The Design of MnOx Based Catalyst in Post-Plasma Catalysis Configuration for Toluene Abatement

 ,
,  ,
, 

Abstract
:1. Introduction
2. The Properties, Applications and Hazardous Effects of Toluene
- Inadvertent sources (65%), such as emissions from motor vehicles and aircraft exhaust, losses during gasoline marketing activities, chemical spills, cigarette smoke, and household products.
- Industrial processes in which toluene is used (33%).
- Toluene production (2%).
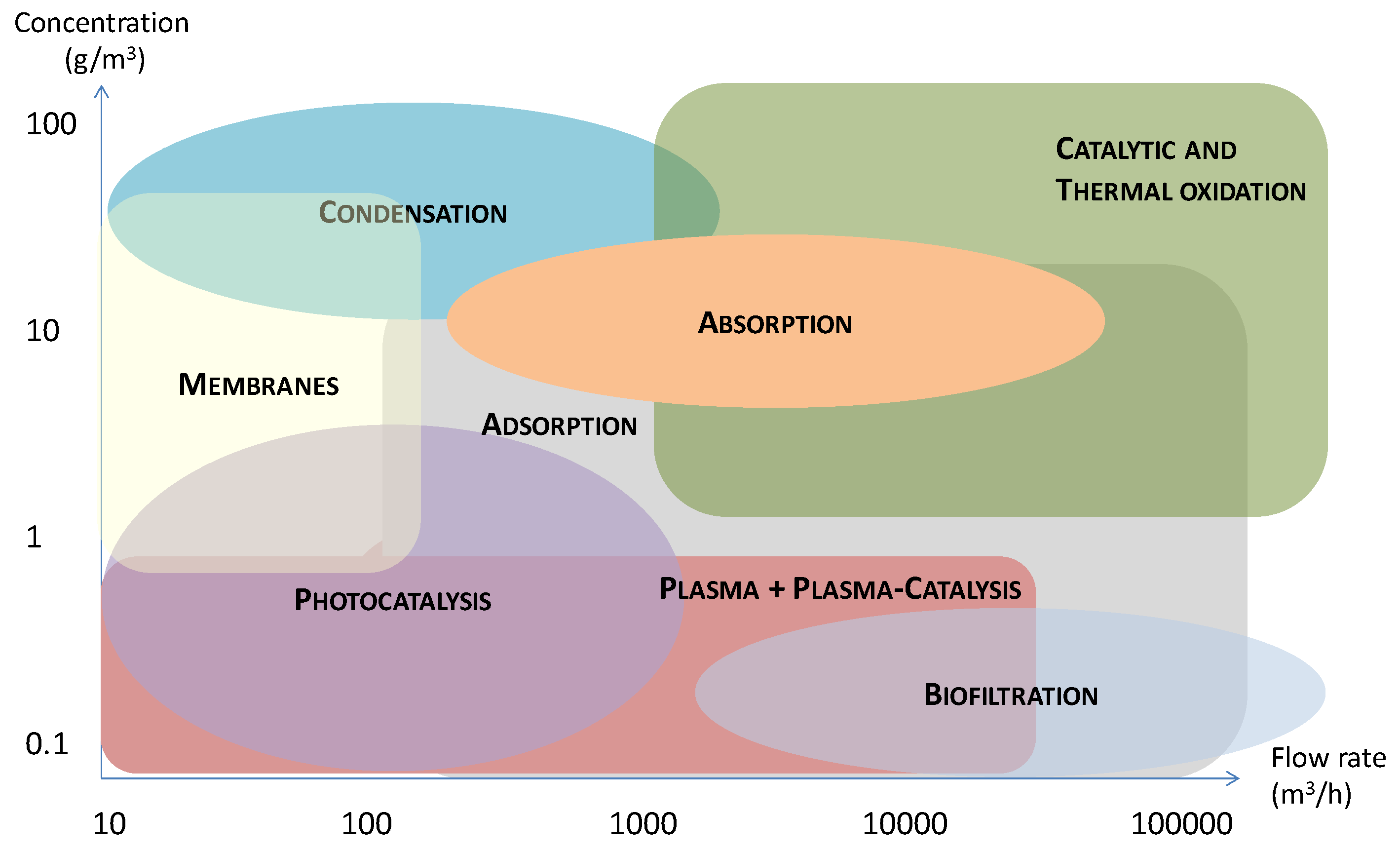
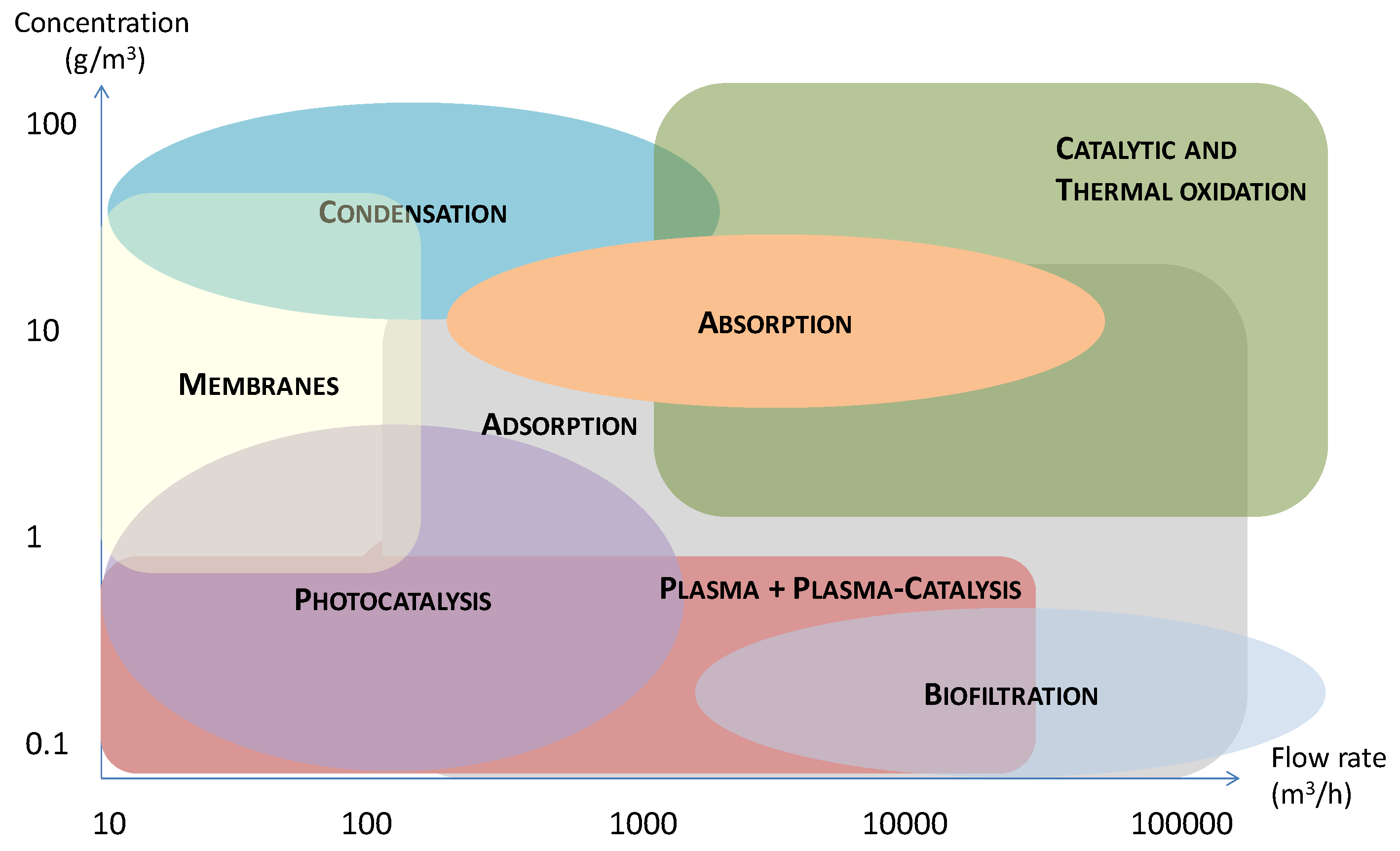
3. The Treatment Techniques for the Removal of Toluene
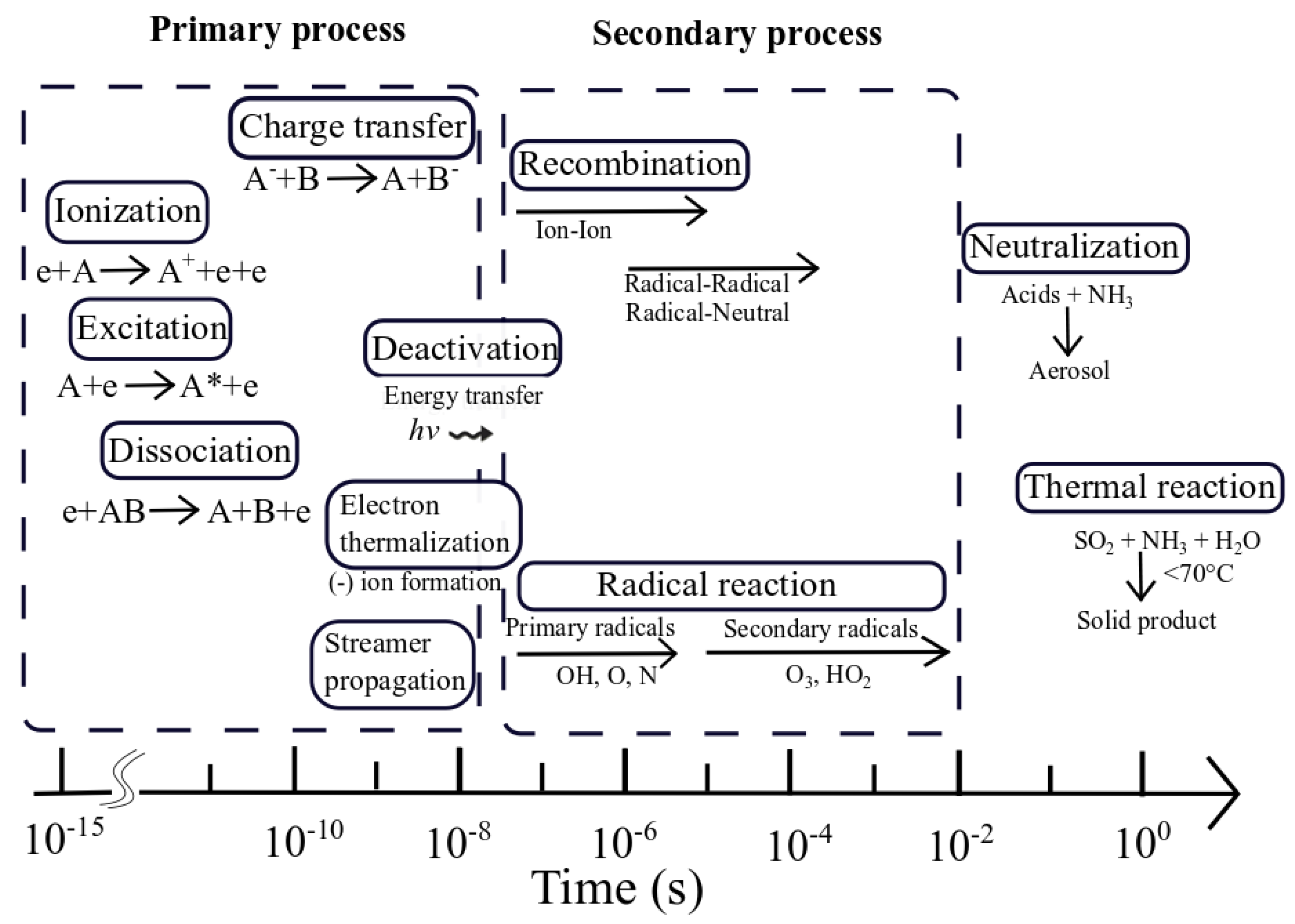
4. Design of Catalysts for Toluene Abatement in PPC Process
4.1. Introduction
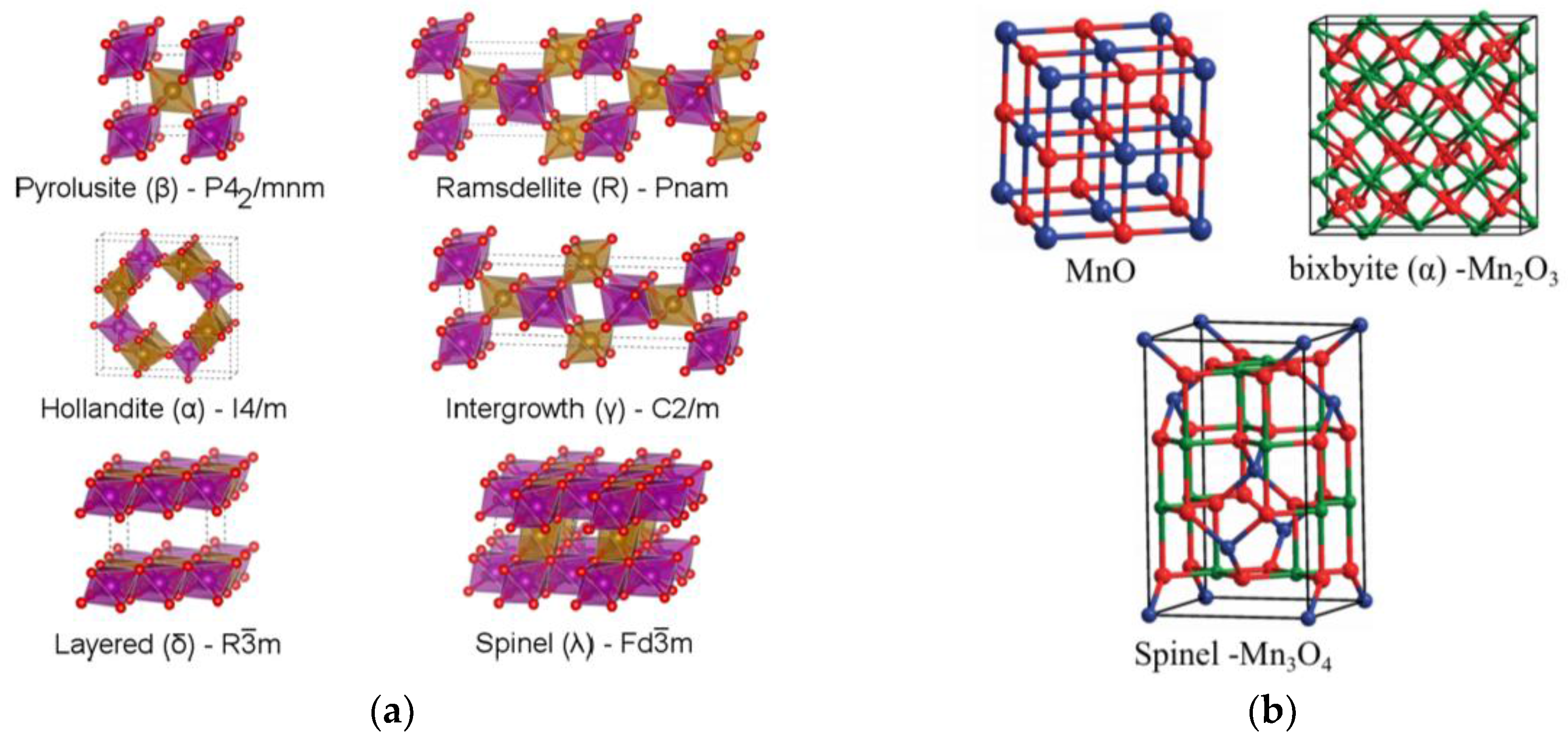
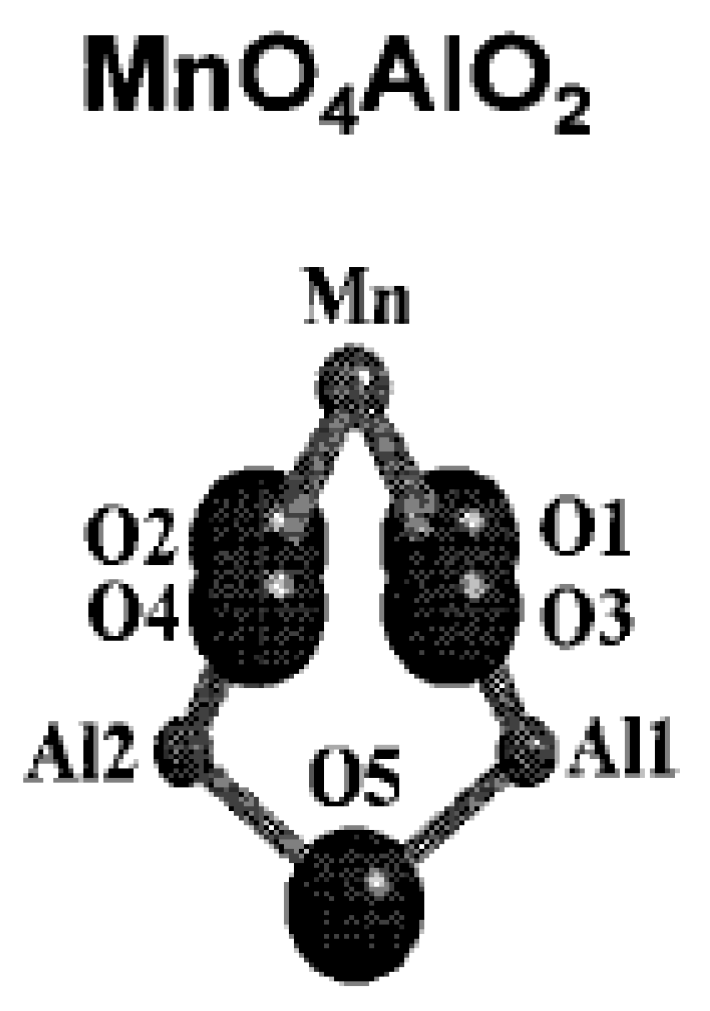
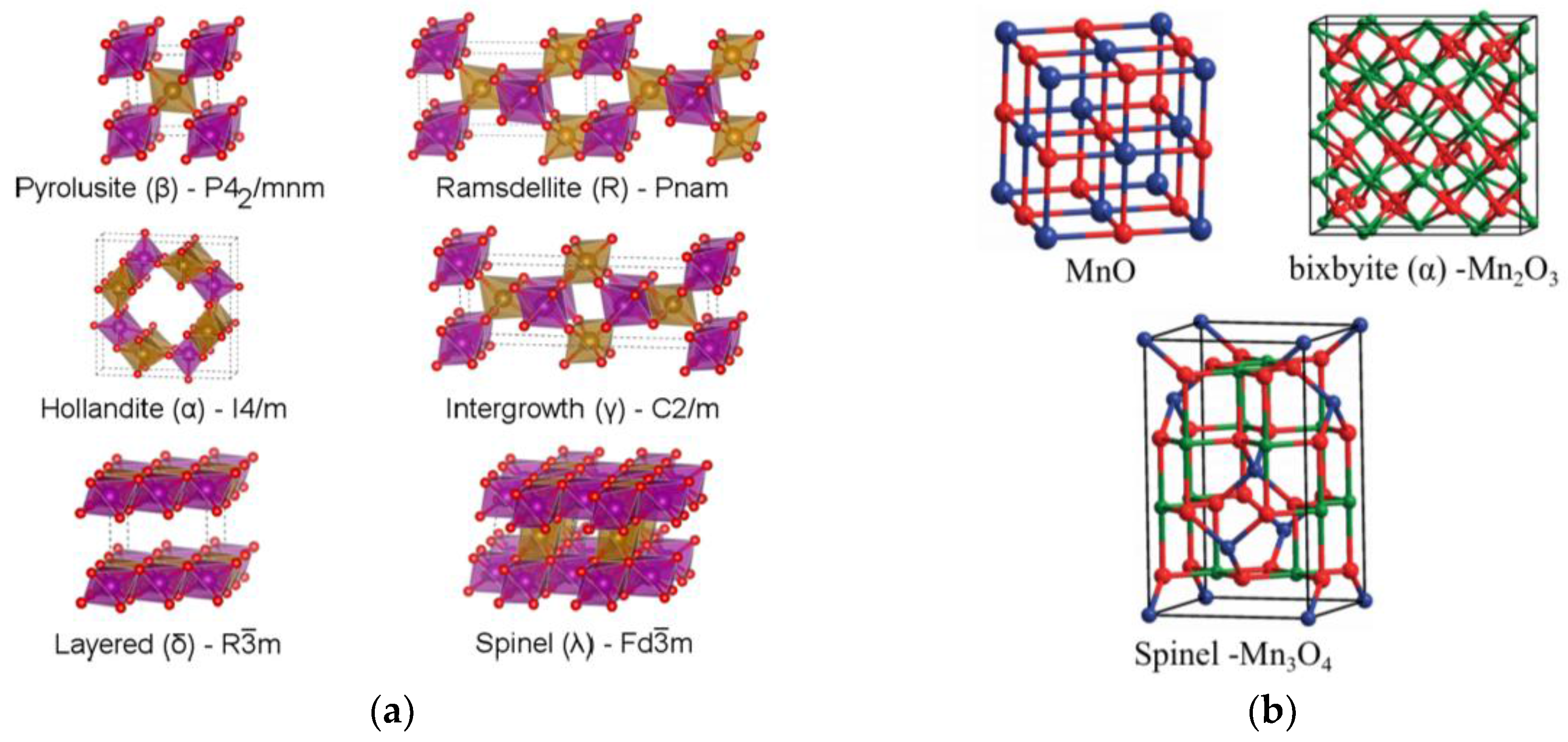
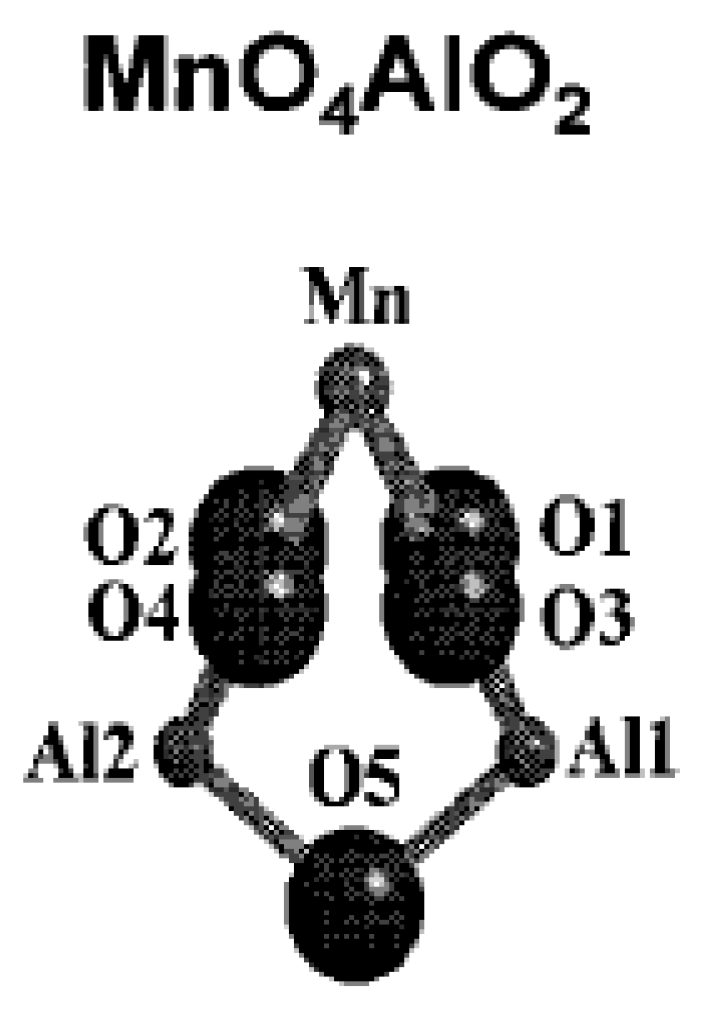
4.2. Presentation of the Main Manganese Oxide Structures
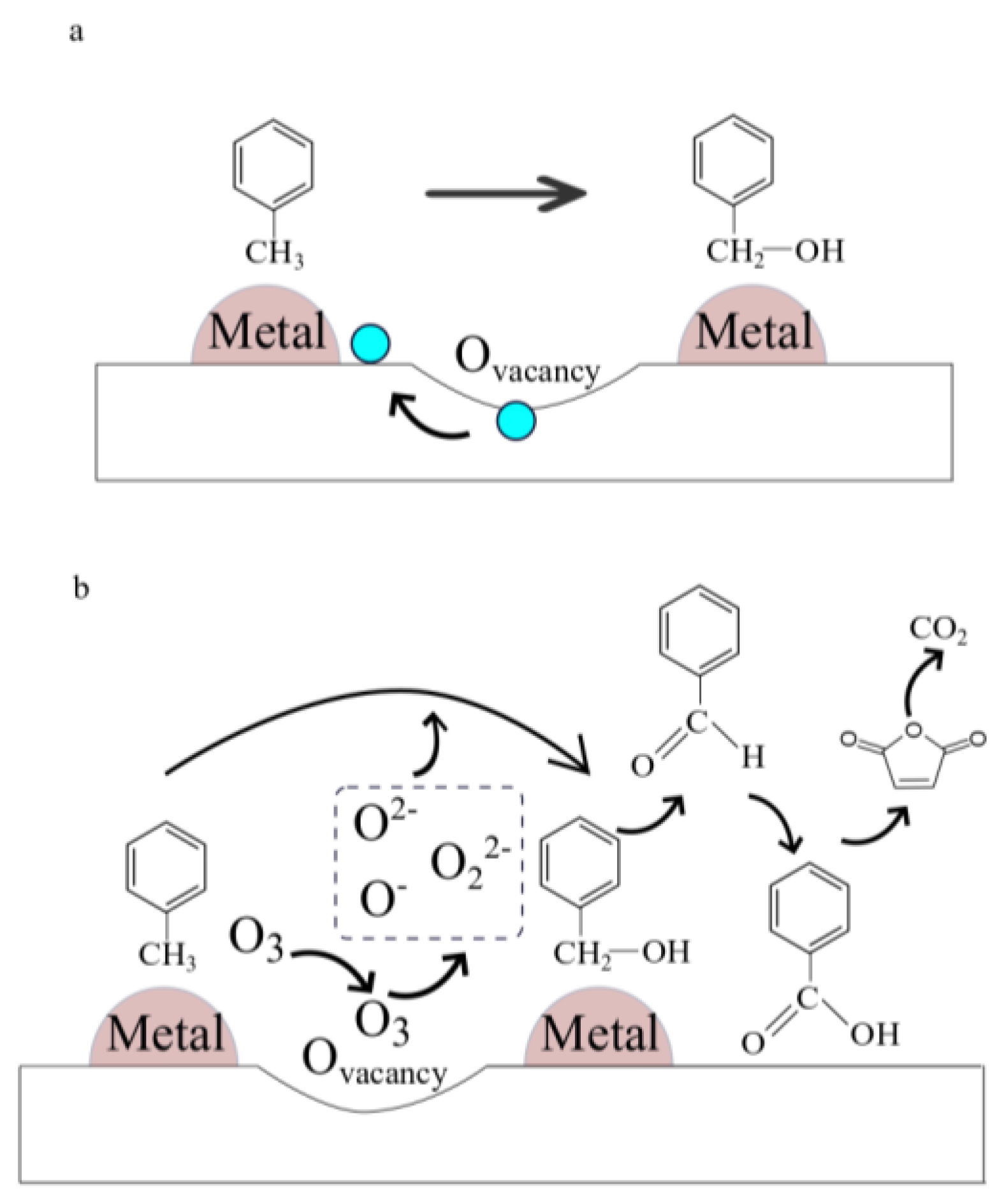
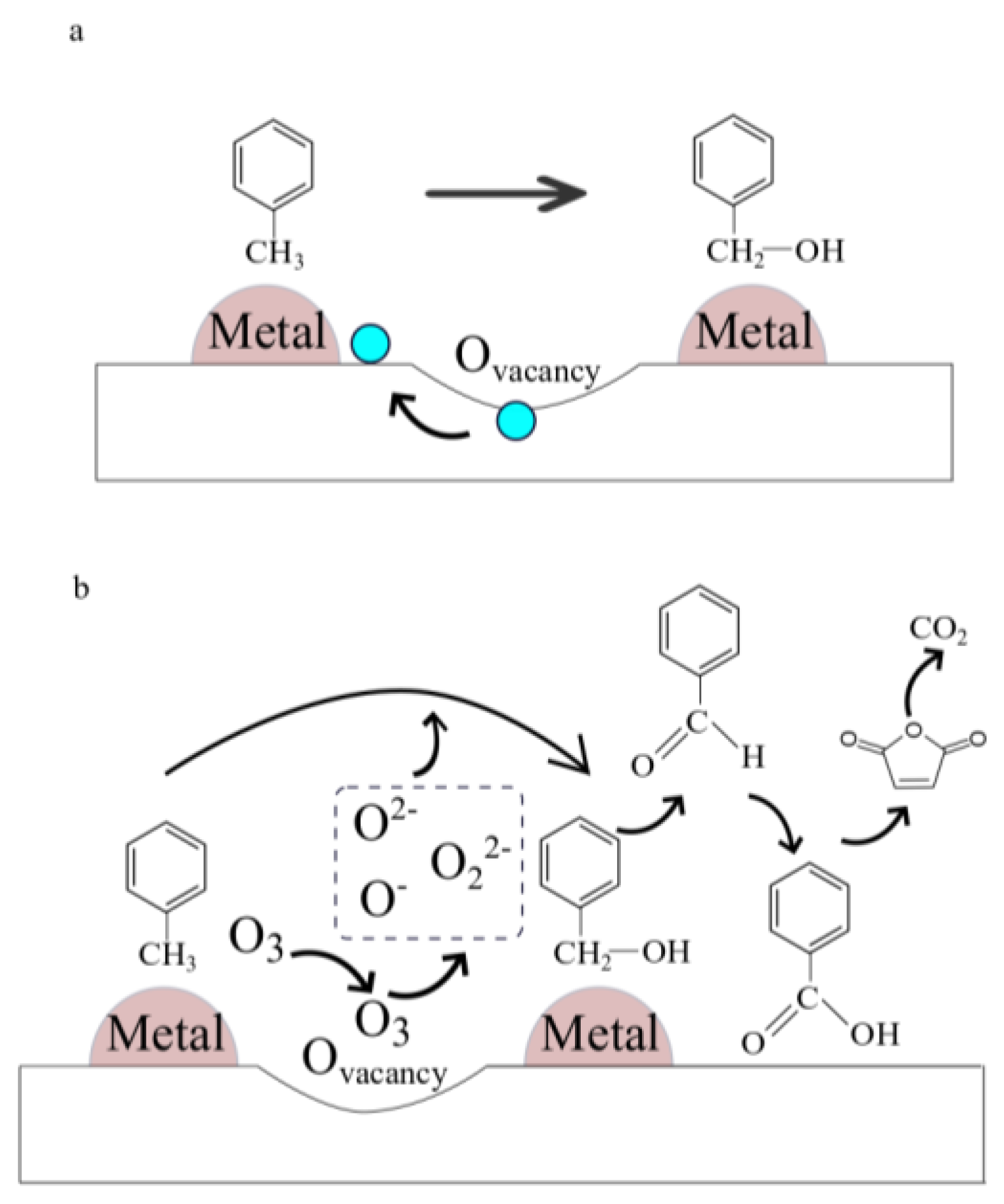
4.3. MnOx in Catalytic Oxidation of Toluene
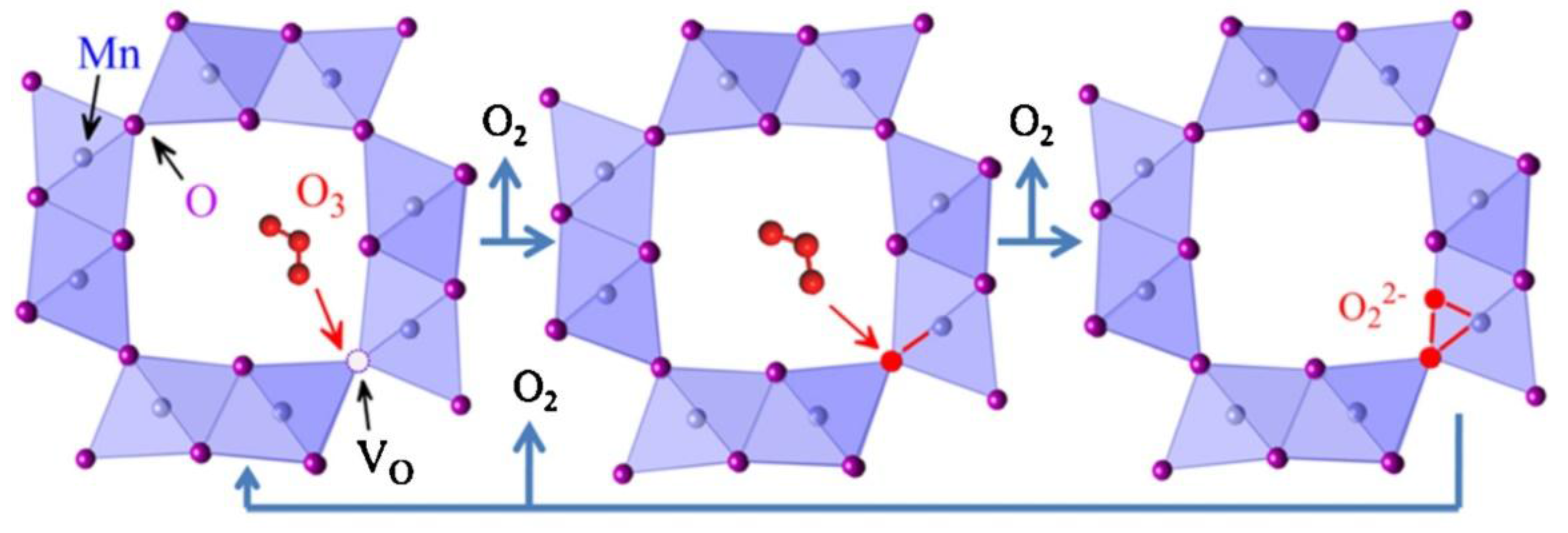
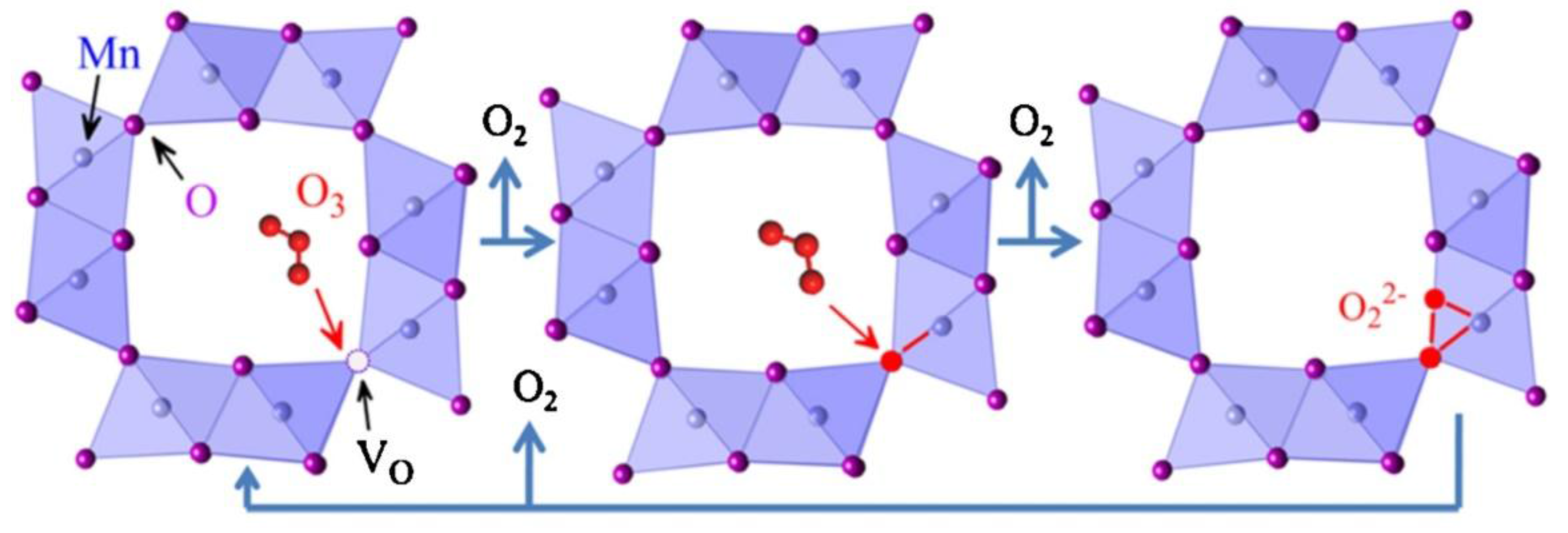
4.4. MnOx in Ozone Decomposition
4.5. MnOx in Ozonation Reactions
4.6. The Ability of MnOx for CO Removal
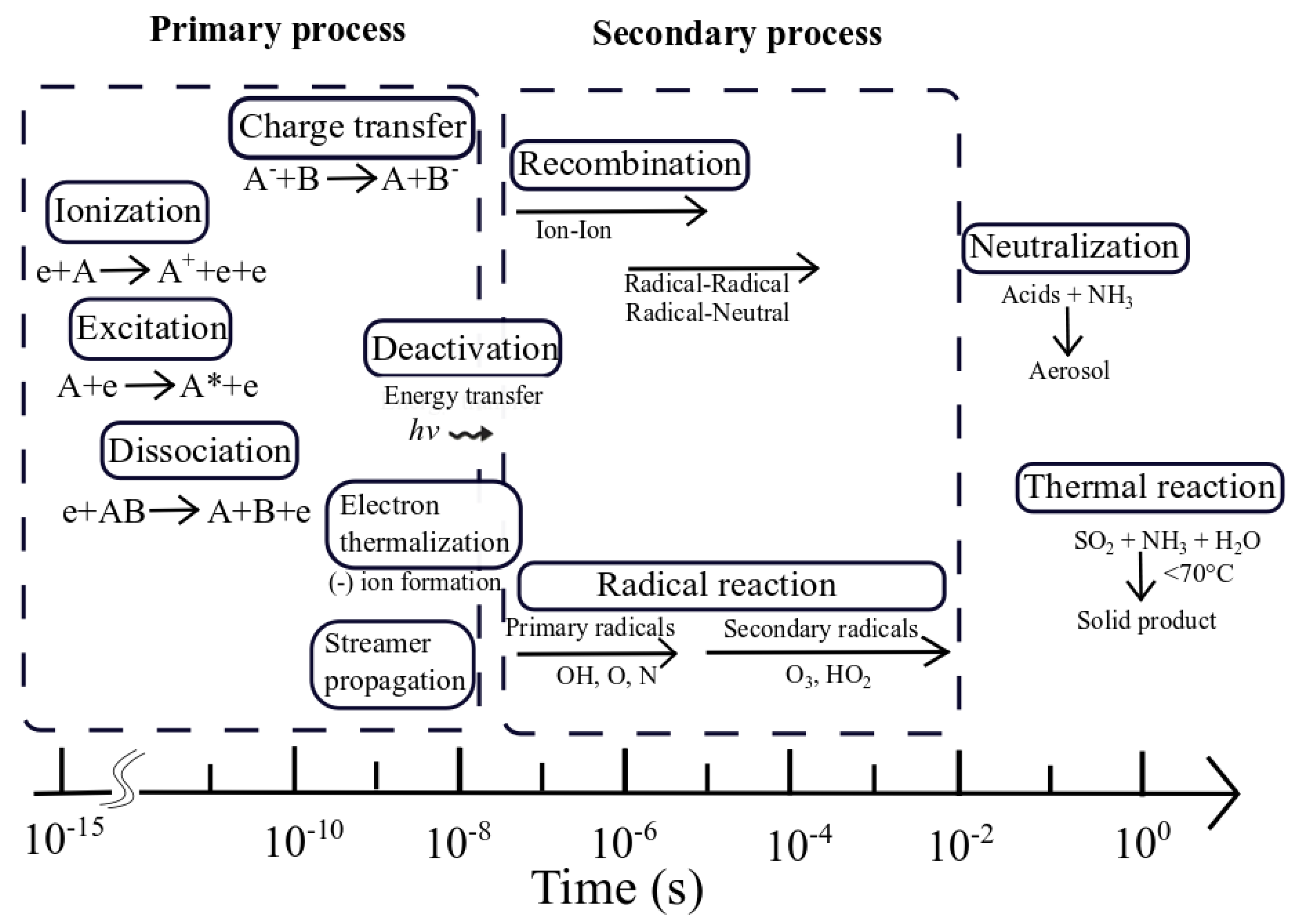
5. MnOx in PPC for Toluene Abatement
5.1. Current Applications of MnOx Catalysts in PPC Systems for Toluene Abatement
5.1.1. The Characteristics of MnOx in PPC
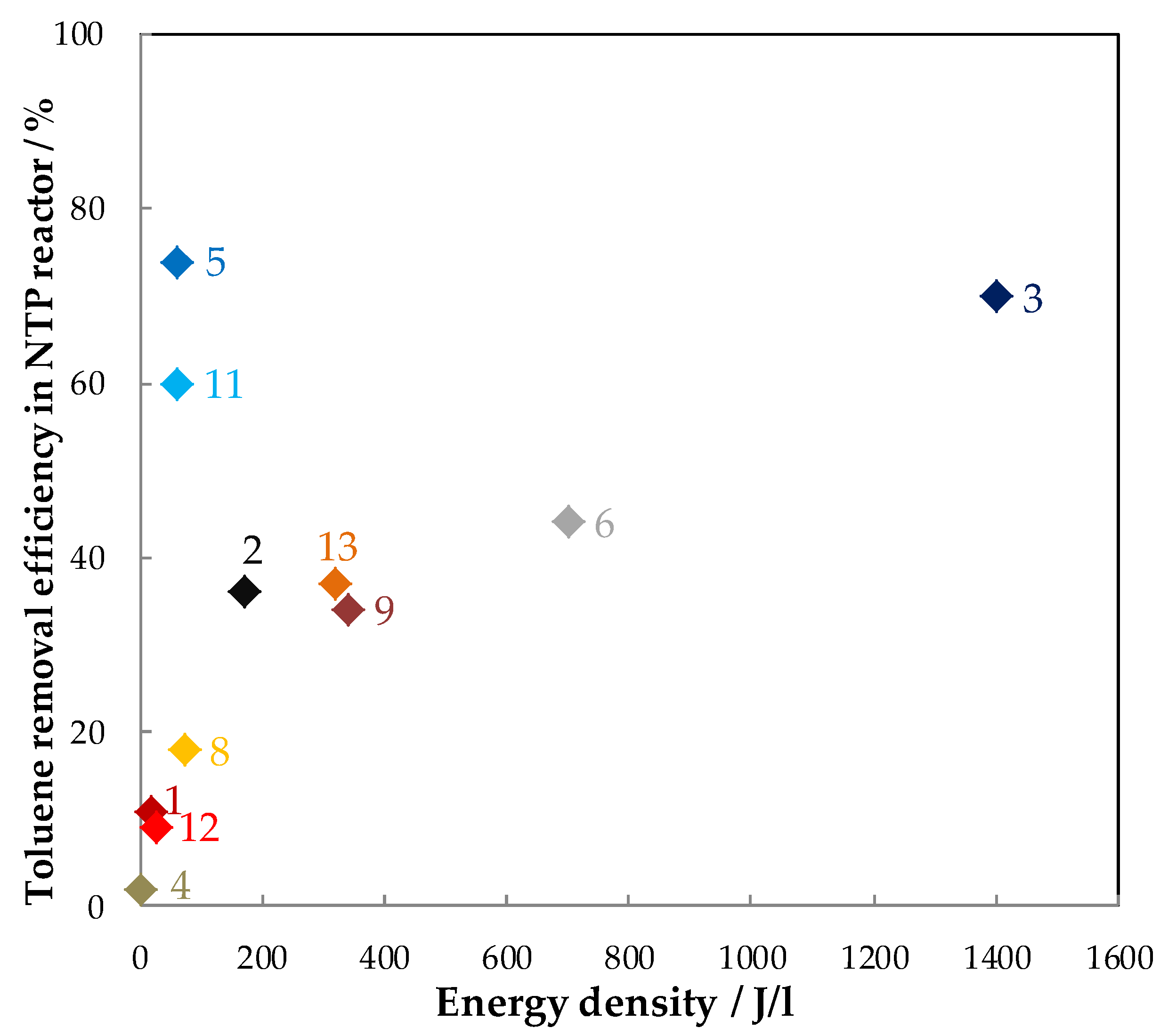
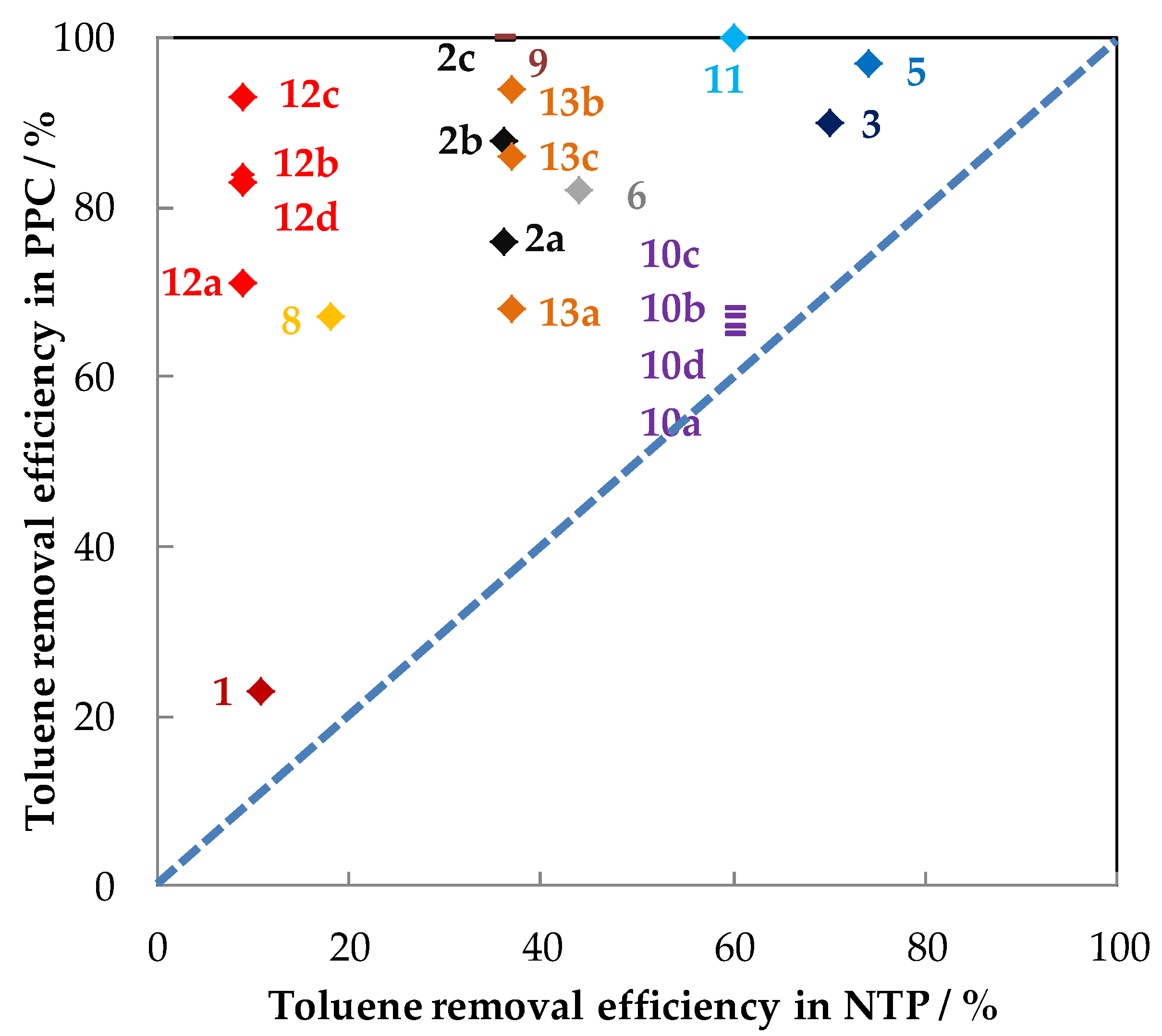
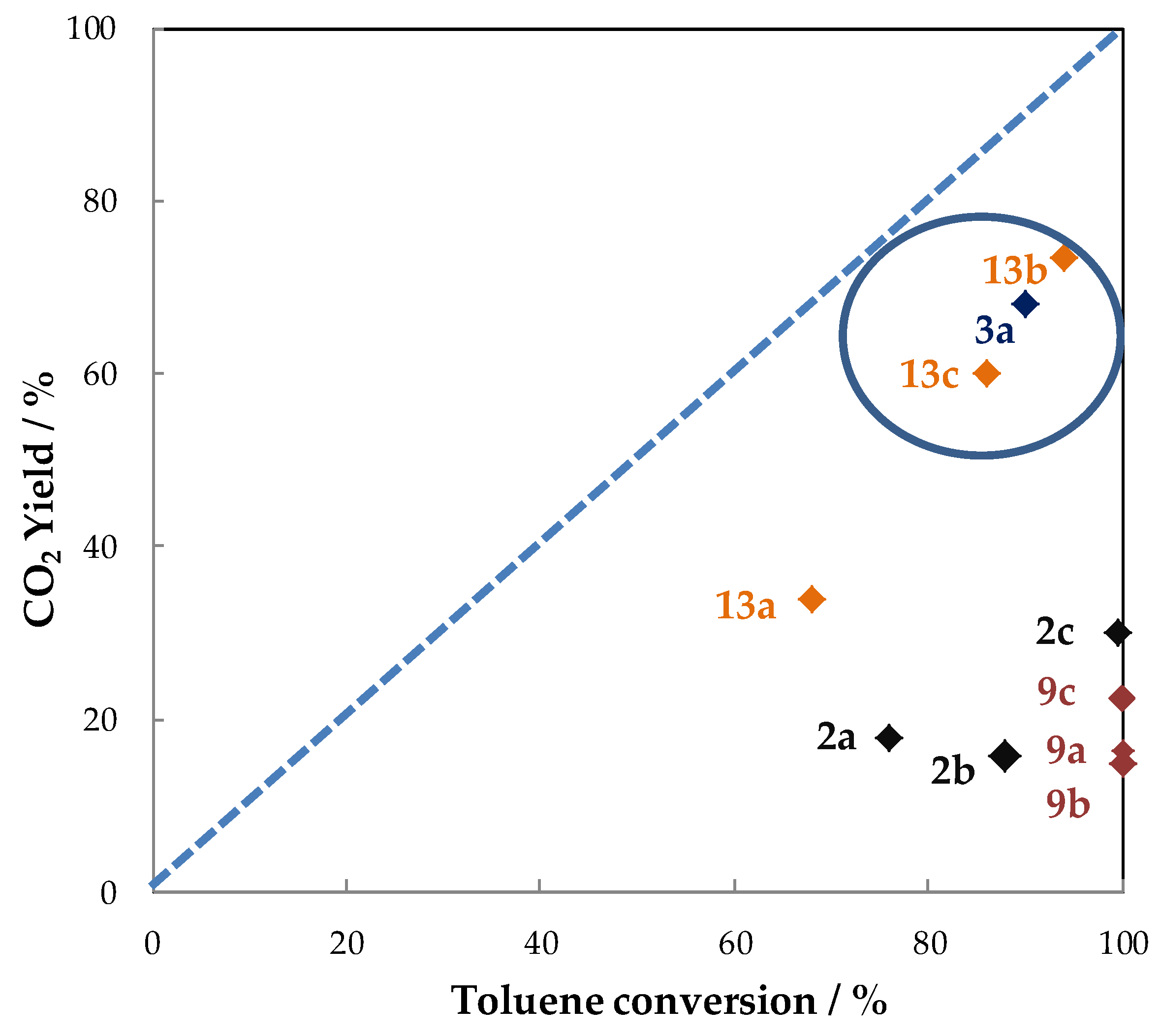
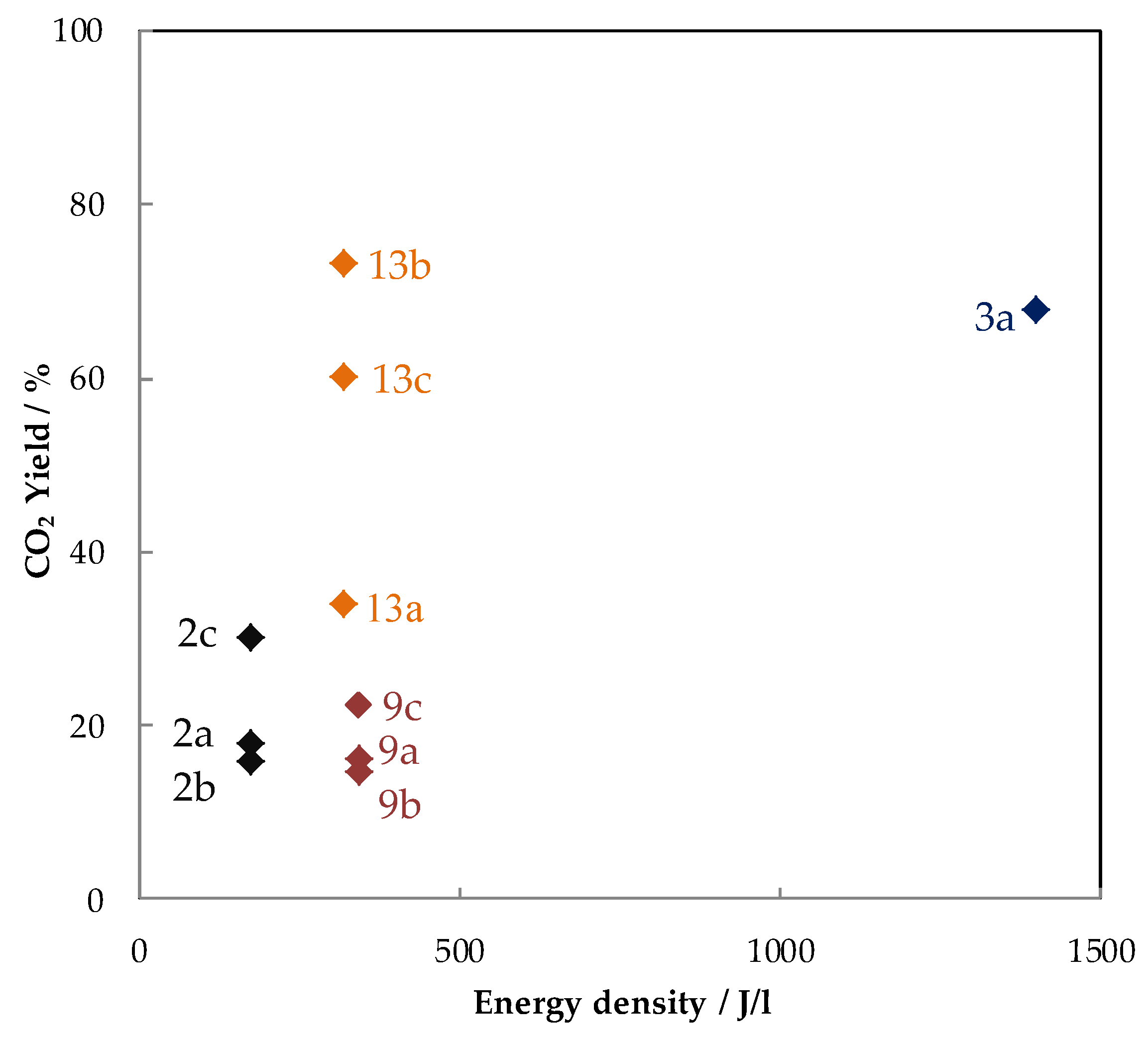
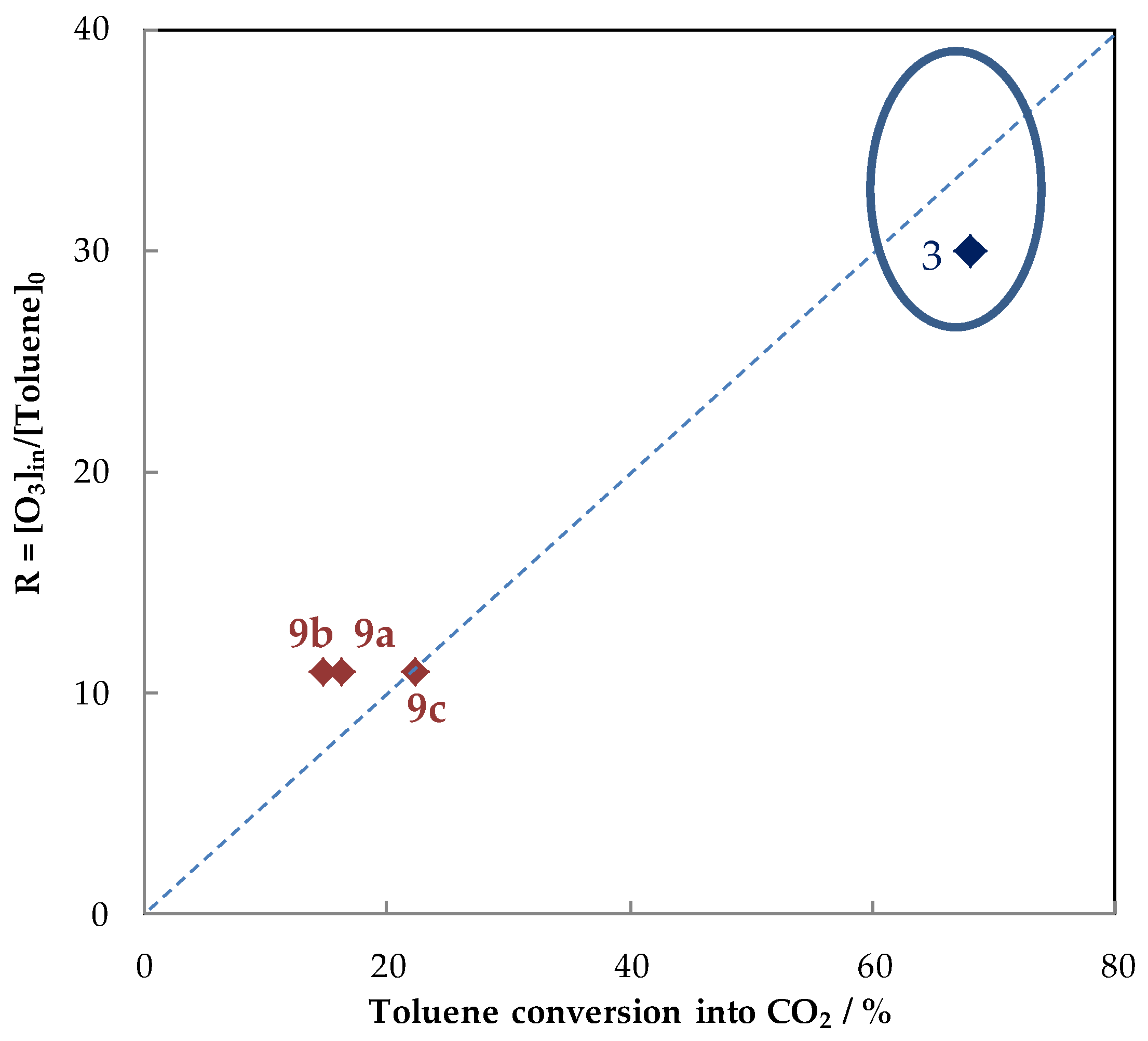
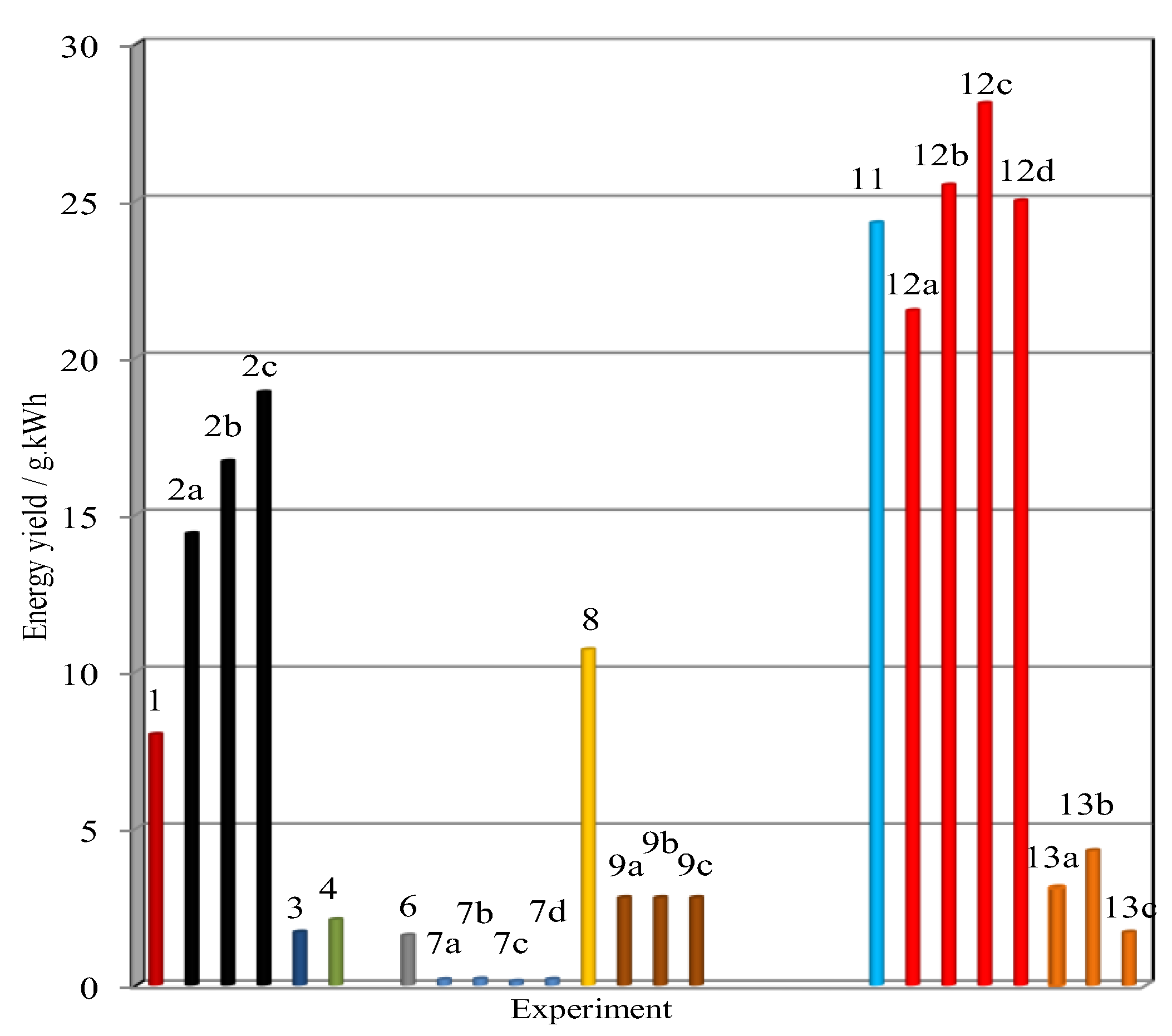
5.1.2. The Performances in Toluene Abatement Using MnOx in PPC
6. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. 5.14 Toluene; WHO Regional Office for Europe, Ed.; WHO: Copenhagen, Denmark, 2000. [Google Scholar]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Toluene; U.S. Department of Health and Human Services, U.S. Public Health Service: Atlanta, GA, USA, 2000.
- World Health Organization. Environmental Health Criteria 52: Toluene; World Health Organization: Geneva, Switzerland, 1985. [Google Scholar]
- Guo, Y.; Liao, X.; Fu, M.; Huang, H.; Ye, D. Toluene decomposition performance and NOx by-product formation during a DBD-catalyst process. J. Environ. Sci. 2015, 28, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-H.; Teramoto, Y.; Ogata, A.; Takagi, H.; Nanba, T. Plasma catalysis for environmental treatment and energy applications. Plasma Chem. Plasma Process. 2016, 36, 45–72. [Google Scholar] [CrossRef]
- Whitehead, J.C. Plasma–catalysis: The known knowns, the known unknowns and the unknown unknowns. J. Phys. D Appl. Phys. 2016, 49, 243001. [Google Scholar] [CrossRef]
- Furniss, B.S. Vogel’s Textbook of Practical Organic Chemistry; Longman/Wiley: New York, NY, USA, 1989. [Google Scholar]
- Genuino, H.C.; Dharmarathna, S.; Njagi, E.C.; Mei, M.C.; Suib, S.L. Gas-Phase Total Oxidation of Benzene, Toluene, Ethylbenzene, and Xylenes Using Shape-Selective Manganese Oxide and Copper Manganese Oxide Catalysts. J. Phys. Chem. C 2012, 116, 12066–12078. [Google Scholar] [CrossRef]
- Market Study: Toluene. Available online: http://www.ceresana.com/en/market-studies/chemicals/toluene/ (accessed on 13 February 2018).
- Toluene Uses and Market Data. Available online: http://www.icis.com/resources/news/2007/11/07/9076550/toluene-uses-and-market-data/ (accessed on 13 February 2018).
- Corporation, T.E. Locating and Estimating Air Emissions from Soruces of Toluene; EPA: Chapel Hill, NC, USA, 1994; p. 207.
- Hobara, T.; Okuda, M.; Gotoh, M.; Oki, K.; Segawa, H.; Kunitsugu, I. Estimation of the lethal toluene concentration from the accidental death of painting workers. Ind. Health 2000, 38, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Zhang, X.; Yu, X.; Feng, T.; Yao, S. Catalytic oxidation of benzene using DBD corona discharges. J. Hazard. Mater. 2006, 137, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Van Durme, J.; Dewulf, J.; Leys, C.; Van Langenhove, H. Combining non-thermal plasma with heterogeneous catalysis in waste gas treatment: A review. Appl. Catal. B Environ. 2008, 78, 324–333. [Google Scholar] [CrossRef]
- Kim, H.H. Nonthermal plasma processing for air-pollution control: A historical review, current issues, and future prospects. Plasma Process. Polym. 2004, 1, 91–110. [Google Scholar] [CrossRef]
- McAdams, R. Pulsed corona treatment of gases: System scaling and efficiency. Plasma Sources Sci. Technol. 2007, 16, 703. [Google Scholar] [CrossRef]
- Chen, H.L.; Lee, H.M.; Chen, S.H.; Chang, M.B.; Yu, S.J.; Li, S.N. Removal of volatile organic compounds by single-stage and two-stage plasma catalysis systems: A review of the performance enhancement mechanisms, current status, and suitable applications. Environ. Sci. Technol. 2009, 43, 2216–2227. [Google Scholar] [CrossRef] [PubMed]
- Magureanu, M.; Mandache, N.B.; Eloy, P.; Gaigneaux, E.M.; Parvulescu, V.I. Plasma-assisted catalysis for volatile organic compounds abatement. Appl. Catal. B Environ. 2005, 61, 12–20. [Google Scholar] [CrossRef]
- Xiao, G.; Xu, W.; Wu, R.; Ni, M.; Du, C.; Gao, X.; Luo, Z.; Cen, K. Non-thermal plasmas for VOCs abatement. Plasma Chem. Plasma Process. 2014, 34, 1033–1065. [Google Scholar] [CrossRef]
- Kim, H.H.; Prieto, G.; Takashima, K.; Katsura, S.; Mizuno, A. Performance evaluation of discharge plasma process for gaseous pollutant removal. J. Electrost. 2002, 55, 25–41. [Google Scholar] [CrossRef]
- Vandenbroucke, A.M.; Morent, R.; De Geyter, N.; Leys, C. Non-thermal plasmas for non-catalytic and catalytic VOC abatement. J. Hazard. Mater. 2011, 195, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, A.M.; Morent, R.; Geyter, N.D.; Leys, C. Decomposition of toluene with plasma-catalysis: A review. J. Adv. Oxid. Technol. 2012, 15, 232–241. [Google Scholar] [CrossRef]
- Tian, Z.-Y.; Mountapmbeme Kouotou, P.; Bahlawane, N.; Tchoua Ngamou, P.H. Synthesis of the catalytically active Mn3O4 spinel and its thermal properties. J. Phys. Chem. C 2013, 117, 6218–6224. [Google Scholar] [CrossRef]
- Kitchaev, D.A.; Peng, H.; Liu, Y.; Sun, J.; Perdew, J.P.; Ceder, G. Energetics of MnO2 polymorphs in density functional theory. Phys. Rev. B 2016, 93, 045132. [Google Scholar] [CrossRef]
- Menezes, P.W.; Indra, A.; Littlewood, P.; Schwarze, M.; Göbel, C.; Schomäcker, R.; Driess, M. Nanostructured manganese oxides as highly active water oxidation catalysts: A boost from manganese precursor chemistry. ChemSusChem 2014, 7, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Craciun, R.; Nentwick, B.; Hadjiivanov, K.; Knözinger, H. Structure and redox properties of MnOx/Yttrium-stabilized zirconia (YSZ) catalyst and its used in CO and CH4 oxidation. Appl. Catal. A Gen. 2003, 243, 67–79. [Google Scholar] [CrossRef]
- Kim, S.C.; Shim, W.G. Catalytic combustion of VOCs over a series of manganese oxide catalysts. Appl. Catal. B Environ. 2010, 98, 180–185. [Google Scholar] [CrossRef]
- Li, X.; Xu, J.; Wang, F.; Gao, J.; Zhou, L.; Yang, G. Direct oxidation of toluene to benzoic acid with molecular oxygen over manganese oxides. Catal. Lett. 2006, 108, 137–140. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, H.; Deng, J.; Zhang, L.; Zhao, Z.; Li, X.; Wang, Y.; Xie, S.; Yang, H.; Guo, G. Controlled generation of uniform spherical LaMnO3, LaCoO3, Mn2O3, and Co3O4 nanoparticles and their high catalytic performance for carbon monoxide and toluene oxidation. Inorg. Chem. 2013, 52, 8665–8676. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; He, S.; Xie, S.; Yang, H.; Liu, Y.; Guo, G.; Dai, H. Ultralow loading of silver nanoparticles on Mn2O3 nanowires derived with molten salts: A high-efficiency catalyst for the oxidative removal of toluene. Environ. Sci. Technol. 2015, 49, 11089–11095. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Dai, H.; Deng, J.; Bai, G.; Ji, K.; Liu, Y. Manganese oxides with rod-, wire-, tube-, and flower-like morphologies: Highly effective catalysts for the removal of toluene. Environ. Sci. Technol. 2012, 46, 4034–4041. [Google Scholar] [CrossRef] [PubMed]
- Lahousse, C.; Bernier, A.; Grange, P.; Delmon, B.; Papaefthimiou, P.; Ioannides, T.; Verykios, X. Evaluation of γ-MnO2 as a VOC removal catalyst: Comparison with a noble metal catalyst. J. Catal. 1998, 178, 214–225. [Google Scholar] [CrossRef]
- Si, W.; Wang, Y.; Peng, Y.; Li, X.; Li, K.; Li, J. A high-efficiency γ-MnO2-like catalyst in toluene combustion. Chem. Commun. 2015, 51, 14977–14980. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Zhang, X.; Peng, R.; Zhao, M.; Ye, D. Catalytic properties of manganese oxide polyhedra with hollow and solid morphologies in toluene removal. Appl. Surf. Sci. 2017, 405, 20–28. [Google Scholar] [CrossRef]
- Herbschleb, C.T. ReactorSTM: Imaging Catalysts under Realistic Conditions; Faculty of Science, Leiden University: Leiden, The Netherlands, 2011. [Google Scholar]
- Aguero, F.N.; Scian, A.; Barbero, B.P.; Cadús, L.E. Influence of the support treatment on the behavior of MnOx/Al2O3 catalysts used in VOC combustion. Catal. Lett. 2009, 128, 268. [Google Scholar] [CrossRef]
- Védrine, J.C.; Fechete, I. Heterogeneous partial oxidation catalysis on metal oxides. C. R. Chim. 2016, 19, 1203–1225. [Google Scholar] [CrossRef]
- Imamura, S.; Ikebata, M.; Ito, T.; Ogita, T. Decomposition of ozone on a silver catalyst. Ind. Eng. Chem. Res. 1991, 30, 217–221. [Google Scholar] [CrossRef]
- Dhandapani, B.; Oyama, S.T. Gas phase ozone decomposition catalysts. Appl. Catal. B Environ. 1997, 11, 129–166. [Google Scholar] [CrossRef]
- Li, W.; Gibbs, G.; Oyama, S.T. Mechanism of ozone decomposition on a manganese oxide catalyst. 1. In situ Raman spectroscopy and ab initio molecular orbital calculations. J. Am. Chem. Soc. 1998, 120, 9041–9046. [Google Scholar] [CrossRef]
- Li, W.; Oyama, S.T. Mechanism of ozone decomposition on a manganese oxide catalyst. 2. Steady-state and transient kinetic studies. J. Am. Chem. Soc. 1998, 120, 9047–9052. [Google Scholar] [CrossRef]
- Einaga, H.; Harada, M.; Futamura, S. Structural changes in alumina-supported manganese oxides during ozone decomposition. Chem. Phys. Lett. 2005, 408, 377–380. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Oyama, S.T.; Ohminami, Y.; Asakura, K. Structure of MnOx/Al2O3 catalyst: A study using EXAFS, in situ laser Raman spectroscopy and ab initio calculations. J. Phys. Chem. B 2001, 105, 9067–9070. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, P.; Chen, L. Catalytic decomposition of gaseous ozone over manganese dioxides with different crystal structures. Appl. Catal. B Environ. 2016, 189, 210–218. [Google Scholar] [CrossRef]
- Kameya, T.; Urano, K. Catalytic decomposition of ozone gas by a Pd impregnated MnO2 catalyst. J. Environ. Eng. 2002, 128, 286–292. [Google Scholar] [CrossRef]
- Ma, J.; Wang, C.; He, H. Transition metal doped cryptomelane-type manganese oxide catalysts for ozone decomposition. Appl. Catal. B Environ. 2017, 201, 503–510. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Oyama, S.T.; Chen, J.G.; Asakura, K. Electron transfer effects in ozone decomposition on supported manganese oxide. J. Phys. Chem. B 2001, 105, 4245–4253. [Google Scholar] [CrossRef]
- Setvín, M.; Aschauer, U.; Scheiber, P.; Li, Y.-F.; Hou, W.; Schmid, M.; Selloni, A.; Diebold, U. Reaction of O2 with subsurface oxygen vacancies on TiO2 anatase (101). Science 2013, 341, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, E.; Soltan, J.; Chen, N. Catalytic oxidation of toluene by ozone over alumina supported manganese oxides: Effect of catalyst loading. Appl. Catal. B Environ. 2013, 136, 239–247. [Google Scholar] [CrossRef]
- Rezaei, E.; Soltan, J. EXAFS and kinetic study of MnOx/γ-alumina in gas phase catalytic oxidation of toluene by ozone. Appl. Catal. B Environ. 2014, 148, 70–79. [Google Scholar] [CrossRef]
- Rezaei, E.; Soltan, J. Low temperature oxidation of toluene by ozone over MnOx/γ-alumina and MnOx/MCM-41 catalysts. Chem. Eng. J. 2012, 198, 482–490. [Google Scholar] [CrossRef]
- Hu, M.; Hui, K.; Hui, K. Role of graphene in MnO2/graphene composite for catalytic ozonation of gaseous toluene. Chem. Eng. J. 2014, 254, 237–244. [Google Scholar] [CrossRef]
- Li, J.; Na, H.; Zeng, X.; Zhu, T.; Liu, Z. In situ DRIFTS investigation for the oxidation of toluene by ozone over Mn/HZSM-5, Ag/HZSM-5 and Mn–Ag/HZSM-5 catalysts. Appl. Surf. Sci. 2014, 311, 690–696. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Kurita, Y.; Sankoda, K.; Namiki, N.; Yasui, F.; Tamura, H. Ozone Catalytic Oxidation of Gaseous Toluene over MnO2-Based Ozone Decomposition Catalysts Immobilized on a Nonwoven Fabric. Aerosol Air Qual. Res. 2017, 17, 2110–2118. [Google Scholar] [CrossRef]
- Liping, L.; Jianguo, Z.; Lixian, Y.; Mingli, F.; Junliang, W.; Huang, B.; Daiqi, Y. Room temperature catalytic ozonation of toluene over MnO2/Al2O3. Chin. J. Catal. 2011, 32, 904–916. [Google Scholar]
- Ramesh, K.; Chen, L.; Chen, F.; Liu, Y.; Wang, Z.; Han, Y.-F. Re-investigating the CO oxidation mechanism over unsupported MnO, Mn2O3 and MnO2 catalysts. Catal. Today 2008, 131, 477–482. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D. Catalytic oxidation of carbon monoxide over transition metal oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Wang, L.-C.; Liu, Q.; Huang, X.-S.; Liu, Y.-M.; Cao, Y.; Fan, K.-N. Gold nanoparticles supported on manganese oxides for low-temperature CO oxidation. Appl. Catal. B Environ. 2009, 88, 204–212. [Google Scholar] [CrossRef]
- Imamura, S.; Tsuji, Y.; Miyake, Y.; Ito, T. Cooperative action of palladium and manganese (III) oxide in the oxidation of carbon monoxide. J. Catal. 1995, 151, 279–284. [Google Scholar] [CrossRef]
- Liang, S.; Teng, F.; Bulgan, G.; Zong, R.; Zhu, Y. Effect of phase structure of MnO2 nanorod catalyst on the activity for CO oxidation. J. Phys. Chem. C 2008, 112, 5307–5315. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y. Selected-control hydrothermal synthesis of α-and β-MnO2 single crystal nanowires. J. Am. Chem. Soc. 2002, 124, 2880–2881. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y. Synthesis and formation mechanism of manganese dioxide nanowires/nanorods. Chemistry 2003, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Iablokov, V.; Frey, K.; Geszti, O.; Kruse, N. High catalytic activity in CO oxidation over MnOx nanocrystals. Catal. Lett. 2010, 134, 210–216. [Google Scholar] [CrossRef]
- Gervasini, A.; Vezzoli, G.; Ragaini, V. VOC removal by synergic effect of combustion catalyst and ozone. Catal. Today 1996, 29, 449–455. [Google Scholar] [CrossRef]
- Quoc An, H.T.; Huu, T.P.; Le Van, T.; Cormier, J.M.; Khacef, A. Application of atmospheric non thermal plasma-catalysis hybrid system for air pollution control: Toluene removal. Catal. Today 2011, 176, 474–477. [Google Scholar] [CrossRef]
- Ye, L.; Feng, F.; Liu, J.; Tang, X.; Zhang, X.; Huang, Y.; Liu, Z.; Yan, K. Toluene decomposition by a two-stage hybrid plasma catalyst system in dry air. IEEE Trans. Plasma Sci. 2014, 42, 3529–3538. [Google Scholar] [CrossRef]
- Magureanu, M.; Mandache, N.; Gaigneaux, E.; Paun, C.; Parvulescu, V. Toluene oxidation in a plasma-catalytic system. J. Appl. Phys. 2006, 99, 123301. [Google Scholar] [CrossRef]
- Van Durme, J.; Dewulf, J.; Demeestere, K.; Leys, C.; Van Langenhove, H. Post-plasma catalytic technology for the removal of toluene from indoor air: Effect of humidity. Appl. Catal. B Environ. 2009, 87, 78–83. [Google Scholar] [CrossRef]
- Harling, A.M.; Glover, D.J.; Whitehead, J.C.; Zhang, K. The role of ozone in the plasma-catalytic destruction of environmental pollutants. Appl. Catal. B Environ. 2009, 90, 157–161. [Google Scholar] [CrossRef]
- Grossmannova, H.; Neirynck, D.; Leys, C. Atmospheric discharge combined with Cu-Mn/Al2O3 catalyst unit for the removal of toluene. Czechoslov. J. Phys. 2006, 56, B1156–B1161. [Google Scholar] [CrossRef]
- Van Durme, J.; Dewulf, J.; Sysmans, W.; Leys, C.; Van Langenhove, H. Efficient toluene abatement in indoor air by a plasma catalytic hybrid system. Appl. Catal. B Environ. 2007, 74, 161–169. [Google Scholar] [CrossRef]
- Huang, Y.; Dai, S.; Feng, F.; Zhang, X.; Liu, Z.; Yan, K. A comparison study of toluene removal by two-stage DBD-catalyst systems loading with MnOx, CeMnOx, and CoMnOx. Environ. Sci. Pollut. Res. 2015, 22, 19240–19250. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Yasui, H.; Tanaka, M.; Futamura, S.; Kurita, S.; Aoyagi, K. Temperature dependence of toluene decomposition behavior in the discharge–catalyst hybrid reactor. IEEE Trans. Ind. Appl. 2009, 45, 1553–1558. [Google Scholar] [CrossRef]
- Delagrange, S.; Pinard, L.; Tatibouët, J.-M. Combination of a non-thermal plasma and a catalyst for toluene removal from air: Manganese-based oxide catalysts. Appl. Catal. B Environ. 2006, 68, 92–98. [Google Scholar] [CrossRef]
- Tang, X.; Feng, F.; Ye, L.; Zhang, X.; Huang, Y.; Liu, Z.; Yan, K. Removal of dilute VOCs in air by post-plasma catalysis over Ag-based composite oxide catalysts. Catal. Today 2013, 211, 39–43. [Google Scholar] [CrossRef]
- Huang, H.; Ye, D.; Guan, X. The simultaneous catalytic removal of VOCs and O3 in a post-plasma. Catal. Today 2008, 139, 43–48. [Google Scholar] [CrossRef]
- Demidyuk, V.; Whitehead, J.C. Influence of temperature on gas-phase toluene decomposition in plasma-catalytic system. Plasma Chem. Plasma Process. 2007, 27, 85–94. [Google Scholar] [CrossRef]
- Chlala, D.; Giraudon, J.-M.; Nuns, N.; Lancelot, C.; Vannier, R.-N.; Labaki, M.; Lamonier, J.-F. Active Mn species well dispersed on Ca2+ enriched apatite for total oxidation of toluene. Appl. Catal. B Environ. 2016, 184, 87–95. [Google Scholar] [CrossRef]
- Spasova, I.; Nikolov, P.; Mehandjiev, D. Ozone decomposition over alumina-supported copper, manganese and copper-manganese catalysts. Ozone 2007, 29, 41–45. [Google Scholar] [CrossRef]
- Vepřek, S.; Cocke, D.; Kehl, S.; Oswald, H. Mechanism of the deactivation of Hopcalite catalysts studied by XPS, ISS, and other techniques. J. Catal. 1986, 100, 250–263. [Google Scholar] [CrossRef]
- Puckhaber, L.S.; Cheung, H.; Cocke, D.L.; Clearfield, A. Reactivity of copper manganese oxides. Solid State Ion. 1989, 32, 206–213. [Google Scholar] [CrossRef]
- Li, W.; Zhuang, M.; Wang, J. Catalytic combustion of toluene on Cu-Mn/MCM-41 catalysts: Influence of calcination temperature and operating conditions on the catalytic activity. Catal. Today 2008, 137, 340–344. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Xia, Q.; Liu, Z.; Li, Z. Catalytic oxidation of toluene over copper and manganese-based catalysts: Effect of water vapor. Catal. Commun. 2011, 14, 15–19. [Google Scholar] [CrossRef]
- Aguilera, D.A.; Perez, A.; Molina, R.; Moreno, S. Cu–Mn and Co.–Mn catalysts synthesized from hydrotalcites and their use in the oxidation of VOCs. Appl. Catal. B Environ. 2011, 104, 144–150. [Google Scholar] [CrossRef]
- Li, W.; Zhuang, M.; Xiao, T.; Green, M. MCM-41 supported Cu–Mn catalysts for catalytic oxidation of toluene at low temperatures. J. Phys. Chem. B 2006, 110, 21568–21571. [Google Scholar] [CrossRef] [PubMed]
- Behar, S.; Gonzalez, P.; Agulhon, P.; Quignard, F.; Świerczyński, D. New synthesis of nanosized Cu–Mn spinels as efficient oxidation catalysts. Catal. Today 2012, 189, 35–41. [Google Scholar] [CrossRef]
- Saqer, S.M.; Kondarides, D.I.; Verykios, X.E. Catalytic oxidation of toluene over binary mixtures of copper, manganese and cerium oxides supported on γ-Al2O3. Appl. Catal. B Environ. 2011, 103, 275–286. [Google Scholar] [CrossRef]
- Zimowska, M.; Michalik-Zym, A.; Janik, R.; Machej, T.; Gurgul, J.; Socha, R.; Podobiński, J.; Serwicka, E. Catalytic combustion of toluene over mixed Cu–Mn oxides. Catal. Today 2007, 119, 321–326. [Google Scholar] [CrossRef]
- Ye, Z.; Giraudon, J.-M.; Nuns, N.; Simon, P.; De Geyter, N.; Morent, R.; Lamonier, J.-F. Influence of the preparation method on the activity of copper-manganese oxides for toluene total oxidation. Appl. Catal. B Environ. 2018, 223, 154–166. [Google Scholar] [CrossRef]
- Lian, Z.; Ma, J.; He, H. Decomposition of high-level ozone under high humidity over Mn–Fe catalyst: The influence of iron precursors. Catal. Commun. 2015, 59, 156–160. [Google Scholar] [CrossRef]
- Khacef, A.; Huu, T.P.; An, H.T.Q.; Le Van, T.; Cormier, J.-M. Removal of Toluene in Air by Non Thermal Plasma-Catalysis Hybrid System. In Proceedings of the 20th International Symposium of Plasma Chemistry, Philadelphia, PA, USA, 24–29 July 2011; pp. 1–4. [Google Scholar]
- Konelschatz, U.; Eliasson, B.; Egli, W. Dielectric-Barrier Discharges. Principle and Applications. J. Phys. IV Colloq. 1997, 7, C4-47. [Google Scholar]
- Chang, J.; Kostov, K.; Urashima, K.; Yamamoto, T.; Okayasu, Y.; Kato, T.; Iwaizumi, T.; Yoshimura, K. Removal of NF3 from semiconductor process flue gases by tandem packed bed plasma-adsorbent hybrid systems. In Proceedings of the 1998 IEEE Industry Applications Conference. Thirty-Third IAS Annual Meeting, St. Louis, MO, USA, 12–15 October 1998; IEEE: Piscataway, NJ, USA, 1998; pp. 1845–1852. [Google Scholar]
- Veerapandian, S.K.; Leys, C.; De Geyter, N.; Morent, R. Abatement of VOCs Using Packed Bed Non-Thermal Plasma Reactors: A Review. Catalysts 2017, 7, 113. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Value |
|---|---|
| Chemical formula | C7H8 |
| No. CAS | 108-88-3 |
| Molar mass (g/mol) | 92.14 |
| Density (g/mL) | 0.87 (20 °C) |
| Melting point (°C) | −95 |
| Boiling point (°C) | 111 |
| Solubility in water (g/L) | 0.52 (20 °C) |
| Vapor pressure (kPa) | 2.8 (20 °C) |
| Odor threshold (ppmv, Parts Per Million by volume) | 0.17 |
| Auto ignition temperature (°C) | 480 |
| Conversion factor | 1 ppmv = 3.76 mg/m3 (in air, 25 °C) |
| Health Effects | Oral | Dermal |
|---|---|---|
| Death | ● | |
| Acute | ● | ● |
| Intermediate | ||
| Chronic | ||
| Immunologic/Lyphoretic | ||
| Neurologic | ||
| Reproductive | ||
| Developmental | ||
| Genotoxic | ● | |
| Cancer |
| Catalyst | Active Phase | Flow Rate (mL/min) | GHSV (mL·h−1·g−1) | Temperature (°C) | CC7H8 (ppm) | CO3 (ppm) | O3/C7H8 | ηToluene (%) | ηOzone (%) | By-Products | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 20% MnO2/Al2O3 | MnO2 | 100 (dry air) | 60,000 | 20 | 177 | 98 | 0.55 | ~25 (160 min) then deactivation | From 100 to 80% in 160 min | Benzoic acid, benzaldehyde, and benzyl alcohol. | [55] |
| 10% MnOx/γ-Al2O3 | MnO2 and Mn2O3 | 1000 (dry air) | 300,000 | 20 | 120 | 1050 | 8.75 | From 100 to 40% in 330 min | From 100 to 20% in 330 min | Acetic acid, oxalic acid, benzene and maleic anhydride Carbon balance: ~20% | [49] |
| 65 wt % MnO2/graphene | MnO2 | 150 (dry air) | 112,500 | 20 | 200 | 400 | 2 | ~33.6 (400 min) then deactivation | ~84.3 (400 min), then deactivation | Organic byproducts | [52] |
| MnO2/TiO2/SiO2 honeycomb | MnO2 | 1000 (dry air) | - | 20 | 10 | 38 | 3.8 | ~72 (100 min) | - | Carbon balance: ~22% | [54] |
| MnO2 | 1000 (RH: 70%) | - | 20 | 10 | 38 | 3.8 | ~35 (100 min) | - | Carbon balance: ~50% | ||
| Mn/HZSM-5 | MnOx | 100 (dry air) | 150,000 | 30 | 20 | 200 | 10 | 78 | 70 | benzaldehyde, benzoic acid and maleic anhydride | [53] |
| Exp. | Catalyst | The Shape of Catalyst | The State of Catalyst | The Support | The Nature of Catalyst | Structure(s) (XRD) | The Synthesis Method/Commercial Company | SBET (m²/g) | Dp 1 (nm) | Added Characterizations | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Cu–Mn/Al2O3 | Powder | Supported | Al2O3 | Cu-Mn | - | Heraeus, Hanau, Germany | - | - | - | [70] |
| 2 | (a) N 150 (MnO2–Fe2O3) | Pellet | Bulk | - | 60 wt % Fe2O3 & 40 wt % MnO2 | MnO2 | Süd-Chemie (Munchen, Germany) | 219 | - | - | [74] |
| (b) MnO2/γ-Al2O3 | Powder | Supported | γ-Al2O3 | 9 wt % MnO2 | Incipient wetness impregnation, Precursor: Mn nitrate, calcined at 350 °C for 2 h under 100 mL/min air | 169 | |||||
| (c) MnO/AC | AC 2 | 3 wt % MnO2 | Wet impregnation, Precursor: Mn acetate, calcined at 300 °C for 2 h under N2 | 1024 | |||||||
| 3 | (a) Mn-1 (Mn–P–O) | Powder | Bulk | - | Mn–P–O | Amorphous phase | Hydrothermal: MnO2 with H3PO4 (85 wt %) (Mn:P 1:1.5) in 50 mL distilled water + dimethylaminoalcohol, 170 °C for 48 h | 4.5 | - | XPS: Mn 2p3/2: oxidation state of Mn < 4; P 2p3/2: inorganic phosphates | [67] |
| (b) Mn-2 (Mn–P–O) | Mn–P–O | Hydrothermal treatment under microwaves: starting gel: 1.00 P: 0.04 Mn: 0.48 HDTMABr 3: 0.48 TMAOH 4: 174.00 H2O, 60 °C (1 h), 80 °C (1 h), and 300 °C (3 h) then calcination at 550 °C | 21 | 1.1 | |||||||
| (c) Mn-3 (M–P–O) | Mn–P–O | Starting gel: 1.00 P: 1.00 Mn: 0.48 HDTMABr : 0.48 TMAOH: 174.00 H2O, same operating mode than above | 85 | 3.4, 50 | |||||||
| 4 | CuMn/TiO2 | Pellets | Supported | TiO2 | 3% Cu, 6.8% MnO2 | - | Heraeus, Hanau, Germany | 50 | - | - | [71] |
| 5 | MnO2/Al2O3 | Pellets | Supported | Al2O3 5 | 7 wt % MnO2 | - | Wetness impregnation Precursor: Mn nitrate, calcined at 400 °C | - | - | - | [77] |
| 6 | Mn2O3/Ni foam | Foam | Supported | Ni foam | Mn2O3 | Mn2O3 | Impregnation Precursor: Mn nitrate, calcined at 600 °C | 10.8 | - | XRD: before 400 °C, ultra-fine or amorphous features of phase. After 500 °C, Mn2O3 | [76] |
| 7 | (a) Cu–Mn/TiO2 (a) | Pellet (1.5 mm) | Supported | TiO2 | CuO (3 wt %) MnO2 (6.8 wt %) | - | Hereaus Hanau, Germany | 32 | - | - | [68] |
| (b) Cu–Mn/TiO2 (b) | - | 50 | - | - | |||||||
| (c) Fe2O3-MnO2 (N 150) | Pellet (6 mm) | Bulk | - | >40 wt % Fe2O3 >25 wt % MnO2 | - | Süd-Chemie (Munchen, Germany) | 100 | - | - | ||
| (d) CuO-MnO2 (N 140) | Pellet (5 mm) | >15 wt % CuO >25 wt % MnO2 | - | 100 | - | - | |||||
| 8 | Fe2O3-MnO2 | Honeycomb 6 | Bulk | - | 60 wt % Fe2O3, 30 wt % MnO2 | - | Süd-Chemie (Japan) | - | - | - | [73] |
| 9 | (a) MnO2/Aluminum honeycomb | Honeycomb | Supported | Aluminum honeycomb | MnO2 | - | Honeycle ZA (Nichias Corporation) | - | - | - | [69] |
| (b) MnO2-CuO | Pellet (4–6 mesh) | Bulk | - | MnO2-CuO | - | Moleculite (Molecular Products Limited) | 20–30 | - | - | ||
| 10 | (a) OMS-2 | Powder | Bulk | - | α-MnO2 | - | Hydrothermal treatment, Redox method, Precursors: KMnO4 + MnSO4 100 °C for 24 h, pH: 1.2, calcined in static air using a range of time and temperature | - | - | - | [65,91] |
| (b) Cu-OMS-2 | Supported | OMS-2 7 | 5 wt % Cu | - | Wetness impregnation, Precursor: Cu(NO3)2 Calcined at 500 °C for 4 h | - | - | - | |||
| (c) Cu-OMS-2 | 10 wt % Cu | - | - | - | - | ||||||
| (d) Cu-OMS-2 | 15 wt % Cu | - | - | - | - | ||||||
| 11 | Ag–Mn–O | Powder | Bulk | - | 10 wt % Ag-MnOx | Mn3O4 + Ag | Ag–Mn–O: Co-precipitation, pH 10, Precursors: Mn(NO3)2 + AgNO3 dried at 110 °C for 12 h | - | - | - | [75] |
| 12 | (a) MnOx | Powder | Bulk | MnOx | Mn3O4 + Mn5O8 | Precipitation, pH 9–10 Precursor: Mn nitrate, then calcined at 400 °C for 2 in air. | 27.9 | - | XPS: lattice oxygen 53.1% | [66] | |
| (b) Co3O4-MnO2 | 1:6 = Co:Mn | Mn3O4 | Coprecipitation at pH 9–10, Precursor: Mn and Co nitrates calcined at 400 °C for 2 h under in air. | 29.5 | XPS: lattice oxygen 56.2% | ||||||
| (c) Co3O4-MnO2 | 1:1 = Co:Mn | Co3Mn3O4 + Co3O4 | 53.4 | - | |||||||
| (d) Co3O4-MnO2 | 6:1 = Co:Mn | Co3O4 | 61.6 | - | |||||||
| 13 | (a) MnOx/ZSM-5 | Powder | Supported | ZSM-5 | 10wt %t MnOx | MnO2,Mn2O3, and Mn3O4 | Precipitation, pH 9–10, calcined at 400 °C for 2 h in under air. Then wet impregnation with ZSM-5, calcined at 400 °C for 5 h | 290.7 | 3.4 | XPS: lattice O at 532.9 eV: 55.2% | [72] |
| (b) CoMnOx/ZSM-5 | 10wt %t CoMnOx (Co/Mn = 1) | Co3Mn3O4, MnO2 and Mn3O4 | Coprecipitation. Same experimental procedure than above | 302.8 | 2.9 | XPS: lattice O at 532.9 eV: 59.2% | |||||
| (c) CeMnOx/ZSM-5 | 10wt %t CeMnOx (Ce/Mn = 1) | CeO2, Ce2O3, MnO2, and Mn3O4 | 305.6 | 2.9 | XPS: lattice O at 532.9 eV: 58.3% |
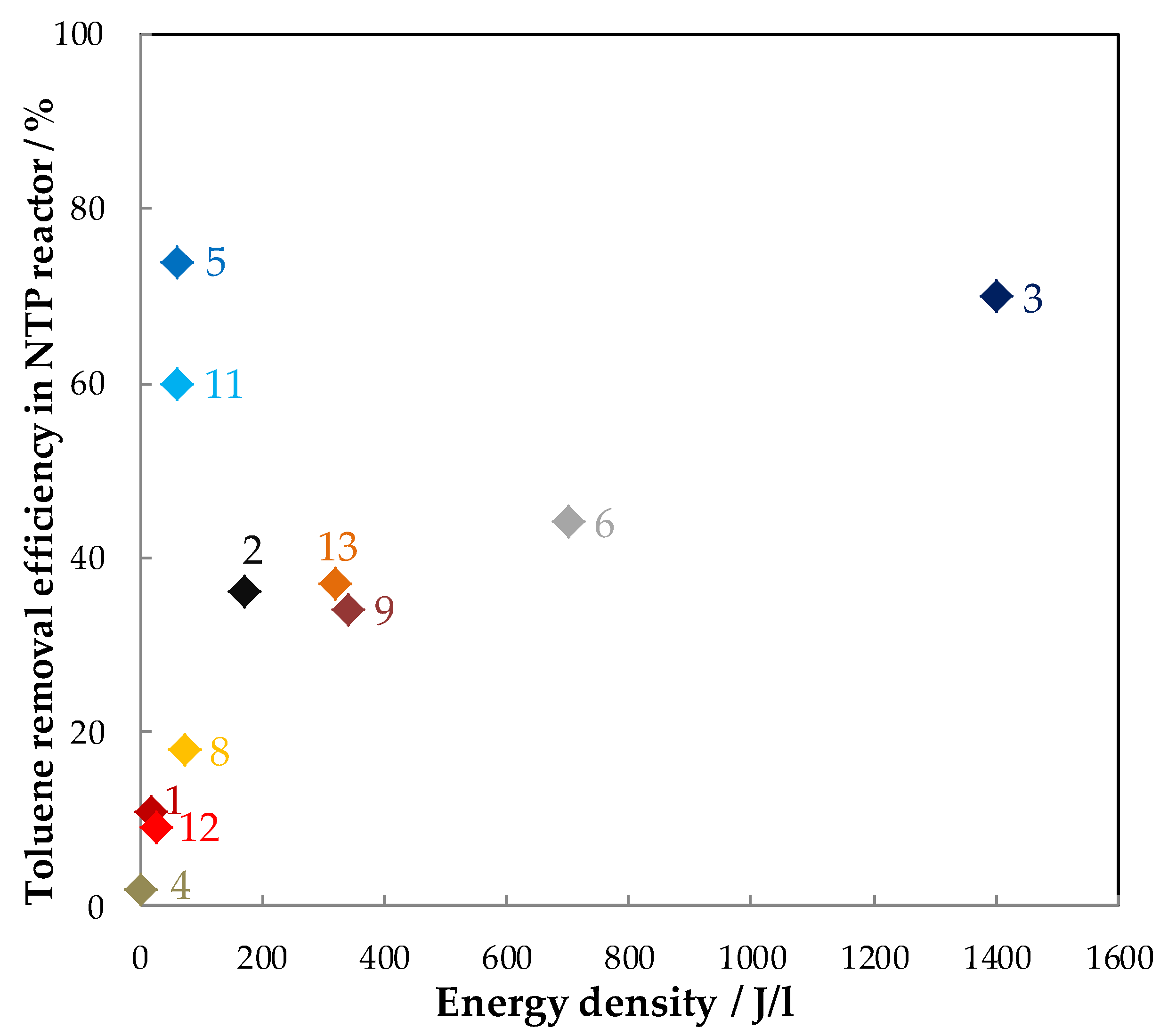
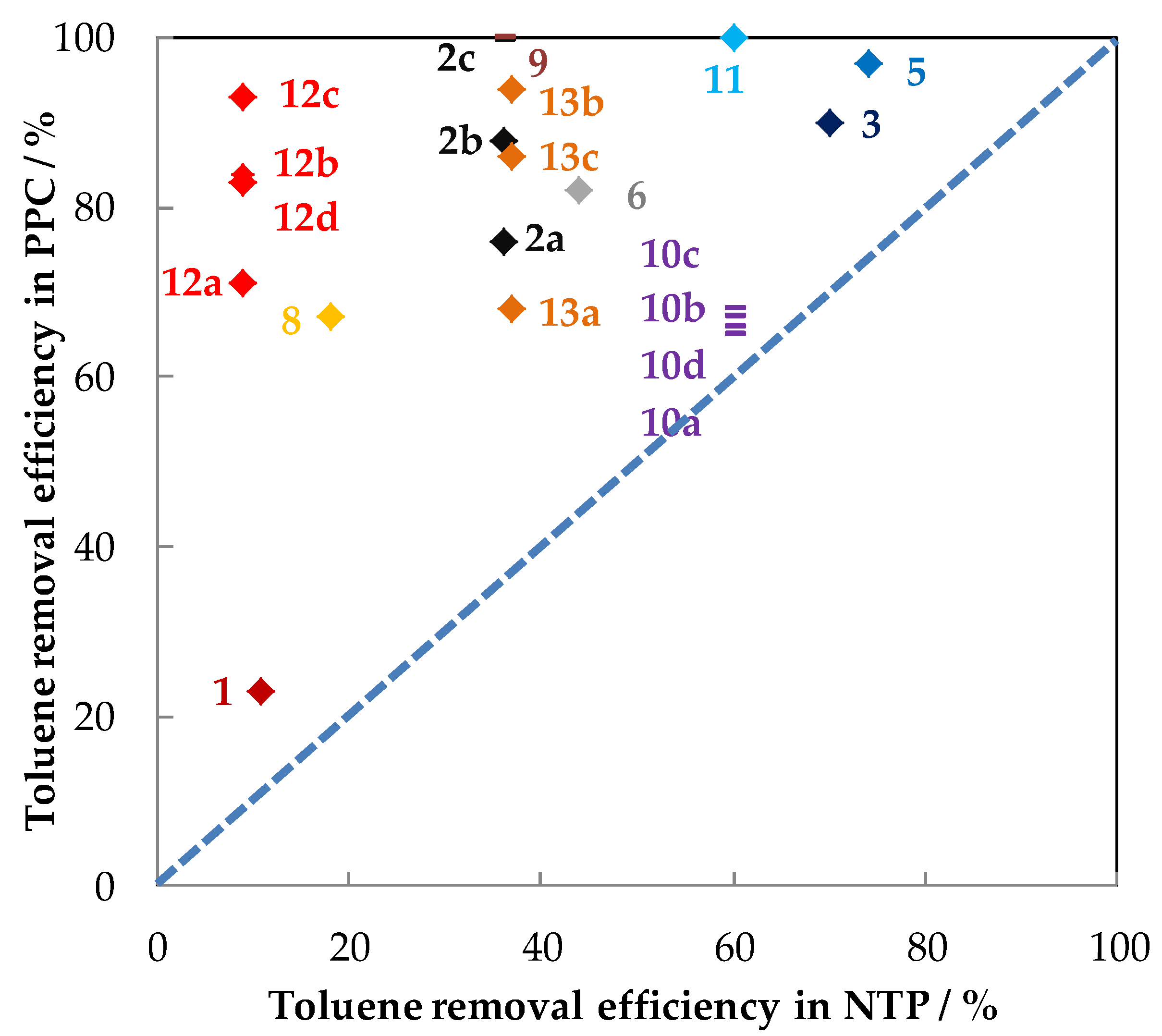
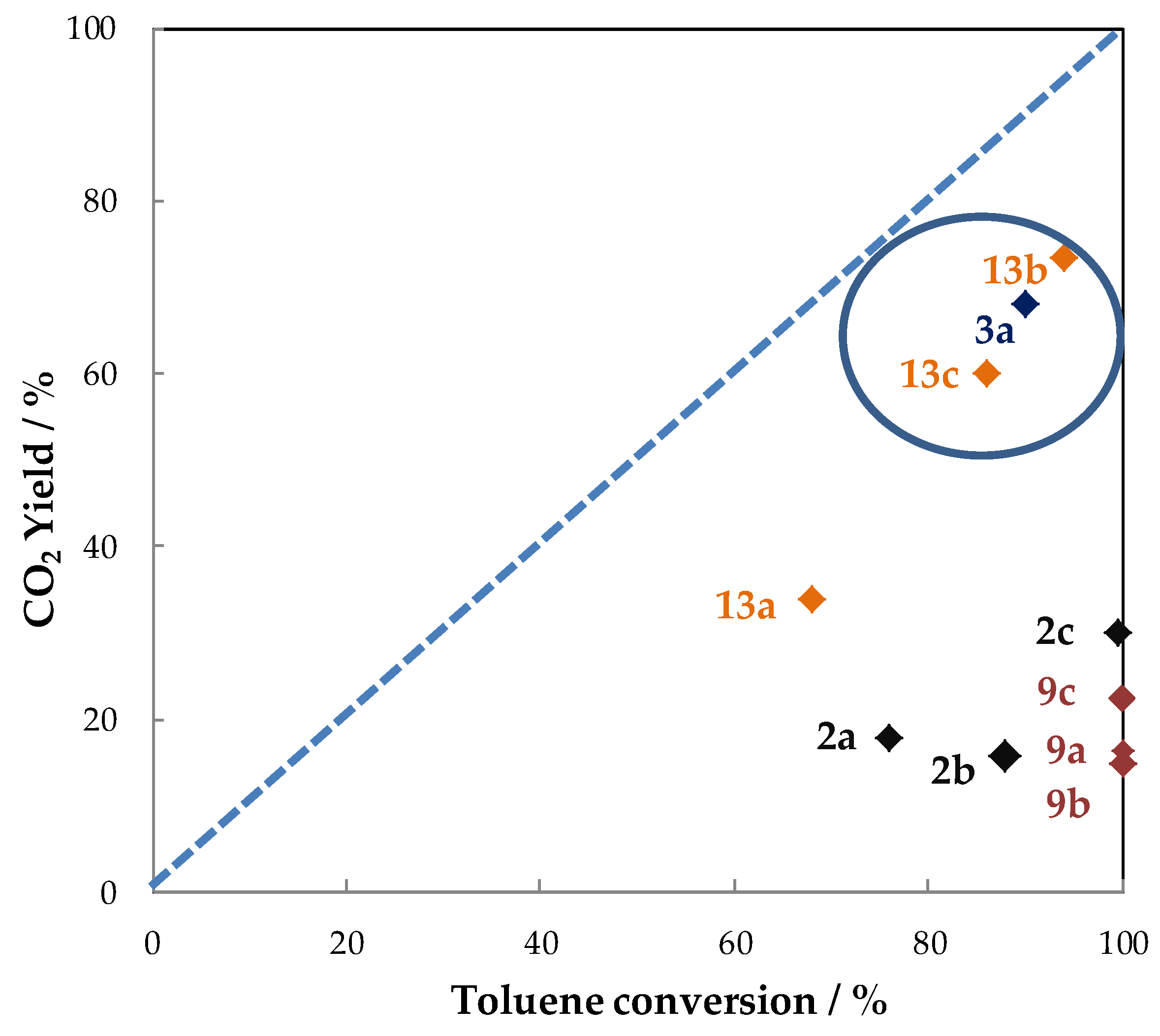
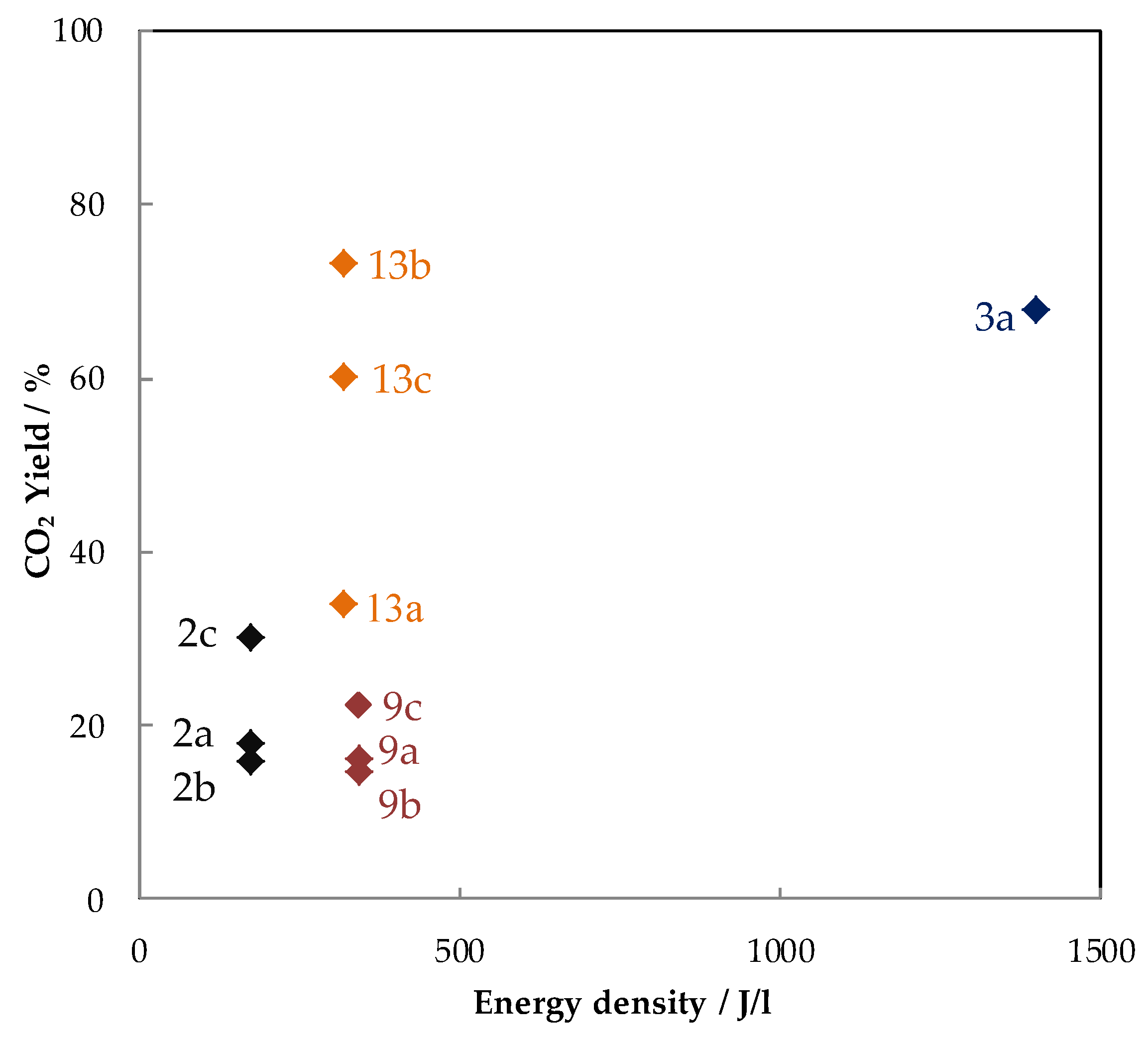
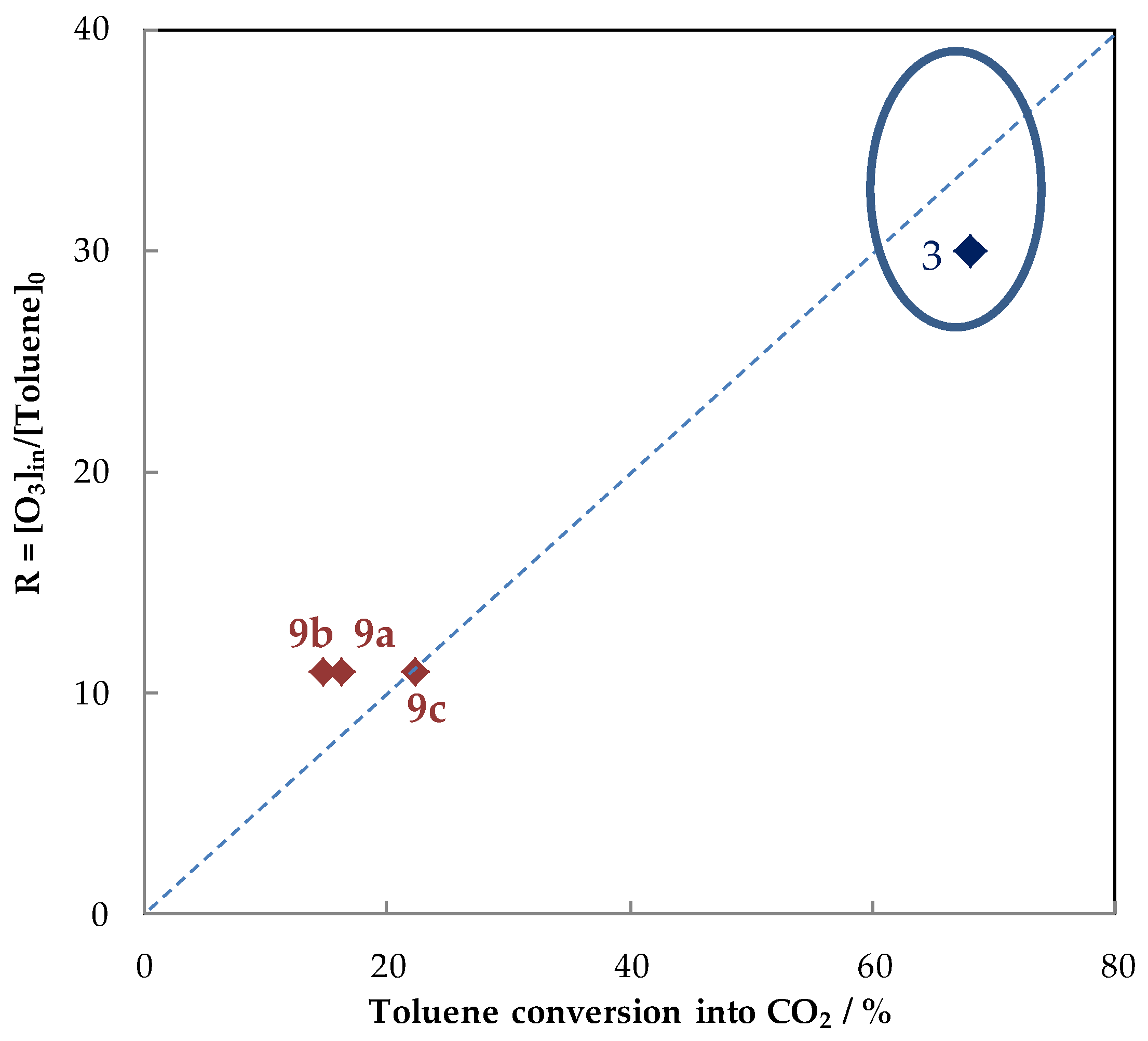
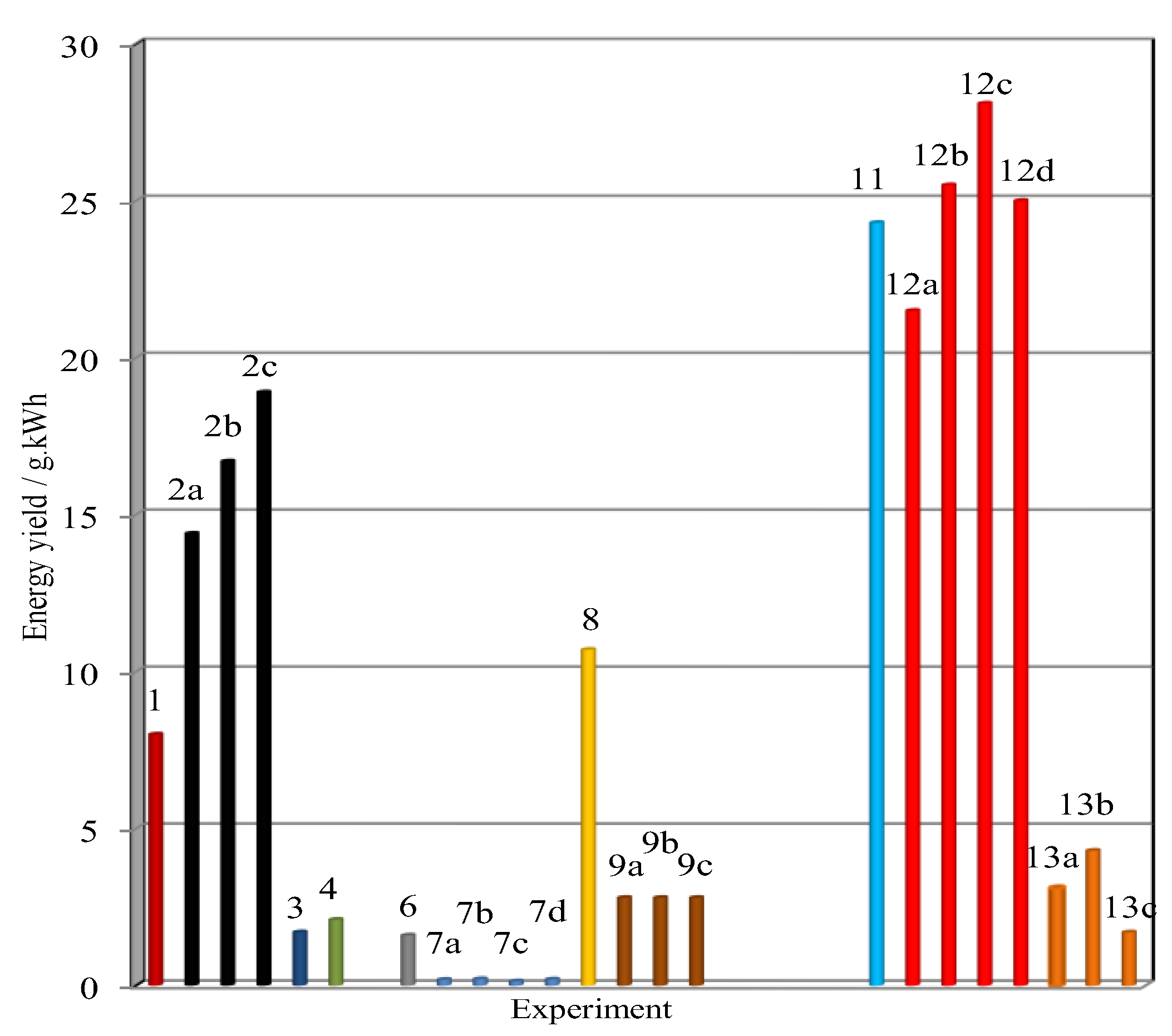
| Exp. | Discharge Type (NTP Reactor) | Catalyst (weight/g) | Configuration | Carrier Gas Flow Rate (mL/min) | Toluene (ppm) | RH (%) | Inlet Ozone (ppm) | Inlet Ozone/Inlet Toluene | GHSV (mL·g−1·h−1) | MRE 1 (%) | ED 2 (J/L) | EY 3 (g/kWh) | SCO2/(SCO) (%) | YCO2 (%) | By-Products CB 4 | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Positive Corona discharge (pin-plate) | Cu–Mn/Al2O3 (-) | PPC | Air 133,600 | 45 | - | 60 | 1.3 | - | 22.8 | 18.7 | 8 | - | - | - | [70] |
| - | NTP | Air 133,600 | 45 | - | - | - | - | 10.8 | 18.7 | 4 | - | - | - | |||
| 2 | Glass beads packed bed DBD (cylindrical reactor) | (a) N 150 (MnO2–Fe2O3) (1) | PPC | Air 315 | 240 | - | - | - | 18,900 | 76 | 172 | 14.4 | 23.5 (16.5) | 17.9 | CB 2: 52.6%; O3: 3.9 ppm | [74] |
| (b) MnO2/γ-Al2O3 (1) | - | - | - | 88 | 16.7 | 18 (14) | 15.8 | CB: 36.3% O3: 14.6 ppm | ||||||||
| (c) MnO/AC (1) | - | - | - | 99.7 | 18.9 | 30.2 (24.8) | 30.1 | CB: 55%. O3: 8 ppm | ||||||||
| - | NTP | Air 315 | 240 | - | - | - | 18,900 | 36 | 172 | 6.9 | 6 | 2.2 | SCO: 8%, CB: 38.8% O3: 8 ppm | |||
| 3 | DBD (Cylindrical reactor) | (a) Mn-1 (Mn–P–O) (0.2) | PPC | Air 300 | 200 | - | 6000 | 30 | 90,000 | 90–95 | 1400 | 1.7–1.8 | 73 (27) | 65.7–69.4 | CB: ~100% | [67] |
| (b) Mn-2 (Mn–P–O) (0.2) | - | 30 | 72 (28) | 64.8–68.4 | CB: ~100% | |||||||||||
| (c) Mn-3 (Mn–P–O) (0.2) | - | 30 | 68 (32) | 61.2–64.6 | CB: ~100% | |||||||||||
| - | NTP | Air 300 | 200 | - | - | - | 90,000 | 70–75 | 1400 | 1.3–1.4 | - | - | - | |||
| 4 | Positive DC corona discharge (Pin-plate) | CuOMnO2/TiO2 (10) | PPC | Air 10,000 | 0.5 | - | ~18 | 36 | 60,000 | 78 | 2.5 | 2.1 | - | - | NO2: ~550 ppm at 15 J/L | [71] |
| - | NTP | Air 10,000 | 0.5 | 27 | - | - | 60,000 | 4 | 2.5 | 0.1 | - | - | - | |||
| 5 | BaTiO3 packed bed DBD (Cylindrical reactor) | 7 wt % MnO2/Al2O3 (7 mL) | PPC | Air 1000 | 500 | - | - | - | 60,000 | 97 5 | 60 | - | - | - | CO: ~300 ppm | [77] |
| - | NTP | Air 1000 | 500 | - | - | - | 8600 h−1 | 74 5 | 60 | - | - | - | - | |||
| 6 | DBD (wire-plate) | Mn2O3/Ni foam (-) | PPC | Air 200 | 100 | - | 150 | 1.5 | - | 82 | 700 | 1.6 | - | - | Ozone: 9 ppm | [76] |
| - | NTP | - | - | - | - | 44 | 700 | 0.8 | - | - | - | |||||
| 7 | Positive DC corona discharge (pin-plate) | (a) Cu–Mn/TiO2(a) (15) | PPC | Air 10,000 | 0.5 | 50 | ~70 | 140 | 40,000 | 40 | 14 | 0.19 | - | - | - | [68] |
| (b) Cu–Mn/TiO2(b) (15) | ~100 | 200 | 63 | 20 | 0.21 | - | - | - | ||||||||
| (c) Fe2O3-MnO2 (N 150) (15) | ~82 | 164 | 34 | 15.8 | 0.14 | - | - | - | ||||||||
| (d) CuO-MnO2 (N 140) (15) | - | - | 47 | - | 0.20 | - | - | - | ||||||||
| 8 | DBD (tubular reactor) | Fe2O3-MnO (-) | PPC | Air 2500 | 85 | - | 330 | 3.9 | - | 67 | 72 | 10.7 | - | 13 4 | Gas: Benzene Solid: benzaldehyde, methylbenzoquinone, benzyl alcohol, benzoic acid, and benzyl benzoate | [73] |
| - | NTP | Air 2500 | 85 | - | - | - | - | 18 | 72 | 2.8 | - | 7 4 | Formic acid, Acetic acid and benzene | |||
| 9 | Multistage reactor | (a) MnO2/Aluminum honeycomb (-) | PPC | Air +10,000 | 70 | - | 766 | 10.9 | - | 100 | 342 | 2.8 | 16.3 | 16.3 | CO: 48 ppm; N2O:18 ppm | [69] |
| (b) MnO2-CuO (-) | 10.9 | - | 100 | 14.7 | 14.7 | CO: 8 ppm; O3: 117 ppm; N2O: 21 ppm | ||||||||||
| (c) MnO2/Aluminum honeycomb+ MnO2-CuO (-) | 10.9 | - | 100 | 22.4 | 22.4 | CO: 10 ppm; N2O: 18 ppm | ||||||||||
| - | NTP | Air 10,000 | 70 | - | - | - | - | ~36 | 342 | 1.0 | 10.8 | 3.9 | CO: 16 ppm; O3: 1327 ppm; N2O: 23 ppm | |||
| 10 | DBD (Cylindrical reactor) | (a) OMS-2 (0.2) | PPC | 80% N2 +20% O2 + 66.67 | 800 | - | - | - | 20,000 | 65 | 18 kV and 50 Hz | - | - | - | - | [65,91] |
| (b) 5 wt % Cu-OMS-2 (0.2) | - | - | - | 67 | - | - | - | - | ||||||||
| (c) 10 wt % Cu-OMS-2 (0.2) | - | - | - | 68 | - | - | - | - | ||||||||
| (d) 15 wt % Cu-OMS-2 (0.2) | - | - | - | 66 | - | - | - | - | ||||||||
| - | NTP | 80% N2 + 20% O2 + 66.67 | 800 | - | - | - | 20,000 | 60 | - | - | - | - | ||||
| 11 | DBD (tubular reactor) | Ag–Mn–O (0.2) | PPC | Air + 498 | 108 | - | 650 | 6.0 | 150,000 | 100 | 60 | 24.3 | - | - | N2O and CO | [75] |
| - | NTP | Air + 498 | 108 | - | - | - | 150,000 | 60 | 60. | 14.6 | - | - | O3:650 ppm N2O and CO | |||
| 12 | DBD (tubular reactor,) | (a) MnOx | PPC | Air + 500 | 107 | - | 580 | 5.4 | 32,233 h−1 | 71 | 48 | 21.5 | - | - | O3: 280 ppm N2O, CO | [66] |
| (b) (1:6) Co3O4-MnO2 | 5.4 | 84 | 25.5 | - | - | O3: 180 ppm N2O, CO | ||||||||||
| (c) (1:1) Co3O4-MnO2 | 5.4 | 93 | 28.1 | - | - | O3: 90 ppm N2O, CO | ||||||||||
| (d) (6:1) Co3O4-MnO2 | 5.4 | 83 | 25.0 | - | - | O3: 110 ppm N2O, CO | ||||||||||
| - | NTP | Air + 500 | 107 | - | - | 32,233 h−1 | ~24 | 48 | 7.3 | - | - | - | ||||
| 13 | DBD (tubular reactor) | (a) MnOx/ZSM-5 | PPC | Air + 1000 | 107 | 12 mL/min air bubbling water | - | - | - | 68 | 320 | 3.1 | 50 (8) | 34 | N2O, O3: 370 ppm | [72] |
| (b) CoMnOx/ZSM-5 | - | - | 94 | 4.3 | 78 (8) | 73.3 | N2O, O3: 200 ppm | |||||||||
| (c) CeMnOx/ZSM-5 | - | - | 86 | 3.9 | 70 (8) | 60.2 | N2O, O3: 260 ppm | |||||||||
| - | NTP | Air + 1000 | 107 | - | - | - | ~37 | 320 | 1.7 | - | - | - |
| Exp. | 1 [70] | 2 [74] | 3 [67] | 4 [71] | 5 [77] | 6 [76] | 7 [68] |
| Positive DC Corona discharge (pin-plate) | Glass beads packed bed DBD (cylindrical reactor) | DBD (cylindrical reactor) | positive DC Corona (pin-plate) | BaTiO3 packed bed DBD (cylindrical reactor) | DBD (wire-plate) | Positive DC corona discharge (pin-plate) | |
| Ctol (ppmv) | 45 | 240 | 200 | 0.5 | 500 | 100 | 0.5 |
| Gap (mm) | 9 | 6 | 2 | 20 | - | 10 | - |
| VR 1/mL (tr 2/s) | 72 (4.5 × 10−4) | 13.2 (2.5) | 8.5 (1.7) | 250 (1.5) | - | - | 250 (1.5) |
| Exp. | 8 [73] | 9 [69] | 10 [65] | 11 [75] | 12 [66] | 13 [72] | |
| DBD (tubular reactor) | Multistage reactor | DBD (tubular reactor) | DBD (tubular reactor) | DBD (tubular reactor) | DBD (tubular reactor) | ||
| Ctol (ppmv) | 85 | 70 | 800 | 109 | 107 | 107 | |
| Gap (mm) | 5.65 | - | - | 2 | 2.5 | 4 | |
| VR 1/L (tr 2/s) | - | - | - | 11.9 (1.4) | 14.9 (1.8) | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Z.; Giraudon, J.-M.; De Geyter, N.; Morent, R.; Lamonier, J.-F. The Design of MnOx Based Catalyst in Post-Plasma Catalysis Configuration for Toluene Abatement. Catalysts 2018, 8, 91. https://doi.org/10.3390/catal8020091
Ye Z, Giraudon J-M, De Geyter N, Morent R, Lamonier J-F. The Design of MnOx Based Catalyst in Post-Plasma Catalysis Configuration for Toluene Abatement. Catalysts. 2018; 8(2):91. https://doi.org/10.3390/catal8020091
Chicago/Turabian StyleYe, Zhiping, Jean-Marc Giraudon, Nathalie De Geyter, Rino Morent, and Jean-François Lamonier. 2018. "The Design of MnOx Based Catalyst in Post-Plasma Catalysis Configuration for Toluene Abatement" Catalysts 8, no. 2: 91. https://doi.org/10.3390/catal8020091
APA StyleYe, Z., Giraudon, J.-M., De Geyter, N., Morent, R., & Lamonier, J.-F. (2018). The Design of MnOx Based Catalyst in Post-Plasma Catalysis Configuration for Toluene Abatement. Catalysts, 8(2), 91. https://doi.org/10.3390/catal8020091