, the introduction of even a small amount of aluminum to Co
3O
4 can drastically reduce the extent of catalyst sintering, whilst still achieving a high degree of reduction. The alumina islands on the surface of cobalt oxide [
18] may act here as a spacer preventing direct contact between cobalt particles and thereby reducing the extent of sintering. This also indicates that contact between alumina and the cobalt phase remains intact throughout the reduction of Co
3O
4 to metallic cobalt.
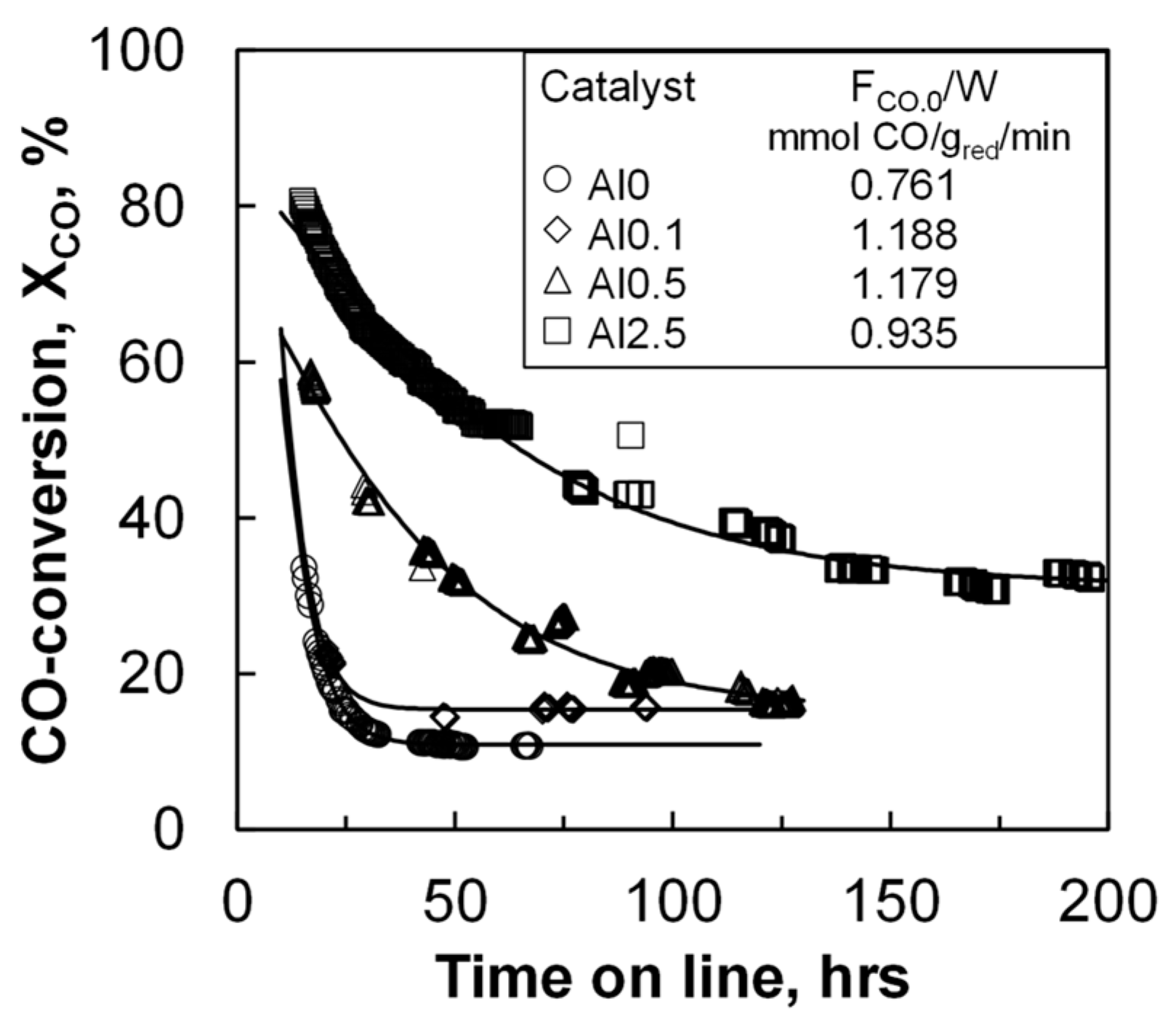
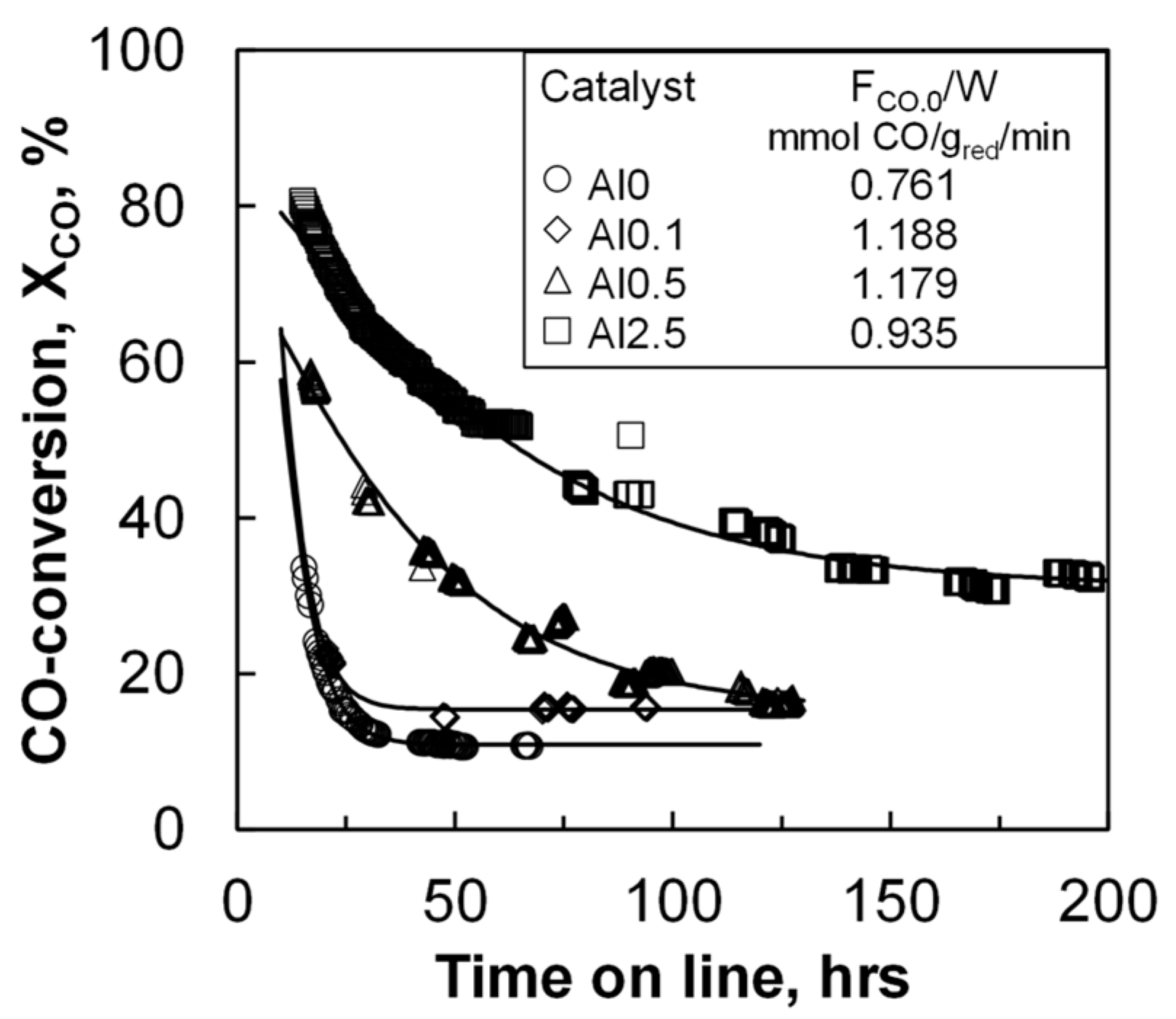
The catalyst activity of the materials was evaluated in a slurry reactor at a pressure of 20 bar and at 220 °C. The initial space velocity was so that the conversion as a function of time could be followed. The CO conversion for all catalysts decreases as a function of time on stream (TOS) during the start-up of the reaction (see
Figure 1) due to loss in the activity of the catalyst. Catalyst deactivation has been reported to take place in two distinct stages [
21]. The first stage of deactivation is typically ascribed to catalyst sintering. This is often followed by a second stage, in which slower deactivation processes take place, such as catalyst poisoning, catalyst re-oxidation, or build-up of polymeric carbon deposits. The conversion of CO drops rather quickly as a function of time with the samples containing none or a very small amount of aluminum. A steady conversion level was achieved with these samples after less than 50 h on-line. The samples with more aluminum require longer times to stabilize.
The initial activity was evaluated after 16 h on-line (see
Table 2), which corresponds to four times the residence time. The initial activity increased with increasing aluminum loading from 0.24 to 0.70 mmol
CO/g
red. cat/min, corresponding to a cobalt time yield (CTY) of 2.3 × 10
−4 s
−1 and 6.9 × 10
−4 s
−1 for the samples Al0 and Al2.5, respectively. However, the initial turnover frequency (TOF) was found to decrease with increasing alumina loading from 0.21 s
−1 to 0.04 s
−1 for catalyst Al0 and Al2.5, respectively. The obtained turnover frequencies for the sample without aluminum (Al0) resembles the turnover frequency obtained over model catalyst supported on alumina with minimal metal support interactions [
6], whereas the obtained turnover frequency for the sample Al2.5 resembles the turnover frequency obtained with a typical impregnated cobalt on alumina catalyst [
22]. The decrease in the turnover frequency with increasing aluminum content despite the increase in the rate of reaction per unit mass is a consequence of the strong effect of the increasing aluminum content on the cobalt dispersion (see
Table 1). The activity after ca. 95 h on stream as a function of the aluminum content shows the same trend.
The CO-conversion as a function of time on-line was modeled using the rate expression proposed by Botes et al. [
23] (see Equation (1)). The loss in activity with time on-line due to deactivation, a, was modeled based on a first-order generalized power-law expression (GPLE) [
21,
24], taking into account a finite, residual activity (see Equation (2)):
where
FCO,inlet is the molar inlet flow of CO, W is the mass of reduced cobalt,
XCO is the conversion of CO,
A0 is the rate constant per unit mass of reduced catalyst for the fresh catalyst,
B is the kinetic inhibition factor,
a is the relative activity, and
a∞ is the final residual activity relative to the initial activity. The partial pressures of hydrogen and carbon monoxide within the reactor are functions of the conversion of CO as well. The conversion of CO as a function of time on-line was fitted to this set of equations, and the obtained kinetic parameters are shown in
Table 2.
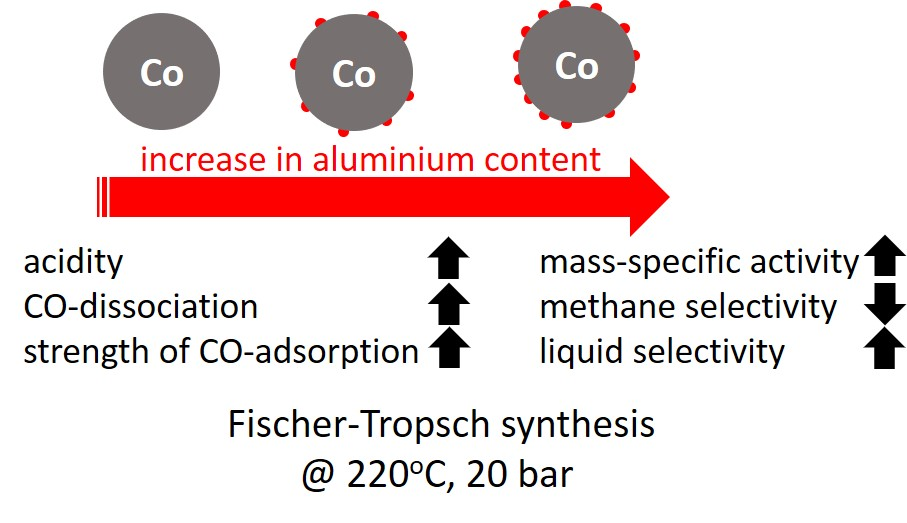
The mass-specific, kinetic constant
A0 increases quite strongly with increasing aluminum content in the catalyst. Correcting the kinetic constant for the change in metal dispersion
A0‘ (see
Table 2) shows an increase in the initial kinetic constant with increasing aluminum loading, implying that alumina on the surface facilitates the activity of the catalyst beyond increasing the dispersion (sample Al0.1 seems to be the exception here, possibly due to the difficulty in adequately determining the initial activity). The kinetic inhibition factor,
B, also increases with increasing aluminum loading, implying that the inhibition of the rate of the Fischer–Tropsch synthesis by carbonaceous species on the surface becomes stronger upon increasing the aluminum loading. These observations are consistent with our DFT (density functional theory) calculations [
26], which indicate that alumina near the metallic cobalt surface (e.g., as a ligand) would facilitate CO dissociation by providing an alternative pathway on the dense Co(111) surface.
The rate constant for deactivation is ca. 10 times smaller for the samples containing 0.5 and 2.5 wt.% aluminum than the rate constants for deactivation for the samples containing none or a very small amount of aluminum, indicating that the presence of small alumina islands on cobalt [
18] retards the fast initial deactivation process. Furthermore, the final activity, a
∞, increases with increasing aluminum content. Aluminum modification was found to prevent sintering during reduction, as evident from the increased metal dispersion of the activated catalyst determined by H
2 chemisorption (see
Table 1). The modification of the cobalt surface with alumina may thus also retard and prevent further sintering during the Fischer–Tropsch synthesis.
The CO hydrogenation over cobalt-based catalysts results mainly in the formation of organic product compounds. Some CO2 is formed, but typically with a selectivity of less than 1–2% C. The origin of CO2 in cobalt-based Fischer–Tropsch synthesis has not been established beyond doubt and may be attributed to the water–gas shift reaction (i.e., with the co-generation of hydrogen), further reduction of the catalyst using CO as the reductant, carbon deposition via the Boudouard reaction, or as an alternative pathway to remove surface oxygen.
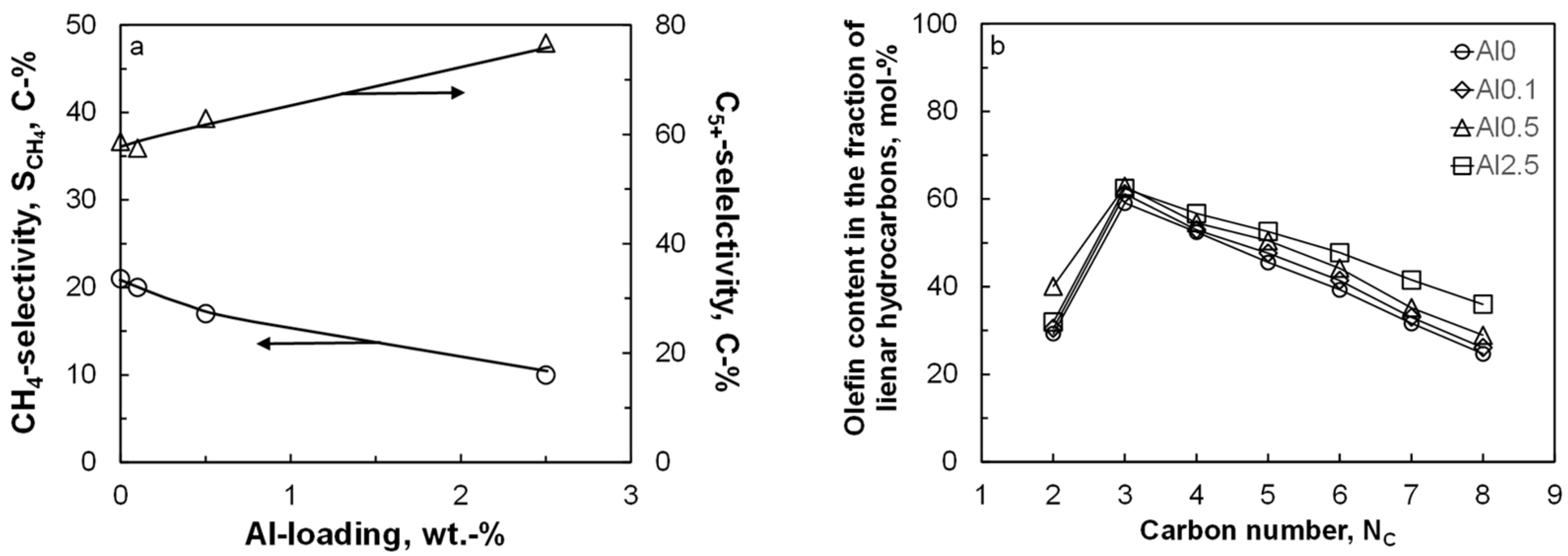
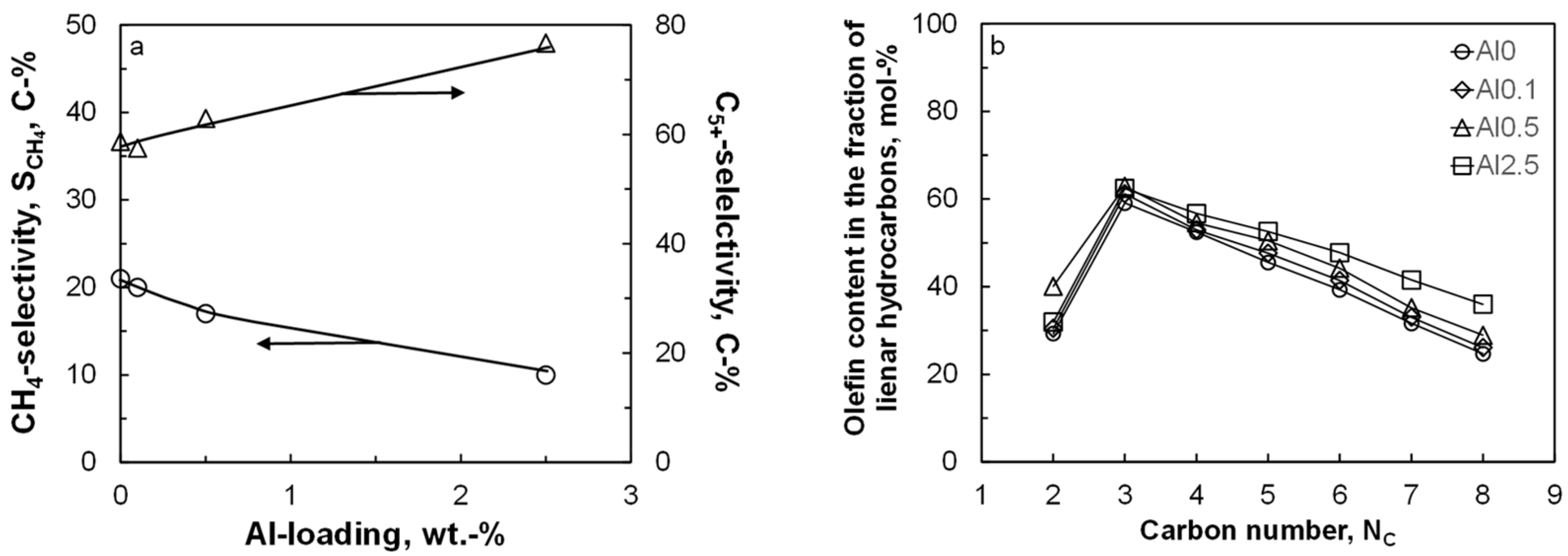
Figure 2 shows the selectivity for the formation of methane and for the formation of the desired liquid product, C
5+ as a function of the aluminum content in the catalyst after ca. 250 h on-line. The conversion level was kept constant at ca. 14.5 ± 2% by varying the space velocity. The methane selectivity and C
5+ selectivity show as expected, an opposite trend. The seesaw behavior for these product groups is expected, due to the normalization of the product selectivity to 100% and the relatively small amount of C
2–C
4 formed in cobalt-based Fischer–Tropsch synthesis [
27]. The methane selectivity compared at the same time on-line shows a clear decrease with increasing aluminum content. The observed change in the methane selectivity cannot be attributed to a particle size effect [
5,
6,
28] seeing the large metallic cobalt particles, which are obtained after reduction (see
Table 1). Hence, the change in product selectivity must be attributed to the presence of alumina islands on the cobalt surface. The methane selectivity is known to decrease with an increase in the ‘surface’ inventory of active carbon [
29,
30]. An increase in the coverage with surface carbon will decrease the availability of surface hydrogen [
31,
32]; this may (in a limited region [
33]) result in a decrease in the methane selectivity. Alumina in close proximity to the catalytically active, metallic cobalt surface may facilitate CO dissociation [
26], thus resulting in an increase in the coverage with surface carbon and reduced hydrogen availability on the surface. This could thus explain the observed decrease in the methane selectivity with increasing aluminum content. The seesaw relationship between the C
5+ selectivity and the methane selectivity implies an increase in the C
5+ selectivity with increasing carbon coverage on the surface.
Olefins are the major, primary product of the Fischer–Tropsch synthesis, even when cobalt is used as the catalytically active material [
27]. The primarily formed olefins can re-adsorb and undergo secondary reactions, such as hydrogenation, double-bond isomerization, and reincorporation in the chain growth scheme [
31].
Figure 2 shows the expected behavior for the olefin content in the fraction of linear hydrocarbons as a function of carbon number: a rather low olefin content in C
2 due to the high reactivity of ethene, and a decrease in the olefin content in the fraction of linear hydrocarbons with increasing carbon numbers from C
3 onwards due to the increased concentration of these compounds in the liquid phase [
34]. The olefin content in the fraction of linear hydrocarbons increases with increasing aluminum content (except for the olefin content in the fraction of C
2 hydrocarbons). This indicates a reduced extent of secondary hydrogenation of the primarily formed long-chain olefins in the Fischer–Tropsch synthesis possibly due to an increased coverage of the surface with carbonaceous species and thus lower hydrogen availability on the surface.
The changes in the initial kinetic parameters,
A0 and
B (see
Table 2), and the change in the selectivity due to the modification of the catalytically active material with aluminum are thought to be related to the facilitation of the CO dissociation and the resulting increase in the coverage of the surface with carbon. The ease of CO dissociation may be monitored by CO treatment of these materials, or by reducing Co
3O
4 in CO rather than H
2 [
35,
36]. CO activation may initially result in the formation of cobalt carbide [
36,
37,
38]. The reduction of Co
3O
4 using carbon monoxide has been reported to proceed sequentially [
36]:
Cobalt carbide is thermodynamically not stable at a low chemical potential of carbon (e.g., in the absence of CO), and will thus decompose, yielding metallic cobalt. In hydrogen, cobalt carbide decomposes rapidly at temperatures as low as 150–200 °C [
36,
38]. The decomposition of cobalt carbide is retarded in inert atmosphere, but it will decompose into metallic cobalt and graphitic carbon at temperatures between 300–350 °C [
38]:
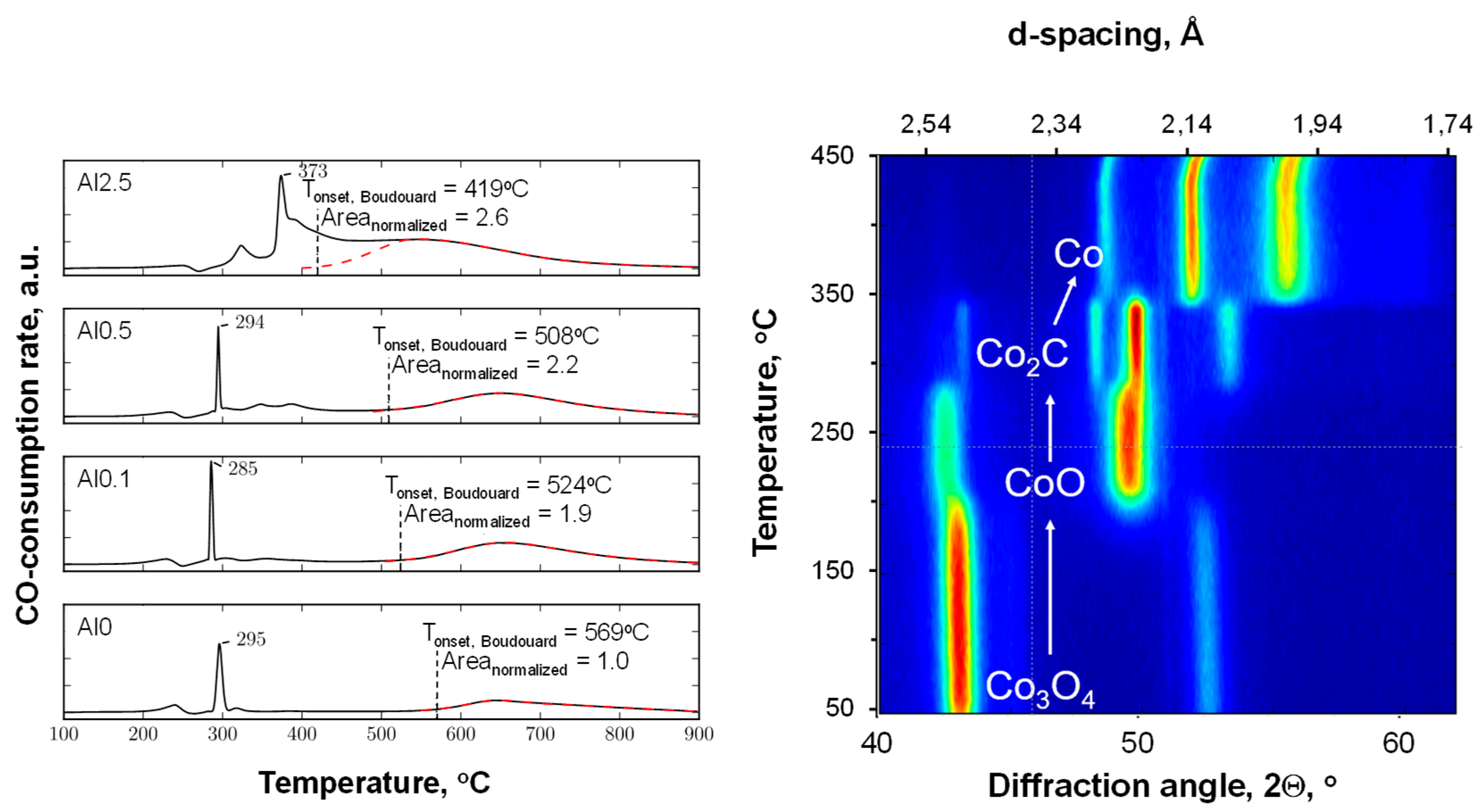
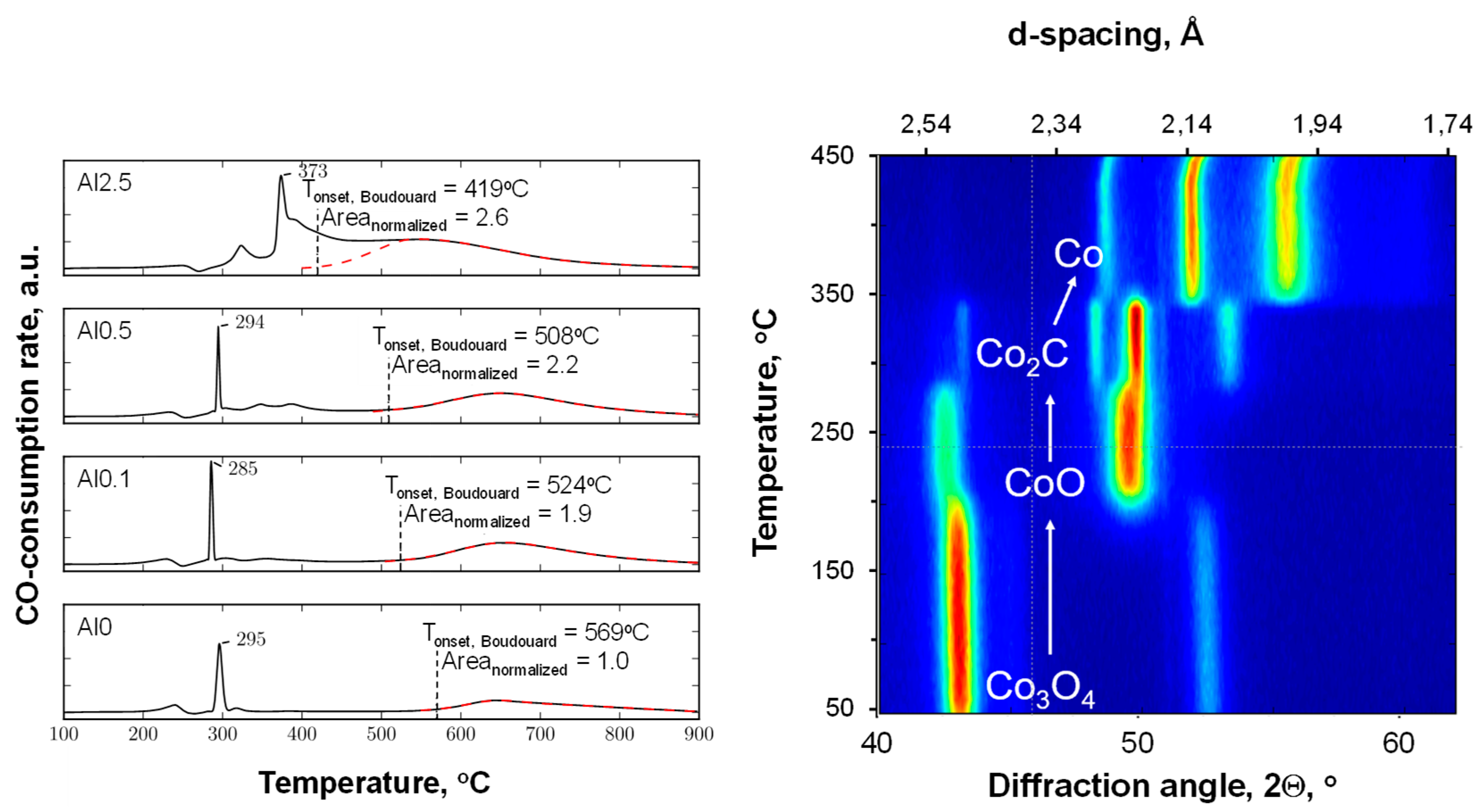
Figure 3 shows the profiles obtained from the temperature-programmed reduction in the presence of CO (CO-TPR) for the alumina-modified catalysts. The profiles for the samples with an aluminum content of 0.5 wt.% or less are characterized by a minor peak at ca. 240 °C, a sharp peak at 295 °C, a small feature at 320 °C, and a broad feature above 500 °C. The CO-TPR was followed using in situ XRD (see
Figure 3, right). It can be seen that the reduction of Co
3O
4 in the presence of carbon monoxide starts at ca. 210 °C with the formation of CoO, showing that the first feature in the CO-TPR can be ascribed to CO consumption for the reduction of Co
3O
4 to CoO. This is subsequently transformed into Co
2C (at ca. 295 °C). This will be accompanied by a large amount of CO consumed, thus appearing as a large peak in the CO-TPR at these temperatures. It can thus be concluded that the transformation of Co
3O
4 into Co
2C proceeds sequentially, Co
3O
4 → CoO → Co
2C, as discussed before [
36]. At temperatures around 350 °C, the transformation of Co
2C to metallic cobalt can be observed in the in situ XRD experiment. This does not appear to be associated with any additional consumption of carbon monoxide. It is thus concluded that at these temperatures, in the presence of carbon monoxide, cobalt carbide transforms into metallic cobalt (with the co-generation of carbon and the simultaneous disappearance of cobalt carbide). The decomposition of Co
2C in the presence of carbon monoxide into metallic cobalt and carbon is observed with this sample at ca. 365 °C, i.e., at slightly higher temperatures than in the presence of argon [
38]. It has been argued that cobalt carbide can be observed if the decomposition of the meta-stable Co
2C phase is kinetically hindered [
38]. The presence of CO in these experiments would have resulted in an increase in the chemical potential of carbon and thus an increase in the decomposition temperature.
Exposure of metallic cobalt to CO-containing gases at elevated temperatures will result in carbon formation [
39] via the Boudouard reaction:
This will be seen at temperatures higher than 400 °C, when metallic cobalt is available.
The CO-TPR profile of sample containing 0.1 wt.% aluminum (Al0.1) shows essentially the same features as the sample containing no aluminum (Al0). However, the onset of the high-temperature peak, which is associated with carbon deposition due to the Boudouard reaction, has shifted to a lower temperature. At the same time, the area under this peak is almost twice as large as the area under the high temperature peak in the CO-TPR of Al0. The on-set for the high temperature peak in the CO-TPR for the sample containing 0.5 wt.% aluminum (Al0.5) was shifted to an even lower temperature. The amount of CO consumed for the Boudouard reaction is larger for the sample Al0.5 than for the sample Al0.1. The observed shift in the on-set temperature for the Boudouard reaction implies that carbon deposition becomes more facile upon increasing the aluminum content in the sample. This could be ascribed to a change in the degree of reduction within the catalyst, but it was shown (see
Table 1) that the reducibility of samples in hydrogen reduces with increasing aluminum content. Hence, it is proposed [
26] that the interface between metallic cobalt and alumina islands on the surface [
18] may be the origin of the higher propensity for CO dissociation. Carbon deposition from CO is thermodynamically limited at high temperatures, and thus the variation in the amount of carbon deposition upon changing the aluminum content in the sample, as visualized by the differences in the area associated with the high-temperature feature in the CO-TPR, may be a consequence of the observed shift in the on-set of the Boudouard reaction to lower temperatures with increasing aluminum content.
The CO-TPR profile of the sample containing 0.5 wt.% aluminum contains additional features with maxima at ca. 345 °C and 380 °C, which are not observed in the CO-TPR profiles of the samples containing less aluminum. This may be considered CO consumption due to the retarded reduction of cobalt, e.g., of cobalt covered by the alumina islands. The reduction of this cobalt will yield directly metallic cobalt at 380 °C, as Co
2C does not seem to be stable at this temperature, as evidenced by the in situ XRD study (see
Figure 3).
The CO-TPR profile of the sample with the highest weight loading, 2.5 wt.% aluminum (Al2.5), differed strongly from the CO-TPR profiles of the samples containing less aluminum. The sharp peak that appeared at ca. 290 °C for the samples with lower aluminum contents was shifted toward 373 °C. This may be attributed to the presence of alumina islands covering the cobalt oxide surface and hindering the overall reduction process. The high-temperature feature, which was separated by almost 300 °C for the other samples, overlaps with the reduction peaks around 400 °C.
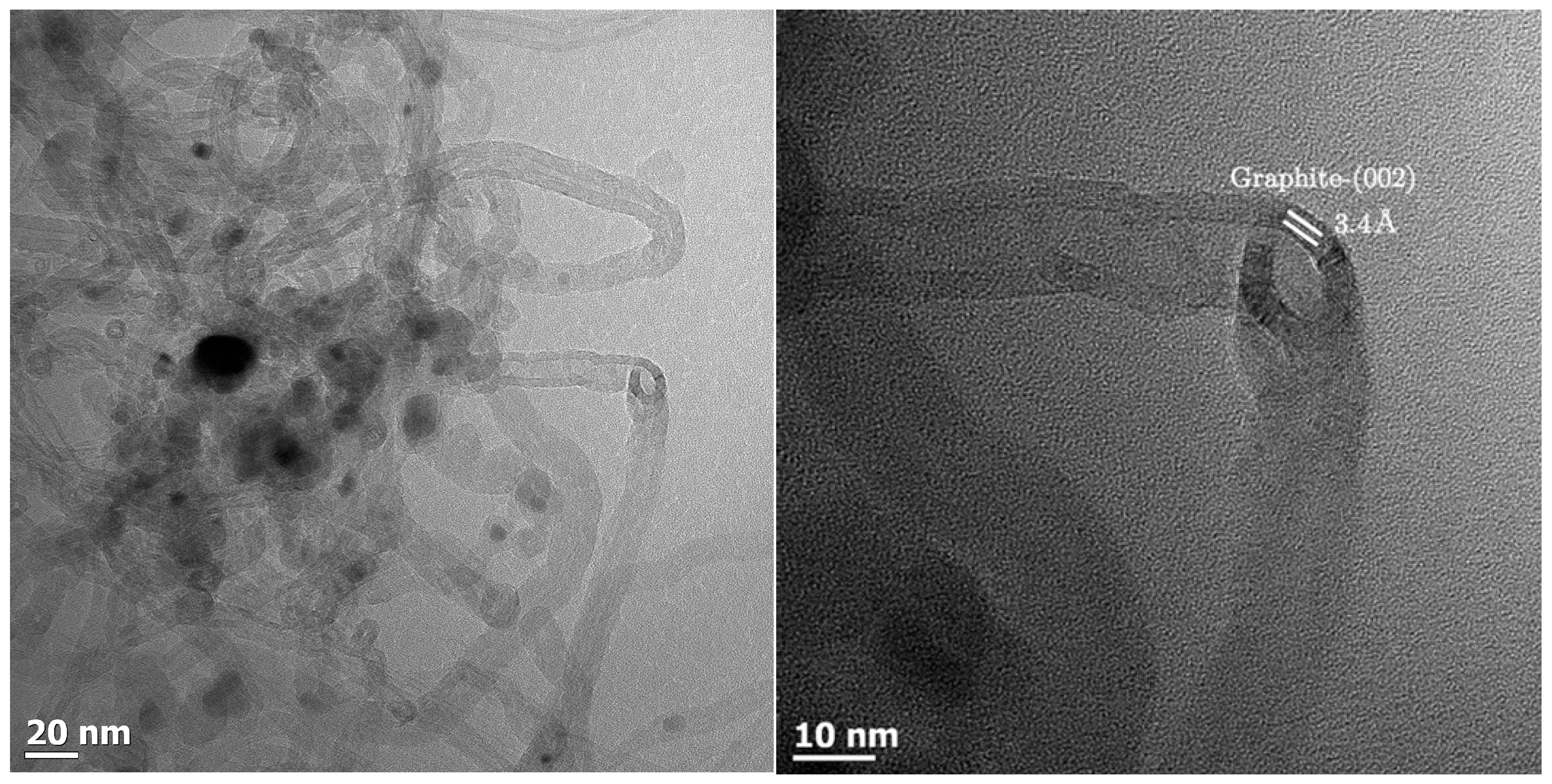
Carbon deposition by the Boudouard reaction could be visualized using TEM imaging of the sample Al0.5 after CO-TPR (see
Figure 4). The image shows metal particle sizes within the expected range if only minimal sintering has taken place. It appears that a reduction in CO inhibits the sintering process observed for H
2 reduction (see
Table 1). The metal particles are encapsulated by material appearing such as multi-wall carbon nanotubes. The d-spacing of this material was determined to be 3.4 Å, which is consistent with graphitic carbon [
40,
41]. The internal diameter of the carbon nanotubes appears to be in the range of 9–11 nm. The internal diameter is thought to be linked to the metal particle size [
42] (the reduction of Co
3O
4 particles with a size of 14 nm is expected to yield in the absence of sintering metal particles with a size of 10–11 nm).
The enhanced activity for CO dissociation as indicated by the shift in the on-set of the Boudouard reaction to lower temperatures upon increasing the aluminum content in the samples and as argued to be the origin of the change in the kinetic coefficients and the change in the selectivity has been linked to the Lewis acid property of the promoter [
25,
42,
43]. Carbon monoxide may act as a weak Lewis base, which can donate electrons from the 4σ molecular orbital to a Lewis acid [
15]. The 4σ molecular orbital is a bonding orbital, and electron withdrawal results in weakening of the C–O bond.
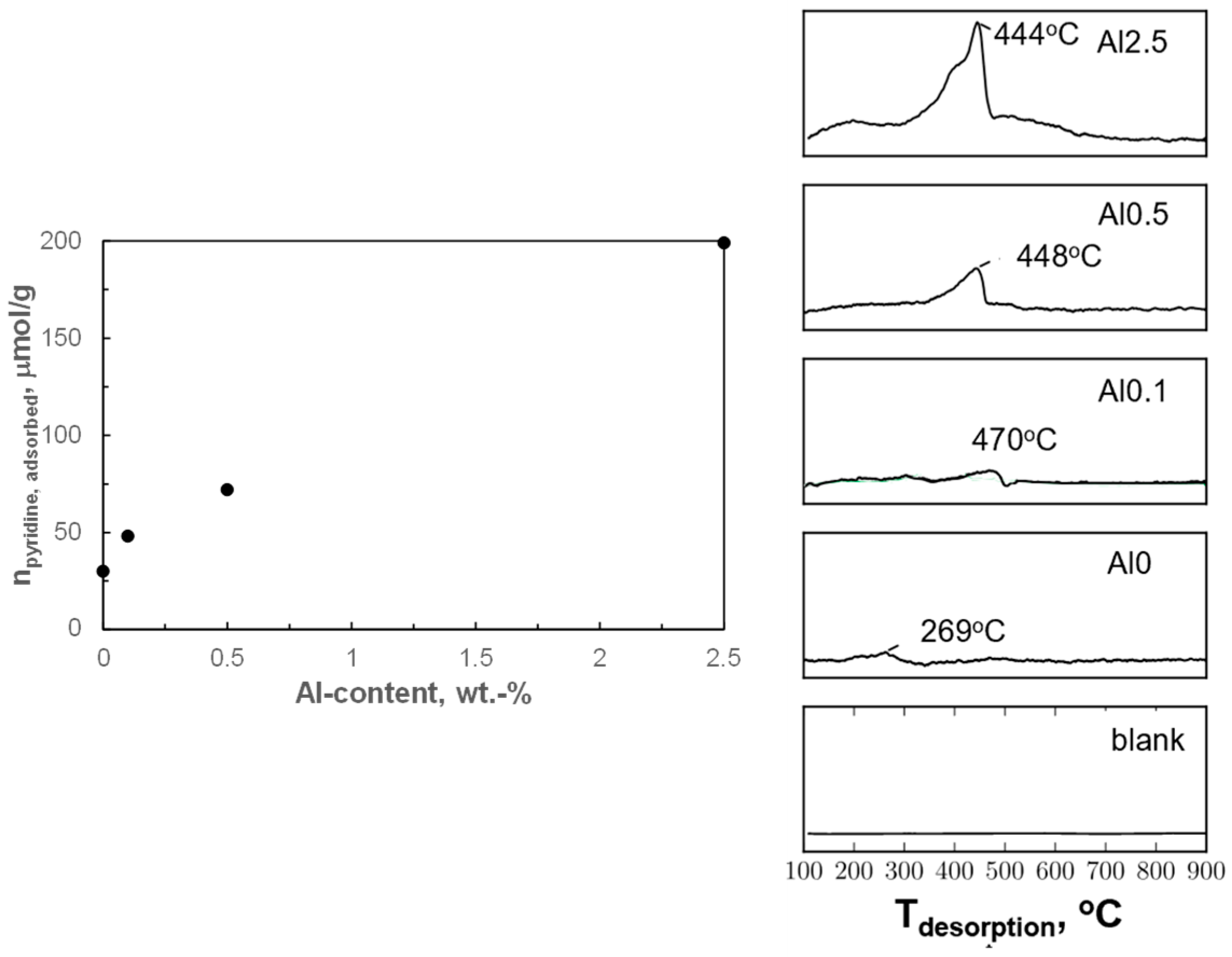
Alumina is known to exhibit Lewis acidity, although the presence of Brønsted acid sites cannot be ruled out [
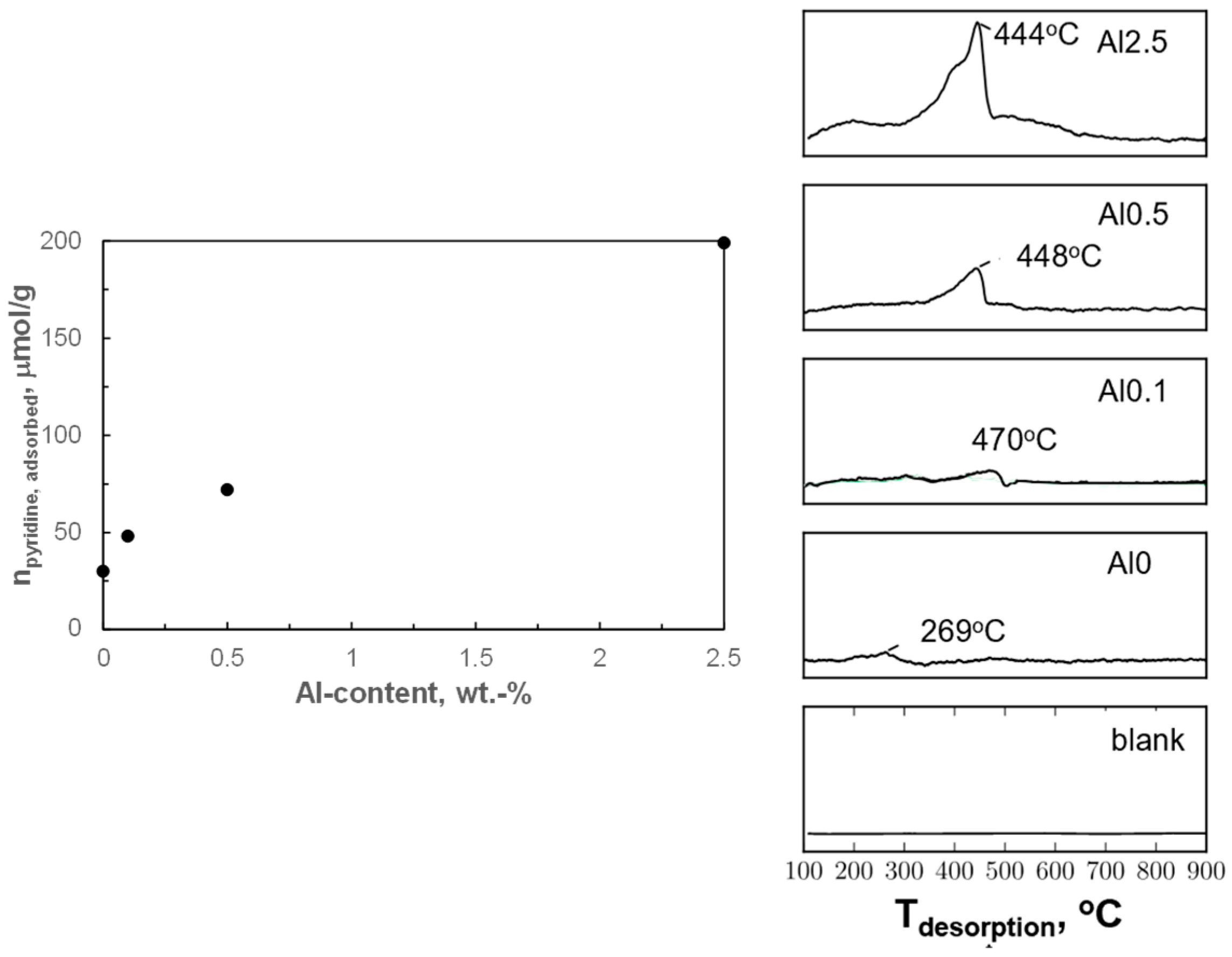
44]. Hence, alumina modification may have introduced acidity in these samples. The acidity of the activated catalysts was investigated by pyridine uptake and by the temperature-programmed desorption of pyridine (py-TPD; see
Figure 5). The pyridine uptake of the activated sample containing no aluminum (Al0) during pulse chemisorption was 30 μmol/g. The pyridine uptake on the sample containing aluminum increases roughly linearly with increasing alumina loading over the activated catalyst, and the pyridine uptake relative to the aluminum loading was determined to be 0.2 mol pyridine/mol Al.
Pyridine desorbed at a relatively low temperature of 269 °C from the sample containing no aluminum (Al0), indicating rather weakly adsorbed pyridine. A sharp peak between 350–500 °C was observed during pyridine TPD for the alumina-modified samples. Sharp peaks above 350 °C have been reported to correspond to the decomposition of pyridine to N
2 and CO
2 [
45,
46]. The catalyst had also darkened after the experiment, which may suggest carbon deposition [
45] indicating that pyridine decomposition may have occurred during the pyridine TPD. Hence, the position of the peak is expected to correlate with the rate of pyridine decomposition rather than the rate of pyridine desorption (and thus the peak area with the thermal conductivity of the decomposition products). The decomposition of pyridine at temperatures below 500 °C, in an inert atmosphere, is only catalyzed by strong acid sites [
46,
47]. The pyridine TPD experiments indicate that the acid sites on the activated modified samples were strong enough to catalyze the decomposition of pyridine at temperatures below 500 °C.
It can be concluded that pyridine adsorption is associated with the presence of strong acid sites upon modification of the samples with alumina, and that the amount of pyridine adsorbed increases roughly linearly with the aluminum content. Thus, it seems plausible that these strong acid sites are associated with the presence of alumina islands on the surface of the metallic cobalt [
18]. The relative constant amount of pyridine adsorbed per aluminum may indicate that the strong acid site is associated with a cluster of ca. 5 Al atoms. Alternatively, these acid sites are located at the interface between the alumina islands and metallic cobalt, and an increase in the aluminum content is associated with an increase in the number of alumina islands, but not necessarily their size.
4. Materials and Methods
Cobalt oxide, Co
3O
4, with an average crystallite size of 14 nm, was prepared by the calcination of a cobalt carbonate precursor at 300 °C. The detailed synthesis procedure has been elsewhere [
18]. The resulting Co
3O
4 crystallites were impregnated with aluminum sec-butoxide in dry n-hexane to achieve a weight loading of aluminum of 0.1, 0.5, or 2.5 wt.%, respectively. The obtained materials were calcined at 300 °C for 4 h. This results in the formation of highly dispersed, island-like alumina structures on cobalt oxide [
18]. The catalysts are labeled according to their loading with aluminum as Al0, Al0.1, Al0.5, and Al2.5.
Hydrogen chemisorption was performed using an ASAP 2020 (Micromeritics, Norcross, GA, USA). The catalyst (100 mg) was dried for 12 h at 120 °C and reduced in hydrogen by increasing the temperature at a heating rate of 1 °C/min to 350 °C and holding for 10 h. After reduction, the chamber was evacuated for 30 min both at 330 °C and 140 °C. The hydrogen uptake was measured at a temperature of 120 °C.
The degree of reduction (DoR) was determined in the temperature-programmed reduction (TPR) cell (Autochem 2950; Micromeritics, Norcross, GA, USA) by activation the catalyst (50 mg) in a flow of 10 mln/min pure H2 at 350 °C by (heating rate: 1 °C/min) for 10 h. Afterwards, the temperature was decreased to 60 °C in flowing hydrogen, and the catalyst bed was purged for 60 min in a flow of 50 mln/min of 5 vol% H2 in Ar. The catalyst bed temperature was increased to 900 °C at 10 °C/min whilst monitoring the hydrogen consumption with a thermal conductivity detector (TCD). The degree of reduction was determined from the ratio of the hydrogen consumption in the TPR after reduction at 350 °C for 10 h (the hydrogen consumption was calibrated using the reduction of Ag2O) with the amount of Co3O4 loaded in the TPR cell.
Temperature programmed reduction studies using CO (CO-TPR) were performed in an Autochem 2950 (Micromeritics, Norcross, GA, USA). The temperature of the catalyst bed (50 mg) was increased to 120 °C at a heating rate of 10 °C/min in a flow of 50 mln/min Ar, and the catalyst was dried for 1 h. The catalyst temperature was decreased to 60 °C, and the gas was switched to CO (10 mln/min). Subsequently, the temperature was increased to 900 °C (heating rate 10 °C /min), and the TCD signal was recorded.
X-ray diffraction (XRD) was performed using a Bruker AXS D8 advance diffractometer (Karlsruhe, Germany) with Co-K
α radiation (1.78897 Å) operating at 40 kV and 30 mA between 20–120° 2θ with a scanning speed of 0.0562° 2θ/s. The phase transitions occurring during the temperature-programmed reduction experiment were explored using in situ reduction with CO (4 ml
n/min) as the reducing agent. The catalyst was loaded into a borosilicate capillary. The capillary was fitted into an in situ cell, which was developed in house [
48,
49]. The capillary was heated with infrared heaters. The temperature was increased at a heating rate of 1 °C/min from 50 to 450 °C, and X-ray diffractograms were taken every 5 min.
Transmission electron microscopy (TEM) was performed using a FEI Tecnai T20 (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a LaB6 filament operated at 200 kV. Catalysts were dispersed in methanol, ultrasonicated for 2 min, and transferred to a carbon coated copper grid.
Pyridine temperature-programmed desorption (Py-TPD) was performed using an Autochem 2920 (Micromeritics, Norcross, GA, USA) equipped with a vapor. The catalyst (100 mg) was activated in a stream of 30 mln/min of pure H2 by increasing the temperature to 350 °C at a heating rate of 10 °C/min and reducing the catalyst for 30 min. The temperature was decreased to 120 °C at a heating rate of 10 °C/min under a flow of 10 mln/min He and kept there for 1 h. The sample temperature was decreased to 110 °C, where pyridine pulse adsorption was performed. Helium saturated with pyridine at 60 °C was pulsed 15 times over the activated catalyst in 5-min intervals. Each dose contained 27 μmol pyridine. The pyridine uptake was monitored using a TCD. Weakly adsorbed pyridine was removed by flowing 50 mln/min of pure helium over the catalyst for 1 h. Py-TPD was carried out by increasing the temperature to 900 °C at a heating rate of 10 °C/min in a stream of 50 mln/min He, whilst monitoring the tail gas using a TCD detector.
The Fischer–Tropsch synthesis was performed in a 1-L slurry reactor. The catalyst (3–5 g) was activated ex situ in a flow of hydrogen with a space velocity of 12 l
n/h/g
cat. The temperature was increased to 350 °C at a constant heating rate of 1 °C/min, and subsequently, the catalyst was reduced for 10 h. The activated catalyst was cooled to room temperature, and then transferred to 30 g of molten wax under an argon atmosphere. The wax was cooled to room temperature, and the mass of the reduced catalyst (m
cat,red) was determined by differential weighing of the wax tablet before and after addition of the catalyst. The reactor was filled with 250 g of molten wax, and the catalyst wax tablet was loaded into the reactor. The reactor was pressurized with argon to 20 bar, and the temperature was increased to 220 °C at a stirring speed of 350 rpm. When the desired temperature and pressure were reached, diluted syngas (H
2:CO:Ar = 2:1:1) was introduced. The permanent gases were analyzed on-line with a Varian CP-4900 micro GC equipped with a TCD detector to determine the CO conversion and methane selectivity using argon as an internal standard. Gaseous samples were collected in pre-evacuated ampoules [
50] at regular intervals, and the samples were analyzed off-line by GC-FID.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}