N,S Co-Doped Carbon Nanofibers Derived from Bacterial Cellulose/Poly(Methylene blue) Hybrids: Efficient Electrocatalyst for Oxygen Reduction Reaction

Abstract
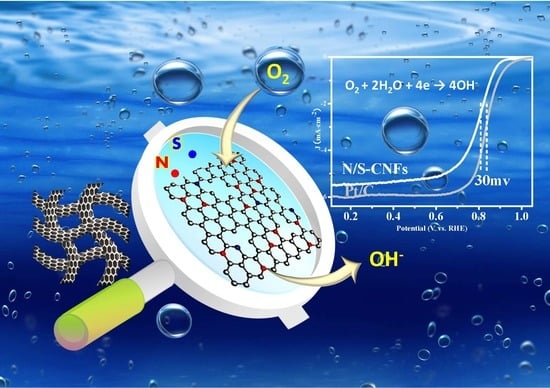
1. Introduction
2. Results
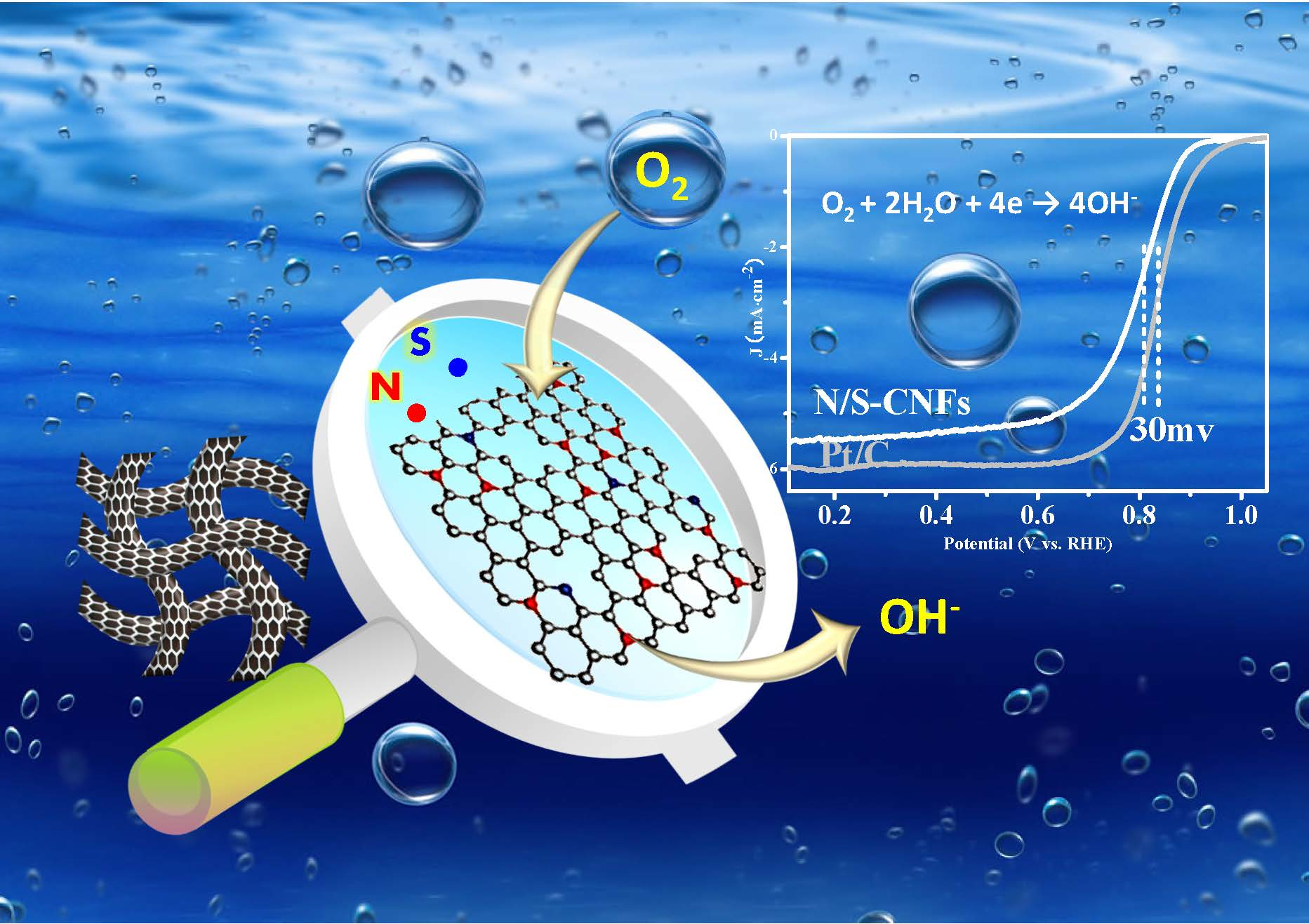
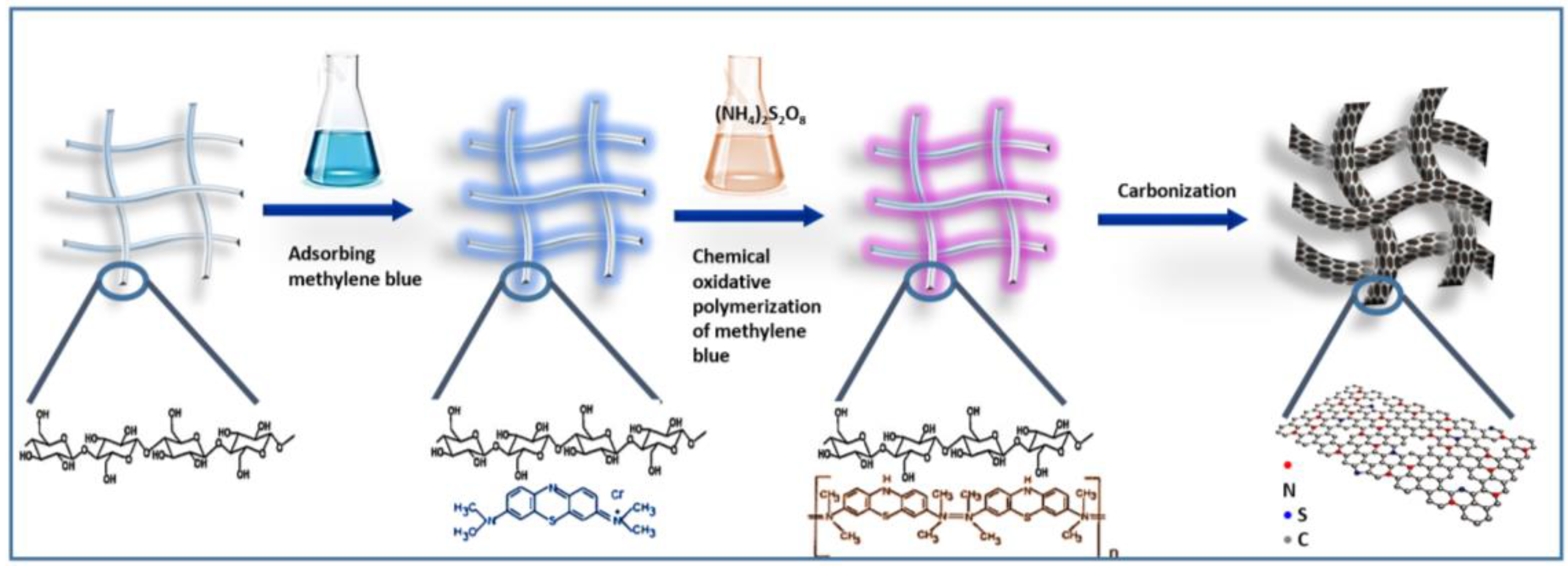
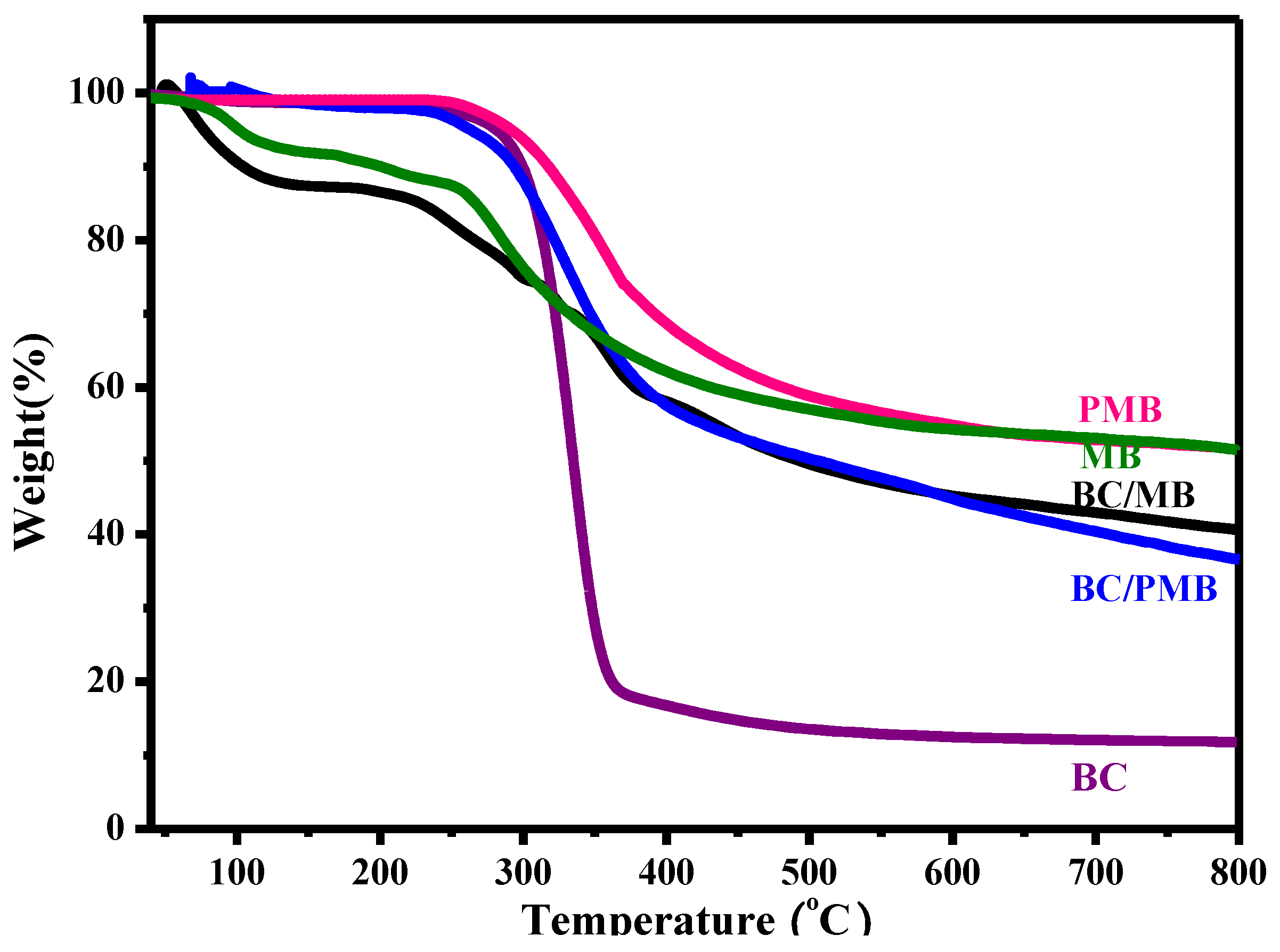
2.1. Characterization of the BC/MB and BC/PMB
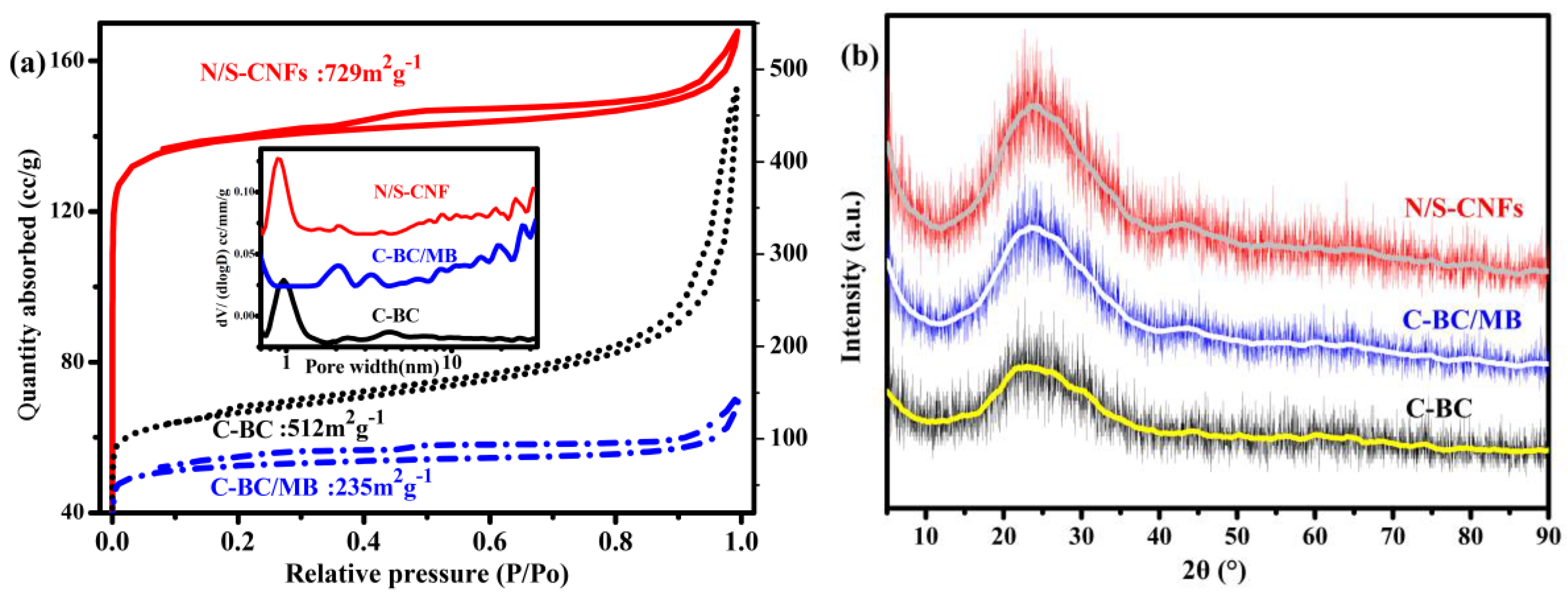
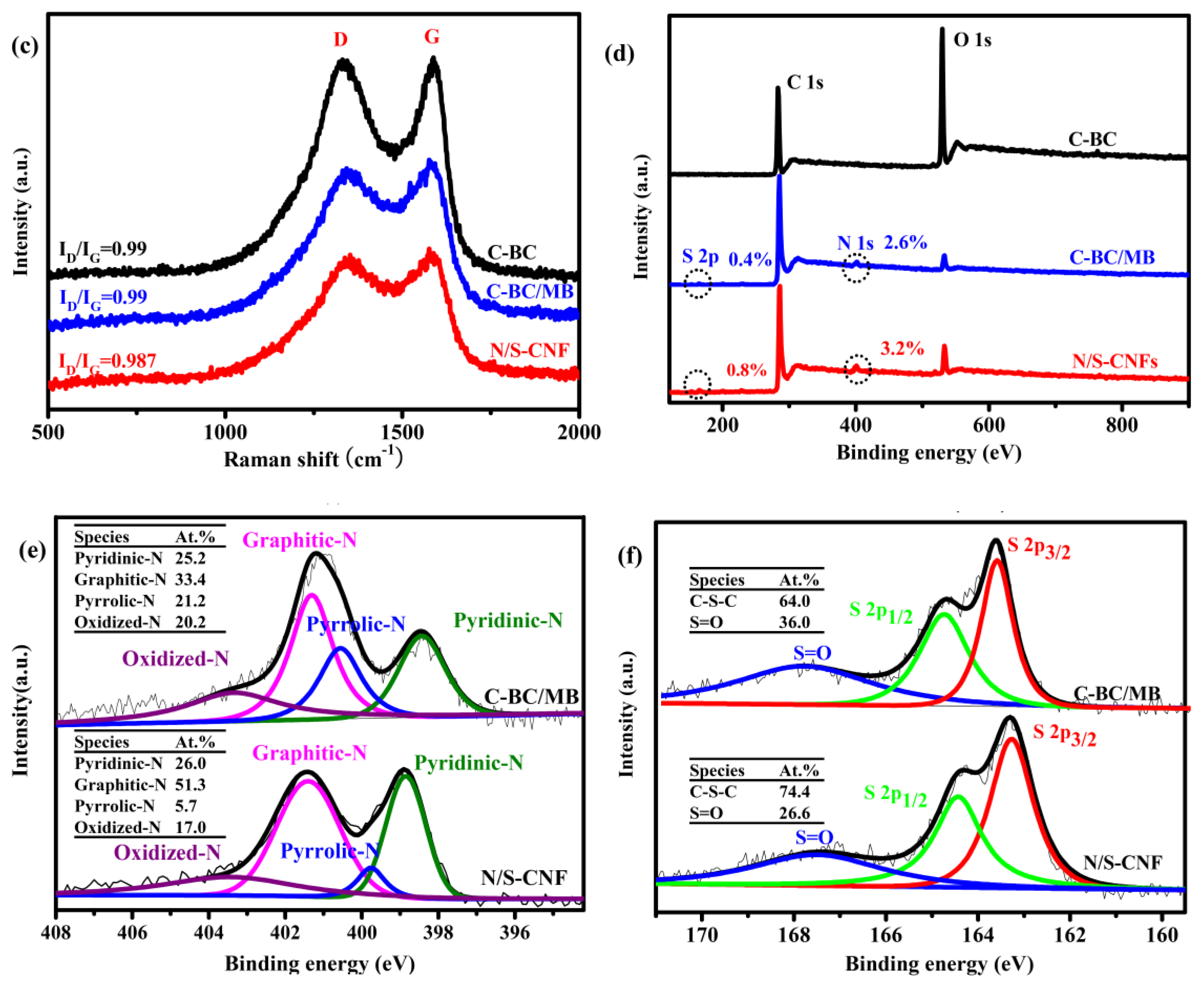
2.2. Characterization of the Carbonization Products of BC, BC/MB and BC/PMB
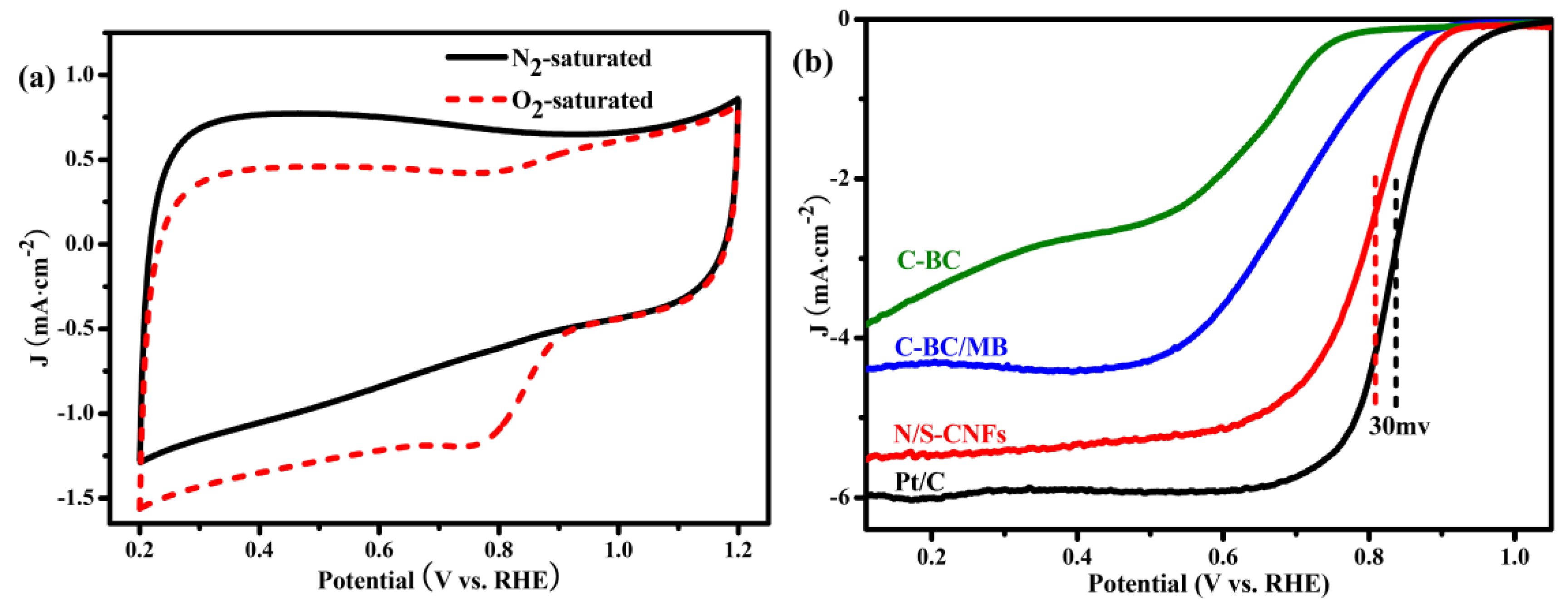
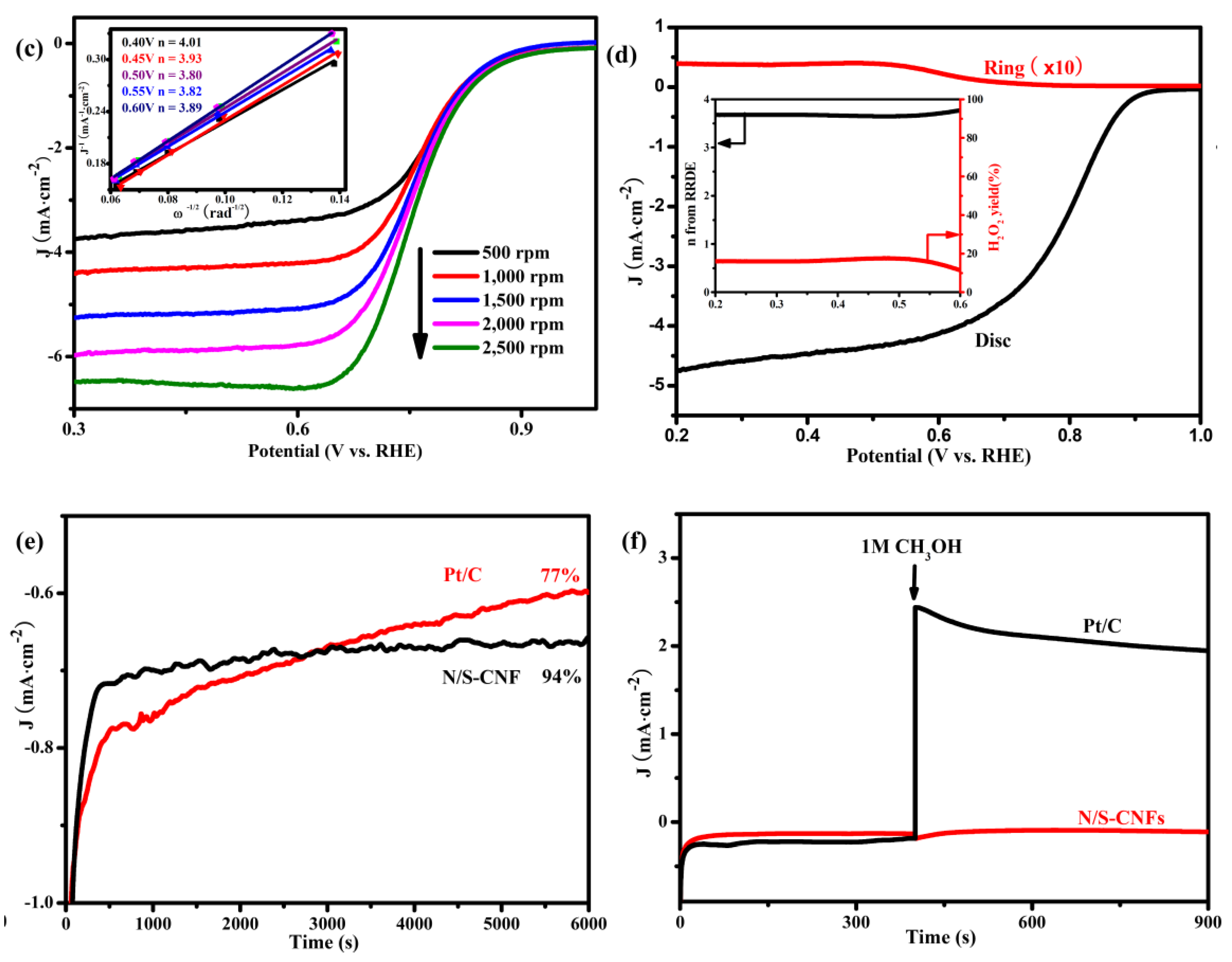
2.3. Electrocatalytic Activity of the N/S-CNF for the ORR
3. Materials and Methods
3.1. Materials
3.2. Preparation of BC/MB and BC/PMB
3.3. Electrochemical Measurements
3.4. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Fu, J.; Cano, Z.P.; Park, M.G.; Yu, A.; Fowler, M.; Chen, Z. Electrically rechargeable zinc-air batteries: Progress, challenges, and perspectives. Adv. Mater. 2017, 29, 1604685. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Zhang, Q. Nanocarbon for oxygen reduction electrocatalysis: Dopants, edges, and defects. Adv. Mater. 2017, 29, 1604103. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent advances in electrocatalysts for oxygen reduction reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Xu, Y.Y.; Yang, H.; Dong, Z.; Liu, H.; Xia, B.Y. Advanced architectures and relatives of air electrodes in Zn-air batteries. Adv. Sci. 2018, 5, 1700691:1–1700691:30. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yan, B.; Li, S.; Wang, J.; Wang, C.; Guo, J.; Du, Y. Facile construction of N-doped graphene supported hollow ptag nanodendrites as highly efficient electrocatalysts toward formic acid oxidation reaction. ACS Sustain. Chem. Eng. 2018, 6, 609–617. [Google Scholar] [CrossRef]
- Cheng, H.; Li, M.L.; Su, C.Y.; Li, N.; Liu, Z.Q. Cu-Co bimetallic oxide quantum dot decorated nitrogen-doped carbon nanotubes: A high-efficiency bifunctional oxygen electrode for Zn-air batteries. Adv. Funct. Mater. 2017, 27, 1701833. [Google Scholar] [CrossRef]
- Liu, X.; Amiinu Ibrahim, S.; Liu, S.; Pu, Z.; Li, W.; Ye, B.; Tan, D.; Mu, S. H2O2-assisted synthesis of porous N-doped graphene/molybdenum nitride composites with boosted oxygen reduction reaction. Adv. Mater. Interfaces 2017, 4, 1601227. [Google Scholar] [CrossRef]
- Su, C.Y.; Cheng, H.; Li, W.; Liu, Z.Q.; Li, N.; Hou, Z.; Bai, F.Q.; Zhang, H.X.; Ma, T.Y. Atomic modulation of FeCo-nitrogen-carbon bifunctional oxygen electrodes for rechargeable and flexible all-solid-state zinc-air battery. Adv. Energy Mater. 2017, 7, 1602420. [Google Scholar] [CrossRef]
- Cui, H.; Zhou, Z.; Jia, D. Heteroatom-doped graphene as electrocatalysts for air cathodes. Mater. Horiz. 2017, 4, 7–19. [Google Scholar] [CrossRef]
- Zhu, C.; Li, H.; Fu, S.; Du, D.; Lin, Y. Highly efficient nonprecious metal catalysts towards oxygen reduction reaction based on three-dimensional porous carbon nanostructures. Chem. Soc. Rev. 2016, 45, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Lee Won, J.; Lim, J.; Kim Sang, O. Nitrogen dopants in carbon nanomaterials: Defects or a new opportunity? Small Methods 2016, 1, 1600014. [Google Scholar]
- Xiang, Z.; Cao, D.; Huang, L.; Shui, J.; Wang, M.; Dai, L. Nitrogen-doped holey graphitic carbon from 2D covalent organic polymers for oxygen reduction. Adv. Mater. 2014, 26, 3315–3320. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Shang, L.; Bian, T.; Shi, R.; Waterhouse Geoffrey, I.N.; Zhao, Y.; Zhou, C.; Wu, L.Z.; Tung, C.H.; Zhang, T. Nitrogen-doped porous carbon nanosheets templated from g-C3N4 as metal-free electrocatalysts for efficient oxygen reduction reaction. Adv. Mater. 2016, 28, 5080–5086. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, K.; Ashokkumar, M.; Gullapalli, H.; Gong, Y.; Vajtai, R.; Thanikaivelan, P.; Ajayan, P.M. Nitrogen-rich carbon nano-onions for oxygen reduction reaction. Carbon 2018, 130, 645–651. [Google Scholar] [CrossRef]
- Geng, D.; Chen, Y.; Chen, Y.; Li, Y.; Li, R.; Sun, X.; Ye, S.; Knights, S. High oxygen-reduction activity and durability of nitrogen-doped graphene. Energy Environ. Sci. 2011, 4, 760–764. [Google Scholar] [CrossRef]
- Li, L.; Dai, P.; Gu, X.; Wang, Y.; Yan, L.; Zhao, X. High oxygen reduction activity on a metal-organic framework derived carbon combined with high degree of graphitization and pyridinic-N dopants. J Mater. Chem. A 2017, 5, 789–795. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, Y.; Quan, X.; Chen, S.; Yu, H.; Zhao, H. Nitrogen-doped carbon with a high degree of graphitization derived from biomass as high-performance electrocatalyst for oxygen reduction reaction. Appl. Surf. Sci. 2017, 396, 986–993. [Google Scholar] [CrossRef]
- Zhan, Y.; Yu, X.; Cao, L.; Zhang, B.; Wu, X.; Xie, F.; Zhang, W.; Chen, J.; Xie, W.; Mai, W.; et al. The influence of nitrogen source and doping sequence on the electrocatalytic activity for oxygen reduction reaction of nitrogen doped carbon materials. Int. J. Hydrogen Energy 2016, 41, 13493–13503. [Google Scholar] [CrossRef]
- Higgins, D.; Chen, Z.; Chen, Z. Nitrogen doped carbon nanotubes synthesized from aliphatic diamines for oxygen reduction reaction. Electrochim. Acta 2011, 56, 1570–1575. [Google Scholar] [CrossRef]
- Lin, C.Y.; Zhang, D.; Zhao, Z.; Xia, Z. Covalent organic framework electrocatalysts for clean energy conversion. Adv. Mater. 2017, 30, 1703646. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Tang, Y.; Zhang, X.; Oshima, Y.; Chen, Q.; Jiang, D. Template conversion of covalent organic frameworks into 2d conducting nanocarbons for catalyzing oxygen reduction reaction. Adv. Mater. 2018, 30, 1706330. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, J.; Amiinu, I.S.; Zhang, C.; Liu, X.; Tu, W.; Pan, M.; Mu, S. Transforming waste biomass with an intrinsically porous network structure into porous nitrogen-doped graphene for highly efficient oxygen reduction. Phys. Chem. Chem. Phys. 2016, 18, 10392–10399. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Li, X.; Li, L.; Wei, X. A versatile biomass derived carbon material for oxygen reduction reaction, supercapacitors and oil/water separation. Nano Energy 2017, 33, 334–342. [Google Scholar] [CrossRef]
- Zheng, X.; Cao, X.; Li, X.; Tian, J.; Jin, C.; Yang, R. Biomass lysine-derived nitrogen-doped carbon hollow cubes via a nacl crystal template: An efficient bifunctional electrocatalyst for oxygen reduction and evolution reactions. Nanoscale 2017, 9, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Ma, R.; Zhou, Y.; Liu, Q.; Dong, X.; Wang, J. Koh activation of biomass-derived nitrogen-doped carbons for supercapacitor and electrocatalytic oxygen reduction. Electrochim. Acta 2018, 261, 49–57. [Google Scholar] [CrossRef]
- Chen, W.; Yu, H.; Lee, S.-Y.; Wei, T.; Li, J.; Fan, Z. Nanocellulose: A promising nanomaterial for advanced electrochemical energy storage. Chem. Soc. Rev. 2018, 47, 2837–2872. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-Y.; Liang, H.-W.; Chen, L.-F.; Hu, B.-C.; Yu, S.-H. Bacterial cellulose: A robust platform for design of three dimensional carbon-based functional nanomaterials. Acc. Chem. Res. 2016, 49, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.-W.; Wu, Z.-Y.; Chen, L.-F.; Li, C.; Yu, S.-H. Bacterial cellulose derived nitrogen-doped carbon nanofiber aerogel: An efficient metal-free oxygen reduction electrocatalyst for zinc-air battery. Nano Energy 2015, 11, 366–376. [Google Scholar] [CrossRef]
- Pei, Z.; Li, H.; Huang, Y.; Xue, Q.; Huang, Y.; Zhu, M.; Wang, Z.; Zhi, C. Texturing in situ: N,S-enriched hierarchically porous carbon as a highly active reversible oxygen electrocatalyst. Energy Environ. Sci. 2017, 10, 742–749. [Google Scholar] [CrossRef]
- Rafatullah, M.; Sulaiman, O.; Hashim, R.; Ahmad, A. Adsorption of methylene blue on low-cost adsorbents: A review. J. Hazard. Mater. 2010, 177, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-Y.; Liang, H.-W.; Li, C.; Hu, B.-C.; Xu, X.-X.; Wang, Q.; Chen, J.-F.; Yu, S.-H. Dyeing bacterial cellulose pellicles for energetic heteroatom doped carbon nanofiber aerogels. Nano Res. 2014, 7, 1861–1872. [Google Scholar] [CrossRef]
- Guo, D.; Shibuya, R.; Akiba, C.; Saji, S.; Kondo, T.; Nakamura, J. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 2016, 351, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Wang, L.; Ma, Z. Triple sensitivity amplification for ultrasensitive electrochemical detection of prostate specific antigen. Biosens. Bioelectron. 2017, 92, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Zhang, P.; Li, M.; Guo, Z.; Zheng, X. Synthesis of polymethylene blue nanoparticles and their application to label-free DNA detection. Anal. Lett. 2016, 49, 2728–2740. [Google Scholar] [CrossRef]
- Li, X.; Zhong, M.; Sun, C.; Luo, Y. A novel bilayer film material composed of polyaniline and poly(methylene blue). Mater. Lett. 2005, 59, 3913–3916. [Google Scholar] [CrossRef]
- Shan, J.; Wang, L.; Ma, Z. Novel metal-organic nanocomposites: Poly(methylene blue)-Au and its application for an ultrasensitive electrochemical immunosensing platform. Sensor. Actuators B Chem. 2016, 237, 666–671. [Google Scholar] [CrossRef]
- Xiao, X.; Zhou, B.; Tan, L.; Tang, H.; Zhang, Y.; Xie, Q.; Yao, S. Poly(methylene blue) doped silica nanocomposites with crosslinked cage structure: Electropolymerization, characterization and catalytic activity for reduction of dissolved oxygen. Electrochim. Acta 2011, 56, 10055–10063. [Google Scholar] [CrossRef]
- Yang, S.; Li, G.; Zhao, J.; Zhu, H.; Qu, L. Electrochemical preparation of ag nanoparticles/poly(methylene blue) functionalized graphene nanocomposite film modified electrode for sensitive determination of rutin. J. Electroanal. Chem. 2014, 717–718, 225–230. [Google Scholar] [CrossRef]
- Rincón Rosalba, A.; Artyushkova, K.; Mojica, M.; Germain Marguerite, N.; Minteer Shelley, D.; Atanassov, P. Structure and electrochemical properties of electrocatalysts for nadh oxidation. Electroanalysis 2010, 22, 799–806. [Google Scholar] [CrossRef]
- Zheng, F.; Yang, Y.; Chen, Q. High lithium anodic performance of highly nitrogen-doped porous carbon prepared from a metal-organic framework. Nat. Commun. 2014, 5, 5261. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-F.; Lu, Y.; Yu, L.; Lou, X.W. Designed formation of hollow particle-based nitrogen-doped carbon nanofibers for high-performance supercapacitors. Energy Environ. Sci. 2017, 10, 1777–1783. [Google Scholar] [CrossRef]
- Klingele, M.; Pham, C.; Vuyyuru, K.R.; Britton, B.; Holdcroft, S.; Fischer, A.; Thiele, S. Sulfur doped reduced graphene oxide as metal-free catalyst for the oxygen reduction reaction in anion and proton exchange fuel cells. Electrochem. Commun. 2017, 77, 71–75. [Google Scholar] [CrossRef]
- Li, Y.; Yang, J.; Huang, J.; Zhou, Y.; Xu, K.; Zhao, N.; Cheng, X. Soft template-assisted method for synthesis of nitrogen and sulfur co-doped three-dimensional reduced graphene oxide as an efficient metal free catalyst for oxygen reduction reaction. Carbon 2017, 122, 237–246. [Google Scholar] [CrossRef]
- Chan, C.H.; Chia, C.H.; Zakaria, S.; Sajab, M.S.; Chin, S.X. Cellulose nanofibrils: a rapid adsorbent for the removal of methylene blue. RSC Adv. 2015, 5, 18204–18212. [Google Scholar] [CrossRef]
- Wu, M.; Liu, Y.; Zhu, Y.; Lin, J.; Liu, J.; Hu, H.; Wang, Y.; Zhao, Q.; Lv, R.; Qiu, J. Supramolecular polymerization-assisted synthesis of nitrogen and sulfur dual-doped porous graphene networks from petroleum coke as efficient metal-free electrocatalysts for the oxygen reduction reaction. J. Mater. Chem. A 2017, 5, 11331–11339. [Google Scholar] [CrossRef]
- Xu, L.; Fan, H.; Huang, L.; Xia, J.; Li, S.; Li, M.; Ding, H.; Huang, K. Chrysanthemum-derived n and s co-doped porous carbon for efficient oxygen reduction reaction and aluminum-air battery. Electrochim. Acta 2017, 239, 1–9. [Google Scholar] [CrossRef]
- Yang, S.; Mao, X.; Cao, Z.; Yin, Y.; Wang, Z.; Shi, M.; Dong, H. Onion-derived n, s self-doped carbon materials as highly efficient metal-free electrocatalysts for the oxygen reduction reaction. Appl. Surf. Sci. 2018, 427, 626–634. [Google Scholar] [CrossRef]
- You, C.; Jiang, X.; Han, L.; Wang, X.; Lin, Q.; Hua, Y.; Wang, C.; Liu, X.; Liao, S. Uniform nitrogen and sulphur co-doped hollow carbon nanospheres as efficient metal-free electrocatalysts for oxygen reduction. J. Mater. Chem. A 2017, 5, 1742–1748. [Google Scholar] [CrossRef]
- Jiang, T.; Wang, Y.; Wang, K.; Liang, Y.; Wu, D.; Tsiakaras, P.; Song, S. A novel sulfur-nitrogen dual doped ordered mesoporous carbon electrocatalyst for efficient oxygen reduction reaction. Appl. Catal. B-Environ. 2016, 189, 1–11. [Google Scholar] [CrossRef]
- Su, Y.; Yao, Z.; Zhang, F.; Wang, H.; Mics, Z.; Cánovas, E.; Bonn, M.; Zhuang, X.; Feng, X. Sulfur-enriched conjugated polymer nanosheet derived sulfur and nitrogen co-doped porous carbon nanosheets as electrocatalysts for oxygen reduction reaction and zinc-air battery. Adv. Funct. Mater. 2016, 26, 5893–5902. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, R.; Wang, J.; Zhu, J.; Xiao, W.; Xuan, C.; Lei, W.; Wang, D. Nitrogen and sulfur co-doping of 3D hollow-structured carbon spheres as an efficient and stable metal free catalyst for the oxygen reduction reaction. Nanoscale 2016, 8, 19086–19092. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Duan, Y.; Zhang, X.; Zhang, J. A facile synthesis of nitrogen/sulfur co-doped graphene for the oxygen reduction reaction. ChemCatChem 2015, 8, 163–170. [Google Scholar] [CrossRef]
- Qu, K.; Zheng, Y.; Dai, S.; Qiao, S.Z. Graphene oxide-polydopamine derived n, s-codoped carbon nanosheets as superior bifunctional electrocatalysts for oxygen reduction and evolution. Nano Energy 2016, 19, 373–381. [Google Scholar] [CrossRef]
- Zehtab Yazdi, A.; Roberts, E.P.L.; Sundararaj, U. Nitrogen/sulfur co-doped helical graphene nanoribbons for efficient oxygen reduction in alkaline and acidic electrolytes. Carbon 2016, 100, 99–108. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ΔE½ (mV) | Ref. |
|---|---|---|
| N/S-CNFs | 30 | This work |
| N-S-CNF-800 (MB) | 200 | [31] |
| NS-3DrGO-950 | 64 | [43] |
| N,S-PGN-800 | 30 | [45] |
| NSC800 | 35.4 | [46] |
| NSC-A2 | 80 | [47] |
| PAC-5S | 48 | [48] |
| S1N5-OMC | 65 | [49] |
| N/S-2DPC-60 | 27 | [50] |
| N,S-hcs-900 °C | 40 | [51] |
| NS-G | 72 | [52] |
| N, S-CN | 50 | [53] |
| CNx/CSx-GNRs 1000° for 2.5 h | 50 | [54] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Ji, Y.-G.; Qiao, B.; Zhao, F.; Gao, H.; Chen, P.; An, Z.; Chen, X.; Chen, Y. N,S Co-Doped Carbon Nanofibers Derived from Bacterial Cellulose/Poly(Methylene blue) Hybrids: Efficient Electrocatalyst for Oxygen Reduction Reaction. Catalysts 2018, 8, 269. https://doi.org/10.3390/catal8070269
Liu J, Ji Y-G, Qiao B, Zhao F, Gao H, Chen P, An Z, Chen X, Chen Y. N,S Co-Doped Carbon Nanofibers Derived from Bacterial Cellulose/Poly(Methylene blue) Hybrids: Efficient Electrocatalyst for Oxygen Reduction Reaction. Catalysts. 2018; 8(7):269. https://doi.org/10.3390/catal8070269
Chicago/Turabian StyleLiu, Jing, Yi-Gang Ji, Bin Qiao, Fengqi Zhao, Hongxu Gao, Pei Chen, Zhongwei An, Xinbing Chen, and Yu Chen. 2018. "N,S Co-Doped Carbon Nanofibers Derived from Bacterial Cellulose/Poly(Methylene blue) Hybrids: Efficient Electrocatalyst for Oxygen Reduction Reaction" Catalysts 8, no. 7: 269. https://doi.org/10.3390/catal8070269
APA StyleLiu, J., Ji, Y.-G., Qiao, B., Zhao, F., Gao, H., Chen, P., An, Z., Chen, X., & Chen, Y. (2018). N,S Co-Doped Carbon Nanofibers Derived from Bacterial Cellulose/Poly(Methylene blue) Hybrids: Efficient Electrocatalyst for Oxygen Reduction Reaction. Catalysts, 8(7), 269. https://doi.org/10.3390/catal8070269