Controlling Redox Enzyme Orientation at Planar Electrodes


 , and
, and 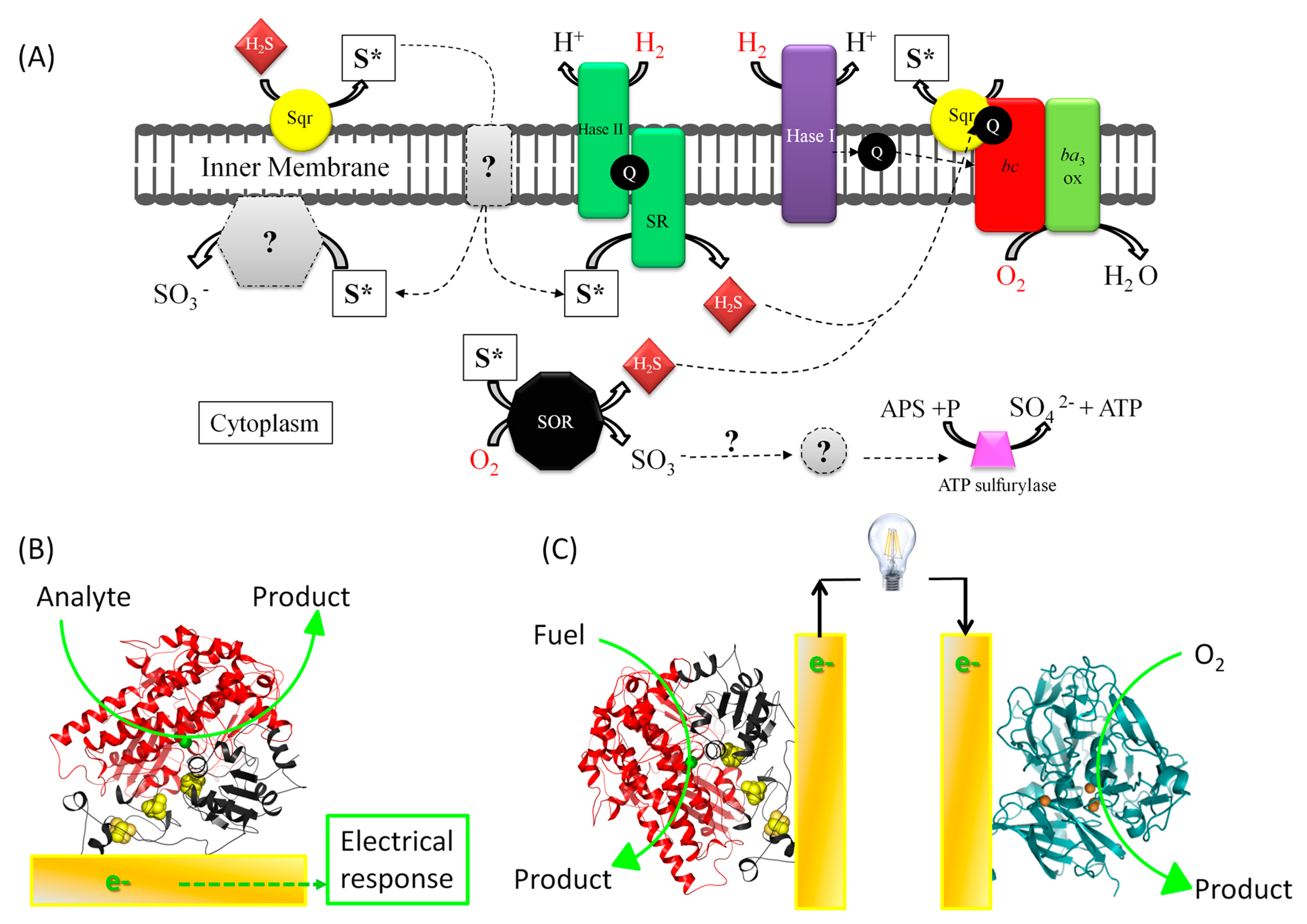{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: Interest in and Limitation of Bioelectrocatalysis Based on Immobilized Redox Enzymes
1.1. Fundamental Issue: Understanding Energy Chains, and the Mechanism of Biocatalysis Involving Redox Enzymes
1.2. The Applicative Issue: Use of Redox Enzymes Immobilized on Solid Conductive Supports for Biosensors, Bioreactors, and Biofuel Cells
1.3. Limitations of Bioelectrocatalysis: Stability, Wiring, and Interrelationships
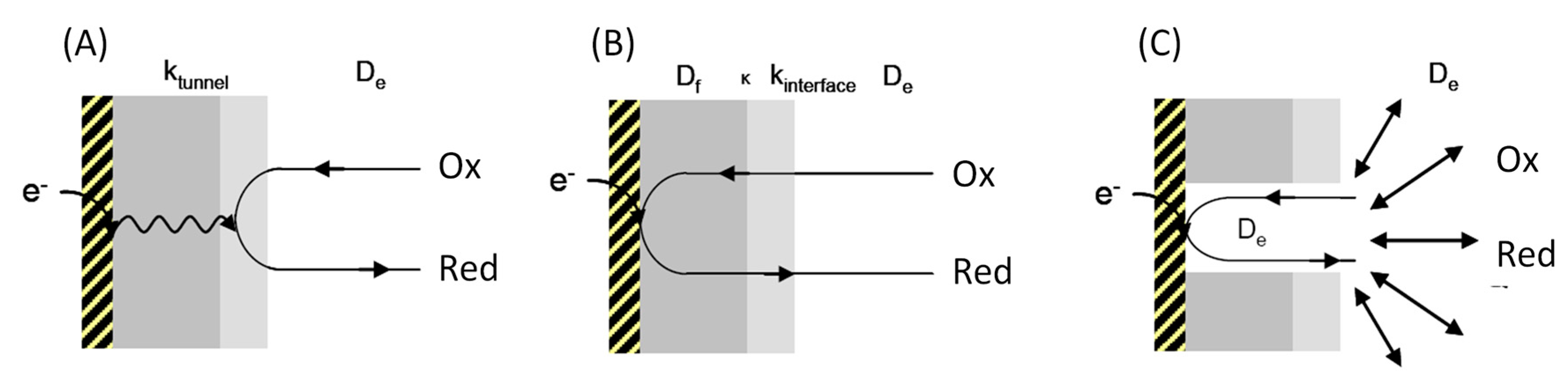
2. Interfacial Electron Transfer: Why Is Orientation a Key Issue?
3. The Key Components
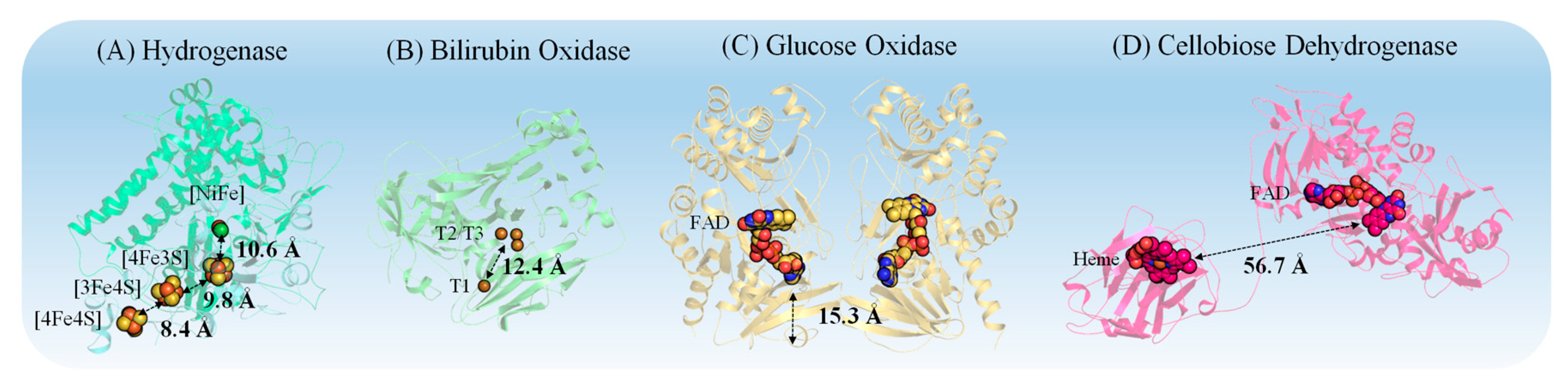
3.1. Properties of Redox Proteins
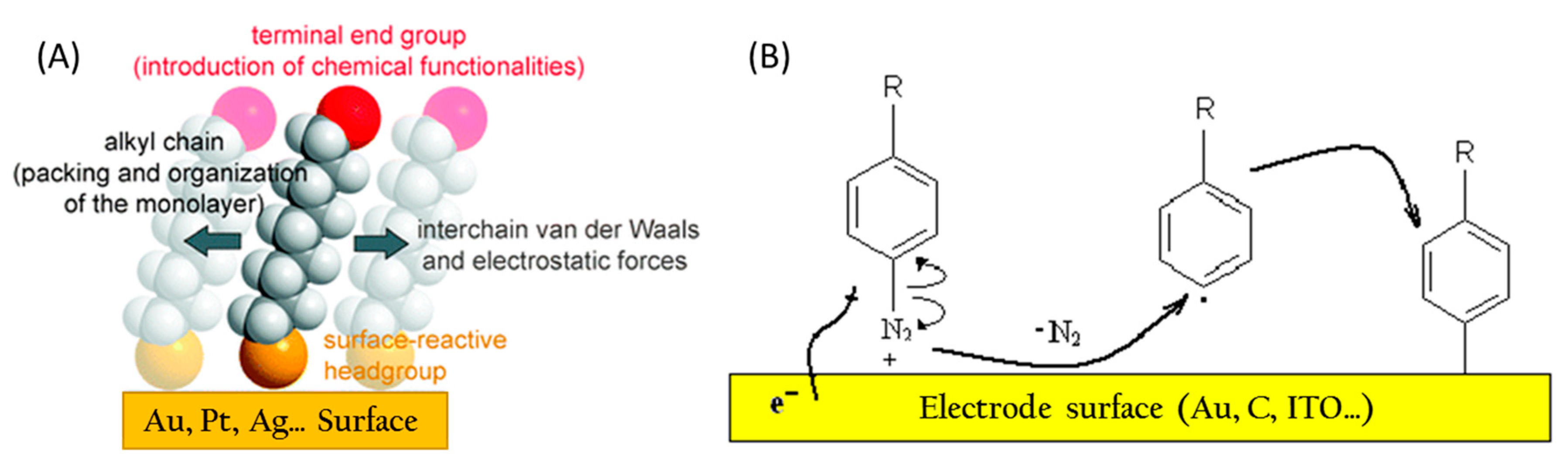
3.2. Conductive Electrode Surfaces
3.3. Interaction between Enzyme and Conductive Surface: How Can It Be Modulated?
3.3.1. Electrode Functionalization
3.3.2. Enzyme Engineering
4. How to Probe Enzyme Orientation on an Electrode
4.1. Electrochemistry
4.2. Spectroscopies
4.3. Microscopy
4.4. Modeling
5. Factors Driving the Oriented Immobilization at an Electrode
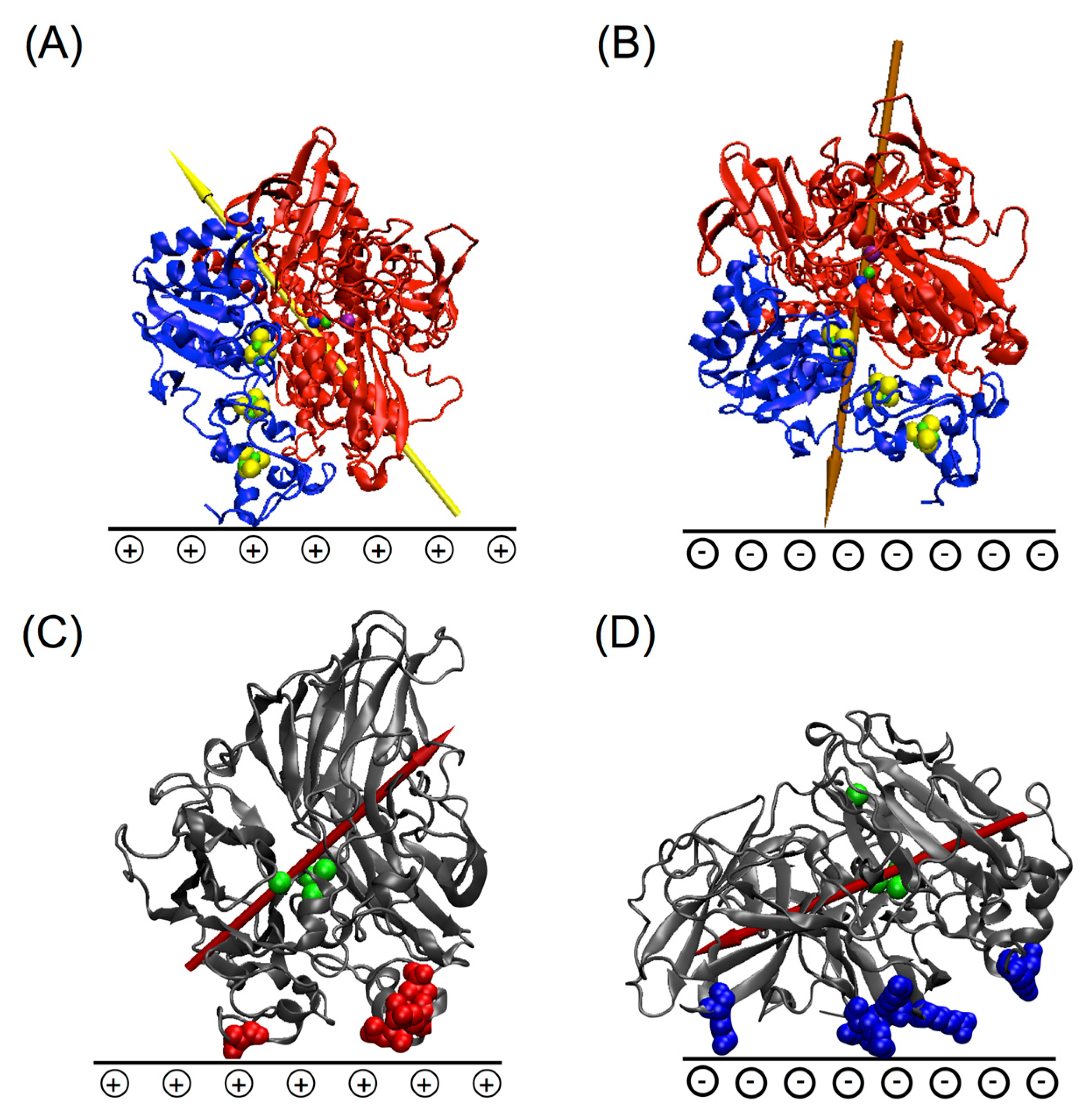
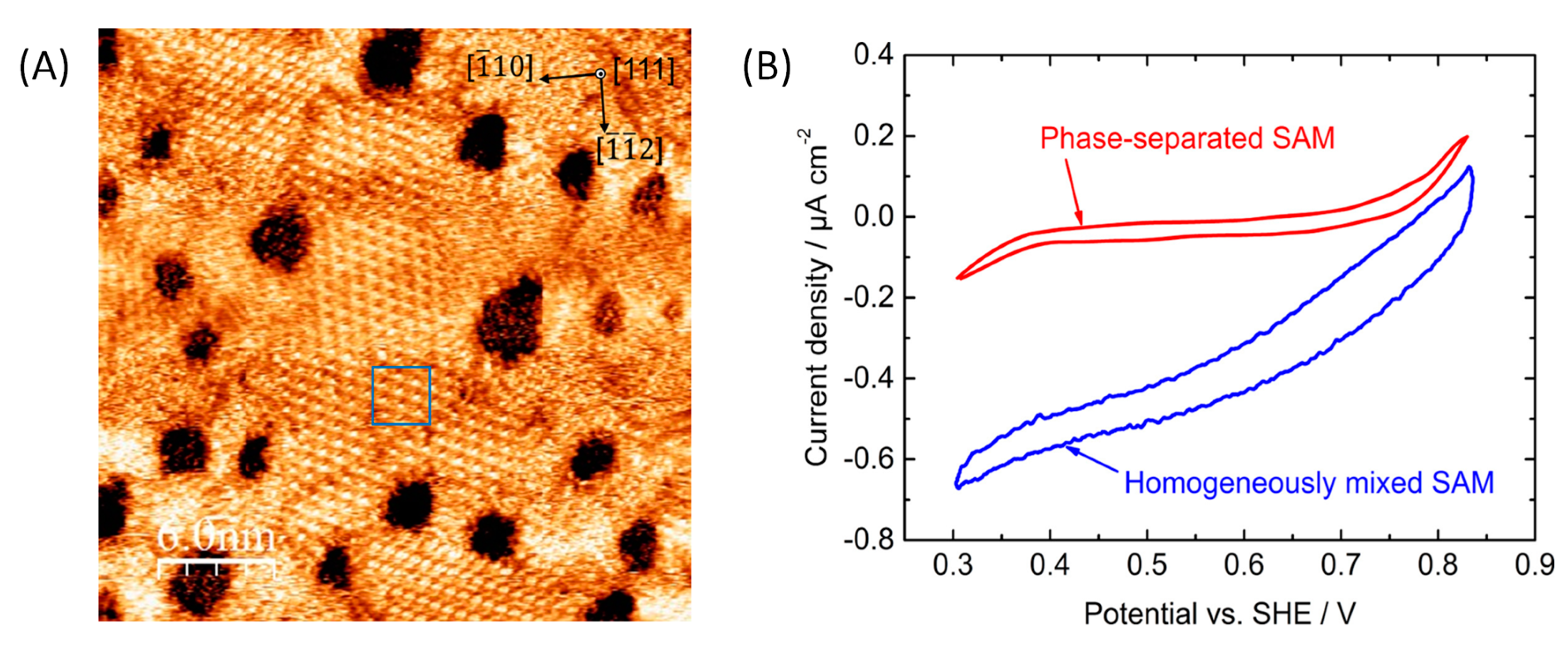
5.1. Importance of Electrostatic Interactions to Drive the Oriented Immobilization for Fast ET
5.2. Effect of Covalent Attachment
5.3. Effect of Enzyme Partition
5.4. Effect of Potential, Electric Field
6. Future Directions: Towards Rational Bioelectrode Design
Acknowledgements
Conflicts of Interest
References
- Mellor, S.B.; Vavitsas, K.; Nielsen, A.Z.; Jensen, P.E. Photosynthetic fuel for heterologous enzymes: The role of electron carrier proteins. Photosynth. Res. 2017, 134, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Ilbert, M.; Bonnefoy, V. Insight into the evolution of the iron oxidation pathways. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Guiral, M.; Aussignargues, C.; Prunetti, L.; Infossi, P.; Ilbert, M.; Giudici-Orticoni, M.T. The energy sulfur metabolism of the hyperthermophilic bacterium Aquifex aeolicus. Biochim. Biophys. Acta Bioenerg. 2012, 1817, S155. [Google Scholar] [CrossRef][Green Version]
- Moehlenbrock, M.J.; Minteer, S.D. Extended lifetime biofuel cells. Chem. Soc. Rev. 2008, 37, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Maduraiveeran, G.; Sasidharan, M.; Ganesan, V. Electrochemical sensor and biosensor platforms based on advanced nanomaterials for biological and biomedical applications. Biosens. Bioelectron. 2018, 103, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Justino, C.I.L.; Duarte, A.C.; Rocha-Santos, T.A.P. Recent Progress in Biosensors for Environmental Monitoring: A review. Sensors 2017, 17, 2918. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.C.; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. Ann. N. Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Bandodkar, A.J.; Imani, S.; Nunez-Flores, R.; Kumar, R.; Wang, C.Y.; Mohan, A.M.V.; Wang, J.; Mercier, P.P. Re-usable electrochemical glucose sensors integrated into a smartphone platform. Biosens. Bioelectron. 2018, 101, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Bruen, D.; Delaney, C.; Florea, L.; Diamond, D. Glucose Sensing for Diabetes Monitoring: Recent Developments. Sensors 2017, 17, 1866. [Google Scholar] [CrossRef] [PubMed]
- Bandodkar, A.J.; Wang, J. Non-invasive wearable electrochemical sensors: A review. Trends Biotechnol. 2014, 32, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Santharaman, P.; Venkatesh, K.A.; Vairamani, K.; Benjamin, A.R.; Sethy, N.K.; Bhargava, K.; Karunakaran, C. ARM-microcontroller based portable nitrite electrochemical analyzer using cytochrome c reductase biofunctionalized onto screen printed carbon electrode. Biosens. Bioelectron. 2017, 90, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Tan, J.; Cui, L.J.; Zhou, Z.D.; Zhou, S.F.; Zhang, Z.H.; Zheng, R.; Xue, Y.W.; Zhang, M.X.; Li, S.S.; et al. Graphene and Au NPs co-mediated enzymatic silver deposition for the ultrasensitive electrochemical detection of cholesterol. Biosens. Bioelectron. 2018, 102, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Jakhar, S.; Pundir, C.S. Preparation, characterization and application of urease nanoparticles for construction of an improved potentiometric urea biosensor. Biosens. Bioelectron. 2018, 100, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Anik, U.; Tepeli, Y.; Diouani, M.F. Fabrication of Electrochemical Model Influenza A Virus Biosensor Based on the Measurements of Neuroaminidase Enzyme Activity. Anal. Chem. 2016, 88, 6151–6153. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Dangi, A.K.; Shukla, P. Contemporary enzyme based technologies for bioremediation: A review. J. Environ. Manag. 2018, 210, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.S.; Armiger, W.B.; Dodds, D.R.; Dordick, J.S.; Koffas, M.A.G. Improved strategies for electrochemical 1,4-NAD(P)H2 regeneration: A new era of bioreactors for industrial biocatalysis. Biotechnol. Adv. 2018, 36, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Bajracharya, S.; Srikanth, S.; Mohanakrishna, G.; Zacharia, R.; Strik, D.; Pant, D. Biotransformation of carbon dioxide in bioelectrochemical systems: State of the art and future prospects. J. Power Sources 2017, 356, 256–273. [Google Scholar] [CrossRef]
- Hadj-Said, J.; Pandelia, M.E.; Leger, C.; Fourmond, V.; Dementin, S. The Carbon Monoxide Dehydrogenase from Desulfovibrio vulgaris. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Gamella, M.; Koushanpour, A.; Katz, E. Biofuel cells—Activation of micro- and macro-electronic devices. Bioelectrochemistry 2018, 119, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Mazurenko, I.; Wang, X.; de Poulpiquet, A.; Lojou, E. H2/O2 enzymatic fuel cells: From proof-of-concept to powerful devices. Sustain. Energy Fuels 2017, 1, 1475–1501. [Google Scholar] [CrossRef]
- Mazurenko, I.; Monsalve, K.; Infossi, P.; Giudici-Orticoni, M.T.; Topin, F.; Mano, N.; Lojou, E. Impact of substrate diffusion and enzyme distribution in 3D-porous electrodes: A combined electrochemical and modelling study of a thermostable H2/O2 enzymatic fuel cell. Energy Environ. Sci. 2017, 10, 1966–1982. [Google Scholar] [CrossRef]
- Hoarau, M.; Badieyan, S.; Marsh, E.N.G. Immobilized enzymes: Understanding enzyme—Surface interactions at the molecular level. Org. Biomol. Chem. 2017, 15, 9539–9551. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.R.; Gray, H.B. Electron flow through metalloproteins. Chem. Rev. 2013, 114, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Moser, C.C.; Keske, J.M.; Warncke, K.; Farid, R.S.; Dutton, P.L. Nature of biological electron-transfer. Nature 1992, 355, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Bostick, C.D.; Mukhopadhyay, S.; Pecht, I.; Sheves, M.; Cahen, D.; Lederman, D. Protein bioelectronics: A review of what we do and do not know. Rep. Prog. Phys. 2018, 81, 026601. [Google Scholar] [CrossRef] [PubMed]
- Page, C.C.; Moser, C.C.; Chen, X.X.; Dutton, P.L. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature 1999, 402, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Ozboyaci, M.; Kokh, D.B.; Corni, S.; Wade, R.C. Modeling and simulation of protein-surface interactions: Achievements and challenges. Q. Rev. Biophys. 2016, 49, e4. [Google Scholar] [CrossRef] [PubMed]
- Léger, C.; Bertrand, P. Direct electrochemistry of redox enzymes as a tool for mechanistic studies. Chem. Rev. 2008, 108, 2379–2438. [Google Scholar] [CrossRef] [PubMed]
- Mazurenko, I.; Monsalve, K.; Rouhana, J.; Parent, P.; Laffon, C.; Goff, A.L.; Szunerits, S.; Boukherroub, R.; Giudici-Orticoni, M.-T.; Mano, N. How the intricate interactions between carbon nanotubes and two bilirubin oxidases control direct and mediated O2 reduction. ACS Appl. Mater. Interfaces 2016, 8, 23074–23085. [Google Scholar] [CrossRef] [PubMed]
- Oteri, F.; Baaden, M.; Lojou, E.; Sacquin-Mora, S. Multiscale simulations give insight into the hydrogen in and out pathways of [NiFe]-hydrogenases from Aquifex aeolicus and Desulfovibrio fructosovorans. J. Phys. Chem. B 2014, 118, 13800–13811. [Google Scholar] [CrossRef] [PubMed]
- Ogorzalek, T.L.; Wei, S.; Liu, Y.; Wang, Q.; Brooks, C.L., III; Chen, Z.; Marsh, E.N.G. Molecular-level insights into orientation-dependent changes in the thermal stability of enzymes covalently immobilized on surfaces. Langmuir 2015, 31, 6145–6153. [Google Scholar] [CrossRef] [PubMed]
- Mano, N.; de Poulpiquet, A. O2 Reduction in Enzymatic Biofuel Cells. Chem. Rev. 2018, 118, 2392–2468. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Kataoka, K. Basic and applied features of multicopper oxidases, CueO, bilirubin oxidase, and laccase. Chem. Rec. 2007, 7, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Augustine, A.J.; Yoon, J. O2 Reduction to H2O by the multicopper oxidases. Dalton Trans. 2008, 30, 3921–3932. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Fontecilla-Camps, J.C.; Volbeda, A.; Cavazza, C.; Nicolet, Y. Structure/function relationships of [NiFe]-and [FeFe]-hydrogenases. Chem. Rev. 2007, 107, 4273–4303. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.-H.; Best, R.B.; Blumberger, J. Multiscale simulation reveals multiple pathways for H2 and O2 transport in a [NiFe]-hydrogenase. J. Am. Chem. Soc. 2011, 133, 3548–3556. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Turner, A. Glucose oxidase: An ideal enzyme. Biosens. Bioelectron. 1992, 7, 165–185. [Google Scholar] [CrossRef]
- Hecht, H.; Schomburg, D.; Kalisz, H.; Schmid, R. The 3D structure of glucose oxidase from Aspergillus niger. Implications for the use of GOD as a biosensor enzyme. Biosens. Bioelectron. 1993, 8, 197–203. [Google Scholar] [CrossRef]
- Zebda, A.; Gondran, C.; Le Goff, A.; Holzinger, M.; Cinquin, P.; Cosnier, S. Mediatorless high-power glucose biofuel cells based on compressed carbon nanotube-enzyme electrodes. Nat. Commun. 2011, 2, 370. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.; Ortiz, R.; Schulz, C.; Harreither, W.; Sygmund, C.; Gorton, L. Cellobiose dehydrogenase modified electrodes: Advances by materials science and biochemical engineering. Anal. Bioanal. Chem. 2013, 405, 3637–3658. [Google Scholar] [CrossRef] [PubMed]
- Secundo, F. Conformational changes of enzymes upon immobilisation. Chem. Soc. Rev. 2013, 42, 6250–6261. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Nishikawa, K.; Higuchi, Y.; Yamamoto, M.; Kano, K. Electrostatic roles in electron transfer from NiFe hydrogenase to cytochrome c3 from Desulfovibrio vulgaris Miyazaki F. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Shinzawa-Itoh, K.; Baba, J.; Aoe, S.; Shimada, A.; Yamashita, E.; Kang, J.Y.; Tateno, M.; Yoshikawa, S.; Tsukihara, T. Complex structure of cytochrome c-cytochrome c oxidase reveals a novel protein-protein interaction mode. EMBO J. 2017, 36, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kinoshita, M.; Kume, S.; Gt, H.; Sugiki, T.; Ladbury, J.E.; Kojima, C.; Ikegami, T.; Kurisu, G.; Goto, Y.; et al. Non-covalent forces tune the electron transfer complex between ferredoxin and sulfite reductase to optimize enzymatic activity. Biochem. J. 2016, 473, 3837–3854. [Google Scholar] [CrossRef] [PubMed]
- Andralojc, W.; Hiruma, Y.; Liu, W.M.; Ravera, E.; Nojiri, M.; Parigi, G.; Luchinat, C.; Ubbink, M. Identification of productive and futile encounters in an electron transfer protein complex. Proc. Natl. Acad. Sci. USA 2017, 114, E1840–E1847. [Google Scholar] [CrossRef] [PubMed]
- Kollipara, S.; Tatireddy, S.; Pathirathne, T.; Rathnayake, L.K.; Northrup, S.H. Contribution of Electrostatics to the Kinetics of Electron Transfer from NADH-Cytochrome b5 Reductase to Fe(III)-Cytochrome b5. J. Phys. Chem. B 2016, 120, 8193–8207. [Google Scholar] [CrossRef] [PubMed]
- Cracknell, J.A.; Vincent, K.A.; Armstrong, F.A. Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis. Chem. Rev. 2008, 108, 2439–2461. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.E.; Prilusky, J.; Silman, I.; Sussman, J.L. A server and database for dipole moments of proteins. Nucleic Acids Res. 2007, 35, W512–W521. [Google Scholar] [CrossRef] [PubMed]
- Topin, J.; Rousset, M.; Antonczak, S.; Golebiowski, J. Kinetics and thermodynamics of gas diffusion in a NiFe hydrogenase. Proteins Struct. Funct. Bioinform. 2012, 80, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Narth, C.; Gillet, N.; Cailliez, F.; Lévy, B.; de la Lande, A.L. Electron transfer, decoherence, and protein dynamics: Insights from atomistic simulations. Acc. Chem Res. 2015, 48, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Mazurenko, I.; de Poulpiquet, A.; Lojou, E. Recent developments in high surface area bioelectrodes for enzymatic fuel cells. Curr. Opin. Electrochem. 2017, 5, 74–84. [Google Scholar] [CrossRef]
- Mazurenko, I.; Clément, R.; Byrne-Kodjabachian, D.; de Poulpiquet, A.; Tsujimura, S.; Lojou, E. Pore size effect of MgO-templated carbon on enzymatic H2 oxidation by the hyperthermophilic hydrogenase from Aquifex aeolicus. J. Electroanal. Chem. 2018, 812, 221–226. [Google Scholar] [CrossRef]
- Ash, P.A.; Liu, J.; Coutard, N.; Heidary, N.; Horch, M.; Gudim, I.; Simler, T.; Zebger, I.; Lenz, O.; Vincent, K.A. Electrochemical and Infrared Spectroscopic Studies Provide Insight into Reactions of the NiFe Regulatory Hydrogenase from Ralstonia eutropha with O2 and CO. J. Phys. Chem. B 2015, 119, 13807–13815. [Google Scholar] [CrossRef] [PubMed]
- Thorum, M.S.; Anderson, C.A.; Hatch, J.J.; Campbell, A.S.; Marshall, N.M.; Zimmerman, S.C.; Lu, Y.; Gewirth, A.A. Direct, Electrocatalytic Oxygen Reduction by Laccase on Anthracene-2-methanethiol-Modified Gold. J. Phys. Chem. Lett. 2010, 1, 2251–2254. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Niu, T.; You, X.; Wan, Z.; Kong, Q.; Bi, S. Studies on the effect of electrode pretreatment on the coverage of self-assembled monolayers of dodecanethiol on gold by electrochemical reductive desorption determination. Analyst 2011, 136, 5058–5063. [Google Scholar] [CrossRef] [PubMed]
- Pankratov, D.; Sotres, J.; Barrantes, A.; Arnebrant, T.; Shleev, S. Interfacial behavior and activity of laccase and bilirubin oxidase on bare gold surfaces. Langmuir 2014, 30, 2943–2951. [Google Scholar] [CrossRef] [PubMed]
- Trasatti, S.; Petrii, O. Real surface area measurements in electrochemistry. Pure Appl. Chem. 1991, 63, 711–734. [Google Scholar] [CrossRef]
- Monsalve, K.; Roger, M.; Gutierrez-Sanchez, C.; Ilbert, M.; Nitsche, S.; Byrne-Kodjabachian, D.; Marchi, V.; Lojou, E. Hydrogen bioelectrooxidation on gold nanoparticle-based electrodes modified by Aquifex aeolicus hydrogenase: Application to hydrogen/oxygen enzymatic biofuel cells. Bioelectrochemistry 2015, 106, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Sezer, M.; Millo, D.; Weidinger, I.M.; Zebger, I.; Hildebrandt, P. Analyzing the catalytic processes of immobilized redox enzymes by vibrational spectroscopies. IUBMB Life 2012, 64, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Buividas, R.; Fahim, N.; Juodkazytė, J.; Juodkazis, S. Novel method to determine the actual surface area of a laser-nanotextured sensor. Appl. Phys. A 2014, 114, 169–175. [Google Scholar] [CrossRef]
- Fang, S.-U.; Hsu, C.-L.; Hsu, T.-C.; Juang, M.-Y.; Liu, Y.-C. Surface roughness-correlated SERS effect on Au island-deposited substrate. J. Electroanal. Chem. 2015, 741, 127–133. [Google Scholar] [CrossRef]
- Mendelsohn, R.; Mao, G.; Flach, C.R. Infrared Reflection-Absorption Spectroscopy: Principles and Applications to Lipid-Protein Interaction in Langmuir Films. Biochim. Biophys. Acta 2010, 1798, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Dongmo, S.; Wittstock, G.; Christoffers, J.; Brand, I. In situ determination of potential-driven structural changes in a redox-active plumbagin polymer film on a glassy carbon electrode using PM IRRAS under electrochemical control. Electrochim. Acta 2017, 255, 298–308. [Google Scholar] [CrossRef]
- Heinz, H.; Vaia, R.; Farmer, B.; Naik, R. Accurate simulation of surfaces and interfaces of face-centered cubic metals using 12−6 and 9−6 Lennard-Jones potentials. J. Phys. Chem. C 2008, 112, 17281–17290. [Google Scholar] [CrossRef]
- Iori, F.; Di Felice, R.; Molinari, E.; Corni, S. GolP: An atomistic force-field to describe the interaction of proteins with Au (111) surfaces in water. J. Comput. Chem. 2009, 30, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.; Lin, T.-J.; Kishore Mishra, R.; Emami, F.S. Thermodynamically consistent force fields for the assembly of inorganic, organic, and biological nanostructures: The INTERFACE force field. Langmuir 2013, 29, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Kubiak-Ossowska, K.; Jachimska, B.; Mulheran, P.A. How Negatively Charged Proteins Adsorb to Negatively Charged Surfaces: A Molecular Dynamics Study of BSA Adsorption on Silica. J. Phys. Chem. B 2016, 120, 10463–10468. [Google Scholar] [CrossRef] [PubMed]
- Mücksch, C.; Urbassek, H.M. Molecular dynamics simulation of free and forced BSA adsorption on a hydrophobic graphite surface. Langmuir 2011, 27, 12938–12943. [Google Scholar] [CrossRef] [PubMed]
- Trohalaki, S.; Pachter, R.; Luckarift, H.; Johnson, G. Immobilization of the Laccases from Trametes versicolor and Streptomyces coelicolor on Single-wall Carbon Nanotube Electrodes: A Molecular Dynamics Study. Fuel Cells 2012, 12, 656–664. [Google Scholar] [CrossRef]
- Heinz, H.; Jha, K.C.; Luettmer-Strathmann, J.; Farmer, B.L.; Naik, R.R. Polarization at metal–biomolecular interfaces in solution. J. R. Soc. Interface 2011, 8, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.B.; Rodger, P.M.; Corni, S.; Walsh, T.R. GolP-CHARMM: First-principles based force fields for the interaction of proteins with Au (111) and Au (100). J. Chem. Theory Comput. 2013, 9, 1616–1630. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xia, Z.; Zhang, J.; Best, R.; Wu, C.; Ponder, J.W.; Ren, P. Polarizable atomic multipole-based AMOEBA force field for proteins. J. Chem. Theory Comput. 2013, 9, 4046–4063. [Google Scholar] [CrossRef] [PubMed]
- Akdim, B.; Pachter, R.; Kim, S.S.; Naik, R.R.; Walsh, T.R.; Trohalaki, S.; Hong, G.; Kuang, Z.; Farmer, B.L. Electronic properties of a graphene device with peptide adsorption: Insight from simulation. ACS Appl. Mater. Interfaces 2013, 5, 7470–7477. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Christena, L.R.; Rajaram, Y.R.S. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, N.R.; Marzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.K.; Prusty, A.K.; Singh, S.; Solanki, P.R.; Pandey, M.K.; Datta, M.; Malhotra, B.D. Cholesterol biosensor based on N-(2-aminoethyl)-3-aminopropyl-trimethoxysilane self-assembled monolayer. Anal. Biochem. 2007, 363, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Ulman, A. Formation and structure of self-assembled monolayers. Chem. Rev. 1996, 96, 1533–1554. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Wang, G.-X.; Millo, D.; Hildebrandt, P.; Xia, X.-H. Electric-field control of the pH-dependent redox process of cytochrome c immobilized on a gold electrode. J. Phys. Chem. C 2012, 116, 13038–13044. [Google Scholar] [CrossRef]
- Bryant, M.A.; Crooks, R.M. Determination of surface pKa values of surface-confined molecules derivatized with pH-sensitive pendant groups. Langmuir 1993, 9, 385–387. [Google Scholar] [CrossRef]
- Marmisollé, W.A.; Capdevila, D.A.; de la Llave, E.; Williams, F.J.; Murgida, D.H. Self-Assembled Monolayers of NH2-Terminated Thiolates: Order, pKa, and Specific Adsorption. Langmuir 2013, 29, 5351–5359. [Google Scholar] [CrossRef] [PubMed]
- Gooding, J.J.; Ciampi, S. The molecular level modification of surfaces: From self-assembled monolayers to complex molecular assemblies. Chem. Soc. Rev. 2011, 40, 2704–2718. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.H.; Nuzzo, R.G. Synthesis, structure, and properties of model organic surfaces. Ann. Rev. Phys. Chem. 1992, 43, 437–463. [Google Scholar] [CrossRef]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 2005, 105, 1103–1170. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.K. Mechanisms and kinetics of self-assembled monolayer formation. Ann. Rev. Phys. Chem. 2001, 52, 107–137. [Google Scholar] [CrossRef] [PubMed]
- Porter, M.D.; Bright, T.B.; Allara, D.L.; Chidsey, C.E. Spontaneously organized molecular assemblies. 4. Structural characterization of n-alkyl thiol monolayers on gold by optical ellipsometry, infrared spectroscopy, and electrochemistry. J. Am. Chem. Soc. 1987, 109, 3559–3568. [Google Scholar] [CrossRef]
- Jambrec, D.; Conzuelo, F.; Estrada-Vargas, A.; Schuhmann, W. Potential-Pulse-Assisted Formation of Thiol Monolayers within Minutes for Fast and Controlled Electrode Surface Modification. ChemElectroChem 2016, 3, 1484–1489. [Google Scholar] [CrossRef]
- Samanta, D.; Sarkar, A. Immobilization of bio-macromolecules on self-assembled monolayers: Methods and sensor applications. Chem. Soc. Rev. 2011, 40, 2567–2592. [Google Scholar] [CrossRef] [PubMed]
- Mondal, P.C.; Fontanesi, C. Electrochemistry of Metalloproteins Attached through Functional Self-Assembled Monolayers on Gold and Ferromagnetic Electrodes. ChemPhysChem 2018, 19, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Aydın, E.B.; Sezgintürk, M.K. Indium Tin Oxide (ITO): A promising material in biosensing technology. TrAC Trends Anal. Chem. 2017, 97, 309–315. [Google Scholar] [CrossRef]
- Vashist, S.K.; Lam, E.; Hrapovic, S.; Male, K.B.; Luong, J.H. Immobilization of antibodies and enzymes on 3-aminopropyltriethoxysilane-functionalized bioanalytical platforms for biosensors and diagnostics. Chem. Rev. 2014, 114, 11083–11130. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Kato, D.; Kamata, T.; Niwa, O. Cytochrome P450 modified polycrystalline indium tin oxide film as a drug metabolizing electrochemical biosensor with a simple configuration. Anal. Chem. 2013, 85, 9996–9999. [Google Scholar] [CrossRef] [PubMed]
- Smalley, J.F.; Feldberg, S.W.; Chidsey, C.E.; Linford, M.R.; Newton, M.D.; Liu, Y.-P. The kinetics of electron transfer through ferrocene-terminated alkanethiol monolayers on gold. J. Phys. Chem. 1995, 99, 13141–13149. [Google Scholar] [CrossRef]
- Xu, J.; Li, H.; Zhang, Y. Relationship between electronic tunneling coefficient and electrode potential investigated by using self-assembled alkanethiol monolayers on gold electrodes. J. Phys. Chem. 1993, 97, 11497–11500. [Google Scholar] [CrossRef]
- Mokrani, C.; Fatisson, J.; Guerente, L.; Labbe, P. Structural characterization of (3-mercaptopropyl)sulfonate monolayer on gold surfaces. Langmuir 2005, 21, 4400–4409. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Bard, A.J.; Mirkin, M.V.; Creager, S.E. Electron transfer at self-assembled monolayers measured by scanning electrochemical microscopy. J. Am. Chem. Soc. 2004, 126, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Chi, Q.J.; Zhang, J.D.; Andersen, J.E.T.; Ulstrup, J. Ordered assembly and controlled electron transfer of the blue copper protein azurin at gold (111) single-crystal substrates. J. Phys. Chem. B 2001, 105, 4669–4679. [Google Scholar] [CrossRef]
- Beulen, M.W.; Kastenberg, M.I.; van Veggel, F.C.; Reinhoudt, D.N. Electrochemical stability of self-assembled monolayers on gold. Langmuir 1998, 14, 7463–7467. [Google Scholar] [CrossRef]
- Ovchinnikova, S.; Medvedev, A.Z. Desorption of octanethiol from gold electrode surface during its electrochemical cleaning. Russ. J. Electrochem. 2015, 51, 287–293. [Google Scholar] [CrossRef]
- Stettner, J.; Winkler, A. Characterization of alkanethiol self-assembled monolayers on gold by thermal desorption spectroscopy. Langmuir 2010, 26, 9659–9665. [Google Scholar] [CrossRef] [PubMed]
- Rzeźnicka, I.I.; Lee, J.; Maksymovych, P.; Yates, J.T. Nondissociative chemisorption of short chain alkanethiols on Au (111). J. Phys. Chem. B 2005, 109, 15992–15996. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, J.; Nascimbeni, G.; Żaba, T.; Verwüster, E.; Rysz, J.; Terfort, A.; Zharnikov, M.; Zojer, E.; Cyganik, P. Relative Thermal Stability of Thiolate-and Selenolate-Bonded Aromatic Monolayers on the Au (111) Substrate. J. Phys. Chem. C 2017, 121, 28031–28042. [Google Scholar] [CrossRef]
- Wang, Y.; Solano Canchaya, J.G.; Dong, W.; Alcami, M.; Busnengo, H.F.; Martin, F. Chain-Length and Temperature Dependence of Self-Assembled Monolayers of Alkylthiolates on Au (111) and Ag (111) Surfaces. J. Phys. Chem. A 2014, 118, 4138–4146. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Ma, L.; Zhang, Y.; Cheng, X.; Xu, Y.; Wang, J.; Wang, E.; Peng, Z. Spectroscopic Identification of the Au–C Bond Formation upon Electroreduction of an Aryl Diazonium Salt on Gold. Langmuir 2016, 32, 11514–11519. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Mascia, M.; Rizzardini, S.; Palmas, S.; Mais, L. Coating of gold substrates with polyaniline through electrografting of aryl diazonium salts. Electrochim. Acta 2014, 126, 81–89. [Google Scholar] [CrossRef]
- Olejnik, P.; Palys, B.; Kowalczyk, A.; Nowicka, A.M. Orientation of laccase on charged surfaces. Mediatorless oxygen reduction on amino-and carboxyl-ended ethylphenyl groups. J. Phys. Chem. C 2012, 116, 25911–25918. [Google Scholar] [CrossRef]
- Hetemi, D.; Pinson, J. Surface functionalisation of polymers. Chem. Soc. Rev. 2017, 46, 5701–5713. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Ghilane, J.; Lacroix, J.C. Formation of mixed organic layers by stepwise electrochemical reduction of diazonium compounds. J. Am. Chem. Soc. 2012, 134, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Mattiuzzi, A.; Jabin, I.; Vandencasteele, N.; Reniers, F.O.; Reinaud, O.; Hapiot, P.; Lhenry, S.B.; Leroux, Y.; Lagrost, C. One-Pot Electrografting of Mixed Monolayers with Controlled Composition. J. Phys. Chem. C 2014, 118, 15919–15928. [Google Scholar] [CrossRef]
- Leroux, Y.R.; Hapiot, P. Nanostructured monolayers on carbon substrates prepared by electrografting of protected aryldiazonium salts. Chem. Mater. 2013, 25, 489–495. [Google Scholar] [CrossRef]
- Zhang, X.; Rösicke, F.; Syritski, V.; Sun, G.; Reut, J.; Hinrichs, K.; Janietz, S.; Rappich, J. Influence of the para-substitutent of benzene diazonium salts and the solvent on the film growth during electrochemical reduction. Z. Phys. Chem. 2014, 228, 557–573. [Google Scholar] [CrossRef]
- Bouden, S.; Pinson, J.; Vautrin-Ul, C. Electrografting of diazonium salts: A kinetics study. Electrochem. Commun. 2017, 81, 120–123. [Google Scholar] [CrossRef]
- Pita, M.; Gutierrez-Sanchez, C.; Olea, D.; Velez, M.; Garcia-Diego, C.; Shleev, S.; Fernandez, V.M.; De Lacey, A.L. High Redox Potential Cathode Based on Laccase Covalently Attached to Gold Electrode. J. Phys. Chem. C 2011, 115, 13420–13428. [Google Scholar] [CrossRef]
- Alam, M.T.; Gooding, J.J. Modification of carbon electrode surfaces. In Electrochemistry of Carbon Electrodes; Wiley: Hoboken, NJ, USA, 2016; Volume 16. [Google Scholar]
- Cracknell, J.A.; McNamara, T.P.; Lowe, E.D.; Blanford, C.F. Bilirubin oxidase from Myrothecium verrucaria: X-ray determination of the complete crystal structure and a rational surface modification for enhanced electrocatalytic O2 reduction. Dalton Trans. 2011, 40, 6668–6675. [Google Scholar] [CrossRef] [PubMed]
- Wiebalck, S.; Kozuch, J.; Forbrig, E.; Tzschucke, C.C.; Jeuken, L.J.C.; Hildebrandt, P. Monitoring the Transmembrane Proton Gradient Generated by Cytochrome bo3 in Tethered Bilayer Lipid Membranes Using SEIRA Spectroscopy. J. Phys. Chem. B 2016, 120, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.Y.; Hutchings, C.H.; Lindsay, M.J.; Werner, C.J.; Bundy, B.C. Enhanced enzyme stability through site-directed covalent immobilization. J. Biotechnol. 2015, 193, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Rueda, N.; dos Santos, J.; Ortiz, C.; Torres, R.; Barbosa, O.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R. Chemical modification in the design of immobilized enzyme biocatalysts: Drawbacks and opportunities. Chem. Rec. 2016, 16, 1436–1455. [Google Scholar] [CrossRef] [PubMed]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Mutation of heme c axial ligands in d-fructose dehydrogenase for investigation of electron transfer pathways and reduction of overpotential in direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2016, 67, 43–46. [Google Scholar] [CrossRef]
- Balland, V.; Hureau, C.; Cusano, A.M.; Liu, Y.; Tron, T.; Limoges, B. Oriented Immobilization of a Fully Active Monolayer of Histidine-Tagged Recombinant Laccase on Modified Gold Electrodes. Chemistry 2008, 14, 7186–7192. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, S.; Asahi, M.; Goda-Tsutsumi, M.; Shirai, O.; Kano, K.; Miyazaki, K. Direct electron transfer to a metagenome-derived laccase fused to affinity tags near the electroactive copper site. Phys. Chem. Chem. Phys. 2013, 15, 20585–20589. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, J.; Huang, X.; Wang, T. Construction and direct electrochemistry of orientation controlled laccase electrode. Biochem. Biophys. Res. Commun. 2014, 446, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Care, A.; Bergquist, P.L.; Sunna, A. Solid-binding peptides: Smart tools for nanobiotechnology. Trends Biotechnol. 2015, 33, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Shiba, K. Natural and artificial peptide motifs: Their origins and the application of motif-programming. Chem. Soc. Rev. 2010, 39, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Kim, S.N.; Jones, S.E.; Wissler, L.L.; Naik, R.R.; McAlpine, M.C. Chemical Functionalization of Graphene Enabled by Phage Displayed Peptides. Nano Lett. 2010, 10, 4559–4565. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.Q.; Botyanszki, Z.; Tay, P.K.R.; Joshi, N.S. Programmable biofilm-based materials from engineered curli nanofibres. Nat. Commun. 2014, 5, 4945. [Google Scholar] [CrossRef] [PubMed]
- Hnilova, M.; Oren, E.E.; Seker, U.O.S.; Wilson, B.R.; Collino, S.; Evans, J.S.; Tamerler, C.; Sarikaya, M. Effect of Molecular Conformations on the Adsorption Behavior of Gold-Binding Peptides. Langmuir 2008, 24, 12440–12445. [Google Scholar] [CrossRef] [PubMed]
- Khatayevich, D.; Gungormus, M.; Yazici, H.; So, C.; Cetinel, S.; Ma, H.; Jen, A.; Tamerler, C.; Sarikaya, M. Biofunctionalization of materials for implants using engineered peptides. Acta Biomater. 2010, 6, 4634–4641. [Google Scholar] [CrossRef] [PubMed]
- Al-Lolage, F.A.; Meneghello, M.; Ma, S.; Ludwig, R.; Bartlett, P.N. A flexible method for the stable, covalent immobilization of enzymes at electrode surfaces. ChemElectroChem 2017, 4, 1528–1534. [Google Scholar] [CrossRef]
- Lalaoui, N.M.; Rousselot-Pailley, P.; Robert, V.; Mekmouche, Y.; Villalonga, R.; Holzinger, M.; Cosnier, S.; Tron, T.; Le Goff, A. Direct electron transfer between a site-specific pyrene-modified laccase and carbon nanotube/gold nanoparticle supramolecular assemblies for bioelectrocatalytic dioxygen reduction. ACS Catal. 2016, 6, 1894–1900. [Google Scholar] [CrossRef]
- Gao, X.; Ni, K.; Zhao, C.; Ren, Y.; Wei, D. Enhancement of the activity of enzyme immobilized on polydopamine-coated iron oxide nanoparticles by rational orientation of formate dehydrogenase. J. Biotechnol. 2014, 188, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Kurra, Y.; Liu, W.; Chen, Z. A click chemistry approach to site-specific immobilization of a small laccase enables efficient direct electron transfer in a biocathode. Chem. Commun. 2015, 51, 2522–2525. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, O.; Pasi, M.; Dandela, R.; Meijler, M.M.; Alfonta, L. Electron transfer rate analysis of a site-specifically wired copper oxidase. Phys. Chem. Chem. Phys. 2018, 20, 6159–6166. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.L.; Martin, L.L. Controlling protein orientation at interfaces using histidine tags: An alternative to Ni/NTA. J. Am. Chem. Soc. 2005, 127, 2018–2019. [Google Scholar] [CrossRef] [PubMed]
- Milton, R.D.; Minteer, S.D. Direct enzymatic bioelectrocatalysis: Differentiating between myth and reality. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599. [Google Scholar] [CrossRef]
- Lojou, E.; Cutruzzola, F.; Tegoni, M.; Bianco, P. Electrochemical study of the intermolecular electron transfer to Pseudomonas aeruginosa cytochrome cd1 nitrite reductase. Electrochim. Acta 2003, 48, 1055–1064. [Google Scholar] [CrossRef]
- Dos Santos, M.M.C.; de Sousa, P.M.P.; Goncalves, M.L.S.; Krippahl, L.; Moura, J.J.G.; Lojou, E.; Bianco, P. Electrochemical studies on small electron transfer proteins using membrane electrodes. J. Electroanal. Chem. 2003, 541, 153–162. [Google Scholar] [CrossRef]
- Pedroso, H.A.; Silveira, C.M.; Almeida, R.M.; Almeida, A.; Besson, S.; Moura, I.; Moura, J.J.G.; Almeida, M.G. Electron transfer and docking between cytochrome cd1 nitrite reductase and different redox partners—A comparative study. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Dagys, M.; Laurynėnas, A.; Ratautas, D.; Kulys, J.; Vidžiūnaitė, R.; Talaikis, M.; Niaura, G.; Marcinkevičienė, L.; Meškys, R.; Shleev, S. Oxygen electroreduction catalysed by laccase wired to gold nanoparticles via the trinuclear copper cluster. Energy Environ. Sci. 2017, 10, 498–502. [Google Scholar] [CrossRef]
- Ciaccafava, A.; Infossi, P.; Ilbert, M.; Guiral, M.; Lecomte, S.; Giudici-Orticoni, M.T.; Lojou, E. Electrochemistry, AFM, and PM-IRRA Spectroscopy of Immobilized Hydrogenase: Role of a Hydrophobic Helix in Enzyme Orientation for Efficient H2 Oxidation. Angew. Chem. Int. Ed. 2012, 51, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Rudiger, O.; Gutierrez-Sanchez, C.; Olea, D.; Pereira, I.A.C.; Velez, M.; Fernandez, V.M.; De Lacey, A.L. Enzymatic Anodes for Hydrogen Fuel Cells based on Covalent Attachment of Ni-Fe Hydrogenases and Direct Electron Transfer to SAM-Modified Gold Electrodes. Electroanalysis 2010, 22, 776–783. [Google Scholar] [CrossRef]
- Vaz-Dominguez, C.; Pita, M.; de Lacey, A.L.; Shleev, S.; Cuesta, A. Combined ATR-SEIRAS and EC-STM Study of the Immobilization of Laccase on Chemically Modified Au Electrodes. J. Phys. Chem. C 2012, 116, 16532–16540. [Google Scholar] [CrossRef]
- Gutierrez-Sanchez, C.; Ciaccafava, A.; Blanchard, P.Y.; Monsalve, K.; Giudici-Orticoni, M.T.; Lecomte, S.; Lojou, E. Efficiency of Enzymatic O2 Reduction by Myrothecium verrucaria Bilirubin Oxidase Probed by Surface Plasmon Resonance, PMIRRAS, and Electrochemistry. ACS Catal. 2016, 6, 5482–5492. [Google Scholar] [CrossRef]
- McArdle, T.; McNamara, T.P.; Fei, F.; Singh, K.; Blanford, C.F. Optimizing the Mass-Specific Activity of Bilirubin Oxidase Adlayers through Combined Electrochemical Quartz Crystal Microbalance and Dual Polarization Interferometry Analyses. ACS Appl. Mater. Interfaces 2015, 7, 25270–25280. [Google Scholar] [CrossRef] [PubMed]
- Osawa, M.; Ataka, K.-I.; Yoshii, K.; Nishikawa, Y. Surface-Enhanced Infrared Spectroscopy: The Origin of the Absorption Enhancement and Band Selection Rule in the Infrared Spectra of Molecules Adsorbed on Fine Metal Particles. Appl. Spectrosc. 1993, 47, 1497–1502. [Google Scholar] [CrossRef]
- Jiang, X.; Zaitseva, E.; Schmidt, M.; Siebert, F.; Engelhard, M.; Schlesinger, R.; Ataka, K.; Vogel, R.; Heberle, J. Resolving voltage-dependent structural changes of a membrane photoreceptor by surface-enhanced IR difference spectroscopy. Proc. Natl. Acad. Sci. USA 2008, 105, 12113–12117. [Google Scholar] [CrossRef] [PubMed]
- Heidary, N.; Utesch, T.; Zerball, M.; Horch, M.; Millo, D.; Fritsch, J.; Lenz, O.; von Klitzing, R.; Hildebrandt, P.; Fischer, A.; et al. Orientation-controlled electrocatalytic efficiency of an adsorbed oxygen-tolerant hydrogenase. PLoS ONE 2015, 10, e0143101. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Sanz, O.; Marques, M.; Pereira, I.A.C.; De Lacey, A.L.; Lubitz, W.; Rüdiger, O. Orientation and Function of a Membrane-Bound Enzyme Monitored by Electrochemical Surface-Enhanced Infrared Absorption Spectroscopy. J. Phys. Chem. Lett. 2013, 4, 2794–2798. [Google Scholar] [CrossRef]
- Olejnik, P.; Pawłowska, A.; Pałys, B. Application of Polarization Modulated Infrared Reflection Absorption Spectroscopy for electrocatalytic activity studies of laccase adsorbed on modified gold electrodes. Electrochim. Acta 2013, 110, 105–111. [Google Scholar] [CrossRef]
- Bergkvist, M.; Carlsson, J.; Oscarsson, S. A method for studying protein orientation with atomic force microscopy using relative protein volumes. J. Phys. Chem. B 2001, 105, 2062–2069. [Google Scholar] [CrossRef]
- Traunsteiner, C.; Sek, S.; Huber, V.; Valero-Vidal, C.; Kunze-Liebhaeuser, J. Laccase immobilized on a mixed thiol monolayer on Au (111)—Structure-dependent activity towards oxygen reduction. Electrochim. Acta 2016, 213, 761–770. [Google Scholar] [CrossRef]
- Gwyer, J.D.; Zhang, J.; Butt, J.N.; Ulstrupy, J. Voltammetry and in situ scanning tunneling microscopy of cytochrome c nitrite reductase on Au (111) electrodes. Biophys. J. 2006, 91, 3897–3906. [Google Scholar] [CrossRef] [PubMed]
- Kartashov, A.V.; Serafini, G.; Dong, M.; Shipovskov, S.; Gazaryan, I.; Besenbacher, F.; Ferapontova, E.E. Long-range electron transfer in recombinant peroxidases anisotropically orientated on gold electrodes. Phys. Chem. Chem. Phys. 2010, 12, 10098–10107. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Sanchez, C.; Olea, D.; Marques, M.; Fernandez, V.M.; Pereira, I.A.C.; Velez, M.; De Lacey, A.L. Oriented Immobilization of a Membrane-Bound Hydrogenase onto an Electrode for Direct Electron Transfer. Langmuir 2011, 27, 6449–6457. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Sanz, O.; Natale, P.; Marquez, I.; Marques, M.C.; Zacarias, S.; Pita, M.; Pereira, I.A.C.; Lopez-Montero, I.; De Lacey, A.L.; Velez, M. H-2-Fueled ATP Synthesis on an Electrode: Mimicking Cellular Respiration. Angew. Chem. Int. Ed. 2016, 55, 6216–6220. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Sanz, O.; Olea, D.; Pita, M.; Batista, A.P.; Alonso, A.; Pereira, M.M.; Velez, M.; De Lacey, A.L. Reconstitution of Respiratory Complex I on a Biomimetic Membrane Supported on Gold Electrodes. Langmuir 2014, 30, 9007–9015. [Google Scholar] [CrossRef] [PubMed]
- Matanovic, I.; Babanova, S.; Chavez, M.S.; Atanassov, P. Protein–Support Interactions for Rationally Designed Bilirubin Oxidase Based Cathode: A Computational Study. J. Phys. Chem. B 2016, 120, 3634–3641. [Google Scholar] [CrossRef] [PubMed]
- Ngai, J.C.; Mak, P.-I.; Siu, S.W. ProtPOS: A python package for the prediction of protein preferred orientation on a surface. Bioinformatics 2016, 32, 2537–2538. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, G.; Zhou, J. Ribonuclease A adsorption onto charged self-assembled monolayers: A multiscale simulation study. Chem. Eng. Sci. 2015, 121, 331–339. [Google Scholar] [CrossRef]
- Kutzner, C.; Pall, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmuller, H. Best bang for your buck: GPU nodes for GROMACS biomolecular simulations. J. Comput. Chem. 2015, 36, 1990–2008. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar] [PubMed]
- Makrodimitris, K.; Masica, D.L.; Kim, E.T.; Gray, J.J. Structure prediction of protein−solid surface interactions reveals a molecular recognition motif of statherin for hydroxyapatite. J. Am. Chem. Soc. 2007, 129, 13713–13722. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhou, J.; Jiang, S. Parallel tempering Monte Carlo simulations of lysozyme orientation on charged surfaces. J. Chem. Phys. 2010, 132, 02B602. [Google Scholar] [CrossRef] [PubMed]
- Brancolini, G.; Kokh, D.B.; Calzolai, L.; Wade, R.C.; Corni, S. Docking of ubiquitin to gold nanoparticles. ACS Nano 2012, 6, 9863–9878. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Li, L.; Chen, S.; Jiang, S. Molecular simulation study of water interactions with oligo (ethylene glycol)-terminated alkanethiol self-assembled monolayers. Langmuir 2004, 20, 8931–8938. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ogorzalek, T.L.; Wei, S.; Zhang, X.; Yang, P.; Jasensky, J.; Brooks, C.L.; Marsh, E.N.G.; Chen, Z. Effect of immobilization site on the orientation and activity of surface-tethered enzymes. Phys. Chem. Chem. Phys. 2018, 20, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Cazelles, R.; Lalaoui, N.; Hartmann, T.; Leimkühler, S.; Wollenberger, U.; Antonietti, M.; Cosnier, S. Ready to use bioinformatics analysis as a tool to predict immobilisation strategies for protein direct electron transfer (DET). Biosens. Bioelectron. 2016, 85, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Utesch, T.; Millo, D.; Castro, M.A.; Hildebrandt, P.; Zebger, I.; Mroginski, M.A. Effect of the protonation degree of a self-assembled monolayer on the immobilization dynamics of a [NiFe] hydrogenase. Langmuir 2013, 29, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Oteri, F.; Ciaccafava, A.; De Poulpiquet, A.; Baaden, M.; Lojou, E.; Sacquin-Mora, S. The weak, fluctuating, dipole moment of membrane-bound hydrogenase from Aquifex aeolicus accounts for its adaptability to charged electrodes. Phys. Chem. Chem. Phys. 2014, 16, 11318–11322. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xie, Y.; Peng, C.; Yu, G.; Zhou, J. Molecular Understanding of Laccase Adsorption on Charged Self-Assembled Monolayers. J. Phys. Chem. B 2017, 121, 10610–10617. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Utesch, T.; Yarman, A.; Jeoung, J.H.; Steinborn, S.; Dobbek, H.; Mroginski, M.A.; Tanne, J.; Wollenberger, U.; Scheller, F.W. Surface-Tuned Electron Transfer and Electrocatalysis of Hexameric Tyrosine-Coordinated Heme Protein. Chemistry 2015, 21, 7596–7602. [Google Scholar] [CrossRef] [PubMed]
- Utesch, T.; Sezer, M.; Weidinger, I.M.; Mroginski, M.A. Adsorption of sulfite oxidase on self-assembled monolayers from molecular dynamics simulations. Langmuir 2012, 28, 5761–5769. [Google Scholar] [CrossRef] [PubMed]
- Climent, V.; Zhang, J.; Friis, E.P.; Østergaard, L.H.; Ulstrup, J. Voltammetry and single-molecule in situ scanning tunneling microscopy of laccases and bilirubin oxidase in electrocatalytic dioxygen reduction on Au (111) single-crystal electrodes. J. Phys. Chem. C 2011, 116, 1232–1243. [Google Scholar] [CrossRef]
- Madden, C.; Vaughn, M.D.; Díez-Pérez, I.; Brown, K.A.; King, P.W.; Gust, D.; Moore, A.L.; Moore, T.A. Catalytic turnover of [FeFe]-hydrogenase based on single-molecule imaging. J. Am. Chem. Soc. 2011, 134, 1577–1582. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Xing, Y.; Wang, G.; Feng, Q.; Chen, Q.; Feng, H.; Sun, X.; Liu, L. Probing of EDC/NHSS-mediated covalent coupling reaction by the immobilization of electrochemically active biomolecules. Int. J. Electrochem. Sci. 2013, 8, 2459–2467. [Google Scholar]
- Palazon, F.; Montenegro Benavides, C.; Léonard, D.; Souteyrand, E.l.; Chevolot, Y.; Cloarec, J.-P. Carbodiimide/NHS derivatization of COOH-terminated SAMs: Activation or byproduct formation? Langmuir 2014, 30, 4545–4550. [Google Scholar] [CrossRef] [PubMed]
- Ran, Q.; Peng, R.; Liang, C.; Ye, S.; Xian, Y.; Zhang, W.; Jin, L. Covalent immobilization of horseradish peroxidase via click chemistry and its direct electrochemistry. Talanta 2011, 83, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; McArdle, T.; Sullivan, P.R.; Blanford, C.F. Sources of activity loss in the fuel cell enzyme bilirubin oxidase. Energy Environ. Sci. 2013, 6, 2460–2464. [Google Scholar] [CrossRef]
- Zanetti-Polzi, L.; Daidone, I.; Bortolotti, C.A.; Corni, S. Surface packing determines the redox potential shift of cytochrome c adsorbed on gold. J. Am. Chem. Soc. 2014, 136, 12929–12937. [Google Scholar] [CrossRef] [PubMed]
- Krzemiński, Ł.; Cronin, S.; Ndamba, L.; Canters, G.W.; Aartsma, T.J.; Evans, S.D.; Jeuken, L.J.C. Orientational Control over Nitrite Reductase on Modified Gold Electrode and Its Effects on the Interfacial Electron Transfer. J. Phys. Chem. B 2011, 115, 12607–12614. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, T.; Iida, M.; Gon, N.; Hobara, D.; Imabayashi, S.-I.; Niki, K. Miscibility of adsorbed 1-undecanethiol and 11-mercaptoundecanoic acid species in binary self-assembled monolayers on Au (111). Langmuir 2001, 17, 1599–1603. [Google Scholar] [CrossRef]
- Smith, R.K.; Reed, S.M.; Lewis, P.A.; Monnell, J.D.; Clegg, R.S.; Kelly, K.F.; Bumm, L.A.; Hutchison, J.E.; Weiss, P.S. Phase separation within a binary self-assembled monolayer on Au {111} driven by an amide-containing alkanethiol. J. Phys. Chem. B 2001, 105, 1119–1122. [Google Scholar] [CrossRef]
- Benavidez, T.E.; Torrente, D.; Marucho, M.; Garcia, C.D. Adsorption of Soft and Hard Proteins onto OTCEs under the Influence of an External Electric Field. Langmuir 2015, 31, 2455–2462. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Kitazumi, Y.; Tsujimura, S.; Shirai, O.; Yamamoto, M.; Kano, K. Electrostatic interaction between an enzyme and electrodes in the electric double layer examined in a view of direct electron transfer-type bioelectrocatalysis. Biosens. Bioelectron. 2015, 63, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Yamamoto, M.; Kano, K. Role of 2-mercaptoethanol in direct electron transfer-type bioelectrocatalysis of fructose dehydrogenase at Au electrodes. Electrochim. Acta 2015, 170, 242–247. [Google Scholar] [CrossRef]
- Lopez, F.; Siepenkoetter, T.; Xiao, X.; Magner, E.; Schuhmann, W.; Salaj-Kosla, U. Potential pulse-assisted immobilization of Myrothecium verrucaria bilirubin oxidase at planar and nanoporous gold electrodes. J. Electroanal. Chem. 2018, 812, 194–198. [Google Scholar] [CrossRef]
- Millo, D.; Ranieri, A.; Gross, P.; Ly, H.K.; Borsari, M.; Hildebrandt, P.; Wuite, G.J.L.; Gooijer, C.; van der Zwan, G. Electrochemical Response of Cytochrome c Immobilized on Smooth and Roughened Silver and Gold Surfaces Chemically Modified with 11-Mercaptounodecanoic Acid. J. Phys. Chem. C 2009, 113, 2861–2866. [Google Scholar] [CrossRef]
- Salewski, J.; Batista, A.P.; Sena, F.V.; Millo, D.; Zebger, I.; Pereira, M.M.; Hildebrandt, P. Substrate-Protein Interactions of Type II NADH:Quinone Oxidoreductase from Escherichia coli. Biochemistry 2016, 55, 2722–2734. [Google Scholar] [CrossRef] [PubMed]
- Rivas, L.; Soares, C.M.; Baptista, A.M.; Simaan, J.; Di Paolo, R.E.; Murgida, D.H.; Hildebrandt, P. Electric-field-induced redox potential shifts of tetraheme cytochromes c3 immobilized on self-assembled monolayers: Surface-enhanced resonance Raman spectroscopy and simulation studies. Biophys. J. 2005, 88, 4188–4199. [Google Scholar] [CrossRef] [PubMed]
- Petrey, D.; Honig, B. Structural bioinformatics of the interactome. Annu. Rev. Biophys. 2014, 43, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, K.; Mazurenko, I.; Gutierrez-Sanchez, C.; Ilbert, M.; Infossi, P.; Frielingsdorf, S.; Giudici-Orticoni, M.T.; Lenz, O.; Lojou, E. Impact of Carbon Nanotube Surface Chemistry on Hydrogen Oxidation by Membrane-Bound Oxygen-Tolerant Hydrogenases. Chemelectrochem 2016, 3, 2179–2188. [Google Scholar] [CrossRef]
- Salamon, Z.; Fitch, J.; Cai, M.; Tumati, S.; Navratilova, E.; Tollin, G. Plasmon-waveguide resonance studies of ligand binding to integral proteins in membrane fragments derived from bacterial and mammalian cells. Anal. Biochem. 2009, 387, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Bouffier, L.; Doneux, T. Coupling electrochemistry with in situ fluorescence (confocal) microscopy. Curr. Opin. Electrochem. 2017, 6, 31–37. [Google Scholar] [CrossRef]
- Siepenkoetter, T.; Salaj-Kosla, U.; Xiao, X.X.; Conghaile, P.O.; Pita, M.; Ludwig, R.; Magner, E. Immobilization of Redox Enzymes on Nanoporous Gold Electrodes: Applications in Biofuel Cells. Chempluschem 2017, 82, 553–560. [Google Scholar] [CrossRef]
- Gutierrez-Sanchez, C.; Pita, M.; Vaz-Dominguez, C.; Shleev, S.; De Lacey, A.L. Gold Nanoparticles as Electronic Bridges for Laccase-Based Biocathodes. J. Am. Chem. Soc. 2012, 134, 17212–17220. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Effects of Mesoporous Structures on Direct Electron Transfer-Type Bioelectrocatalysis: Facts and Simulation on a Three-Dimensional Model of Random Orientation of Enzymes. Electrochemistry 2017, 85, 82–87. [Google Scholar] [CrossRef]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hitaishi, V.P.; Clement, R.; Bourassin, N.; Baaden, M.; De Poulpiquet, A.; Sacquin-Mora, S.; Ciaccafava, A.; Lojou, E. Controlling Redox Enzyme Orientation at Planar Electrodes. Catalysts 2018, 8, 192. https://doi.org/10.3390/catal8050192
Hitaishi VP, Clement R, Bourassin N, Baaden M, De Poulpiquet A, Sacquin-Mora S, Ciaccafava A, Lojou E. Controlling Redox Enzyme Orientation at Planar Electrodes. Catalysts. 2018; 8(5):192. https://doi.org/10.3390/catal8050192
Chicago/Turabian StyleHitaishi, Vivek Pratap, Romain Clement, Nicolas Bourassin, Marc Baaden, Anne De Poulpiquet, Sophie Sacquin-Mora, Alexandre Ciaccafava, and Elisabeth Lojou. 2018. "Controlling Redox Enzyme Orientation at Planar Electrodes" Catalysts 8, no. 5: 192. https://doi.org/10.3390/catal8050192
APA StyleHitaishi, V. P., Clement, R., Bourassin, N., Baaden, M., De Poulpiquet, A., Sacquin-Mora, S., Ciaccafava, A., & Lojou, E. (2018). Controlling Redox Enzyme Orientation at Planar Electrodes. Catalysts, 8(5), 192. https://doi.org/10.3390/catal8050192