Carbon-Supported Copper-Based Nitrogen-Containing Supramolecule as an Efficient Oxygen Reduction Reaction Catalyst in Neutral Medium


Abstract
:1. Introduction
2. Results and Discussion
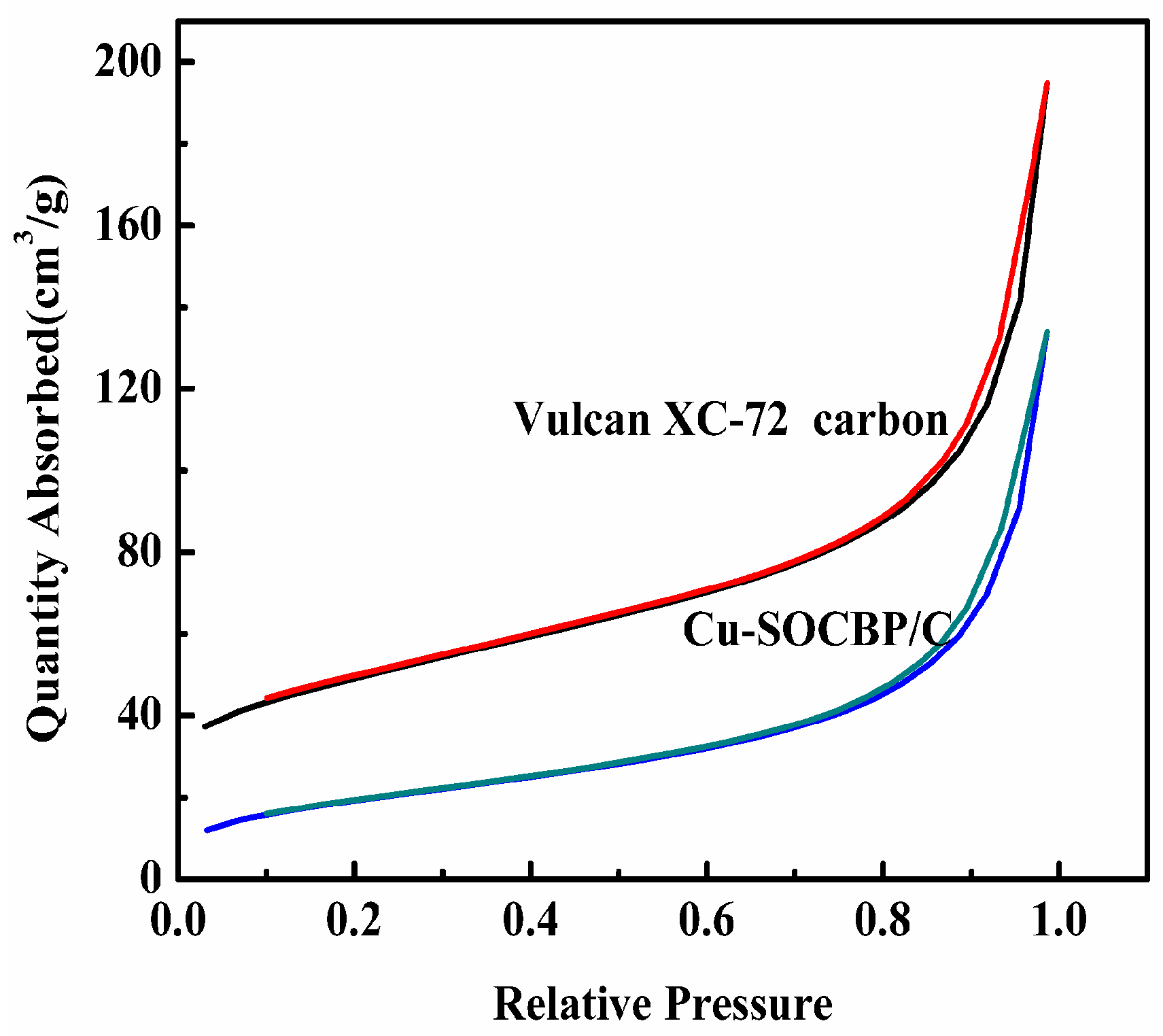
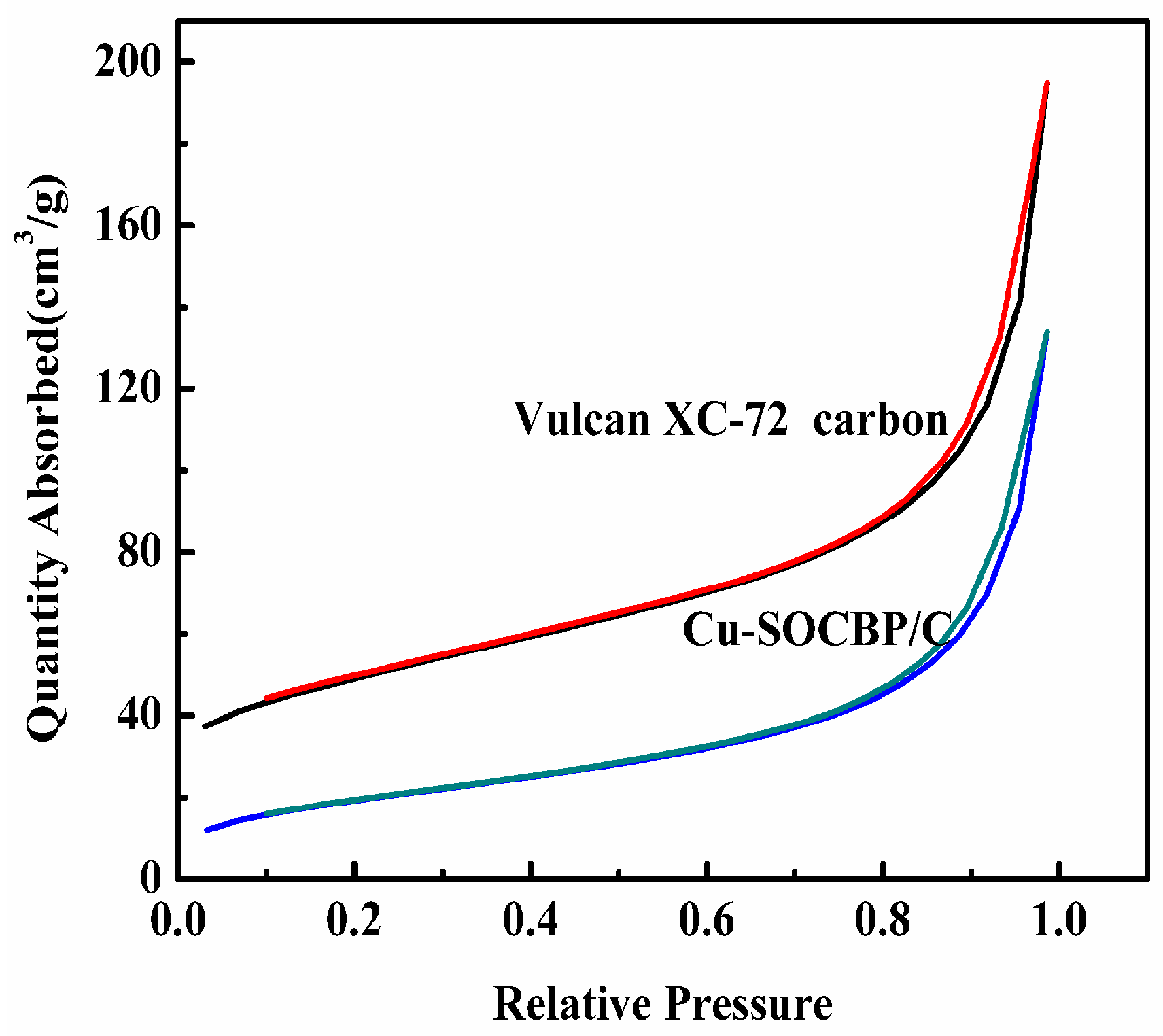
2.1. SEM and Physisorption
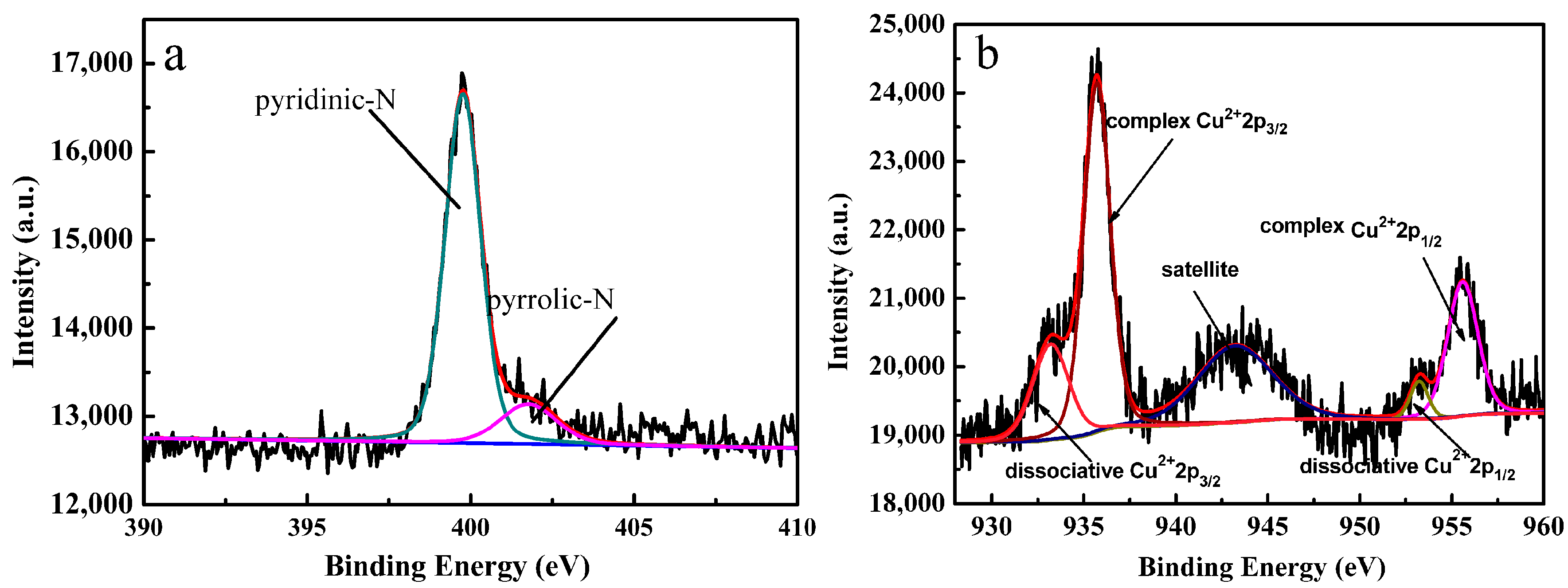
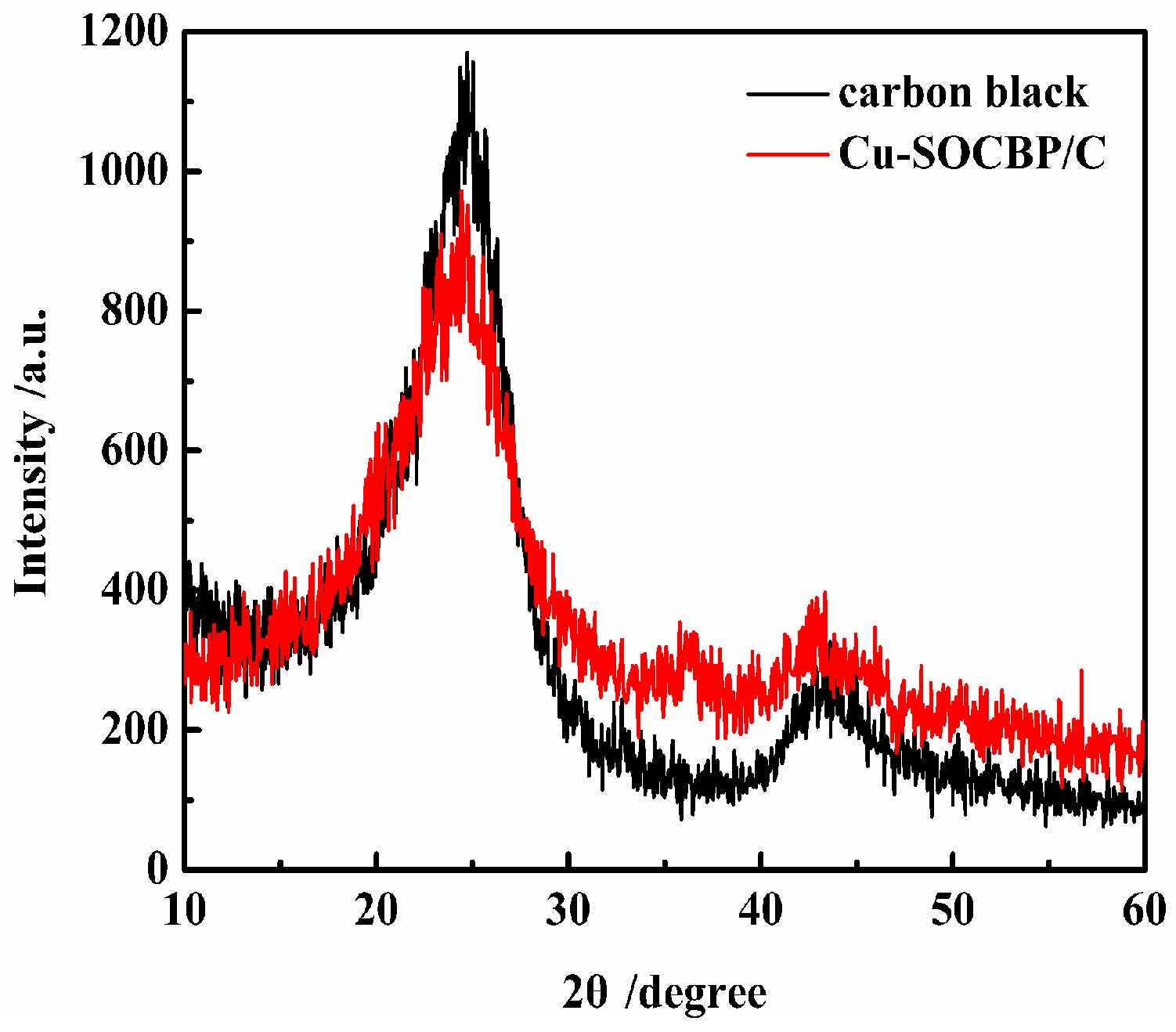
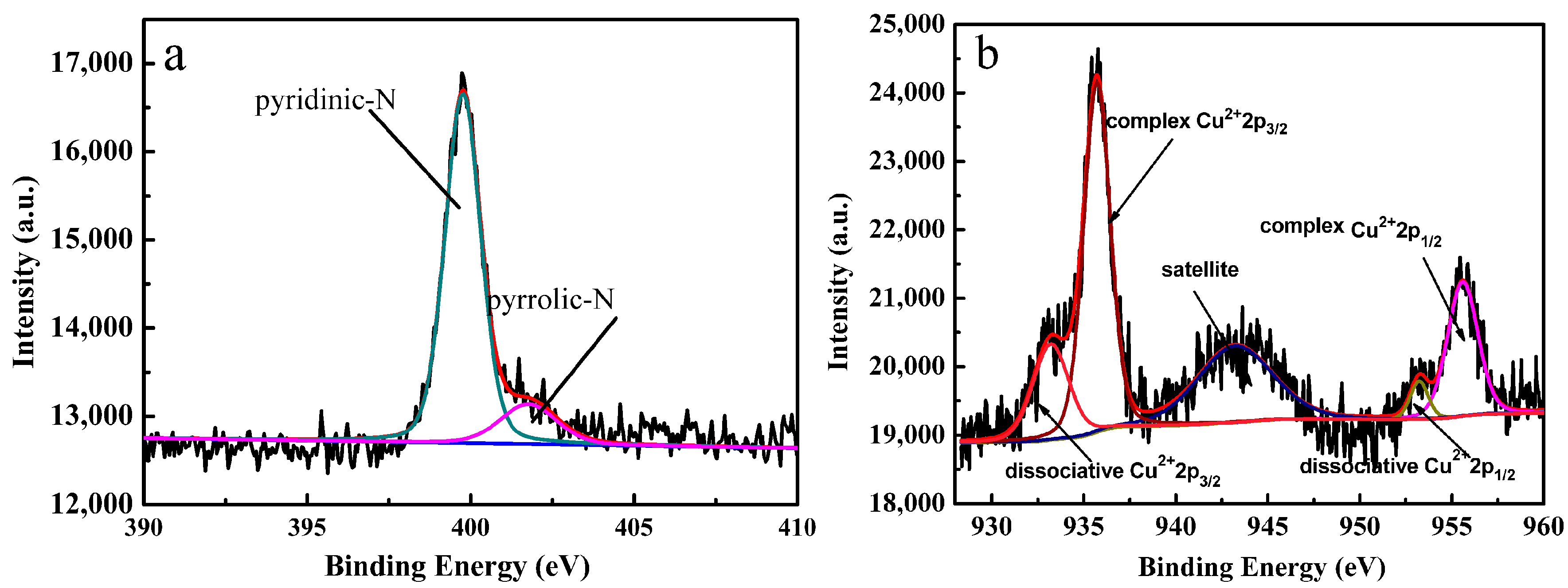
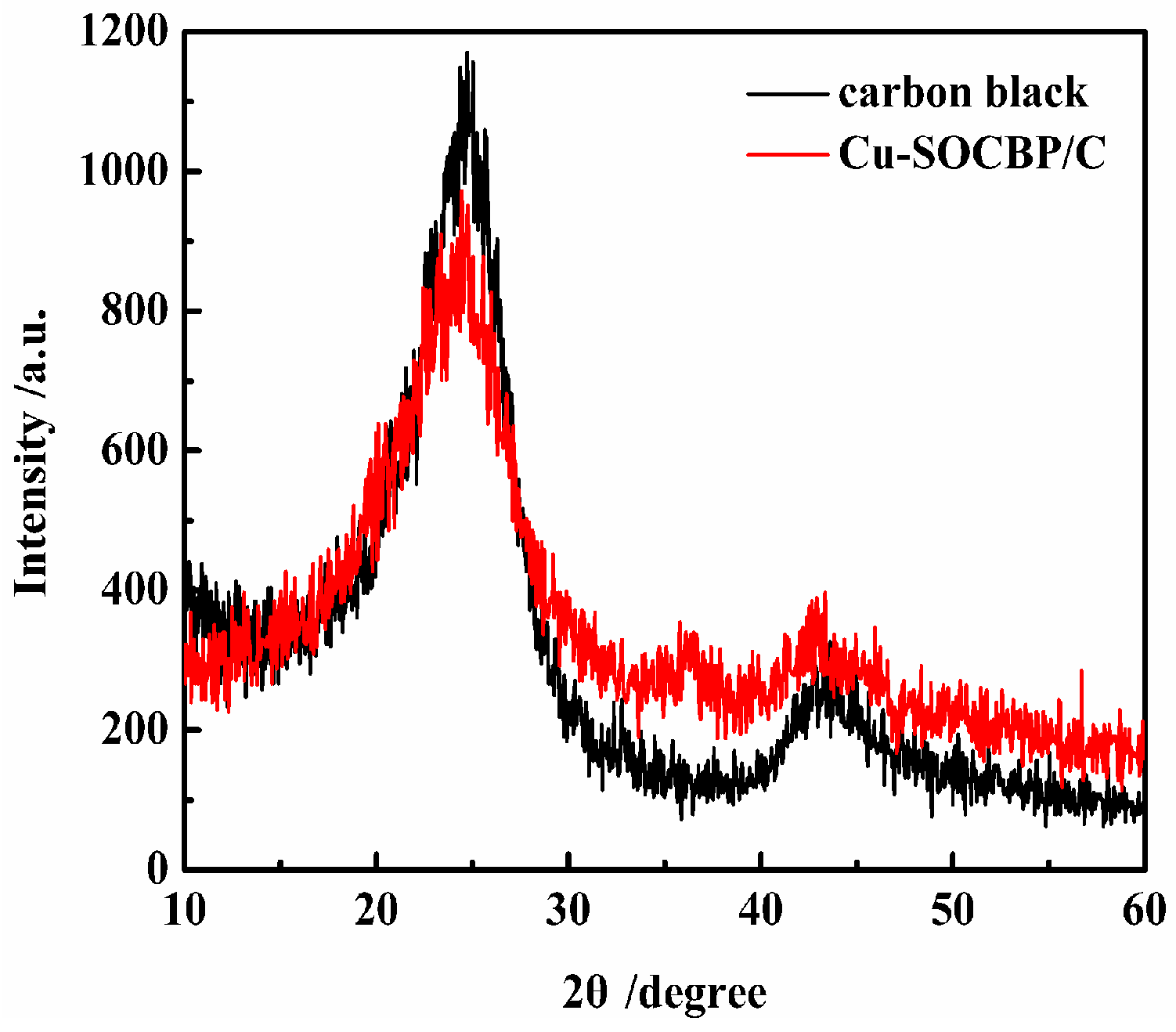
2.2. XPS and Powder X-ray Diffraction of the Catalyst
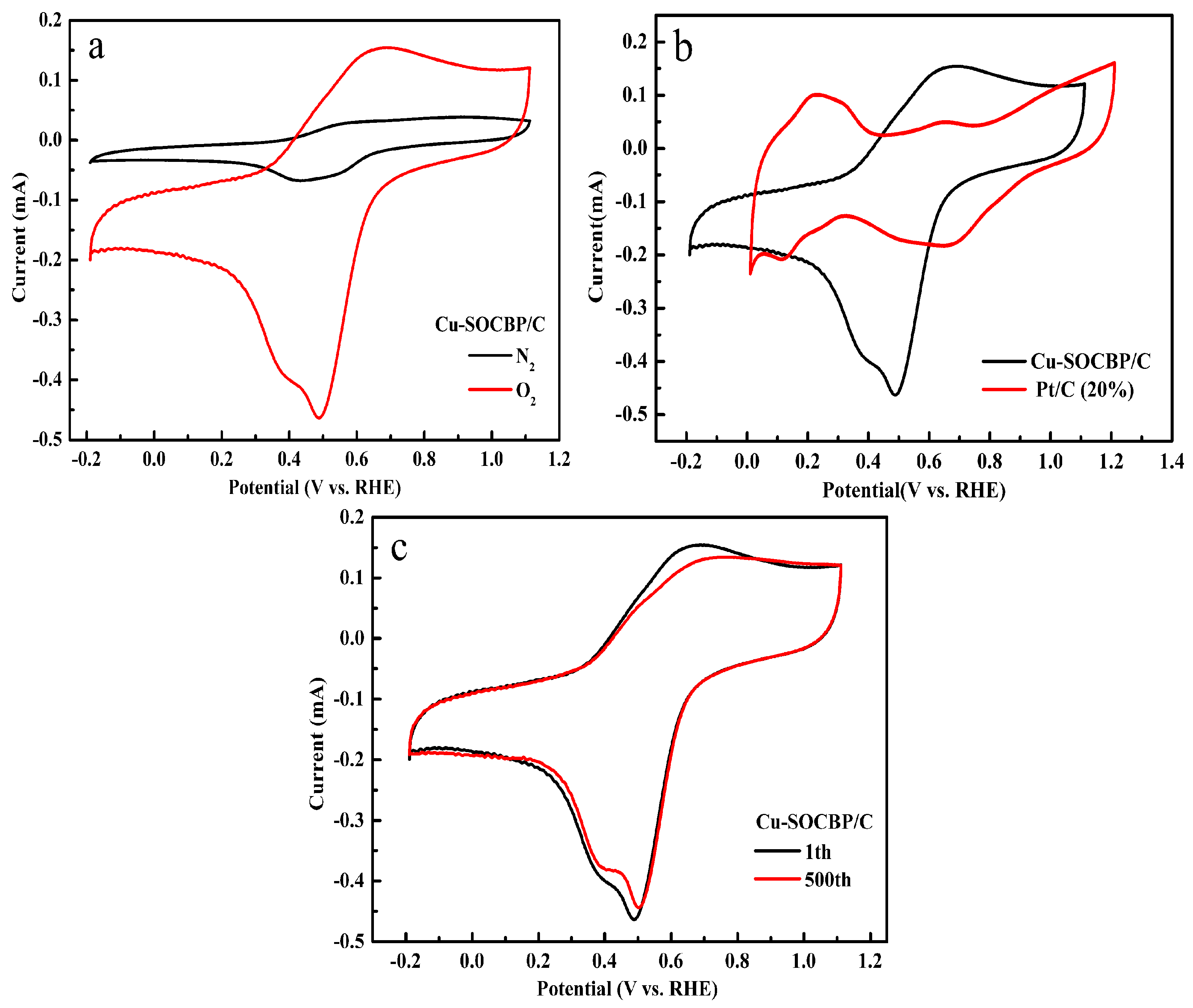
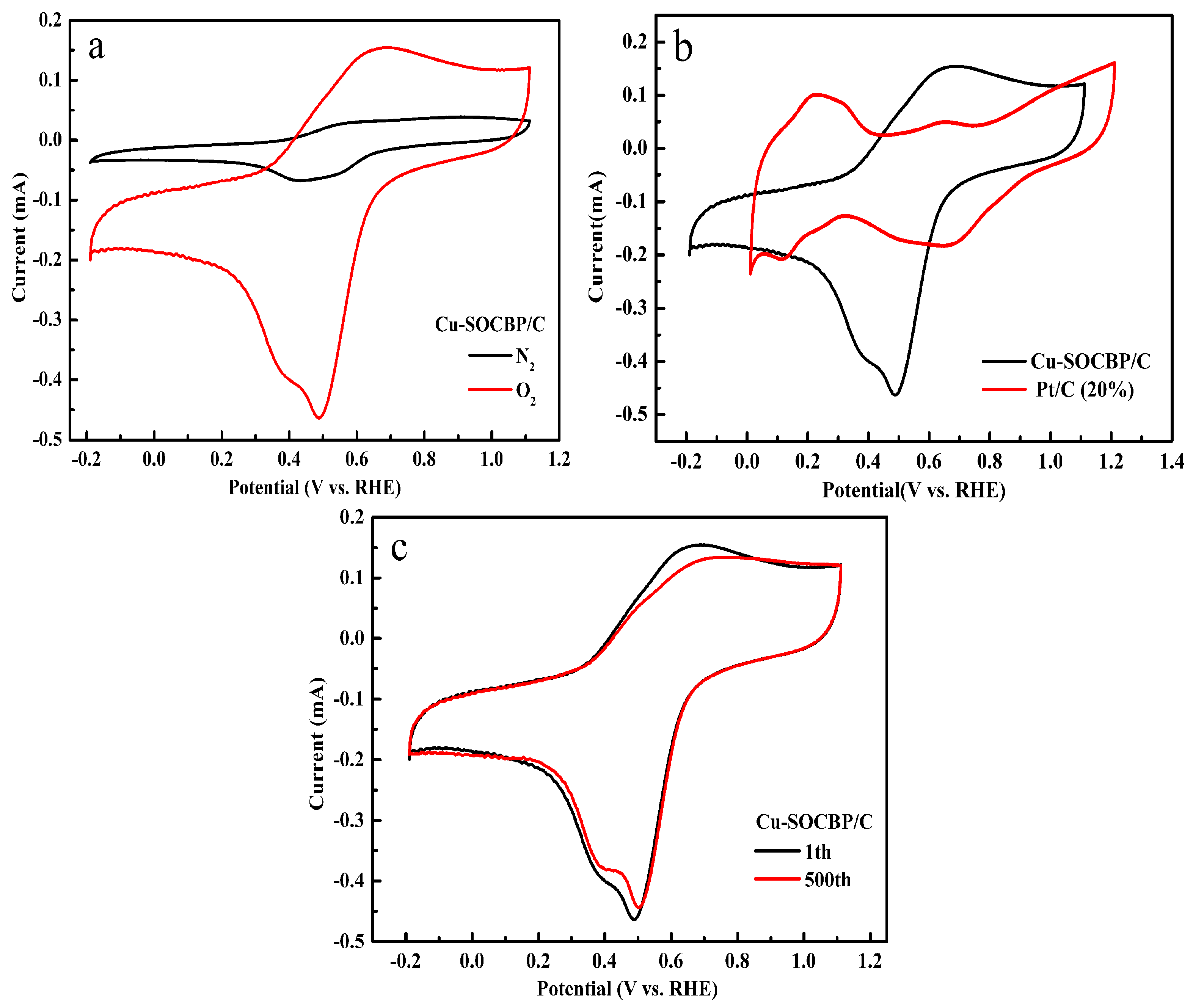
2.3. Cyclic Voltammetry of the Catalyst
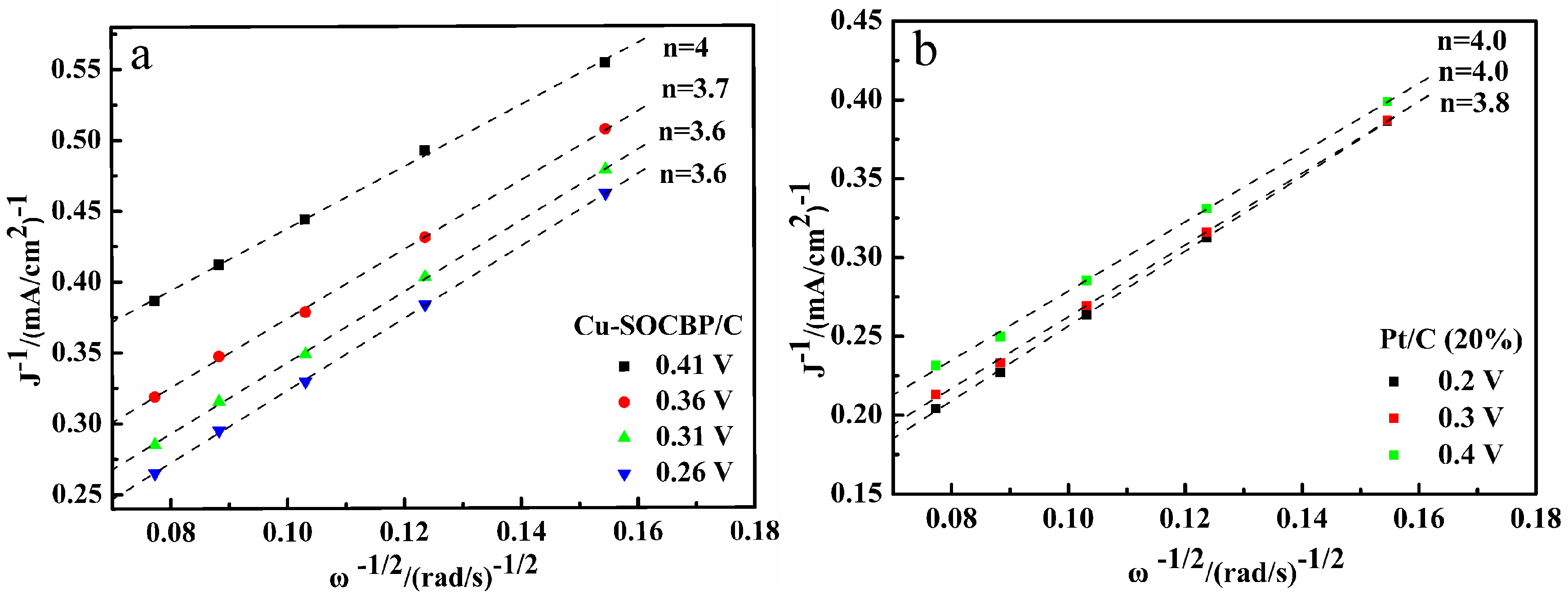
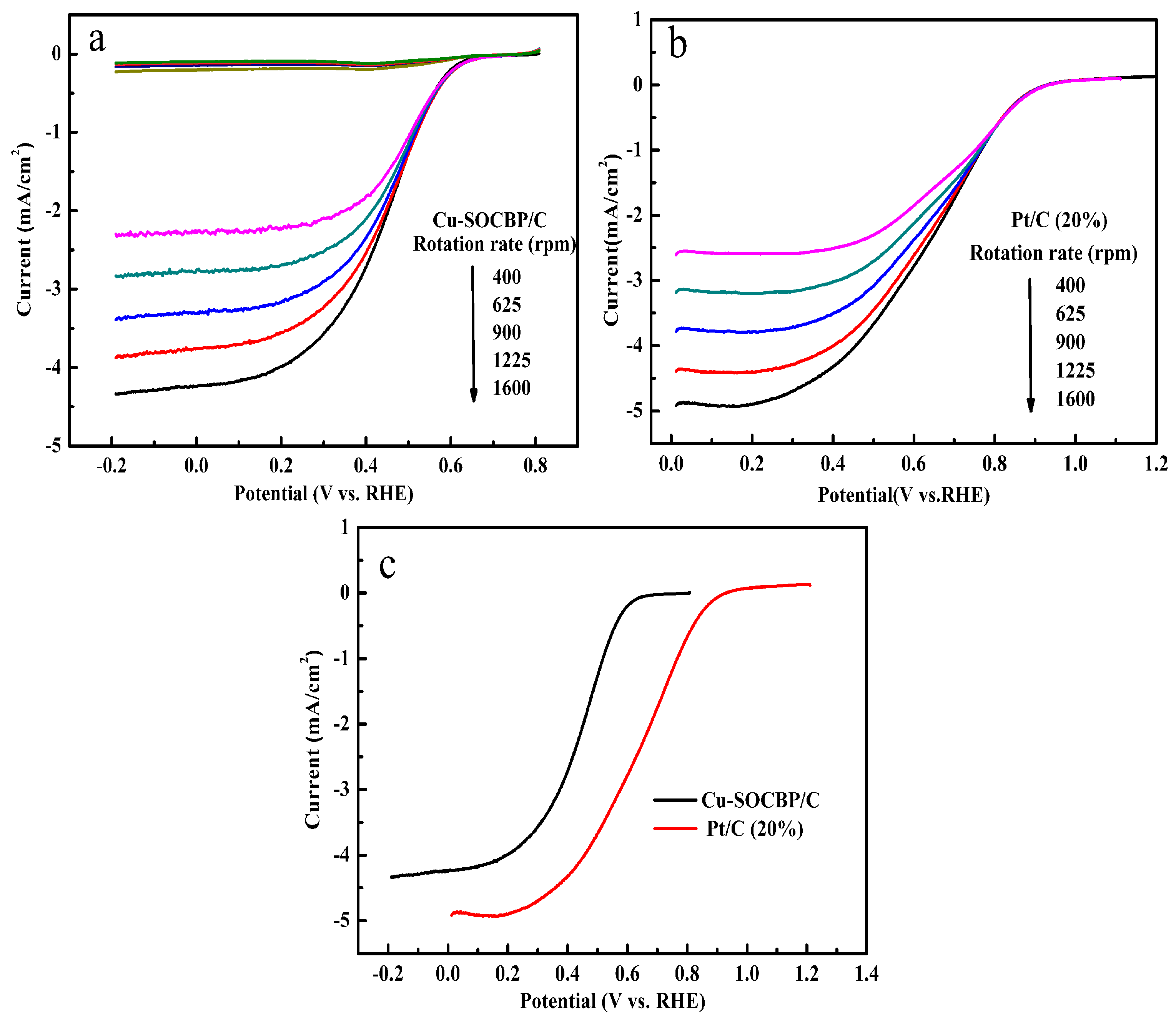
2.4. Linear Sweep Voltammetry
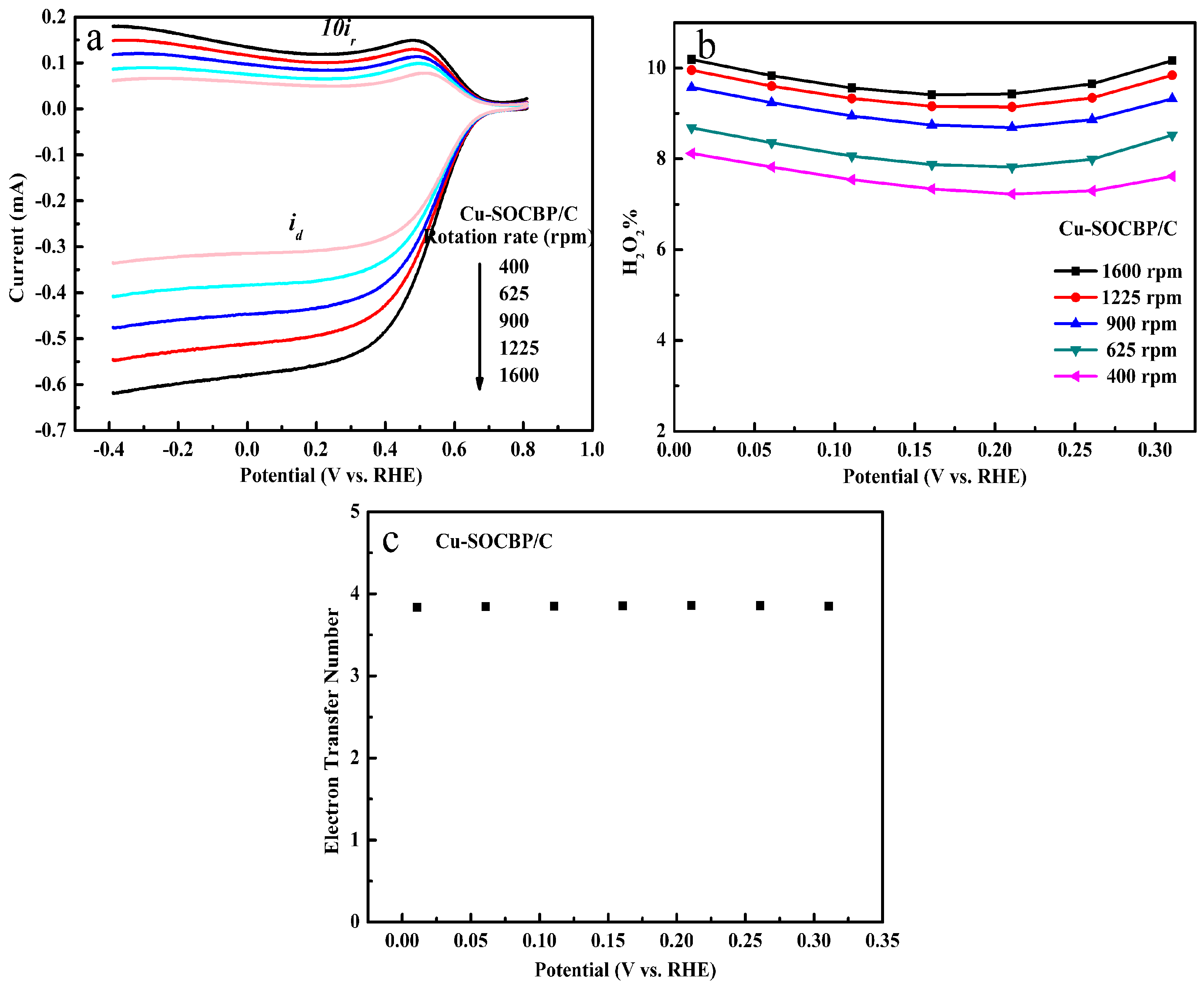
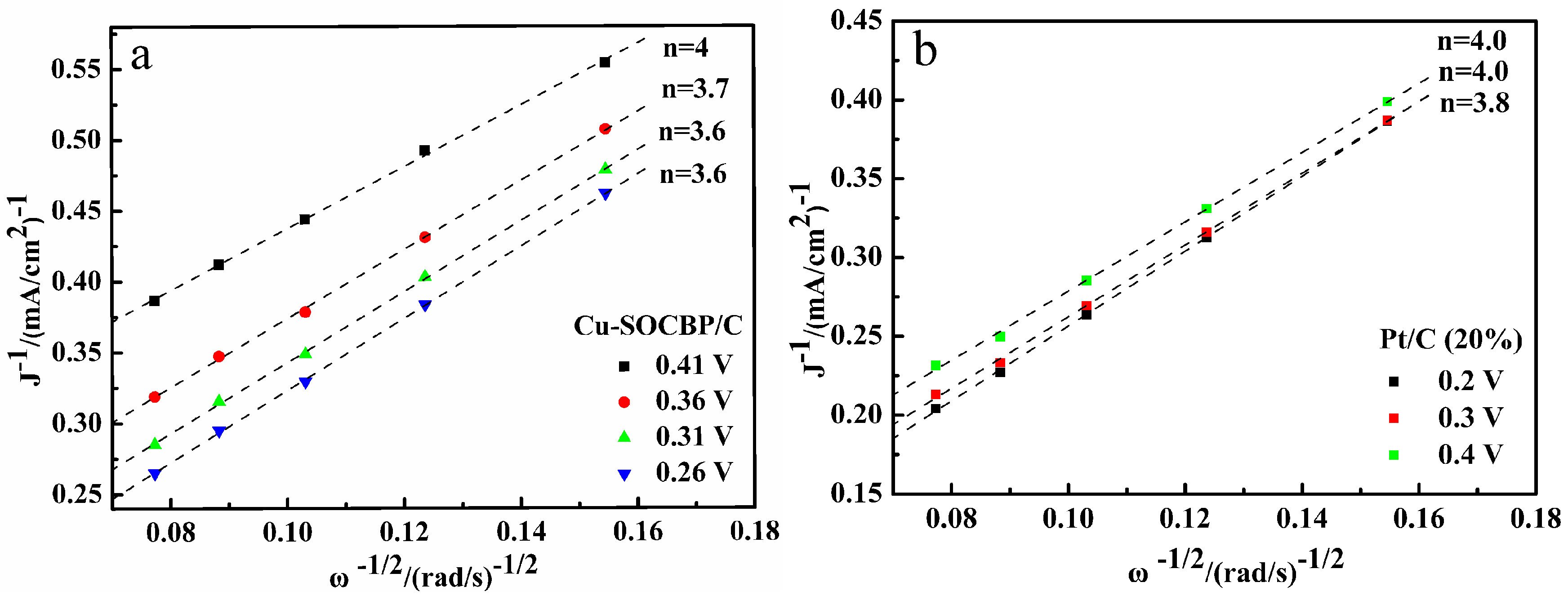
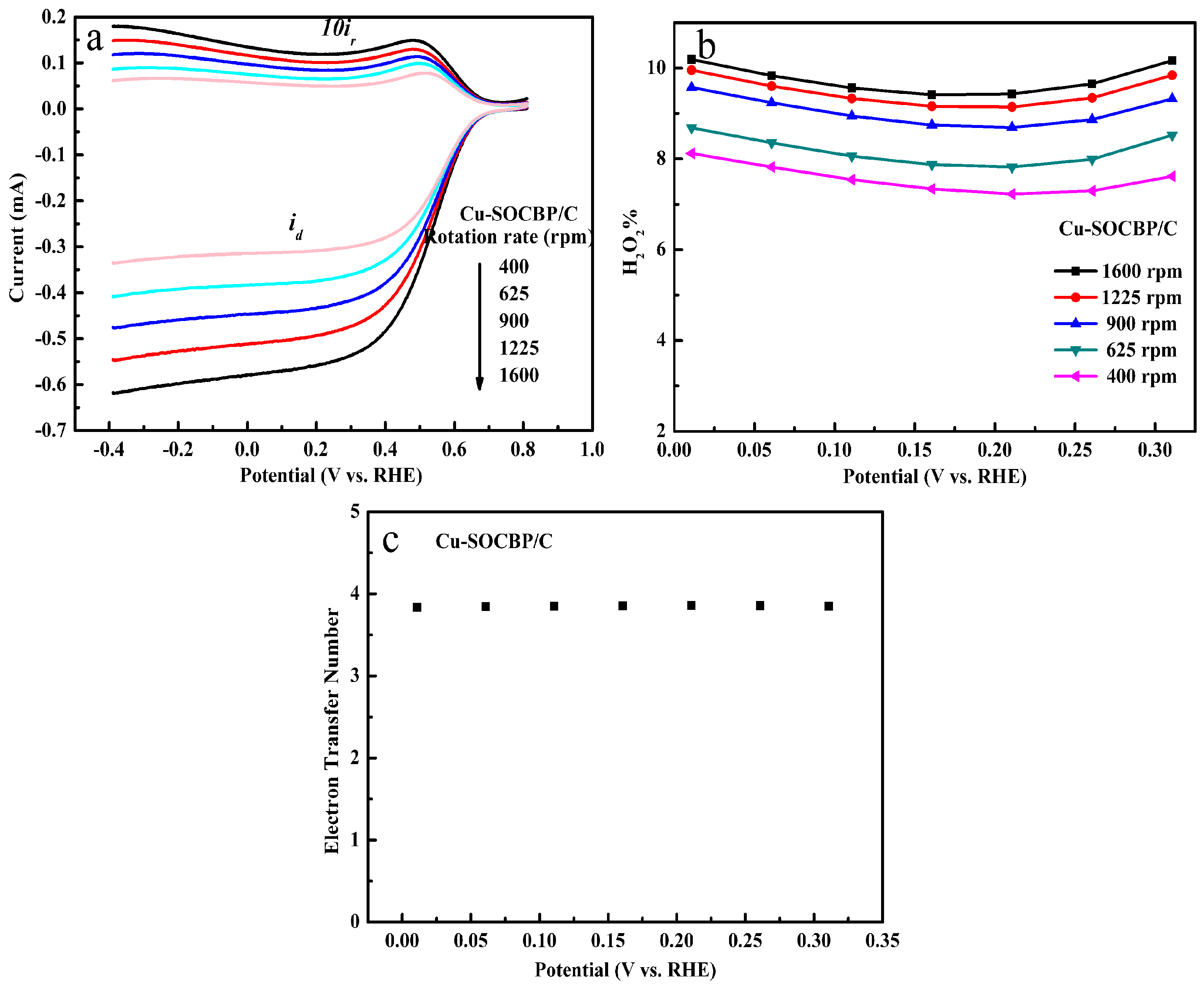
2.5. RRDE Measurements of the Catalysts
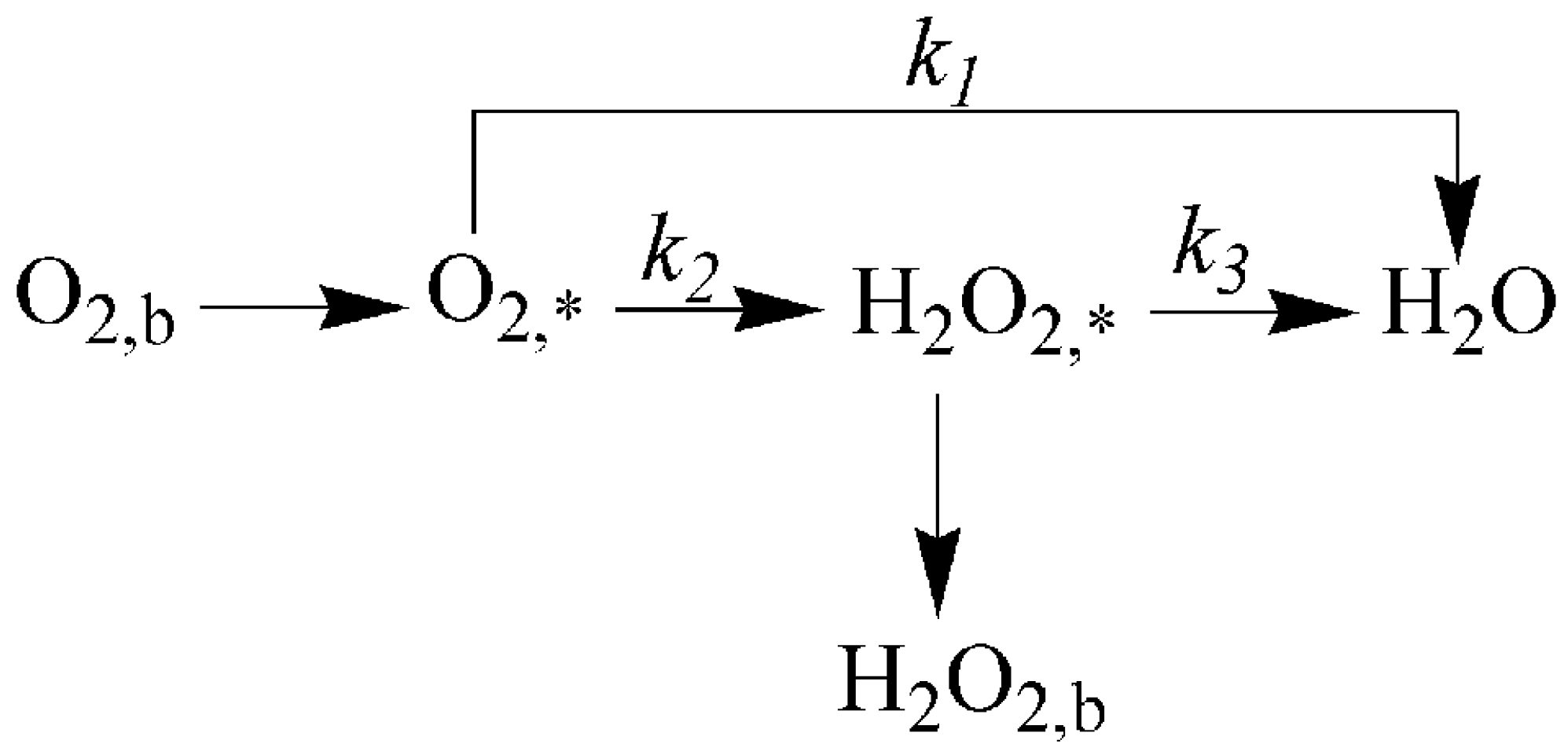
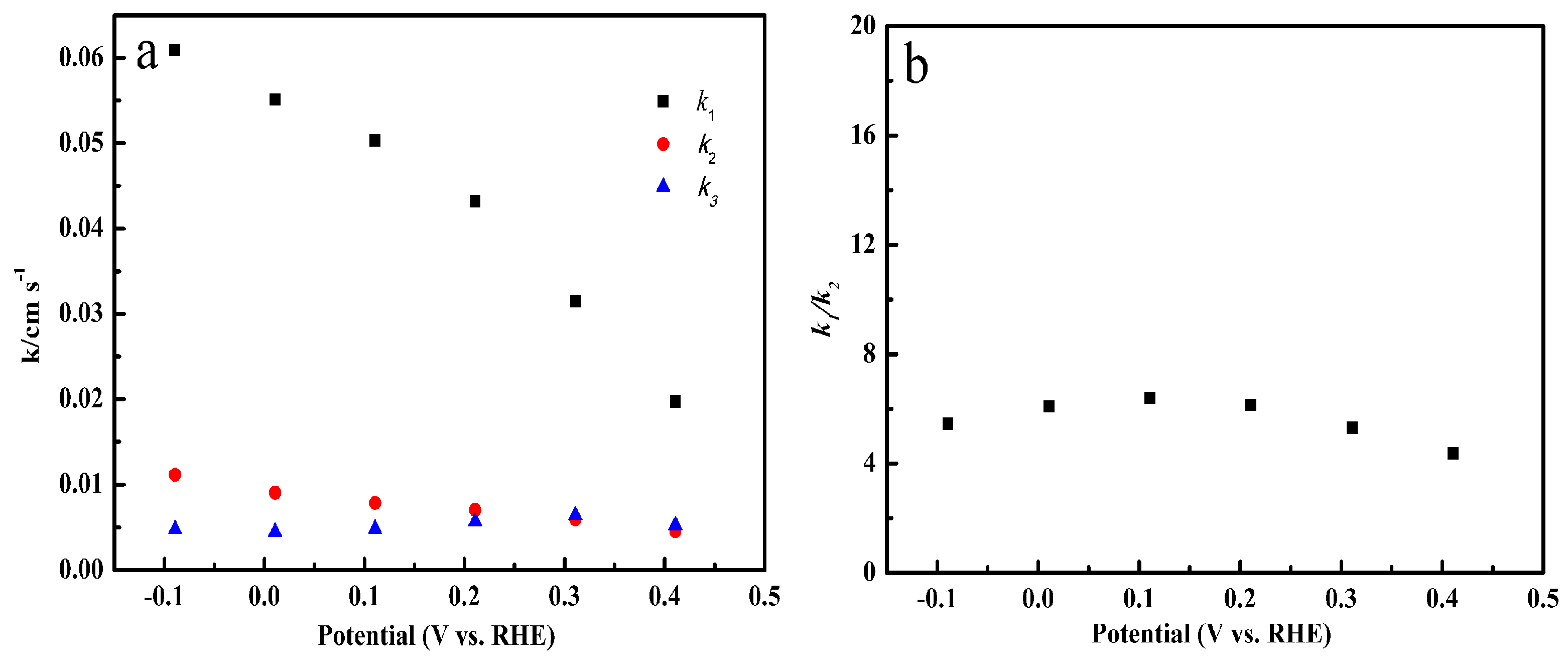
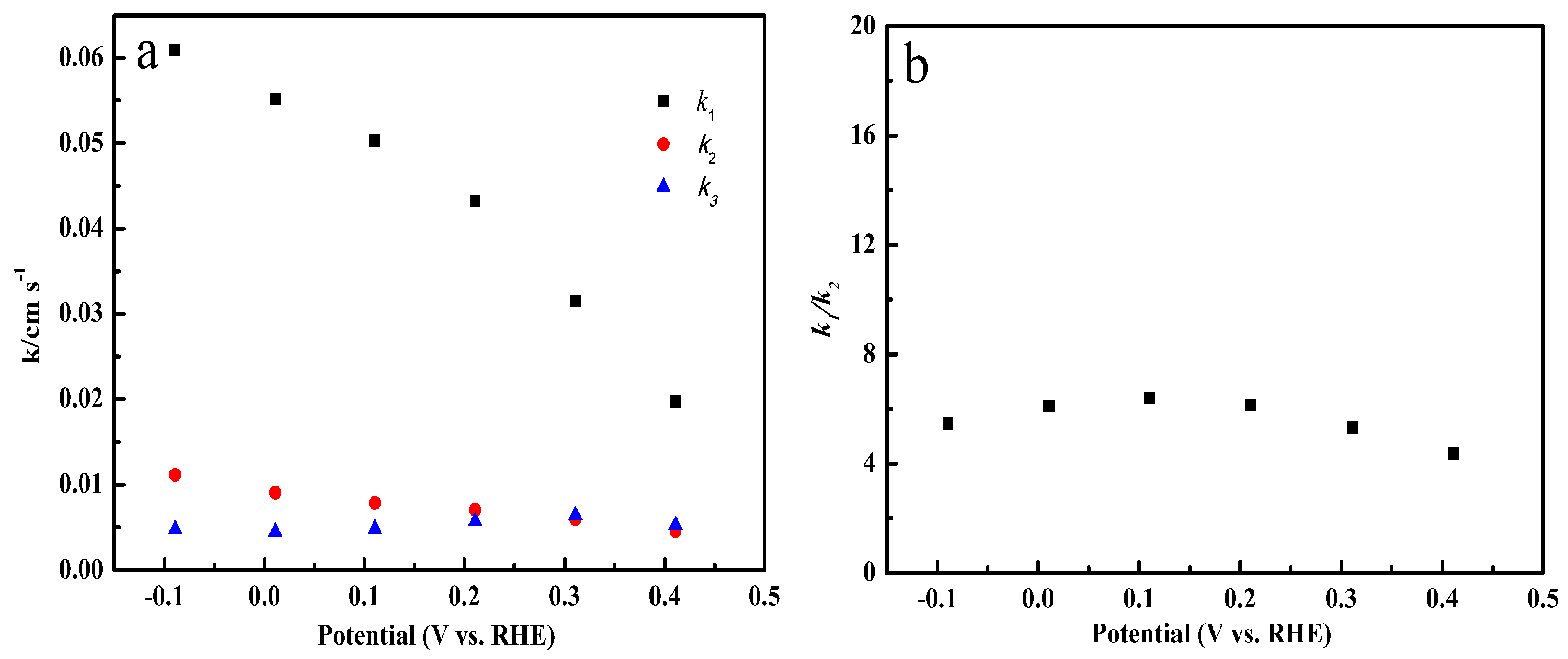
2.6. Specific Reduction Pathway of Cu-SOCBP/C
3. Experimental
3.1. Materials
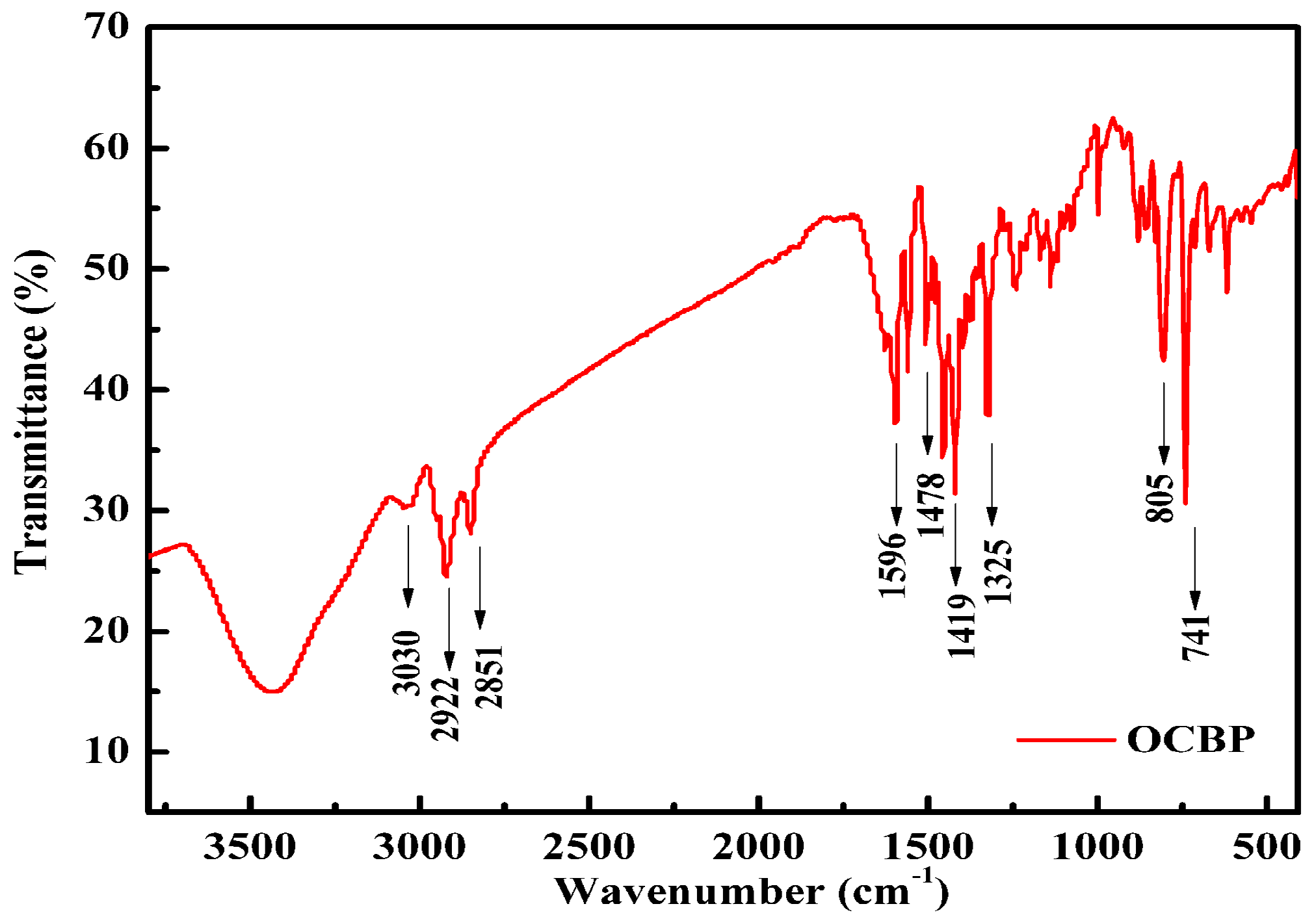
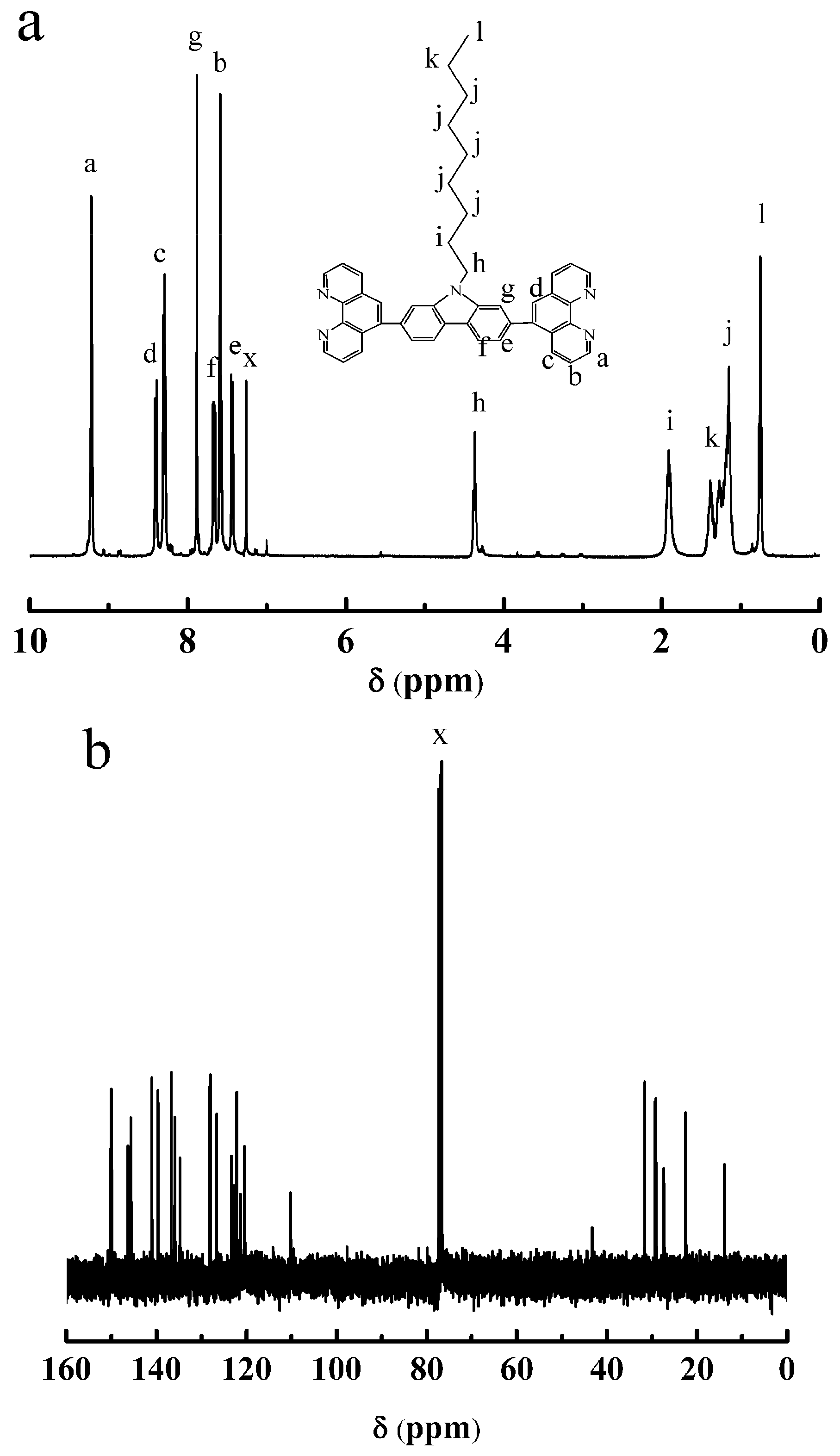
3.2. Synthesis of OCBP and Cu-SOCBP
3.3. Catalyst Preparation
3.4. Physical Characterization and Electrochemical Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kato, M.; Oyaizu, N.; Shimazu, K.; Yagi, I. Oxygen Reduction Reaction Catalyzed by Self-Assembled Monolayers of Copper-Based Electrocatalysts on a Polycrystalline Gold Surface. J. Phys. Chem. C 2016, 120, 15814–15822. [Google Scholar] [CrossRef]
- Wang, J.; Wang, T.T.; Wang, F.B.; Zhang, D.Y.; Wang, K.; Xia, X.H. Exploration of the Copper Active Sites in Electrooxidation of Glucose on a Copper/Nitrogen Doped Graphene Nanocomposite. J. Phys. Chem. C 2016, 120, 15593–15599. [Google Scholar] [CrossRef]
- Gartia, Y.; Parnell, C.M.; Watanabe, F.; Szwedo, P.; Biris, A.S.; Peddi, N.; Ghosh, A. Graphene-enhanced oxygen reduction by MN4 type Cobalt(III) catalyst. ACS Sustain. Chem. Eng. 2014, 3, 97–102. [Google Scholar] [CrossRef]
- Guo, C.Z.; Liao, W.L.; Sun, L.T.; Chen, C.G. Synthesis of non-noble nitrogen-containing catalysts for cathodic oxygen reduction reaction: A critical review. Int. J. Electrochem. Sci. 2015, 10, 2467–2477. [Google Scholar]
- Nie, Y.; Li, L.; Wei, Z.D. Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction. Chem. Soc. Rev. 2015, 44, 2168–2201. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wu, Q.; Che, R.; Bu, Y.; Jiang, Y.; Li, Y.; Yang, L.J.; Wang, X.Z.; Hu, Z. Alloyed Co-Mo Nitride as High-Performance Electrocatalyst for Oxygen Reduction in Acidic Medium. ACS Catal. 2015, 5, 1857–1862. [Google Scholar] [CrossRef]
- Panomsuwan, G.; Saito, N.; Ishizaki, T. Electrocatalytic oxygen reduction on nitrogen-doped carbon nanoparticles derived from cyano-aromatic molecules via a solution plasma approach. Carbon 2016, 98, 411–420. [Google Scholar] [CrossRef]
- Othman, R.; Dicks, A.L.; Zhu, Z. Non precious metal catalysts for the PEM fuel cell cathode. Int. J. Hydrogen Energy 2012, 37, 357–372. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Zhang, Y.X.; Mu, X.M.; Du, J.W.; Wang, H.; Huang, B.Y.; Zhou, J.Y.; Pan, X.J.; Xie, E.Q. The carbonization temperature effect on the electrochemical performance of nitrogen-doped carbon monoliths. Electrochim. Acta 2017, 242, 100–106. [Google Scholar] [CrossRef]
- Chen, Y.C.; Gokhale, R.; Serov, A.; Artyushkova, K.; Atanassov, P. Novel highly active and selective Fe-N-C oxyen reduction electrocatalysts derived from in-situ polymerization pyrolysis. Nano Energy 2017, 38, 201–209. [Google Scholar] [CrossRef]
- Sarapuu, A.; Samolberg, L.; Kreek, K.; Koel, M.; Matisen, L.; Tammeveski, K. Cobalt-and iron-containing nitrogen-doped carbon aerogels as non-precious metal catalysts for electrochemical reduction of oxygen. J. Electroanal. Chem. 2015, 746, 9–17. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, H.; Zhou, J.; Li, Y.; Wang, J.; Regier, T.; Dai, H. Covalent hybrid of spinel manganese-cobalt oxide and graphene as advanced oxygen reduction electrocatalysts. J. Am. Chem. Soc. 2012, 134, 3517–3523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Zhao, L.Z.; Walton, J.; Liu, Z.; Tang, Z. Facile fabrication of PtPd alloyed worm-like nanoparticles for electrocatalytic reduction of oxygen. Int. J. Hydrogen Energy 2017, 42, 17112–17121. [Google Scholar] [CrossRef]
- Kato, M.; Kimijima, K.I.; Shibata, M.; Notsu, H.; Ogino, K.; Inokuma, K.; Ohba, T. Deprotonation of a dinuclear copper complex of 3,5-diamino-1,2,4-triazole for high oxygen reduction activity. Phys. Chem. Chem. Phys. 2015, 17, 8638–8641. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, M.; Lin, L.; Li, Y.; He, X.; Cui, L. Electrocatalytic activity of metalloporphyrins grown in situ on graphene sheets toward oxygen reduction reaction in an alkaline medium. RSC Adv. 2014, 4, 26653–26661. [Google Scholar] [CrossRef]
- Ou, Z.P.; Lü, A.X.; Meng, D.Y.; Huang, S.; Fang, Y.Y.; Lu, G.F.; Kadish, K.M. Molecular oxygen reduction electrocatalyzed by meso-substituted cobalt corroles coated on edge-plane pyrolytic graphite electrodes in acidic media. Inorg. Chem. 2012, 51, 8890–8896. [Google Scholar] [CrossRef] [PubMed]
- He, H.Y.; Wang, M.; Zhao, J.S.; Zhang, Y. Poly(10,12-bis (4-hexylthiophene-2-yl)thieno[3′,4′:5,6]pyrazino[2,3-F][1,10]-phenanthroline-copper(II) complex as an efficient electrocatalyst for oxygen reduction. Chem. Eng. J. 2017, 316, 680–691. [Google Scholar] [CrossRef]
- Ren, C.C.; Li, H.B.; Li, R.; Xu, S.L.; Wei, D.H.; Kang, W.J.; Wang, L.; Jia, L.P.; Yang, B.C.; Liu, J.F. Electrocatalytic study of a 1,10-phenanthroline-cobalt(II) metal complex catalyst supported on reduced graphene oxide towards oxygen reduction reaction. RSC Adv. 2016, 6, 33302–33307. [Google Scholar] [CrossRef]
- Passard, G.; Ullman, A.M.; Brodsky, C.N.; Nocera, D.G. Oxygen reduction catalysis at a dicobalt center: The relationship of faradic efficiency to overpotential. J. Am. Chem. Soc. 2016, 138, 2925–2928. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Zhang, L.; Lui, H.; Hui, R.; Shi, Z.; Zhang, J.J. Oxygen reduction reaction (ORR) catalyzed by carbon-supported cobalt polypyrrole (Co-PPy/C) electrocatalysts. Electrochim. Acta 2009, 54, 4704–4711. [Google Scholar] [CrossRef]
- Lei, H.T.; Liu, C.Y.; Wang, Z.J.; Zhang, Z.Y.; Zhang, M.N.; Chang, X.M.; Zhang, W.; Cao, R. Noncovalent immobilization of a pyrene-modified cobalt corrole on carbon supports for enhanced electrocatalytic oxygen reduction and oxygen evolution in aqueous solution. ACS Catal. 2016, 6, 6429–6437. [Google Scholar] [CrossRef]
- Wang, F.F.; Wei, P.J.; Yu, G.Q.; Liu, J.G. Titanium Dioxide-Grafted Copper Complexes: High-Performance Electrocatalysts for the Oxygen Reduction Reaction in Alkaline Media. Chem. Eur. J. 2016, 22, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.F.; Lu, Y.Q.; Yuan, C.G.; Zhao, J.S.; Wang, M.; Liu, R.M. Carbon supported polyindole-5-carboxylic acid covalently bonded with pyridine-2, 4-diamine copper complex as a non-precious oxygen reduction catalyst. Electrochim. Acta 2014, 143, 1–9. [Google Scholar] [CrossRef]
- Levy, N.; Mahammed, A.; Friedman, A.; Gavriel, B.; Gross, Z.; Elbaz, L. Metallocorroles as non-precious metal electrocatalysts for highly efficient oxygen reduction in alkaline media. ChemCatChem 2016, 8, 2832–2837. [Google Scholar] [CrossRef]
- Levy, N.; Mahammed, A.; Kosa, M.; Major, D.T.; Gross, Z.; Elbaz, L. Metallocorroles as nonprecious metal catalysts for oxygen reduction. Angew. Chem. Int. Ed. 2015, 54, 14080–14084. [Google Scholar] [CrossRef] [PubMed]
- Thorseth, M.A.; Tornow, C.E.; Tse, E.C.M.; Gewirth, A.A. Cu complexes that catalyze the oxygen reduction reaction. Coord. Chem. Rev. 2013, 257, 130–139. [Google Scholar] [CrossRef]
- Zhong, J.P.; Fan, Y.J.; Wang, H.; Wang, R.X.; Fan, L.L.; Shen, X.C.; Shi, Z.J. Copper phthalocyanine functionalization of graphene nanosheets as support for platinum nanoparticles and their enhanced performance toward methanol oxidation. J. Power Sources 2013, 242, 208–215. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Y.; Li, Z.; Yang, Z.; Shen, C.; He, C. Effect of Pore Morphology on the Dielectric Properties of Porous Carbons for Microwave Absorption Applications. J. Phys. Chem. C 2014, 118, 26027–26032. [Google Scholar] [CrossRef]
- Ferrandon, M.; Kropf, A.J.; Myers, D.J.; Artyushkova, K.; Kramm, U.; Bogdanoff, P.; Zelenay, P. Multitechnique characterization of a polyaniline-iron-carbon oxygen reduction catalyst. J. Phys. Chem. C 2012, 116, 16001–16013. [Google Scholar] [CrossRef]
- Martínez, J.M.L.; Rodríguez-Castellón, E.; Sánchez, R.M.T.; Denaday, L.R.; Buldain, G.Y.; Dall’Orto, V.C. XPS studies on the Cu(I, II)-polyampholyte heterogeneous catalyst: An insight into its structure and mechanism. J. Mol. Catal. A Chem. 2011, 339, 43–51. [Google Scholar] [CrossRef]
- Kumar, R.; Yadav, A.; Mahiya, K.; Mathur, P. Copper(II) complexes with box or flower type morphology: Sustainability versus perishability upon catalytic recycling. Inorg. Chim. Acta 2016, 450, 279–284. [Google Scholar] [CrossRef]
- Yang, L.; Su, Y.; Li, W.; Kan, X. Fe/N/C Electrocatalysts for Oxygen Reduction Reaction in PEM Fuel Cells Using Nitrogen-Rich Ligand as Precursor. J. Phys. Chem. C 2015, 119, 11311–11319. [Google Scholar] [CrossRef]
- Huang, H.; Wang, X. Pd nanoparticles supported on low-defect graphene sheets: For use as high-performance electrocatalysts for formic acid and methanol oxidation. J. Mater. Chem. 2012, 22, 22533–22541. [Google Scholar] [CrossRef]
- Fan, T.Y.; Yin, F.X.; Wang, H.; He, X.B.; Li, G.R. A metal-organic-framework/carbon composite with enhanced bifunctional electrocatalytic activities towards oxygen reduction/evolution reactions. Int. J. Hydrogen Energy 2017, 42, 17376–17385. [Google Scholar] [CrossRef]
- Ju, J.; Bo, X.J.; Wang, H.; Zhang, Y.F.; Luhana, C.; Guo, L.P. Poly-o-toluidine cobalt supported on ordered mesoporous carbon as an efficient electrocatalyst for oxygen reduction. Electrochem. Commun. 2012, 25, 35–38. [Google Scholar] [CrossRef]
- Li, T.F.; Peng, Y.X.; Li, K.; Zhang, R.; Zheng, L.R.; Xia, D.G.; Zuo, X. Enhanced activity and stability of binuclear iron(III) phthalocyanine on graphene nanosheets for electrocatalytic oxygen reduction in acid. J. Power Sources 2015, 293, 511–518. [Google Scholar] [CrossRef]
- Wu, J.J.; Zhang, D.; Niwa, H.; Harada, Y.; Oshima, M.; Ofuchi, H.; Nabae, Y.; Okajima, T.; Ohsaka, T. Enhancement in Kinetics of the Oxygen Reduction Reaction on a Nitrogen-Doped Carbon Catalyst by Introduction of Iron via Electrochemical Methods. Langmuir 2015, 31, 5529–5536. [Google Scholar] [CrossRef] [PubMed]
- Parimi, N.S.; Umasankar, Y.; Atanassov, P.; Ramasamy, R.P. Kinetic and mechanistic parameters of laccase catalyzed direct electrochemical oxygen reduction reaction. ACS Catal. 2011, 2, 38–44. [Google Scholar] [CrossRef]
- Hossain, M.D.; Sato, D.T.; Higuchi, D.M. A Green Copper-Based Metallo-Supramolecular Polymer: Synthesis, Structure, and Electrochromic Properties. Chem. Asian J 2013, 8, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Glatfelter, A.; Dybowski, C.; Bai, S.; Kragten, D.; Blake, M.J.; Segarra, S.; Perry, D.L. Infrared studies of lead(II) halide-1,10-phenanthroline photosensitive materials. Sprctrochim. Acta A 2009, 71, 1922–1926. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.H.; Wang, M.; Liu, J.F.; Liu, R.M.; Zhao, J.S. Glycerol-stabilized NaBH4 reduction atroom-temperature for the synthesis of a carbon-supported PtxFe alloy with superior oxygen reduction activity for a microbial fuel cell. Electrochim. Acta 2014, 141, 331–339. [Google Scholar] [CrossRef]


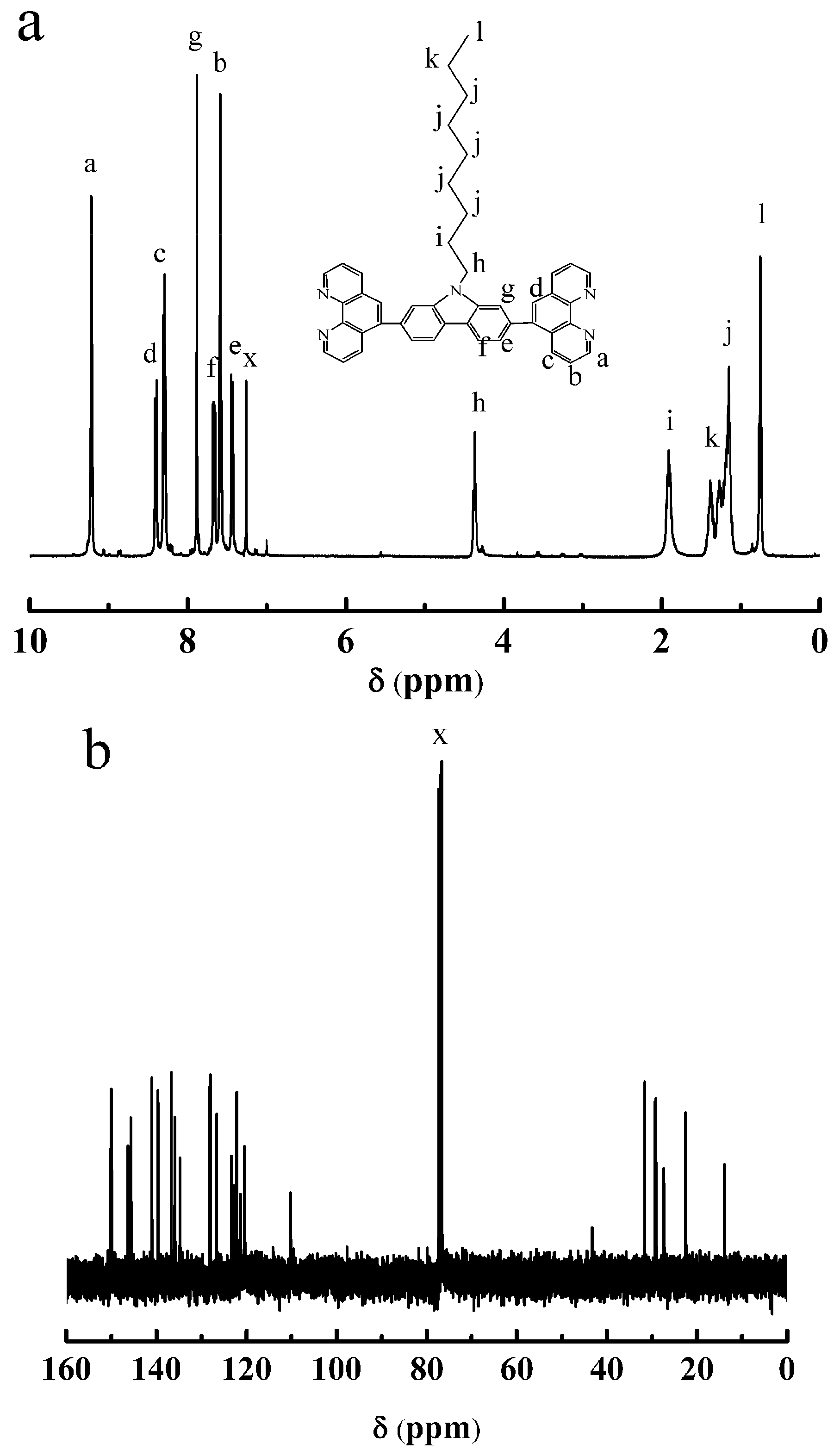


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Eonset (V) | E1/2 (V) | ETN | Average H2O2 (%) | Electrolyte Solution | Referance Electrode | Ref. |
|---|---|---|---|---|---|---|---|
| Cu-SOCBP/C | 0.62 | 0.44 | 3.8 | 9% | PBS (pH = 7) | RHE | This study |
| Pt/C (20%) | 0.90 | 0.63 | 4.0 | 1.6% | PBS (pH = 7) | RHE | This study |
| CoTHPP/PSS-rGO | - | −0.22 | 4.0 | - | 0.1 M KOH | SCE | [15] |
| Co-OBA/C | −0.197 | - | 3.75 | - | 0.1 M KOH | Ag/AgCl | [37] |
| POT-CO-OMC | −0.3 | - | 3.82 | - | 0.1 M KOH | Ag/AgCl | [38] |
| bi-FePc/GNS | 0.12 | 3.7 | - | 0.5 M H2SO4 | Hg/Hg2SO4 | [39] | |
| Phen-Co/rGO | - | −0.25 | 2.4 | - | 0.1 M NaOH | Ag/AgCl | [18] |
| Corrole-Co/MWCNT | 0.75 | 0.40 | 4.0 | - | 0.5 M H2SO4 | NHE | [16] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Chu, Y.; Ju, X.; Zhao, J.; Kong, L.; Zhang, Y. Carbon-Supported Copper-Based Nitrogen-Containing Supramolecule as an Efficient Oxygen Reduction Reaction Catalyst in Neutral Medium. Catalysts 2018, 8, 53. https://doi.org/10.3390/catal8020053
Zhao Y, Chu Y, Ju X, Zhao J, Kong L, Zhang Y. Carbon-Supported Copper-Based Nitrogen-Containing Supramolecule as an Efficient Oxygen Reduction Reaction Catalyst in Neutral Medium. Catalysts. 2018; 8(2):53. https://doi.org/10.3390/catal8020053
Chicago/Turabian StyleZhao, Yuanyuan, Ya Chu, Xiuping Ju, Jinsheng Zhao, Lingqian Kong, and Yan Zhang. 2018. "Carbon-Supported Copper-Based Nitrogen-Containing Supramolecule as an Efficient Oxygen Reduction Reaction Catalyst in Neutral Medium" Catalysts 8, no. 2: 53. https://doi.org/10.3390/catal8020053
APA StyleZhao, Y., Chu, Y., Ju, X., Zhao, J., Kong, L., & Zhang, Y. (2018). Carbon-Supported Copper-Based Nitrogen-Containing Supramolecule as an Efficient Oxygen Reduction Reaction Catalyst in Neutral Medium. Catalysts, 8(2), 53. https://doi.org/10.3390/catal8020053