1. Introduction
The atmosphere is a very complex matrix that, in addition to nitrogen (N
2) and oxygen (O
2) gases, so important to human survival, consists of various other pollutant gases albeit at much lower concentrations: (i) carbonaceous oxides (CO
x); (ii) nitrogen oxides (NO
x); (iii) sulfur oxides (SO
x); (iv) various hydrocarbons (HCs); and (v) particulate matter. These pollutants originate from both natural sources (e.g., volcanic eruptions, wildfires, lightning, and natural degradation of forests among others) and anthropogenic areas (fertilizers and livestock, farms, and urban areas), stationary sources (e.g., industries, power plants, and sewage treatment plants), and mobile sources (e.g., automobiles, trucks, buses, motorcycles, ships, and airplanes) (see
Figure 1) [
1]. The natural sources of chemical pollutants, however, are of lesser concerns as they are part of the natural environment equilibrium, contrary to the anthropogenic sources that keep increasing in number and concentration with the ever increasing global human population and society’s continuous increasing demand for energy and associated technological advances.
Undoubtedly, the most important sources of air pollutants implicate the combustion of fossil fuels to produce energy (residential heating and electricity-generating power plants), together with major metallurgical industries, cement/construction industries, and the transportation sector.
Figure 1 also identifies the primary pollutants from various sources: carbon monoxide, sulfur dioxide, ammonia, volatile organic compounds (VOCs), particulates and, relevant to the present article, the two major NO
x agents (NO and NO
2). Subsequently, through various interacting events that involve the Sun’s radiation, secondary pollutants are generated, among which are sulfur trioxide, ozone, hydrogen peroxide, and sulfuric and nitric acids (the causes of acid rain). Another class of air pollutants generated from internal combustion engines and industrial fumes that react in the atmosphere with sunlight produce secondary pollutants that, in combination with the primary emissions, create photochemical smog [
2].
Pre-industrial concentrations of atmospheric nitrogen oxides have increased steadily from about 280 ppbv to ca. 320 ppbv until a decade ago (2010), with estimated annual emissions of 13.8 Tg of N per year (teragrams; 10
12 g), of which ca. 70% is produced by nitrification and denitrification processes in undisturbed terrestrial environments and world’s oceans, and ca. 3 Tg of N per year (~8%) from agricultural tillage, fertilizer use, and animal wastes [
3].
The NO
x gases are formed in large measure in gasoline/diesel combustion engines and in power plants that use fossil fuels to produce electricity via high-temperature combustion/oxidation of the fuel’s nitrogen with air oxygen. Initially, only NO is formed followed by formation of NO
2 after combustion in the exhaust and in the atmosphere in the presence of more O
2.
Figure 2a reports the 2011 levels of NO
x emissions in the European Union [
4], while
Figure 2b reports the 2005 NO
x emission levels in the United States [
5]. The major anthropogenic sources of nitrogen oxides are combustion engines (transportation sector) and the electricity/heating sectors.
Most of the tropospheric ozone is formed when NO
x, CO and VOCs react in the atmosphere in the presence of sunlight, and, although they might originate in urban areas, airstreams can carry the NO
x far from its sources causing ozone formation in less populated regions. Globally, a VOC whose atmospheric concentration has increased greatly during the last century (viz., methane) contributes to the formation of ozone [
6]. A series of complex reactions that involve a VOC (e.g., CO) in the formation of ozone implicates oxidation of this VOC by a hydroxyl radical (
•OH) [
7] first to yield the radical species HO
•CO (Equation (1)), which subsequently reacts with oxygen to produce the hydroperoxy radical HOO
• (Equation (2)) that later reacts with NO to give NO
2 (Equation (3)); the latter photolyzes in sunlight to NO and atomic O(
3P) (Equation (4)), which by reaction with oxygen yields ozone (O
3; Equation (5)).
While the chemistry involving other VOCs might be more complex, the critical step that leads to ozone formation remains nonetheless the oxidation of NO to NO
2 by HOO
• radicals. Nitrogen dioxide also reacts with hydrocarbon molecules present in VOCs to produce yet another pollutant (peroxyacetyl nitrates; PAN), a component of photochemical smog that is mostly responsible for eye irritation and is more damaging to plants than ozone [
8].
The NO
x family of pollutants (NO, NO
2, N
2O, and their derivatives) causes a wide range of health issues. Nitric oxide (NO) spreads to all parts of the respiratory system because of its low solubility in water, while the health effects of NO
2 are related to its ability to dissolve in moisture to produce HNO
3 acid—a strong mineral acid. Some of the acute health effects include eye irritation (stinging and watering), throat irritation (pungent smell, stinging nose, and coughing), lung irritation (coughing, wheezing and tight chest—difficulty in breathing), and asthma triggered in asthmatics [
9,
10]. The most serious acute effects occur after significant exposure to NO
2 causing: (a)
acute pulmonary edema—fluid from damaged lung tissue pours out into air spaces preventing air from getting to deeper lung thereby causing choking (asphyxia); and (b) other chronic health effects such as
asthma and
obliterative bronchiolitis, in which the smallest air passages (the bronchioles) are seriously scarred and become distorted and blocked. Consequently, no one questions the need for NO
x-free clean air as essential to maintain/enhance an individual’s health, and to maintain the integrity of the surrounding environment. In this regard, transformation of the two major NO
x species (NO and NO
2), indeed their suppression, has become a necessity as they underwrite (with the VOCs) the formation of hazardous secondary air pollutants and the accompanying photochemical smog.
Two cities where photochemical smog is not insignificant are the Greater Los Angeles (LA) area in the United States and Beijing, China. Home to nearly 19 million people and located in a geological basin confined by the Pacific Ocean and mountains, LA is the basin of considerable pollution caused by its car-centric culture, its bustling industries and ports, its sprawling development, and its sunny climate with often stagnant winds. It was only in the 1950s that hydrocarbons and NO
x were recognized as the source of photochemical smog (
Figure 3); however, with the implementation of mandatory catalytic converters in automobiles in the last two decades, smog has been attenuated somewhat.
With its rapid growth and home to nearly 20 million people, and being an important industrial hub, Beijing is a city where poor air quality has been for decades a regrettable fact of everyday life owing to the presence of significant quantities of particulate matter and photochemical smog, as (
Figure 4) experienced by the author in the early 1990s; in subsequent trips several years later, however, this author experienced significant improvements of air quality but by no means have the pollutants and smog been totally eliminated.
There have been many attempts to remediate the occurrence of NO
x and VOC species in polluted urban environments with TiO
2-based photocatalytic cementitious-like materials and photocatalytic coatings (paints) on various supports [
11]. Several studies report on the performance of titania deposited on, or otherwise incorporated into cementitious substrates toward the minimization, if not suppression, of air pollutants (see, for example, Refs. [
12,
13,
14,
15,
16,
17,
18]). Laboratory studies have shown, rather conclusively, that NO
x can be oxidized to nitrate anions [
19,
20], while VOCs can be converted into CO
2 and H
2O [
21]. Of some concern, however, are studies that demonstrate the formation of harmful intermediates (e.g., nitrous acid, HONO), which are far more harmful to human health than either NO or NO
2 during the disposal of NO
x [
22,
23]. Not least is the potential that nitrates (NO
3−) produced and deposited on the TiO
2 particulate surface in the disposal of NO
x may be implicated in
reNOxification reactions; that is, back to NO
x [
24,
25,
26] and formation of ozone [
26] that would forestall the application of TiO
2-based photocatalytic surfaces to improve the quality of urban air environments.
The objective of this review article is to examine the various attempts at eliminating NO
x species in the urban environment produced mostly by vehicular traffic through application of commercially available titania-based photocatalytic materials, coatings and paints in tunnels, highways, highway noise barriers, and urban roads. However, before tackling that discussion, we describe briefly some fundamentals from basic research that underpin this TiO
2-based photocatalytic technology. In its pristine or modified form, TiO
2 has been the most popular and most extensively investigated photocatalyst, and is the primary source of modern third generation composite photoactive materials [
27,
28].
Photocatalytic processes occurring in heterogeneous systems are complex and multifarious starting from the absorption of photons by the solid photocatalyst, and ending with the evolution of reaction products. This complexity is particularly reflected in the terminology used to describe various characteristics of heterogeneous photocatalysis, which, although it has come to some maturity in recent years, continues to undergo extensive developments through efforts of many researchers from the fields of catalysis, photochemistry and materials science, among others. Accordingly, prior to tackling the many deNOxing efforts in cleaning up the atmospheric environment, an important aspect of applied photocatalysis with metal-oxide semiconductor photocatalysts, it is imperative to appreciate and understand some of the fundamentals underlying Heterogeneous Photocatalysis (following
Section 2 and
Section 3)—the primary approach in these efforts.
4. Applied Photocatalysis: Laboratory-Scale deNOxing of NOx Agents (NO & NO2)
As discussed earlier, nitrogen oxides (NO
x) are major atmospheric pollutants that play an important role in atmospheric chemistry, and have been the object of a significant number of investigations toward their minimization, if not complete removal from the environment. The concentration of NO
x in polluted urban air is around 100 ppbv, whereas in the unpolluted troposphere, it ranges from 10 to 500 pptv [
88]. Recall that NO
x are emitted primarily from artificial sources (e.g., traffic, coal burning boilers, thermal power plants, and industries of various sorts) and from natural sources (e.g., biological degradation in soil and from lighting thunder). NO
x participate in various environmental processes: for instance, in the formation of acid rain; in the greenhouse effect in synergy with sulfur oxides; in the formation of photochemical smog in the presence of CO and VOCs; in the depletion of stratospheric ozone; and in the formation of peroxyacetyl nitrates (PAN), all of which have negative effects on ecosystems and lead to non-insignificant human health issues. With regard to the latter, NO
x pollutants cause problems in the respiratory tract that include lung edema and the reduction of the oxygen-caring capacity of blood—e.g., in the transformation of hemoglobin to methemoglobin.
No wonder then that significant efforts have been expended to reduce environmental NO
x agents back to N
2 via a thermal technology using a variety of reductants (e.g., CO, hydrocarbons, H
2 and NH
3) in what is known as Selective Catalytic Reduction (SCR). While reduction of NO occurs around 100 °C in the presence of H
2 and a Pd-supported catalyst [
89], other reactions require significantly greater temperatures. In fact, reduction of NO to N
2 through selective catalytic reduction with NH
3, and thus potentially treat NO
x agents, the costs of the SCR technology for the construction and operation of a facility to treat NO
x pollutants, together with the required consumption of energy, may prove prohibitively high. Nonetheless, despite the many efforts to eliminate the NO
x emitted from the various sources noted earlier by SCR, the fact remains that the concentration of NO in air in Japan was nearly constant throughout the 1980s, and was often higher than the air quality standard set for NO
2, principally along heavily trafficked roads in densely populated areas [
90]. This led to the development of a new technology for the disposal of NO
x at sub-ppm level from air and from trafficked roads and tunnels, and other environmental sources that emit NO
x.
Recognition that plants and micro-organisms can easily consume nitrite (NO
2−) and nitrate (NO
3−) ions as raw materials for nitrogen assimilation provided a further impetus to examine alternative technologies to achieve a practical removal of dilute NO
x agents from the environment using sunlight (UV-Visible) radiation at significantly lower costs. In this regard, Takeuchi and Isubuki [
91] investigated the dry deposition of NO
x onto the ground and found that the rate of adsorption of NO
x on some soil particles was enhanced by photoillumination. Of the metal oxides constituting the soil particles, TiO
2 showed the highest activity for NO
x adsorption under photoillumination with ca. 60% of NO
x being captured as nitric acid (HNO
3) on the surface of TiO
2 particulates. Accordingly, the authors thought that the photocatalytic oxidation of NO
x to HNO
3 by illuminated TiO
2 might be most advantageous to treat dilute environmental NO
x, as any extra reactants such as NH
3 were not required and HNO
3 could be trapped on the surface as nitrates.
One of the first studies to examine the fate of one of the NO
x agents, namely NO, in the presence of (Degussa) P-25 TiO
2 exposed to UV irradiation was reported in 1984 by Courbon and Pichat [
92] who exposed isotopically labeled N
18O at 295 K in the dark to pre-oxidized and pre-reduced TiO
2 powder; subsequent to UV illumination resulted in three phenomena: photoadsoprtion, photoexchange, and photodecomposition of NO to yield N
2O and, to a lesser extent, N
2. The formation of N
2O + N
2 corresponded to a photodecomposition of ca. 15% of the NO pressure (decrease) for a pre-reduced titania sample and ca. 20% for a pre-oxidized titania; N
2 formed only at the beginning and the percent N
216O produced was initially greater for the pre-oxidized titania sample. This early study [
92] confirmed that illumination of TiO
2 with UV light considerably increased the ease of detachment of surface oxygen atoms, as the isotopic hetero-exchange of N
18O occurred at room temperature, while it required higher temperatures in the absence of bandgap (3.2 eV) illumination of the mostly anatase TiO
2. Adsorbed oxygen species were involved, as pre-oxidized titania exhibited higher initial efficiency; however, the instantaneous exchange with a pre-reduced titania sample in H
2 at 723 K showed that detachment also involved surface oxygen atoms that were replenished from NO. Another aspect of this study was the corroboration of the direct involvement of O
− species in photocatalytic oxidations over TiO
2 and other n-type semiconductors, since NO and O
2 played similar roles in yielding dissociated oxygen species active in both oxidation and oxygen isotopic exchange.
A later study (1985) by Hori and coworkers [
93] demonstrated that NO
2− ions are oxidized to NO
3− with or without O
2 in aqueous suspensions of some semiconductor powders (Ag
2O, PbO, anatase TiO
2, Si, ZnO, SnO
2, CdS, and Bi
2O
3) under bandgap illumination; with TiO
2, 96% of nitrite was oxidized to nitrate in the presence of oxygen. Along similar lines, Anpo and coworkers [
94] found that Cu
+ ions on SiO
2 (Cu
+/SiO
2 catalyst) could decompose NO molecules photocatalytically and stoichiometrically into N
2 and O
2 at 275 K, which they attributed to the significant role played by the excited state of the Cu
+ species; the photoreaction involved an electron transfer from the excited state of the Cu
+ ion into an anti-bonding π orbital of the NO molecule within the lifetime of its excited state. The relationship between the local structures of Cu
+ ions in zeolite and their photocatalytic reactivity in the decomposition of NO
x into N
2 and O
2 at 275 K was reviewed by Anpo and coworkers [
95] after which Anpo’s group [
96] reported on the metal ion-implantation of TiO
2 with various transition metal ions that subsequent to calcination in oxygen at ca. 723 K resulted in a large shift of the absorption edge of TiO
2 toward visible light regions depending on the amount and type of metal ions implanted; the resulting metal ion-implanted TiO
2s proved active in the photocatalytic decomposition of NO to N
2, O
2 and N
2O at 275 K under irradiation with visible light at wavelengths longer than 450 nm.
Following their 1989 report [
91], Ibusuki and Takeuchi [
97] examined the photocatalytic destructive oxidation of NO to NO
3− using a mixture (200–250 mg) of TiO
2, activated carbon (AC) and Fe
2O
3 particles located in a flow-type photochemical reactor system (
Figure 15) that was photo-illuminated by a cylindrical bank of 12 black lights (wavelength: 300–400 nm) [
97]. The AC and Fe
2O
3 had a remarkable effect in increasing the catalytic activity for NO
x removal, likely due to their high adsorptive activity for NO and NO
2. The authors inferred that photo-illuminated TiO
2 generated reactive oxygen species that oxidized NO and NO
2, respectively, to NO
2 and NO
3−, while activated carbon trapped NO
2 to allow enough time for TiO
2 to oxidize NO
2 to NO
3−; it appears that Fe
2O
3 acted as a promoter for more NO/NO
2 molecules to be adsorbed on the surface of the titania photocatalyst [
97].
In a further study, the Takeuchi group [
98] examined the use of TiO
2 to eliminate NO
x in open air with the photocatalyst being activated by sunlight, but noted, however, that in so doing desorption of NO
2 occurred during the oxidative removal of NO; the NO
2 also needed to be suppressed as it is also a regulated pollutant. Although NO
x adsorb on activated carbon to be oxidized ultimately to NO
3−, development of an activated carbonaceous photocatalytic material proved difficult. Accordingly, recognizing that thin films have many micropores they designed and prepared TiO
2 thin film photocatalysts by a dip-coating process using titanium alkoxide as the TiO
2 precursor and the polymer additive polyethylene glycol (PEG) of different molecular masses (PEG-300, PEG-600, PEG-1000) to give TiO
2-PX films with thicknesses of 1.0, 0.5 and 0.25 μm after calcination of the films at 450 °C for 1 h deposited on silica-coated glass plates.
Table 1 summarizes the extent of NO removal [
98]. Adsorbed NO was photooxidized to NO
2 by the thin films, while the produced NO
2 was re-photooxidized to NO
3− before it desorbed from the film surface.
Following reports that TiO
2 prepared by high-temperature hydrolysis of titanium tetra-alkoxides, Ti(OR)
4, in a hydrocarbon solvent was very active toward the photocatalytic dehydrogenation of
iso-propanol in aqueous media under deaerated conditions [
99] and mineralization of acetic acid under aerated conditions [
100], Hashimoto et al. [
101] prepared TiO
2 by the hydrolysis of titanium alkoxide in a hydrocarbon solvent, followed by calcination at various temperatures; the titania calcined at 300 °C proved most active for the photocatalytic oxidation of NO (
Table 2), in comparison with P-25 titania. The photocatalytic oxidation was carried out in a fixed bed continuous flow Pyrex-glass reactor under atmospheric pressure with the TiO
2 (0.12 g) UV-irradiated with a 10-Wh black light; air contained 10 ppm of NO; flow rate, 110 mL min
−1. IR spectral results indicated that UV irradiation promoted the oxidation of NO in the presence of oxygen to yield nitrate species, while the data from ESR measurements for oxygen radicals showed that UV irradiation increased the number of O
2−• adsorbed on the surface of titania in the presence of oxygen. These O
2−• species vanished simultaneously with their exposure to NO, whereas the spectral intensity of the radical generated from secondary products of O
− showed no change. The number of O
2−• radical anions generated by UV irradiation reflected the photocatalytic oxidative activity of titania toward the oxidation of NO. The rate of formation of O
2−• and the number of free electrons induced by UV irradiation decreased significantly with an increase in post-calcination temperature (
Table 2) [
101].
A photocatalytic reaction that takes place in a gas/solid reactor necessitates both the exposure of the catalysts to light irradiation and good contact between reactants and catalyst. In this regard, Lim and coworkers [
102] noted that a two-dimensional fluidized-bed photoreactor not only increased the contact of catalyst and gas, but also enhanced UV light penetration compared with a packed bed reactor in which light could not easily penetrate the interior of the catalyst bed, so that it was important to design and fabricate a fluidized-bed photoreactor with higher light throughputs and lower pressure drops. Accordingly, they used: (i) an annular flow-type photoreactor; and (ii) a modified two-dimensional fluidized-bed photoreactor to examine the photocatalytic decomposition of NO. In the first case, two serial annular flow photoreactors were used to increase contact time between the gas and the photocatalyst (
Figure 16).
By comparison, the modified two-dimensional fluidized-bed reactor (
Figure 17) consisted of an annular-type reactor made of a larger quartz glass tube (internal diameter, 30 mm; height, 400 mm) in which a small diameter quartz tube (inner diameter, 20 mm; height, 375 mm) was located at the center of the larger tube such that the thickness of the annulus in the bed was 5 mm [
102]. A quartz filter (100-mesh size) was used to distribute a uniform fluidization of the catalyst; a square mirror box surrounded the photoreactor to minimize loss of light irradiation and to improve utilization of reflected and deflected light.
In their study [
102], the authors examined the effects of gas-residence time, initial NO concentration, reaction temperature and UV light source on the photocatalytic decomposition of NO carried out in the annular flow-type reactor. P-25 titania powder was used to cover a quartz tube (430 mm) by dipping it into a stirred 5% TiO
2 slurry solution and then air-dried for 24 h, after which the TiO
2-coated quartz tube was fired in a high-temperature furnace at 400 °C for 1 h; TiO
2 coating was repeated several times until the amount of TiO
2 deposited on the quartz tube reached 0.10 g. The quartz tube had been sandblasted previously to create a granular texture to anchor the fine TiO
2 powder. In addition, precursor solutions for coating TiO
2 on silica gel were prepared using titanium ethoxide as a precursor to prepare the TiO
2 sample.
A gas stream (200 mL min
−1) of 138 ppmv NO in He in the annular flow-type reactor was irradiated by four UV lamps without TiO
2 photocatalyst at ambient temperature for 140 min with no variation in the NO concentration; in the presence of TiO
2, however, irradiation led to the decomposition of NO with formation of NO
2, N
2O and N
2 products [
102]. The reaction rate followed the power law
R =
Ro In with n = 0.48 and n = 0.87 depending on the UV intensity of the germicidal white lamp (254 nm) and the fluorescent black lamp (365 nm), respectively (gas flow rate, 100 mL min
−1; TiO
2 loading, 0.1 g; reaction temperature, 311 K; initial NO concentration, 50 ppm). Adsorption of nitrate on the surface of the photocatalyst increased with irradiation time leading to the deactivation of the photocatalyst. The decomposition of NO decreased linearly on increasing the initial NO concentration and on decreasing the residence time of gas in the photoreactor, so that it was necessary to increase the residence time of the gaseous reactant to provide effective contact of UV light, gaseous reactant and photocatalyst to obtain higher NO decomposition in the annular photoreactor.
In the modified two-dimensional fluidized-bed photoreactor, four reaction conditions (without TiO
2/SiO
2 and UV lamp on/off, with TiO
2/SiO
2 and UV lamp-on/off) were tested to confirm whether the decomposition of NO really took place by a photocatalytic process. Indeed, in the presence of TiO
2/SiO
2 and UV lamp-on, the NO concentration decreased indicating that it was in fact decomposed [
101]. Decomposition of NO increased with decreasing initial NO concentration and increasing gas-residence time; the reaction rate increased with increasing UV light intensity. Clearly, the modified photoreactor displayed efficient contact between photocatalyst and reactant gas with good transmission of UV-light and, consequently, increased the NO decomposition efficiency (>70%) compared with the annular flow-type photoreactor. Hence, the former photoreactor was an effective tool with which to carry out significant NO decomposition with efficient utilization of photon energy [
102].
Anpo and coworkers [
96] had earlier prepared a TiO
2 photocatalyst that subsequent to the implantation of Cr ion and upon irradiation with visible light (>450 nm) decomposed NO into N
2, O
2, and N
2O under O
2-free conditions. Additionally, the Cr ion-implanted TiO
2 catalyst displayed the exact same photocatalytic efficiency as the original TiO
2 catalyst, albeit under UV irradiation. As a follow-up to this study, Nakamura et al. [
103] examined the role of oxygen vacancies in the removal of NO under an oxidative atmosphere using a commercial TiO
2 (Ishihara ST-01; 100% anatase; crystallite size, 7 nm; nominal specific surface area, 300 m
2 g
−1) and hydrogen plasma-treated TiO
2 powders; the latter was photoactive up to 600 nm without a decrease in UV light activity. Reactions to remove 1.0 ppm of NO were carried out in a Pyrex glass flow reactor (500 cm
3) with irradiation from a 300-W Xe light source; the UVA (315–400 nm) irradiance at the photocatalyst surface was 0.03 mW cm
−2 (flow rate, 1500 mL min
−1; TiO
2 loading, 0.20 g; total pressure, 760 Torr)—no removal of NO occurred without the metal oxide photocatalyst. NO was converted mainly to NO
3− (also less than 2% NO
2− formed) by oxidation over the TiO
2 powder; NO
3− ions accumulated on the catalyst surface. Electrons trapped in oxygen vacancies in the plasma-treated TiO
2 were detected under visible light irradiation (
F-type color centers; ESR measurements displayed a signal at
g = 2.004) with the number being proportional to the percent of NO
x removed, which suggested that the number of trapped electrons determined the activity of the photocatalytic oxidation of NO to NO
3−. The visible-light photocatalytic activity of the plasma-treated TiO
2 was due to photoexcitation of the
F-type color centers with energy levels within the forbidden bandgap of the metal oxide (see
Figure 18).
The Anpo group [
104] investigated the photocatalytic decomposition of NO
x (NO and NO
2) on five well-characterized standard reference ultrafine powdered TiO
2 photocatalysts (grain size, 0.02–1 μm) denoted TiO
2 (JRC-TIO-2, -3, -4, and -5) supplied by the Catalysis Society of Japan (properties summarized in
Table 3) in a large-scale continuous flow reaction system (
Figure 19a) with high efficiency. Special attention was expended on the effects of pretreatment and reaction conditions on the reaction and conversion rates of NO. The authors established that surface hydroxyl groups played a significant role as active sites in the decomposition of NO.
When used on a large scale for long periods in photoreactions, photocatalysts tended to lose, albeit gradually, their photocatalytic activity. In Anpo’s study [
104], after 2 h, conversion of NO for each photocatalyst leveled off and dropped to between 0.25 and 0.20 the photocatalytic activity observed initially, indicating a decline in photocatalytic activity of the TiO
2 in the decomposition of NO in the absence of O
2 and/or H
2O. Reaction products in the flow reaction system were N
2, O
2, and N
2O, just as occurred in a closed reaction system. The JRC-TIO-4 photocatalyst displayed the highest photocatalytic activity for the conversion of NO, while for the other three there were small differences: JRC-TIO-4 (11%) >>> −3 (~2%) > −5 (1.8%) > −2 (1%) (see
Figure 19b). The anatase TiO
2 catalyst with the larger surface area, wider bandgap, and numerous surface –OH groups (
Table 3) exhibited the highest photocatalytic reactivity in the decomposition of NO, which the authors deduced that these were the principal factors that affected photocatalytic efficiency. The increased bandgap of JRC-TiO-4 was accompanied by a shift in the conduction band edge to higher negative energies, thus moving the redox potential to more negative values thereby enhancing photocatalytic reactivity. Moreover, surface –OH groups and/or physisorbed H
2O also played a significant role in the photocatalytic reactions through the facile formation of reactive
•OH radicals. The intensity of the incident light is also an important factor that affects the kinetics of the photocatalytic decomposition. The quantum efficiency of the photocatalytic reaction was higher at the lower intensities of the incident light, and lower at higher intensities of the incident UV light; in addition, the efficiency of conversion of NO increased with increase in the O
2 flow rate.
The activity of the JRC-TiO-4 photocatalyst was also tested by Tanaka and coworkers [
105] in the photoassisted selective catalytic reduction of NO with ammonia (photo-SCR) at low temperature over irradiated TiO
2 in a flow reactor; the process was efficient and the adsorbed ammonia reacted with NO under irradiation of TiO
2 (
Figure 20); note the nearly identical kinetics of formation of both N
2 and N
2O.
The total amount of N
2 formed was 0.23 mmol g
cat−1, consistent with the amount of ammonia (0.24 mmol g
cat−1) adsorbed over TiO
2 in equilibrium at 323 K. The kinetic experiment carried out under differential conditions in the pressure range 300 <
p(NO),
p(NH
3) < 2000 ppm, and the presence of excess O
2 affected the evolution rate of N
2 which depended only on partial pressure of NO; kinetics were first order on NO, and zeroth order on O
2 and NH
3, which strongly suggested that the rate-determining step was adsorption of NO to the irradiated TiO
2 adsorbing ammonia molecules [
105]. To the extent that the selective catalytic reduction (SCR) with ammonia is a downhill reaction, it also proceeded in the dark at low temperature with a 20% conversion of NO. However, photoirradiation caused a remarkable enhancement of the activity: the evolution rate of N
2 gradually increased attaining a steady rate at ca. 80% conversion after 2 h of irradiation.
To achieve a further understanding of surface reactions involved in TiO
2-based photocatalysis, Dalton and coworkers [
106] examined two titania samples (one of unknown source) using X-ray photoelectron spectroscopy and Raman spectroscopy to investigate the NO
x adsorbate reaction at the surface of these two TiO
2 substrates. The NO
x gas was composed of 109 ± 5 ppm of NO
x, 21.0 ± 0.4% O
2, the remaining ca. 79% being N
2; dry air was the mixer gas to dilute the NO
x (NO
x concentration, 10–100 ppm) during the reaction performed under UV exposure for 6 and 48 h in a glass vessel (ca. 3 mm thick;
Figure 21) that allowed > 80% transmission of the radiation at λ = 320 nm. Formation of NO
3− did not vary significantly with either exposure time or NO
x concentration. The authors [
106] proposed a stepwise mechanism (
Figure 22) in which the surface hydroxyls increased the efficacy of the process and participated by reacting with NO
x molecules to yield nitrate ions formed indirectly via initial reductive (formation of O
2−• radical anions by conduction band electrons) and oxidative (formation of
•OH radicals by valence band holes) processes.
Dalton et al. [
106] concluded that TiO
2 was effective at converting NO
x agents to NO
3− and that XPS proved useful in quantifying the efficiency of the reaction, while Raman spectroscopy was a quick and simple way of ascertaining the surface crystal structure of the titania. XPS confirmed only one oxidation state of Ti on the untreated TiO
2 materials; however, the O
1s peak indicated the presence of two additional components of TiO
2: Ti–OH and Ti–OH
2. After exposure to UV radiation, XPS spectra revealed nitrogen peaks attributable to organic species (also present before reaction), to some unreacted NO adsorbed on the surface, and to nitrate anions.
Reactive nitrogen (NO
y) in the atmosphere consists of the sum of the two NO
x oxides (NO + NO
2) and all compounds produced by atmospheric oxidation of NO
x that include the minor species: HNO
3, HNO
2, the nitrate radical NO
3•, N
2O
5, peroxynitric acid HNO
4, peroxyacetyl nitrate (PAN) (CH
3C(O)OONO
2) and its homologs, and peroxyalkyl nitrates (RC(O)OONO
2) [
107]. Such compounds can be regarded as reservoirs of NO
2 but apparently play no critical role in the formation of ozone O
3 that the precursors NO
2 and NO do. The oxidative removal of NO over irradiated TiO
2 catalyst was examined by Devahasdin and coworkers [
107] at source levels (5–60 ppm) in a thin-film photoreactor systems (see
Figure 23); the process involved a series of oxidation steps through the action of photoformed
•OH radicals (NO → HNO
2 → NO
2 → HNO
3). Light intensity increased the capability to oxidize NO (from 0 to 0.8 mW cm
−2); the selectivity for NO
2 increased with light intensity for 5 ppm inlet NO but remained constant for 40 ppm inlet NO. The steady-state conversion of NO increased with relative humidity from 0 to 50% leveling off at higher relative humidity; the ratio of NO
2− to NO
3− from spent catalyst liquor decreased with irradiation time until steady state was reached.
Transient behavior of TiO
2 during the first 2 h of operation with the system setup of
Figure 23 (conditions: space time: 12 s; inlet concentration, 40 ppm; light source, two 8-W black lamps; relative humidity, 50%; TiO
2 loading: 1.07 mg cm
−2) revealed that initially the conversion of NO was very high (ca. 95% after 0.5–3 min of irradiation depending on TiO
2 loading) and decreased approaching steady state after 6 h of operation; all the nitrogen was accounted for in the gas phase: NO out (26 ppm) + NO
2 out (14 ppm) = NO in (40 ppm). Conversion of NO was 35%; gas phase mass balance showed no N
2O formed in the reaction system under steady-state conditions [
106]; NO
2 selectivity remained constant at 100% for 40 ppm inlet NO and increased with light intensity for 5 ppm inlet NO with a 50% relative humidity, which suggested that for 40 ppm inlet NO at steady state, all the NO should have been converted to NO
2. However, the authors [
107] believed that, for the 5 ppm inlet NO, the true steady state had not yet been reached, so that increasing light intensity caused the HNO
3 to dissociate back to NO
2 and
•OH and to promote NO
2 selectivity from 82% to 95%. The latter inference called attention for the first time to the possible reNOxification of the nitrates produced in the deNOxification of the environment (see
Section 5).
Toma and workers [
108] reported using a test chamber built specifically for the TiO
2 (Degussa P-25; in powder or pellet form) photocatalytic decrease of NO
x. The experimental device consisted of three parts: (a) a chamber where gaseous NO
x were prepared in situ by chemical reaction of Cu powder with a dilute solution of HNO
3; (b) an environmental chamber; and (c) a NO
x analyzer. The pollutants were subsequently injected at ambient temperature into the environmental chamber (volume, ~0.4 m
3) until the concentration of NO
x reached 1–2 ppmv. A fan ensured homogenization of the gaseous pollutants in the environmental chamber. A polycarbonate photoreactor (100 mm × 100 mm × 50 mm box) equipped with a 70 × 70 mm Plexiglas window allowed light transmission from a 15-W daylight lamp (30% UVA, 4% UVB) placed inside the environmental chamber and crossed by the NO
x flow (flow rate, 0.6 L min
−1); NO
x concentrations were continuously monitored with a chemiluminescence NO
x analyzer.
For small TiO
2 powder quantities, conversion rates increased proportionally reaching maximal value at 0.2 g loading of TiO
2; at higher quantities of TiO
2 the decrease in NO
x remained constant and independent of TiO
2 powder amount. After 30 min of UV irradiation (surface, 54 cm
2), conversion rates were about 32–35% and 15–18%, respectively, when the mass of the catalyst varied from 0.2 to 1.2 g; maximal conversion was reached at 3.7–4 mg cm
−2 of TiO
2 powder [
108]. Exposing a TiO
2 pellet surface (mass, 0.4 to 1.2 g; 54 cm
2) to UV radiation from one side only led to a photocatalytic conversion of ca. 28–30% of NO and 10–12% of NO
x (
Figure 24); the conversion efficiency increased with the surface area of the pellet. The amount of compressed TiO
2 powder and the thickness of the pellet had little influence on the extent of NO
x decomposition. Anatase TiO
2 showed better efficiency for the photocatalytic decrease of NO
x relative to rutile TiO
2, accounting for only 10% and 5%, respectively, for NO and NO
x removal.
We noted above that the photocatalytic decomposition of NO over TiO
2 reported in some of the literature led to the formation of N
2O as the main reaction product [
92,
94,
98] with minor N
2, NO
2 and O
2 products. Only Anpo’s group [
96] reported the selectivity of NO photodecomposition over a TiO
2 photocatalyst to yield N
2O and N
2, and no other products. According to the views of Bowering and coworkers [
109], use of only TiO
2 as the catalyst is not ideal for removing NO from the atmosphere as N
2O itself is also a regulated pollutant. As such, Tanaka et al. [
105] reported that photoassisted selective catalytic reduction (photo-SCR) of NO over TiO
2 with NH
3 as a reductant was very selective towards N
2 formation, with relatively small amounts of N
2O. As NH
3 is also a pollutant, it would need to be eliminated from the exhaust gas, thereby causing an increase in overall costs of a system.
Car exhaust and industrial emissions are mostly controlled using selective catalytic reduction (SCR) to convert NO
x to N
2. Accordingly, Bowering et al. [
109] used CO as the reductant in eliminating NO photocatalytically with Degussa P-25 TiO
2 in a continuous flow reactor (
Figure 25) with the objective to convert the NO
x preferentially into N
2 gas. The authors added TiO
2 powder (ca. 0.2 g) to acidified triply deionized water (TDW; 6 mL of 0.05 M HNO
3 in 500 mL of TDW) yielding a dispersion that was stirred for 12 h and then dried at 70 °C for 48 h, after which the resulting powders were calcined for 2 h at 120, 200, 450 or 600 °C. Subsequently, 25-mL fractions of the dispersion were evaporated at 70 °C onto degreased borosilicate glass slides; an amount of TiO
2 powder (~1 mg) was deposited on the slides and then calcined following the same methodology as for the powders.
The effect of calcination temperatures on the composition and crystallite sizes of P-25 photocatalysts, and the effect of pretreatment temperature on rate of NO conversion and selectivity for N
2 formation for NO decomposition and reduction reactions are presented in
Table 4 [
109]. Pretreatment (calcination) temperatures caused no appreciable change in phase composition; original composition (ca. 77 vol.% anatase, 23 vol.% rutile) was maintained even after treatment at 600 °C. The photocatalytic activity for both decomposition and reduction reactions decreased with increasing pretreatment temperature, which was attributed to removal of surface hydroxyl species that acted as active sites for reaction. The only products observed in the decomposition reactions were N
2 and N
2O; the selectivity for nitrogen formation remained constant (ca. 23%) regardless of pretreatment temperature. However, the presence of CO in the reaction gas had a dramatic effect on selectivity of the reactions with N
2 selectivity as high as 65%; in addition, an increase in the CO/NO ratio led to increased selectivity for N
2 formation.
It is likely that under UV illumination electron transfer occurred from electron trapped centers into antibonding orbitals of adsorbed NO molecules, resulting in their decomposition and formation of N
(ads) and O
(ads) surface species, which can then scan the TiO
2 surface and react with other surface species (e.g., NO
(ads), N
(ads), O
(ads)) to form N
2O, NO
2, O
2 and N
2. To the extent that neither O
2 nor NO
2 was detected led the authors [
109] to deduce that Reactions (69) and (70) did not occur on P-25 surfaces under decomposition conditions; the main surface reaction was Reaction (71), as N
2O was the major reaction product under these conditions.
In the presence of CO on the photocatalyst surface, and under UV illumination, other reactions are possible between adsorbed CO and NO molecules together with reactions of CO with N
(ads) and O
(ads) atoms. No reaction occurred in the dark and under UV illumination without TiO
2 indicating that both TiO
2 and UV are required for adsorbed NO and CO species to react. Under decomposition conditions, the major reaction product was N
2O (~75%) with N
2 being the minor product (ca. 25%). On the other hand, under reduction conditions selectivity for N
2 formation increased (ca. 48%) at the pretreatment temperatures of 70 and 120 °C. However, at higher pretreatment temperatures, the selectivity was similar to that achieved in the absence of CO suggesting that the surface N
2 forming reaction was favored on a titanium surface rich in hydroxyl groups [
109].
Germane to the previous study [
109], Roy and coworkers [
110] examined a photocatalytic route to destroy NO
x by developing a new Pd ion-substituted TiO
2 system (Ti
1−xPd
xO
2−δ) with which to reduce NO in the presence of CO via creation of redox adsorption sites and using anion oxygen vacancies on titania; the optimal Pd
2+ ion concentration was 1 at.% in TiO
2 (anatase). Apparently, even though both NO and CO competed for the same Pd
2+ adsorption sites, reduction of NO to N
2O was two orders of magnitude higher with the Ti
0.99Pd
0.01O
1.99 photocatalyst under ambient conditions than unsubstituted TiO
2; using UV irradiation with a 125-W high-pressure Hg lamp and an inlet 5000 ppm of NO in a flow-type reactor, the rate of reduction of NO was 0.53 μmol g
−1 s
−1.
The photocatalytic deNOxing activities of TiO
2, N-doped TiO
2, Fe-loaded N-doped TiO
2, and Pt-loaded N-doped TiO
2 exposed to irradiation from monochrome LED lamps at various wavelengths have been investigated in some detail by Yin and coworkers [
111], unlike many studies that have typically used 100–500 Watt high-pressure Hg or otherwise Xe light sources to activate titania-based photocatalysts. Bare TiO
2−xN
x (denoted TiON) powders were prepared by treating 20-nm Ishihara ST-01 anatase TiO
2 in an NH
3 atmosphere at 600 °C for 3 h, followed by annealing at 300 °C for 2 h in humid air (N content, ca. 0.25 at.%); for comparison, the ST-01 anatase TiO
2 powder was heat-treated in air at 500 °C for 1 h (S-TiO
2)—BET specific surface areas were 57.7 m
2 g
−1 and 100.7 m
2 g
−1, respectively, for TiON and S-TiO
2 powders. The Fe- and Pt-loaded N-doped TiO
2 systems were prepared by dispersing TiON powder in a HNO
3 aqueous solution containing either Fe(NO
3)
3 or Pt(NH
3)
2(NO
3)
2 at ambient temperature, followed by stirring for 1 h, heated at 150 °C to remove the water, and then calcined at 300 and 400 °C for 2 h, respectively (loading of Fe and Pt, 0.5 wt.%; systems denoted TiONFe and TiONPt; BET areas were 61.1 m
2 g
−1 and 59.0 m
2 g
−1, respectively).
The specifics of irradiation from the four LED sources were (wavelength, irradiance): (i) red light LED (627 nm, 72.76 μmol m
−2 s
−1); (ii) green light LED (530 nm, 125.12 μmol m
−2 s
−1); (iii) blue light LED (445 nm, 76.22 μmol m
−2 s
−1); and (iv) UV light LED (390 nm, 73.70 μmol m
−2 s
−1). Different samples showed different wavelength dependencies; for instance, S-TiO
2 displayed excellent activity at 390 nm but very weak activity at 445 nm, while TiON showed excellent UV light (390 nm) and visible light-induced photocatalytic activity on exposure to 445 nm and 530 nm irradiation (
Figure 26) [
111]. By comparison, both TiONFe and TiONPt showed excellent deNOxing abilities even under red light irradiation at 627 nm. Specifically, TiONPt showed the highest deNO
x abilities at all light wavelength ranges: about 37.8%, 36.8%, 28.2%, and 16.0% of NO
x was removed, respectively, under continuous irradiation by monochromatic light at 390 nm (UV LED), 445 nm (blue LED), 530 nm (green LED), and 627 nm (red LED).
Because of the newly formed N2p level within the bandgap of titania above the O2p valence band, N-doped titania displayed an extrinsic bandgap smaller than the intrinsic bandgap of titania such that TiON absorbed visible light. In addition, TiON displayed significant chemiluminescence emission attributed to formation of singlet oxygen 1O2 relative to undoped S-TiO2 which failed to display any light emission. For comparison, TiONFe and TiONPt also displayed relatively high chemiluminescence emission, albeit lower than TiON; the latter showed chemiluminescence intensity increases in the order: UV < blue < green < red.
For the undoped S-TiO
2 sample, the study of Yin et al. [
111] showed a correlation between very weak chemiluminescence emission intensities and very low visible-light induced photocatalytic activity that they attributed to its relatively large intrinsic bandgap. By contrast, for TiON, TiONFe, and TiONPt, results demonstrated that the deNO
x ability decreased with an increase in chemiluminescence emission intensity. Nonetheless, the photocatalytic deNO
x activity of TiON was nearly the same as that of S-TiO
2 under 390 nm (UV LED) and 627 nm (red LED) irradiation. However, TiON exhibited greater activity than S-TiO
2 under 445 nm and 530 nm irradiation, but lower than TiONFe and TiONPt samples under every type of LED light irradiation. The authors ascribed this variation to different band structures and to the presence of Fe and Pt loaded onto the surface of the TiON.
Mechanistically, the sequence of events that led to deNOxing by these four titania samples was summarized [
110] by the series of Reactions (72)–(77). Subsequent to irradiation of the titania that yields conduction band electrons (e
−) and valence band holes (h
+), formation of singlet oxygen
1O
2 (Equation (76)) competes with formation of superoxide radical anions (Equation (73)) and hydroxyl radicals (Equation (74)) in air (molecular oxygen; relative humidity, ca 25%). Fe and Pt loading on TiON increased charge transfer and charge separation on the surface of the TiONFe and TiONPt photocatalysts [
111].
In the present context, deNOxing reportedly occurred by oxidation of the NO
x molecules via the active oxygen
•OH and O
2−• species (Equations (78)–(80)), whereby NO is converted to NO
2 and subsequently to NO
3− ions.
In a NO
x atmosphere, the NO
x molecules adsorb onto the photocatalyst’s surface and then interact with the superoxide radical anions O
2−• to form NO
3− (Equation (80)), as a result of which the NO
x molecules consume O
2−• and delay singlet oxygen formation (Equation (76)), thereby causing the chemiluminescence emission intensity in the NO
x atmosphere to be much lower than in air. To recapitulate, Yin and coworkers [
111] deduced that:
Nanosized titania exhibited very low deNOx ability under visible light irradiation, irrespective of their excellent UV light-induced (390 nm) deNOx ability.
N-doped titania displayed excellent photocatalytic activity under 445 nm and 530 nm light irradiation.
Fe and Pt loading improved the photocatalytic activity of N-doped TiO2 under not only UV light but also long-wavelength visible-light irradiation (λ = 530 nm and λ = 627 nm).
Pt-loaded, N-doped titania possessed the best visible light- and UV-induced photocatalytic activity. In addition, Fe- and Pt-loaded N-doped titania exhibited relatively high quantum yields of deNOxing under long-wavelength LED light irradiation.
In their extensive 2009 review article on the catalytic abatement of NO
x in the environment, Roy and coworkers [
112] focused mostly on thermal methods in the presence of suitable reducing agents, and briefly gave a short account of the alternative photocatalytic methodology at ambient conditions; summarized was also some of the earlier work reported by selected researchers noting that direct photocatalytic decomposition of NO would yield N
2 and O
2, which would indeed be the ideal outcome and sole products if that could be realized. Unfortunately, as noted above, different conditions and different titania-based photocatalysts lead to significantly different results that are worth recalling briefly. For instance,
Anpo and coworkers showed that metal ion-implanted TiO
2 decomposed NO photo- catalytically to N
2, O
2 and N
2O at 275 K under irradiation with visible light at wavelengths longer than 450 nm [
96].
Lim et al. [
102] found that the photocatalytic decomposition of NO over Degussa P-25 TiO
2 in an annular flow type reactor produces NO
2, N
2O and N
2, with the efficiency increasing with light intensity and residence time and decreasing with initial NO concentration.
Bowering et al. [
109] showed that the photocatalytic activity of Degussa P-25 TiO
2 toward deNOxing decreased with increasing pretreatment temperature.
Roy and coworkers [
110] reported that reduction of NO over the catalyst Ti
1−xPd
xO
2−δ was two orders of magnitude greater than unsubstituted TiO
2. Direct NO decomposition into N
2 and N
2O occurred via dissociation of NO in the presence of UV radiation at room temperature yielding N
2, N
2O and O
2 with the O
2 evolved reacting with NO to give NO
2 that is adsorbed by the catalyst upon formation. Prolonged NO
2 adsorption makes the surface inactive for NO dissociation; NO dissociation resumed when CO was passed to scavenge the evolved dissociated O
2 [
110].
On the other hand, the seminal review article by Skalska et al. [
113] presented an extensive survey of NO
x emission control technologies for three major anthropogenic emission sources: power plants, vehicles and the chemical industry, and further described new and alternative methods such as a hybrid system of SCR (selective catalytic reduction) and O
3 injection, fast SCR, and electron beam gas treatment, among others. Also described was the influence of NO
x on the environment and human health. The main focus was put on NO
x control methods applied in the combustion of fossil fuels in power stations and mobile vehicles, together with methods used in the chemical industry; the authors emphasized the implementation of ozone and other oxidizing agents in NO
x oxidation.
Following these footsteps, Heo and coworkers [
114] combined photocatalysis and SCR with hydrocarbons as reducing agents (HC/SCR) to improve the activity and durability of deNO
x catalysts. The authors developed a photocatalytic HC/SCR system that exhibited high deNO
x performance (54.0−98.6% NO
x conversion) at low temperatures (150−250 °C) using dodecane as the HC reductant over a hybrid SCR system that included a photocatalytic reactor (PCR) and a dual-bed HC/SCR reactor (
Figure 27). The PCR generated the highly active oxidants O
3 and NO
2 from O
2 and NO in the feed stream, followed by subsequent formation of the highly efficient reducing oxygenated hydrocarbon (OHC), NH
3, and organo-nitrogen compounds. These reductants were key in enhancing the low-temperature deNO
x performance of the dual-bed HC/SCR system containing Ag/Al
2O
3 and CuCoY in the front and rear bed of the reactor, respectively (
Table 5). Moreover, the OHCs proved particularly effective for both NO
x reduction and NH
3 formation over the Ag/Al
2O
3 catalyst, while NH
3 and organo-nitrogen compounds were effective for the reduction of NO
x over CuCoY. The photocatalytic assisted hybrid HC/SCR system demonstrated an overall deNOxing performance comparable to that of the NH
3/SCR, thus its potential as a promising alternative to the current urea/SCR technology [
114].
To the extent that the deNO
x performance of the conventional HC/SCR catalyst was enhanced by both OHCs and NH
x-containing reductants, the two representative PCR catalysts V
2O
5/TiO
2 and Ag/Al
2O
3 were chosen by the authors [
114] for further examination in the PCR + HC/SCR hybrid system: the V
2O
5/TiO
2 PCR for its superior OHC formation and the Ag/Al
2O
3 PCR for its formation of NH
3 and possibly organo-nitrogen compounds as precursors of NH
3.
Table 6 lists the conversions of NO and NO
x, the conversion of NO
x to N
2, and the yields of NO
2, N
2O, and NH
3 during the reduction of NO with the PCR + HC/SCR system [
114]. The NO
x conversion to N
2 was estimated from the conversion of NO
x and the yields of NO
2, N
2O, and NH
3 by the mass balance of nitrogen. The selectivity of the Ag/Al
2O
3 PCR + HC/SCR system for N
2 was 94%, 91%, and 82% at 200, 250, and 300 °C, respectively. The slight decrease N
2 in selectivity of the Ag/Al
2O
3 PCR + HC/SCR system at 300 °C was ascribed to increased formation of NH
3 by reaction of NO with OHCs over the HC/SCR reactor, since the PCR readily converted dodecane to OHCs. At 400 °C, the HC/SCR system alone completely reduced NO
x with high N
2 selectivity up to 96% so that the PCR could be turned off and bypassed to save energy at temperatures above 400 °C.
Key to the successful demonstration of this advanced deNO
x process was the unique design and functionality of the PCR, which led to three major conclusions [
114]: (1) PCR with catalysts was very efficient for both OHC formation and reduction of NO
x because of its dual function: in situ UV-induced formation of OHC and conversion of NO
x over the catalysts; (2) blank PCR (no catalyst) was very efficient for oxidation of NO to NO
2 and HC to OHC, but was inefficient for converting NO
x because of the absence of a catalyst; and (3) Ag/Al
2O
3 PCR (with BaY + Ag/Al
2O
3) produced OHC and NH
3 as intermediates that could be used subsequently to further convert NO
x in a downstream reactor containing a dual-bed catalyst such as Ag/Al
2O
3 (for OHC/SCR) and CuCoY (for NH
3/SCR).
7. Recommendations
In practical applications of photocatalysis, conventional TiO
2-based photocatalytic surfaces have been used to oxidize NO
x to nitrate species; the latter species do not desorb spontaneously and consequently deactivate or block the surface-active centers of the photocatalyst from carrying out the next cycles. To avoid such deactivation, the nitrates (or nitric acid) should typically be washed away by rain [
172]; however, the nitric acid is corrosive and could pollute the soil when its concentration at the site becomes too high. A promising way to resolve this problem, which the users of applied photocatalysis have failed to consider but known to occur in a laboratory setting since the first report by Courbon et al. [
92] in 1984, would be to change the selectivity of the photocatalytic reaction so that the NO
x gases are converted back to N
2 and O
2 by some photoreduction pathway as reported some time ago by Anpo and coworkers [
94,
95], who used Cu
+ ions in SiO
2 or in zeolite to effect the photocatalytic reduction of NO
x. No deactivation of the active sites would occur for this photoreduction reaction since nitrogen and oxygen readily desorb from the surface [
173]. The selectivity toward the photoreduction of NO could be improved greatly by reducing the hexacoordinated Ti
4+ species (TiO
6 octahedra) to tetracoordinated Ti
4+ species (TiO
4 tetrahedra) [
174], as successfully achieved by depositing isolated TiO
4 clusters inside cavities of zeolite-Y using ion beam implantation [
174,
175].
In a more recent article, Wu and van de Krol [
176] proposed a novel strategy to change the photocatalytic selectivity of TiO
2 by creating a large and stable concentration of oxygen vacancies in TiO
2 nanoparticles through thermal reduction in a reducing atmosphere; these oxygen vacancies were stabilized by doping the TiO
2 nanoparticles with an electron acceptor-type dopant such as Fe
3+ which also greatly enhanced the activity of the photoreduction process. The authors [
176] further showed that with this strategy NO was indeed photoreduced to N
2 and O
2 and that photooxidation of NO was largely suppressed. Moreover, photoreducing Fe
3+ to Fe
2+ provided a recombination pathway that suppressed nearly quantitatively the formation of NO
2 and consequently enhanced the selectivity of the reaction for N
2 formation [
176]. The authors also alluded to formation of N
2 and O
2 via two different routes. One route would see a small amount of tetrahedrally coordinated Ti formed in the Fe-doped TiO
2 samples, which Anpo et al. [
175,
177] claimed as the active site for the catalytic decomposition of NO to N
2 and O
2 at Ti-modified zeolites. As most Ti
IV ions at the TiO
2 surface are fivefold-coordinated, a single oxygen vacancy created at or near the surface could lead to a fourfold-coordinated Ti
4+ center; however, this would require a strong reduction of the Ti–O bond length that would be possible only at very high oxygen vacancy concentrations, which the authors [
177] deemed an unlikely pathway and proposed the other route that implicated oxygen vacancies acting as the catalytic centers through the capture of the oxygen side of NO as illustrated in
Figure 59 and summarized in Reactions (101)–(104); the associated experimental data are also displayed in
Figure 59.
Summing Reactions (101)–(104) yields the overall Reaction (105).
Although the conversion efficiency was somewhat modest (ca. 4.5% after 1050 min correspond-ing to a TON of ∼2 NO molecules per O vacancy site), the Fe-doped TiO
2 photocatalyst showed no signs of deactivation as the NO conversion centers were not blocked by nitrate species [
168], contrary to standard deNO
x TiO
2-based photocatalysts that have to be washed away periodically.
In a most recent article, Cao and coworkers [
178] investigated the adsorption of NO and the consequent reactions on differently treated rutile TiO
2(110) surfaces using polarization/azimuth-resolved infrared reflection absorption spectroscopy. Apparently, surface defects (e.g., oxygen vacancies,
Vo) and reconstructions on TiO
2(110) had a strong effect on the reaction pathways of NO → N
2O conversion (N
2O is laughing gas). The pathway proposed involved a defect-free oxidized TiO
2(110) surface in which two NO molecules are adsorbed on adjacent surface-pentacoordinated Ti (Ti
5c) sites first, which then couple to form a cis-(NO)
2/Ti&Ti dimer through the N−N bond of the dimer, and then are converted to N
2O species (or perhaps even to N
2 gas) [
178].
Clearly, much fundamental research in TiO
2-based photocatalysis needs to be undertaken in the optic toward applications to environmental
deNOxification, with special attention and efforts directed at titania doped with Fe, Cr, Co and Ni dopants that may yet prove interesting [
176].
As a case in point, a recent article by Kuznetsov and coworkers [
70] examined possible additional specific channels of photoactivation of solid semiconductors with regard to thermo-/photo-stimulated bleaching of photoinduced Ti
3+ color centers in visible-light-active (VLA) photo-chromic rutile TiO
2, which an optical emission spectroscopic analysis had shown to contain 99.4 at.% Ti and 0.2 at.% Al as the principal impurity, together with 0.09 at.% Fe, 0.05 at.% Sn, 0.04 at.% Nb, and 0.03 at.% Cr as minor impurities. Considering that the prime photophysical process of photostimulated bleaching of Ti
3+ color centers is absorption of light quanta by the Ti
3+ centers, the authors [
70] found that no selectivity of photostimulated bleaching of a certain type of Ti
3+ centers could be ascertained, and that photogenerated holes captured at a set of traps were also participants in the photostimulated bleaching of these color centers. Based on current findings and earlier results, the authors hypothesized that the heat released during nonradiative electron transitions, following the prime photophysical processes of excitation and ionization of Ti
3+ centers, dissipates in the nearest neighborhood of the Ti
3+ centers and that localized nonequilibrated excitation of the phonon subsystem leads to thermal detrapping of the photoholes with different depths up to 1 eV. Subsequent recombination of free holes with trapped electrons from Ti
3+ centers leads to the observable photostimulated bleaching of the color centers [
70]. Based on experimental evidence, the authors further argued that following absorption of vis–NIR light by the color centers, the subsequent release of thermal energy accompanying nonradiative electron transitions provides an additional specific channel to photoactivate the VLA rutile TiO
2, in particular, and possibly other photocolorable metal-oxide semiconductors as well.
Following their interest of the photophysics of color centers in VLA rutile titania ceramics and titania powder resulting from the photoformation and separation of charge carriers, Kuznetsov et al. [
179] noted that the action spectrum of the photoformation of Ti
3+ centers at very low temperatures (90 K) accorded fully with the absorption spectra of intrinsic defects that consisted of a set of individual absorption bands that they attributed to several different Ti
3+ centers. Analysis of the dependencies of the photoformation of separate centers on the wavelength of illumination and light exposure, which provided extraction of specific Ti
3+ centers, led the authors to identify Ti
3+-based centers with excessive negative charge that formed at significantly high concentration upon maximal exposure of the titania specimens to Vis-light illumination: (2Ti
3+ + V
o2+) ←→ (Ti
δ+ + V
o2+) with 3 > δ > 2. They also showed from thermoprogrammed annealing (TPA) that the spectra of Ti
3+ color centers in the range 90–500 K consisted of a set of first-order peaks corresponding to traps, whose depths ranged from ~0.2 eV (peak at 130 K in the powder specimen) to 1.06 eV (peak at 455 K in the ceramics specimen). The highest rate of recombination of holes released to the valence band with Ti
3+ centers—an event attributed to Ti
δ+ centers—provided TPA spectra that clearly manifested the existence of shallow traps. In addition, mass spectrometric experiments on the photoadsorption of molecular oxygen and photodesorption of photoadsorbed oxygen from the surface of powdered VLA titania specimens provided further evidence of the photoformation of electrons and holes in VLA TiO
2 under Vis-light illumination, and allowed the authors [
179] to determine the kinetics of photo- desorption of O
2 under orange light illumination subsequent to photoadsorption of O
2 stimulated by blue light excitation. Those experiments provided further proof of the occurrence of another specific channel toward the photoactivation of VLA TiO
2 via photoexcitation of photoinduced Ti
3+ color centers.
It is important to recognize that Ti
3+-based centers (i.e., Ti
δ+ centers) appeared after many other Ti
3+ centers had already been formed. In other words, such Ti
3+-based centers appeared at high density of Ti
3+ centers (see below). Accordingly, specific properties of Ti
3+-related centers responsible for the (
extrinsic) absorption bands at 1.56 eV and 1.26 eV were postulated to account for the excess negative charge characteristic of such Ti
3+-based centers. In line with the work of Déak and coworkers [
180], the two adjacent Ti
3+ centers located near a single oxygen vacancy forming a (2Ti
3+ + V
o2+) complex (
Figure 60a) were taken by Kuznetsov et al. [
179] as extra charged Ti
3+ centers when compared to isolated Ti
3+ centers (
Figure 60b). The two Ti
3+ species in the (2Ti
3+ + V
o2+) complex can, in principle, disproportionate to (Ti
2+ + Ti
4+ + V
o2+) so that, in accord with the more generally accepted view, these extra-negatively charged Ti
3+ centers are best referred to as Ti
δ+ centers for which 3 > δ > 2. Clearly, the appearance of such Ti
δ+ centers, whether photogenerated or resulting from the removal of the structural oxygen during a reduction event, is of lower probability because of the electrostatic repulsion of the two trapped electrons and the well-known instability of such centers to oxidation. Consequently, the formation and increase in the concentration of such photoinduced Ti
δ+ centers appears to occur only at high density of photogenerated Ti
3+ centers that ensue upon prolonged exposure to Vis-light illumination in the later stages of photocoloration (i.e., formation of Ti
3+ color centers).
Germane to the above, the work of Déak and coworkers [
180] showed that the first case scenario is that two self-trapped electrons in the (V
o2+ + 2
e) complexes are located at two equivalent first neighbors of the oxygen vacancy (extra-negatively charged Ti
3+ → Ti
δ+ centers;
Figure 60a), while in the second scenario both electrons are more remote from the V
o2+ vacancy and are not in the same plane as the vacancy (isolated Ti
3+ centers;
Figure 60b); the energies of the vertical transitions of these self-trapped electrons to the conduction band are ca. 1.1 eV. Following this reasoning, a question arose as to why the growth of the number of such Ti
δ+ centers was observed only under Vis-light illumination. This led Kuznetsov and coworkers [
70] to focus attention on the differences in the spatial photoexcitation events that occur in the microparticle when illuminated in the UV and Vis spectral regions. Such differences had not heretofore been considered in the literature; their views of the events that occur under UV and Visible light illumination are summarized in
Figure 61.
When h
ν >
Eg, light quanta are absorbed spontaneously in solids in an arbitrary manner, each time producing e-h pairs at new spatial sites (black stars in
Figure 61a) for which charge carrier transport and localization in the microparticle are determined by the distribution of charge carrier traps (Processes
2 and
3 in
Figure 61a). At moderate UV-light irradiances, the authors [
70] supposed that since every subsequent photoformation of charge carriers and trapping event occur in other spatial sites, a significant density of Ti
3+ centers would not be reached. However, photoformation of electrons and holes can also be achieved on illumination in the Vis region at h
ν ≤
Eg when light quanta are absorbed by the native point defects (i.e.,
F (or
F+) centers); the latter are limited in number and are located at definite sites in the microparticle (
Figure 61b). Moreover, because photoexcitation of
F (or
F+) centers can produce charge carriers followed by their subsequent decay to their initial electronic states, as proposed in earlier studies [
68,
69], repetitive absorption of light quanta and photogeneration of electrons and holes occurs each time at the same spatial sites (
F or
F+ centers) in the microparticle (blue stars in
Figure 61b). Such considerations then lead to the reasonable inference that transport of carriers and occupation of traps (processes
2 and
3 in
Figure 61b) start repetitively at the same sites in the microparticle. In that case, filling of the nearest neighbor
F (or
F+) center traps facilitates the attainment of a high density of Ti
3+ centers and the consequent formation of the Ti
δ+ centers [
70].
The above notwithstanding regarding the TiO2-based technology, people intending to deNOxify the environment must first come to appreciate and understand the rich chemistry of nitrogen oxides, in general, and NO and NO2, in particular, in a homogeneous phase and in hetero- geneous media.
For instance, NO dimerizes to N
2O
2 upon condensing to a liquid, although the association is weak and reversible [
181]. In addition, to the extent that the enthalpy of formation of NO is endothermic, NO can easily undergo disproportionation back to its constituent elements N
2 and O
2 as might occur in catalytic converters—for example, Reaction (106) occurs over the zeolite Cu
2+-ZSM-5 [
182].
Nitric oxide is also thermodynamically unstable at 25 °C and 1 atm; under pressure, it decomposes readily in the temperature range 30–50 °C to yield NO
2 and N
2O (Reaction (107)) and may react either as NO
2 or as N
2O
3 [
183].
When exposed to atmospheric oxygen, nitric oxide converts instantly to NO
2 (Reaction (108)) [
181], which likely occurs via the intermediate ON–O–O–NO.
In water, NO reacts with oxygen and water to form nitrous acid HONO (Reaction (109)).
Since both NO
2 and NO are radical species, they combine to form the intensely blue dinitrogen trioxide N
2O
3 (Reaction (110)) [
184].
Both the brown gas nitrogen dioxide, NO
2, and the colorless gas dinitrogen tetroxide, N
2O
4, exist in a strongly temperature-dependent equilibrium (Reaction (111)) for which Δ
H = −57.23 kJ mol
−1, with NO
2 being favored at higher temperatures, while N
2O
4 predominates at lower temperatures.
Because of the relatively weak N–O bond in NO
2, nitrogen dioxide is a relatively good oxidizing agent in aqueous media (Reaction (112)
; nearly comparable to Br
2 gas), which makes the mixed oxides NO
2 and N
2O
4—also known as nitrous fumes—react vigorously if not explosively with several compounds, particularly with hydrocarbons via hydrogen abstraction as a first step (Reaction (113)) [
181].
In aqueous media, NO
2 also hydrolyzes to form nitrous acid and nitric acid via Reaction (114), which is one of the steps in the industrial production of nitric acid from ammonia via the Ostwald process [
185].
Although Reaction (114) is negligibly slow at the low concentrations of NO
2 characteristically encountered in the ambient atmosphere, it does proceed upon uptake of NO
2 onto surfaces to produce gaseous HONO in outdoor and indoor environments [
186].
Several studies have examined the interactions of NO
2 on the TiO
2 surface under various experimental conditions, such as different NO
2 partial pressures and various temperatures in the range 323–573 K [
102,
106,
108,
172]. All these studies reported production of NO in the gas phase, and formation of nitrates on the TiO
2 surface, albeit under photocatalytic conditions.
However, other aspects that seem to have been overlooked by many are the potential specific interactions between the two NOx molecules and the TiO2 surface under dark conditions, which need to be re-emphasized constantly.
In this regard, in their 2003 FTIR study carried out in the dark in borosilicate glass vessels, Finlayson-Pitts et al. [
186] discovered that the loss of gaseous NO
2 was accompanied by formation of HONO, NO and N
2O; further FTIR studies also revealed the formation of HNO
3, N
2O
4 and NO
2+ species, which led them to hypothesize that the symmetric form of the NO
2 dimer, N
2O
4, is taken up on the surface and isomerizes to the asymmetric form, ONONO
2, with the latter undergoing autoionization to NO
+NO
3−. Apparently, it is the latter intermediate species that react with water to generate HONO and surface-adsorbed HNO
3. Subsequently, NO is generated by secondary reactions of HONO on the highly acidic surface. The authors further noted that a key aspect of this chemistry is that in the atmospheric boundary layer where human exposure occurs and many measurements of HONO and related atmospheric constituents (e.g., ozone) are made, a major component for this heterogeneous chemistry is the surface of buildings, roads, soils, vegetation and other materials [
186].
A more recent investigation by Sivachandrian and coworkers [
187] on the adsorption of NO and NO
2 molecules on the metal oxide TiO
2 at ambient temperature, specifically carried out under dark experimental conditions to avoid any photocatalytic interference, showed no significant adsorption of NO on TiO
2. By contrast, not only did NO
2 significantly influence the adsorption of VOCs and mineralization on the TiO
2 surface, but once the threshold surface coverage of NO
2 was reached at room temperature, the NO
2 adsorbed reactively on the TiO
2 surface by evolving NO in the gas phase. Quantitative measurements performed downstream of the reactor led the authors [
187] to propose a new mechanism expressed by the Reactions (115)–(118) for the adsorption of NO
2 on TiO
2 at room temperature under dry air conditions:
According to this sequence, the global reaction of NO
2 adsorption is then (Reaction (118)):
Accordingly, the proposed NO
2 adsorption mechanism on TiO
2 at room temperature in the dark [
187], together with the experimental observations, could be summarized thus: (i) three NO
2 molecules adsorb on TiO
2, produce two NO
3− ions on the TiO
2 surface, and evolve one NO molecule in the gas phase; (ii) the ratio between consumed NO
2, TPD desorbed NO
2 subsequent to adsorption, and NO produced during NO
2 adsorption = 3:2:1; (iii) the NO
2 adsorption time (i.e., the TiO
2 surface coverage) significantly modified the nature of the adsorbed species at ambient temperature; (iv) the NO formation time was controlled principally by the surface coverage of NO
2− and NO
3− ions, rather than by the NO
2 inlet concentration; and (v) at higher NO
2 gas phase concentrations (greater than 35 ppm) at room temperature, the total amount of consumed NO
2 decreased as a result of self-poisoning of the sites by adsorbed NO
3− species.
Similarly, Haubrich and coworkers [
188] examined the interaction of NO
2 on rutile TiO
2(110) and discovered that the presence of NO
2 and water led to the formation of multilayers under dark conditions of nitric acid, HNO
3, contrary to exposure of the surface to pure water after saturation of the surface with 200 mTorr of NO
2; no further growth of the AP-XPS (ambient pressure X-ray photoelectron spectroscopy) nitrate signals occurred under the latter conditions. Apparently, formation of HNO
3 requires weakly adsorbed NO
2 molecules, an important finding with important implications in environmental processes since their study [
188] confirmed that metal oxides facilitate the formation of nitric acid under ambient humidity (in the dark) conditions typically encountered in atmospheric environments.
It is evident that there is much more that needs to be investigated to fully understand whatever events occur in and on the TiO2 (and others) semiconductor photocatalyst. Laboratory experiments using models and solar simulators are just the beginning, after which what is needed is to bring the laboratory outdoors using actual environmental quantities of NOx as the reagents and humid air as prevails in the environment being investigated for application of the photocatalytic substrates. A careful study of the levels of NOx or its products at various distances from the photocatalytic surface, both vertically and horizontally, without precluding determination of all other environmental factors is also needed. The deNOx index has shown that metal-doped TiO2 systems are also worth investigating further.

































































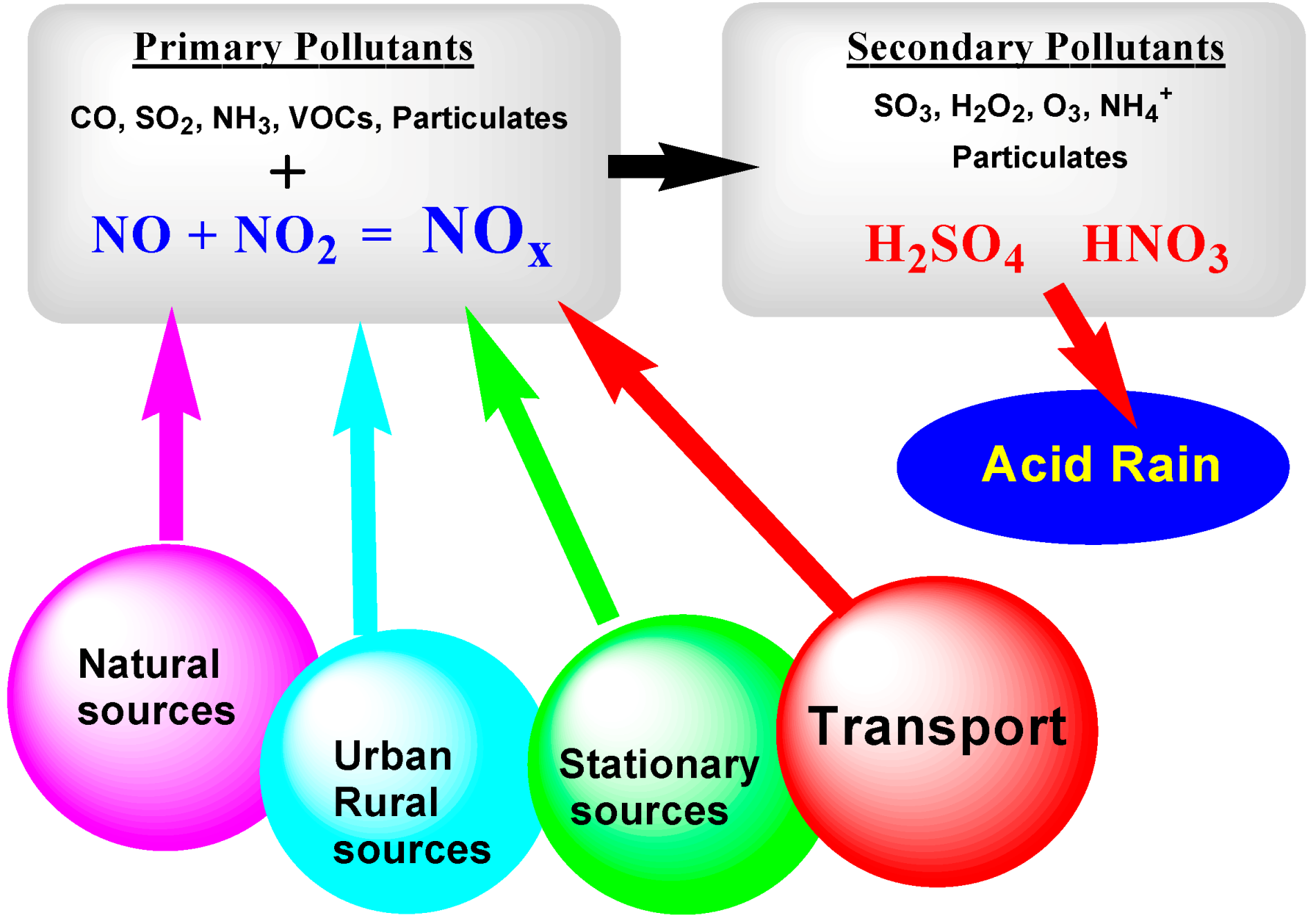{kind=link}
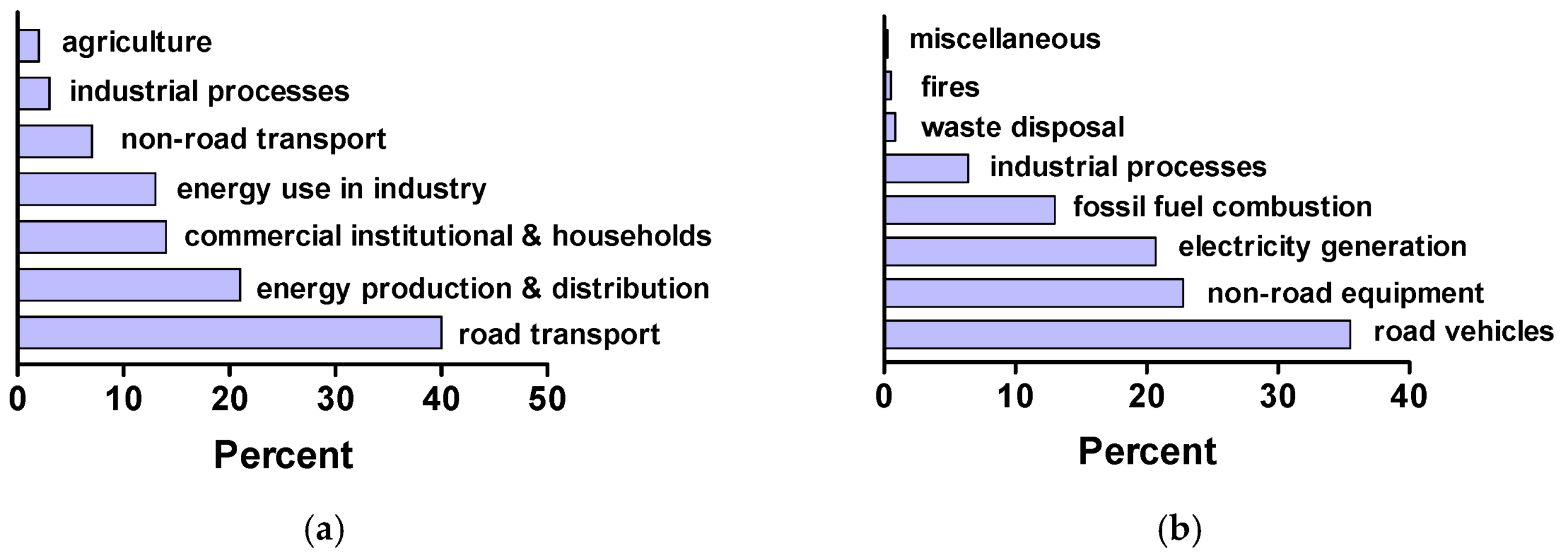{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
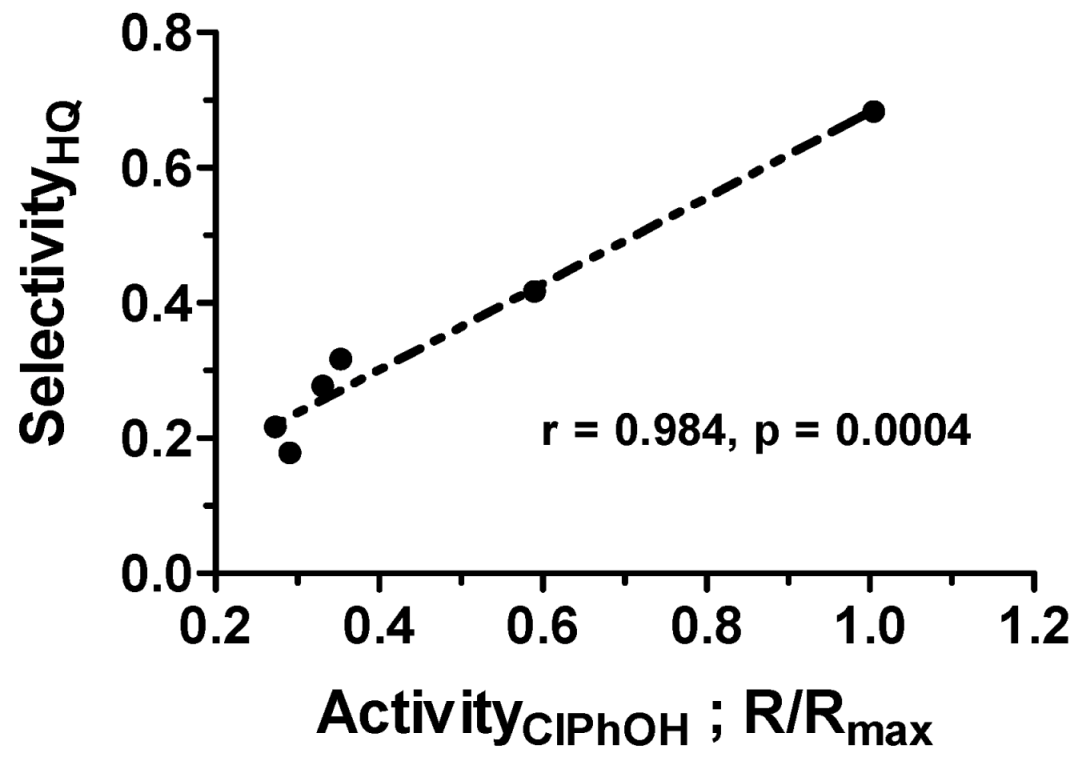{kind=link}
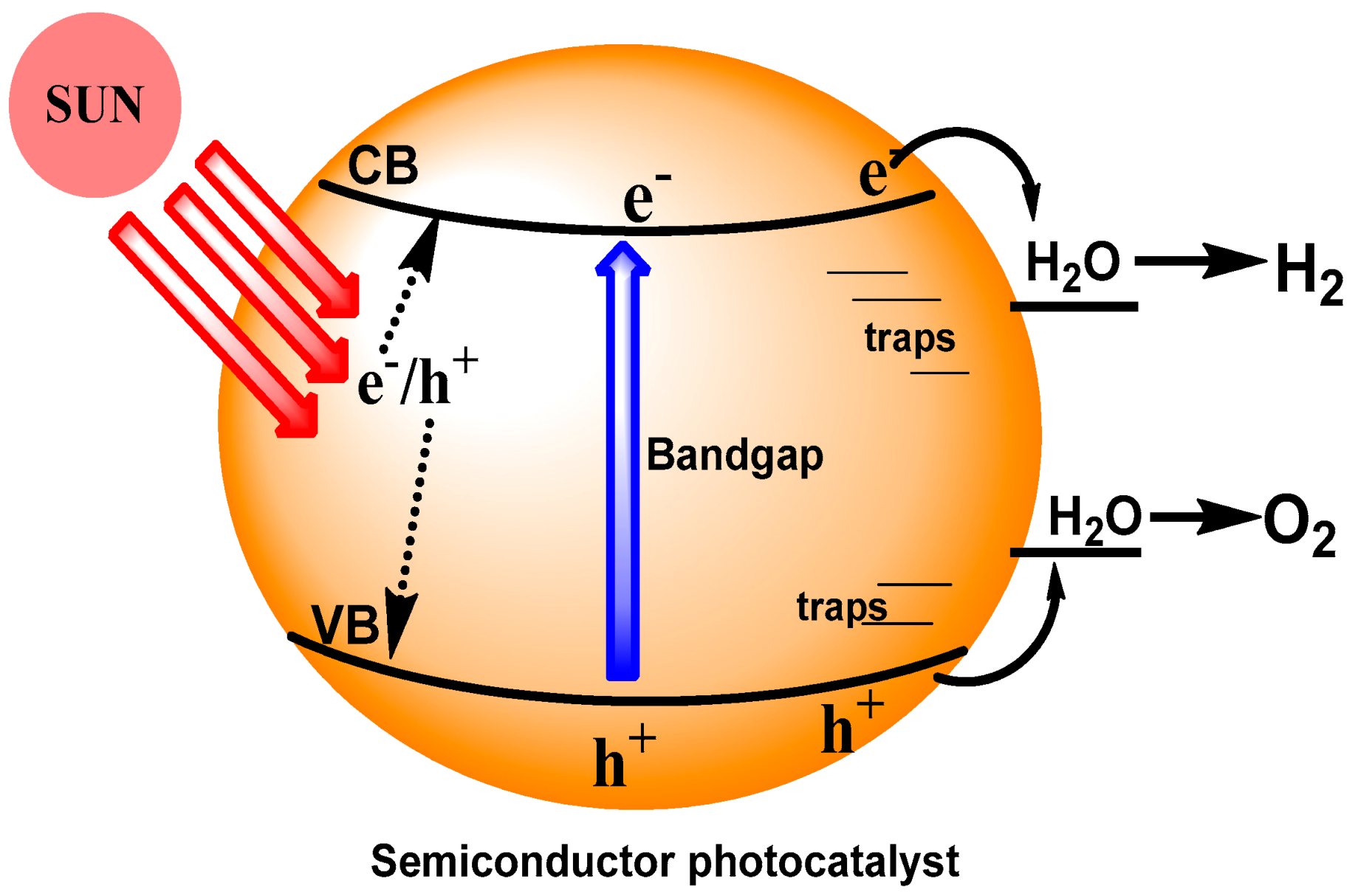{kind=link}
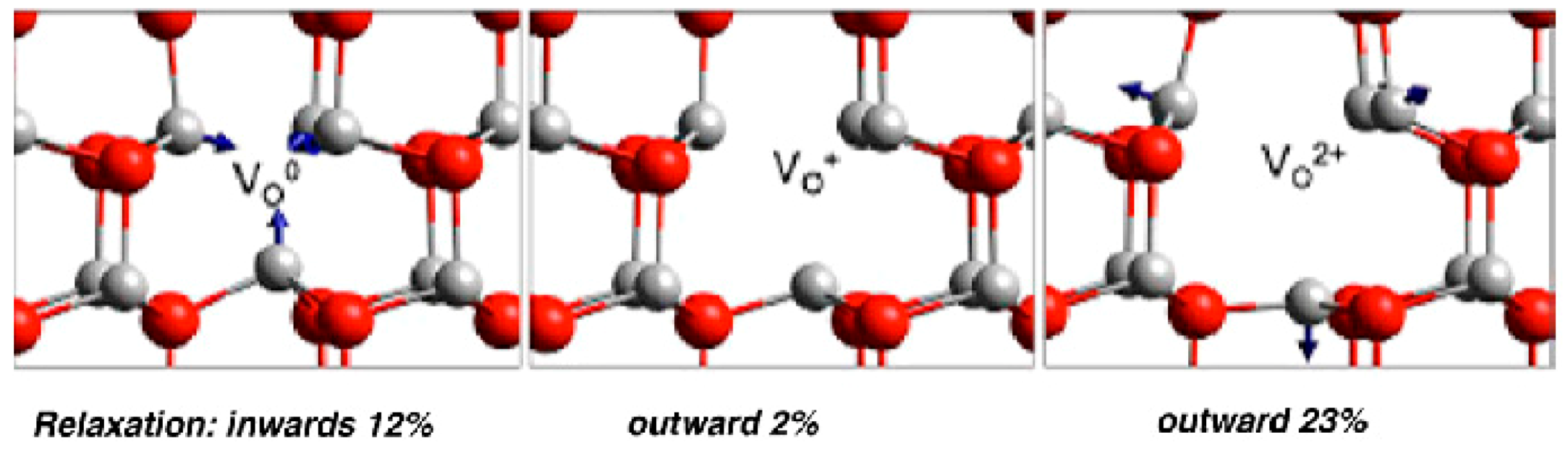{kind=link}
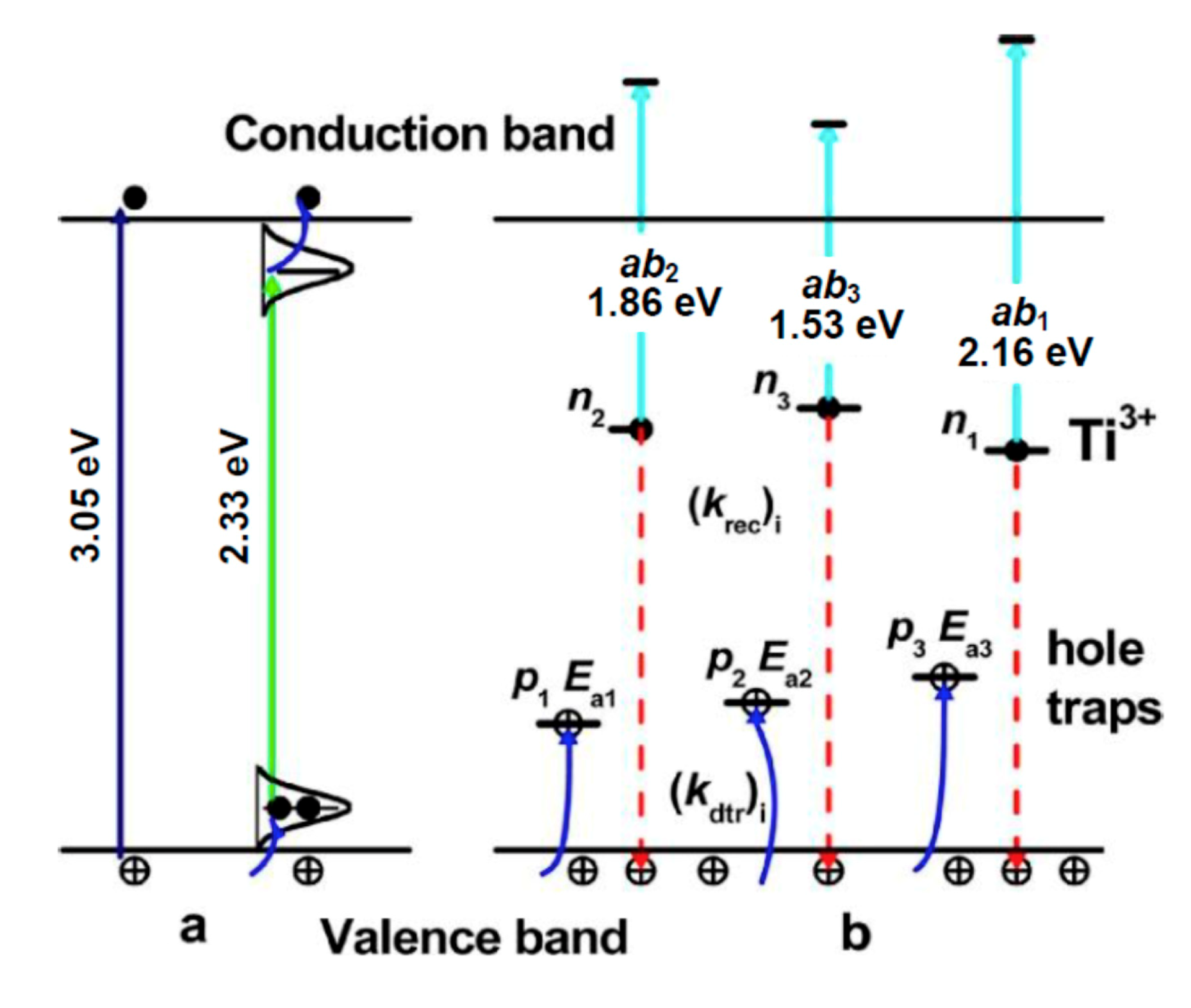{kind=link}
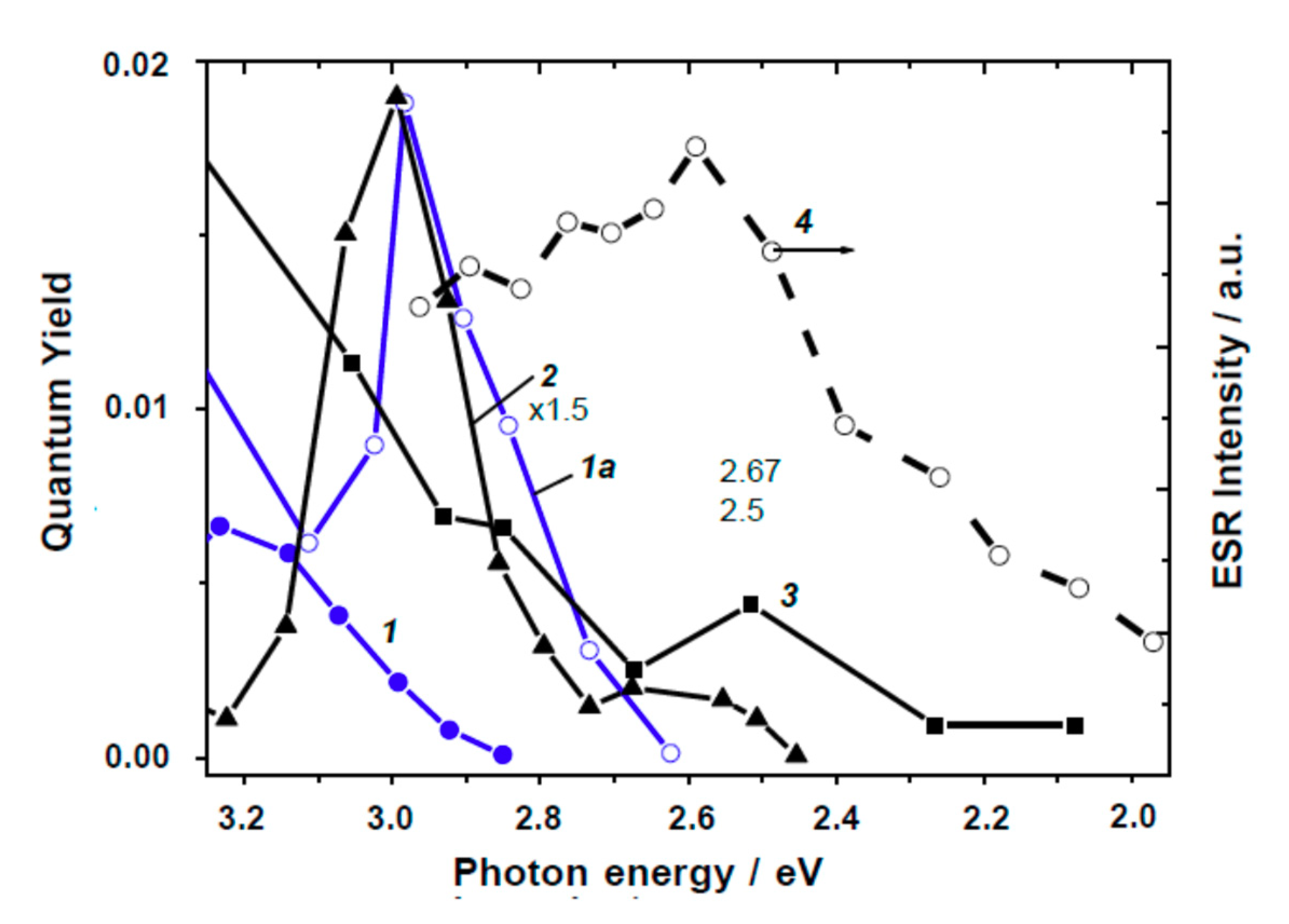{kind=link}
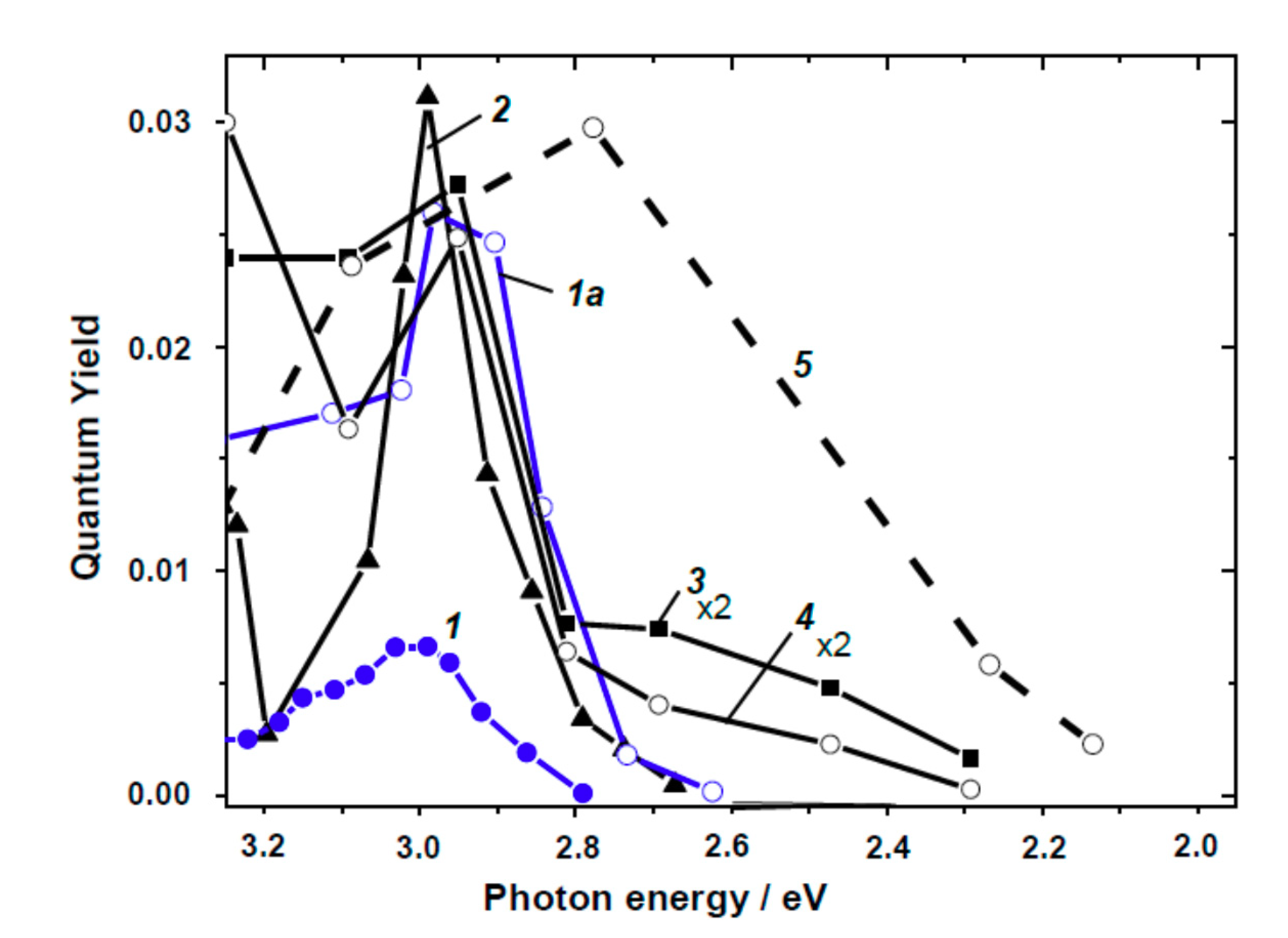{kind=link}
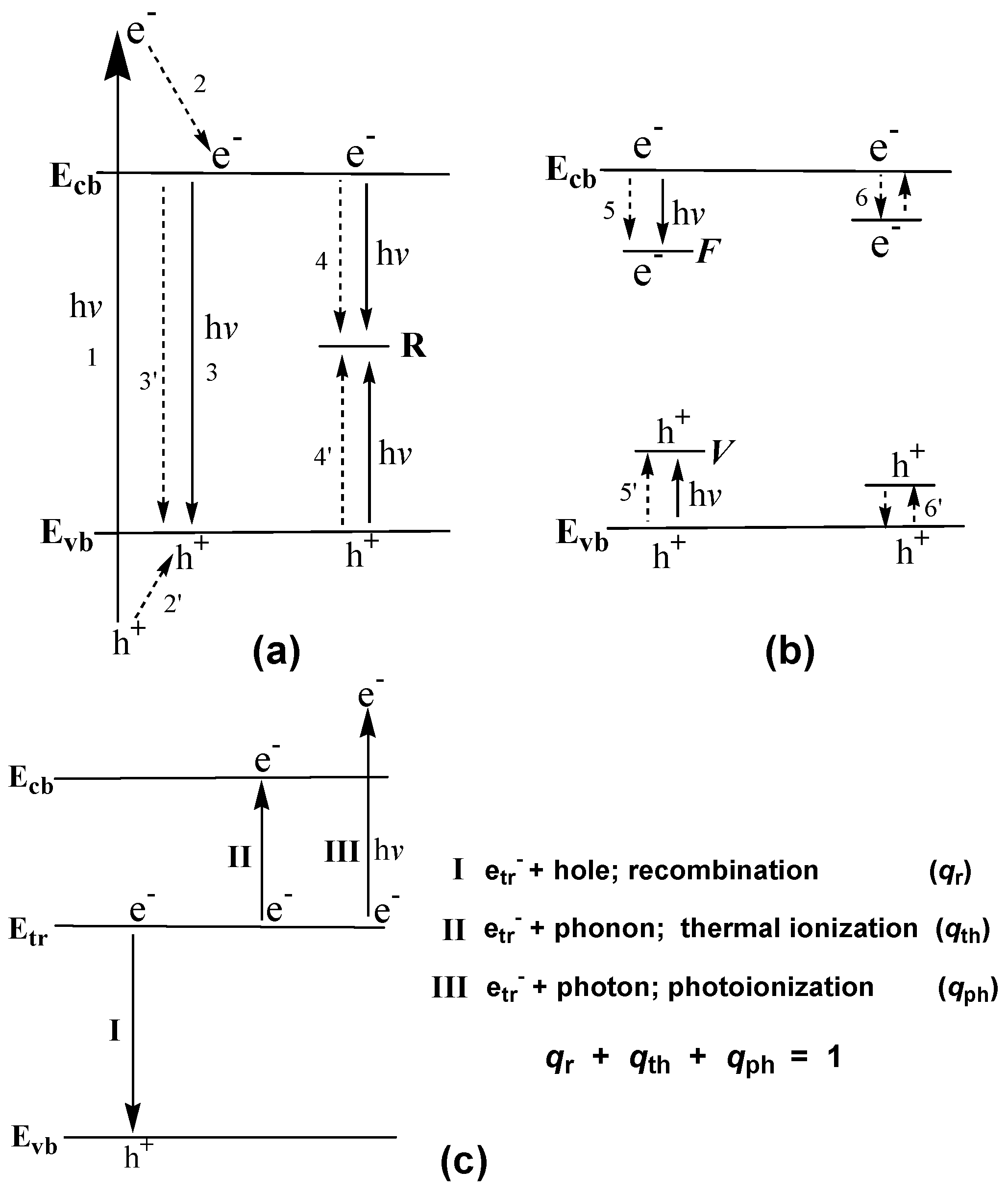{kind=link}
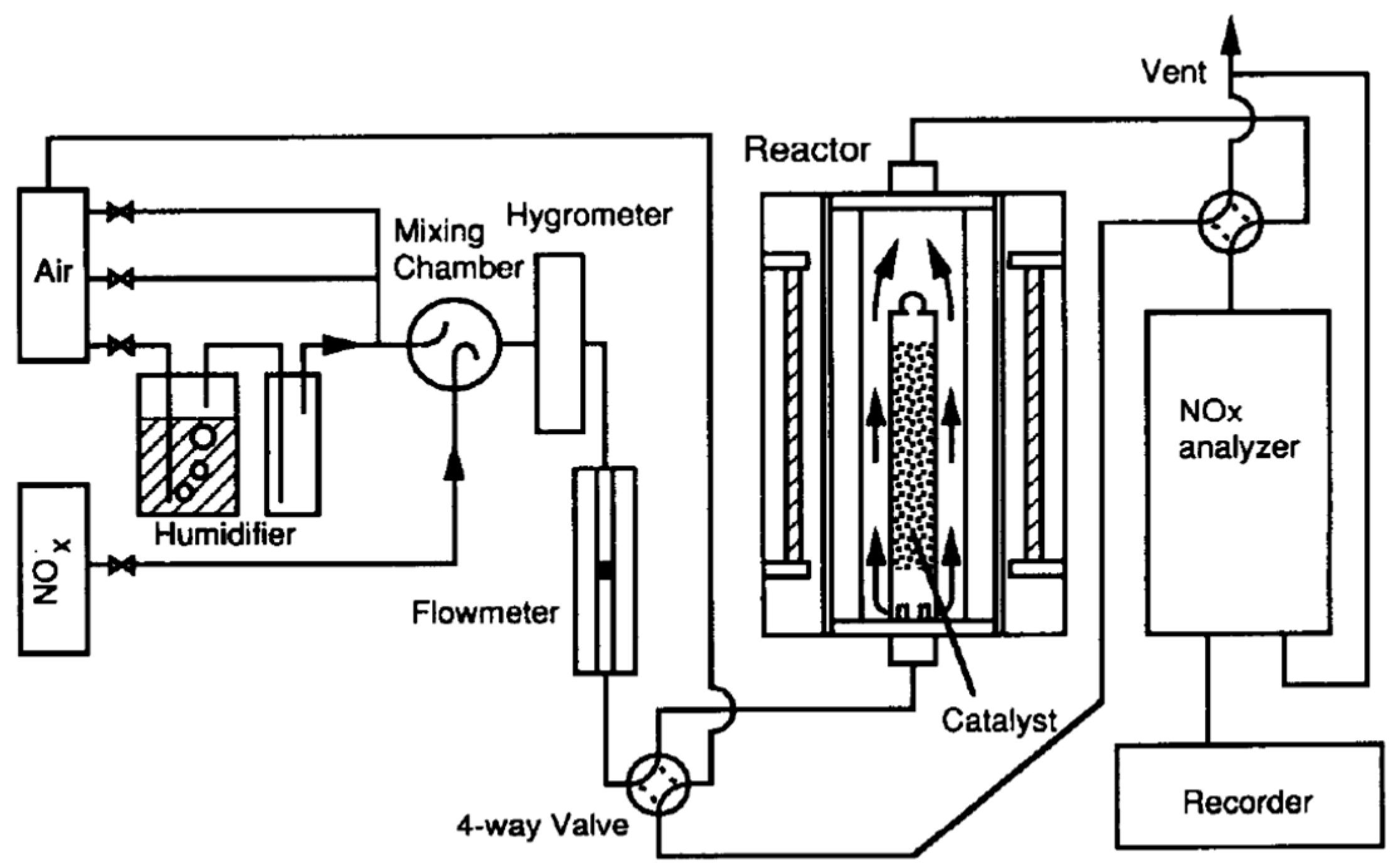{kind=link}
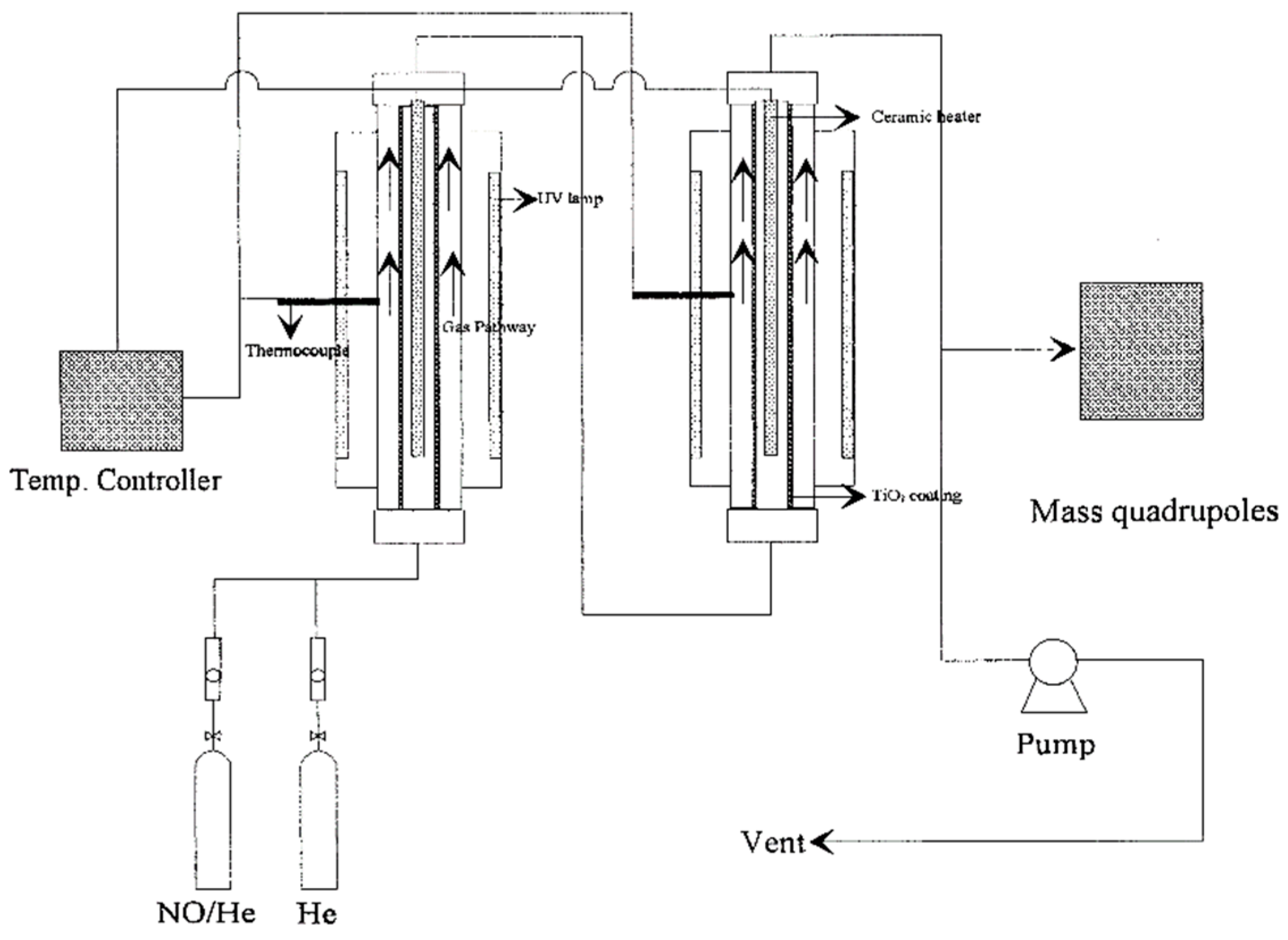{kind=link}
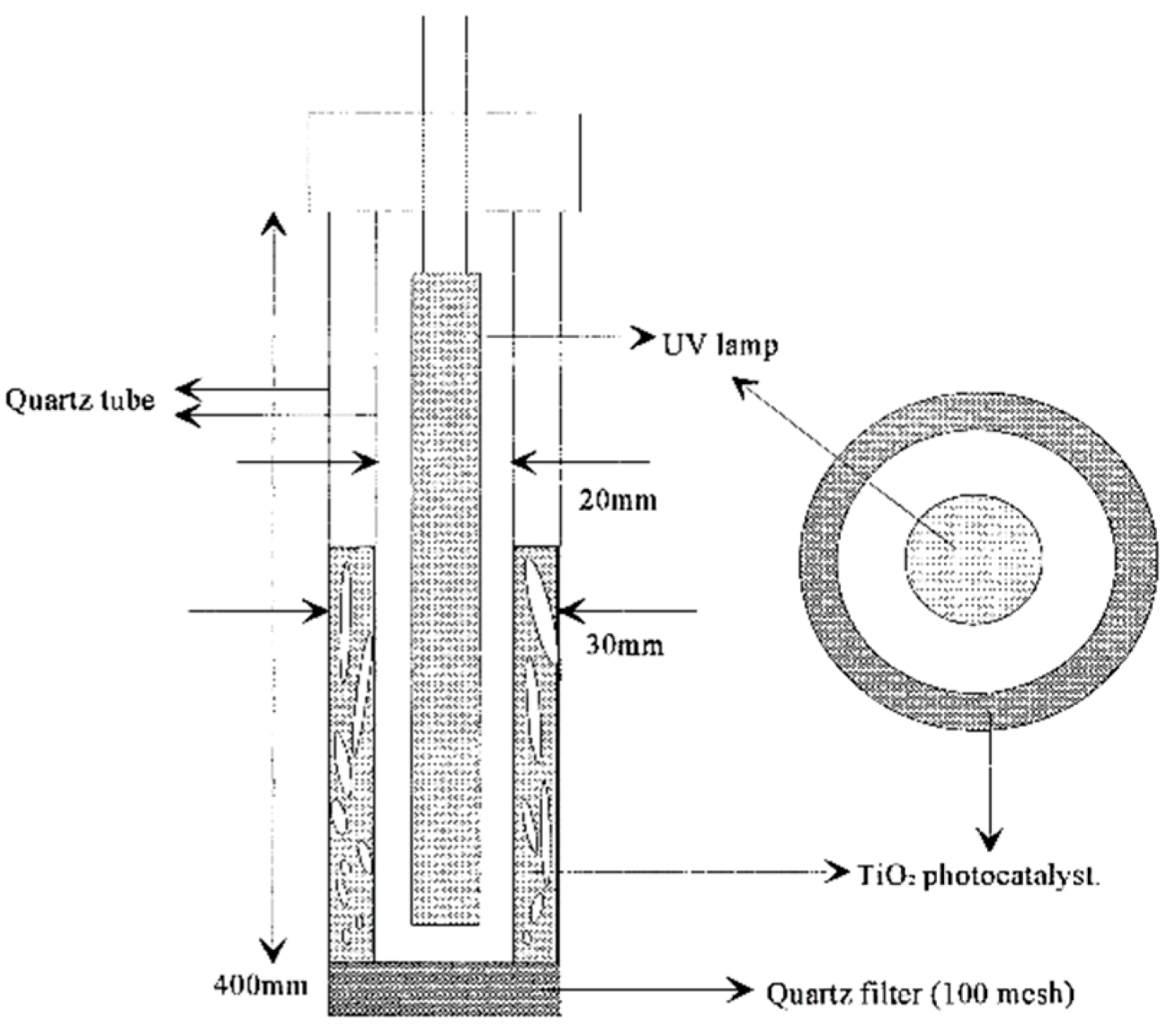{kind=link}
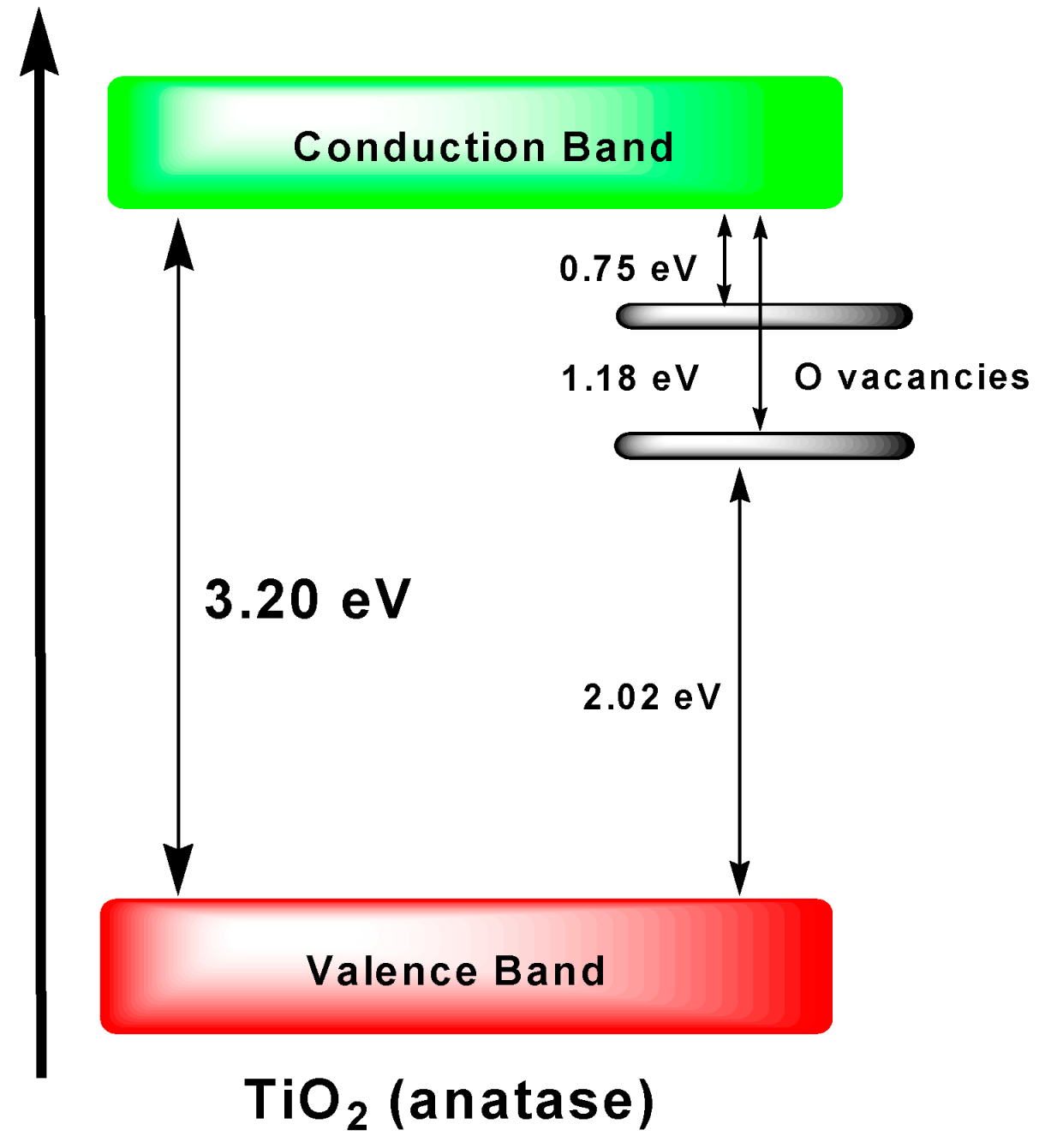{kind=link}
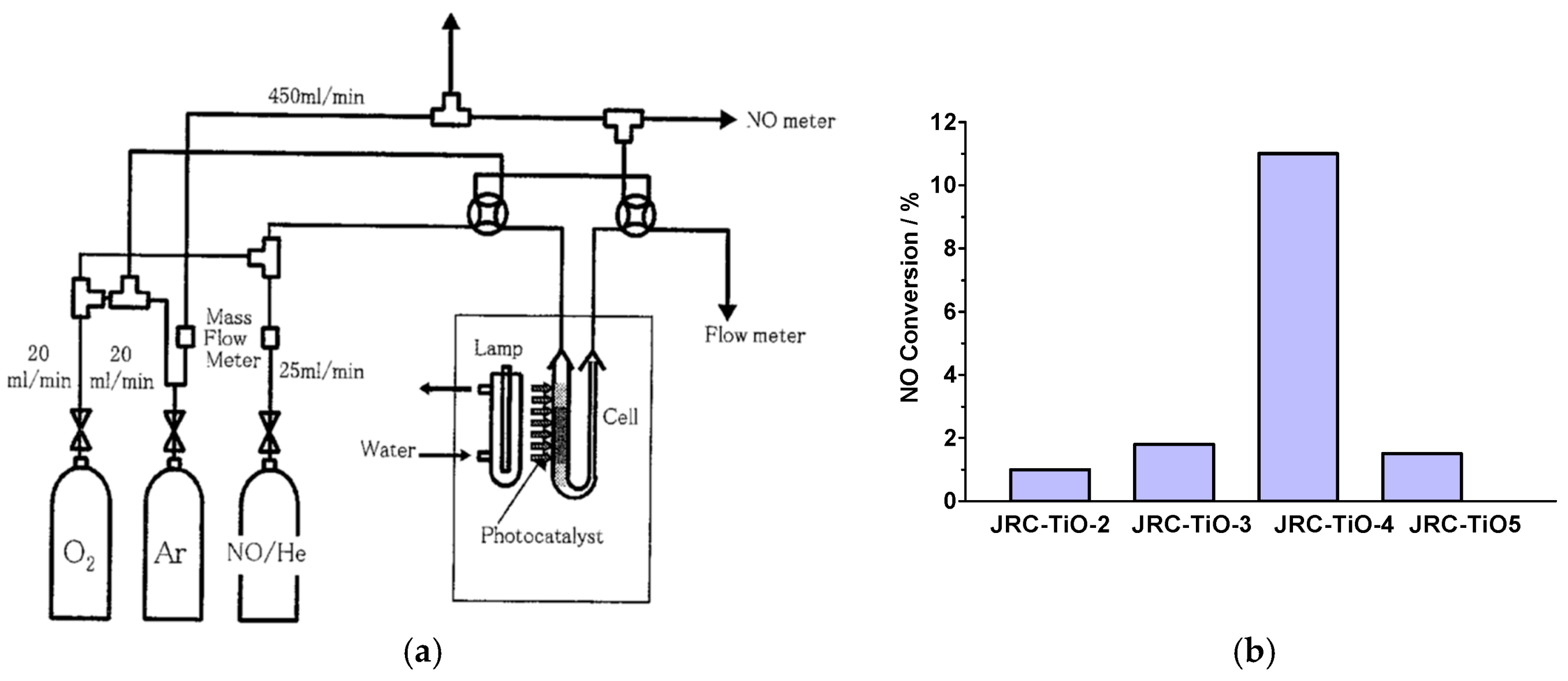{kind=link}
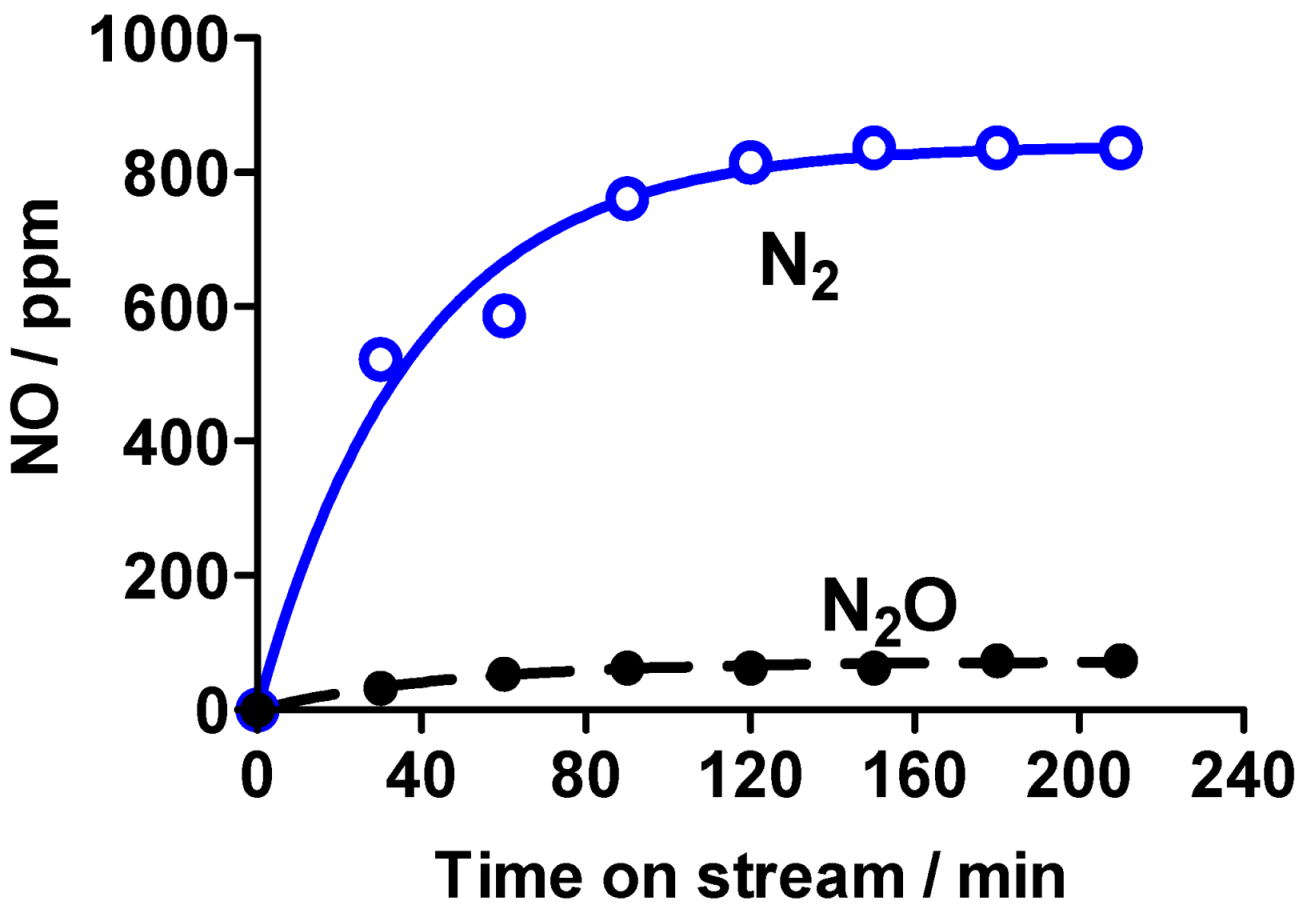{kind=link}
{kind=link}
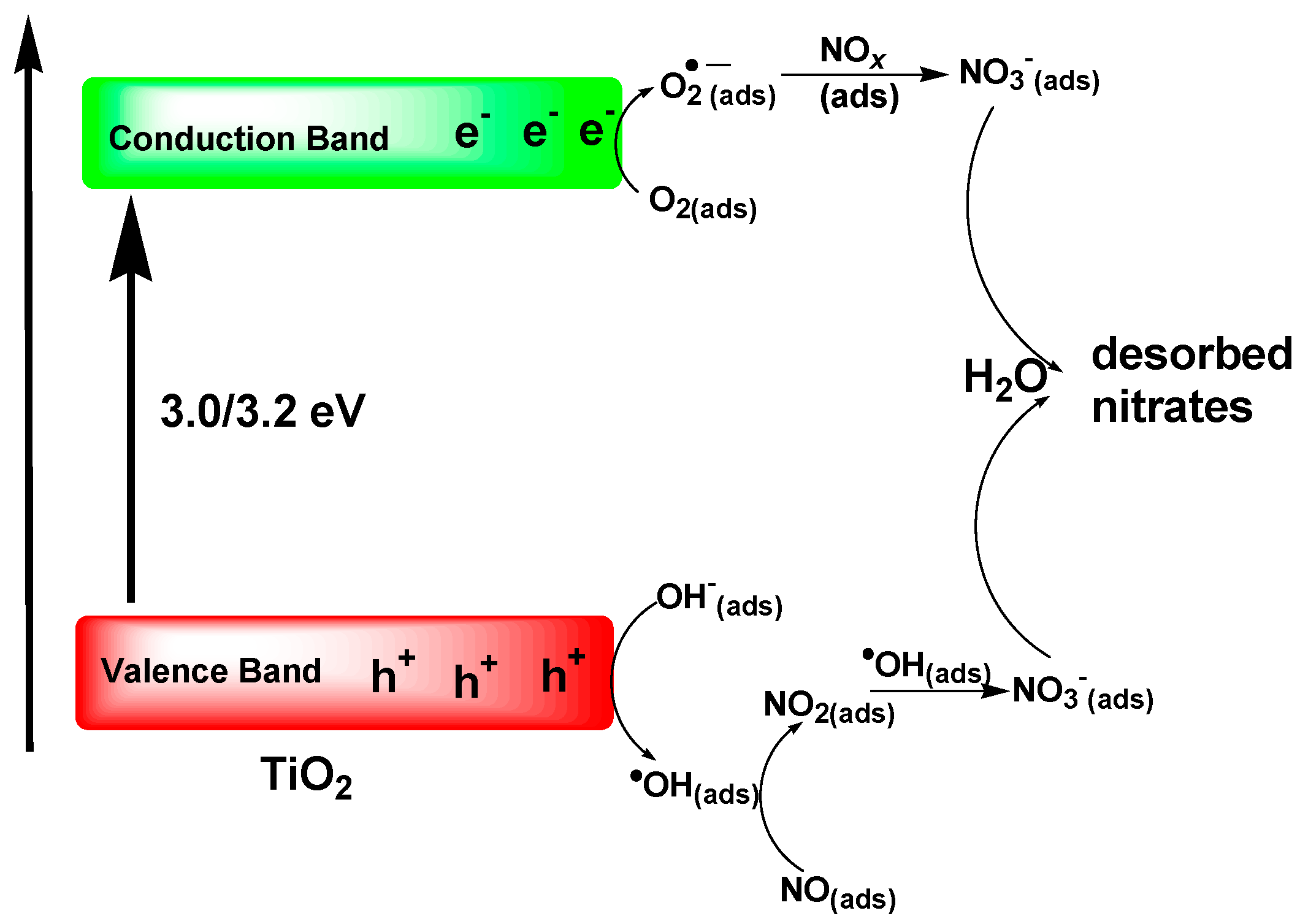{kind=link}
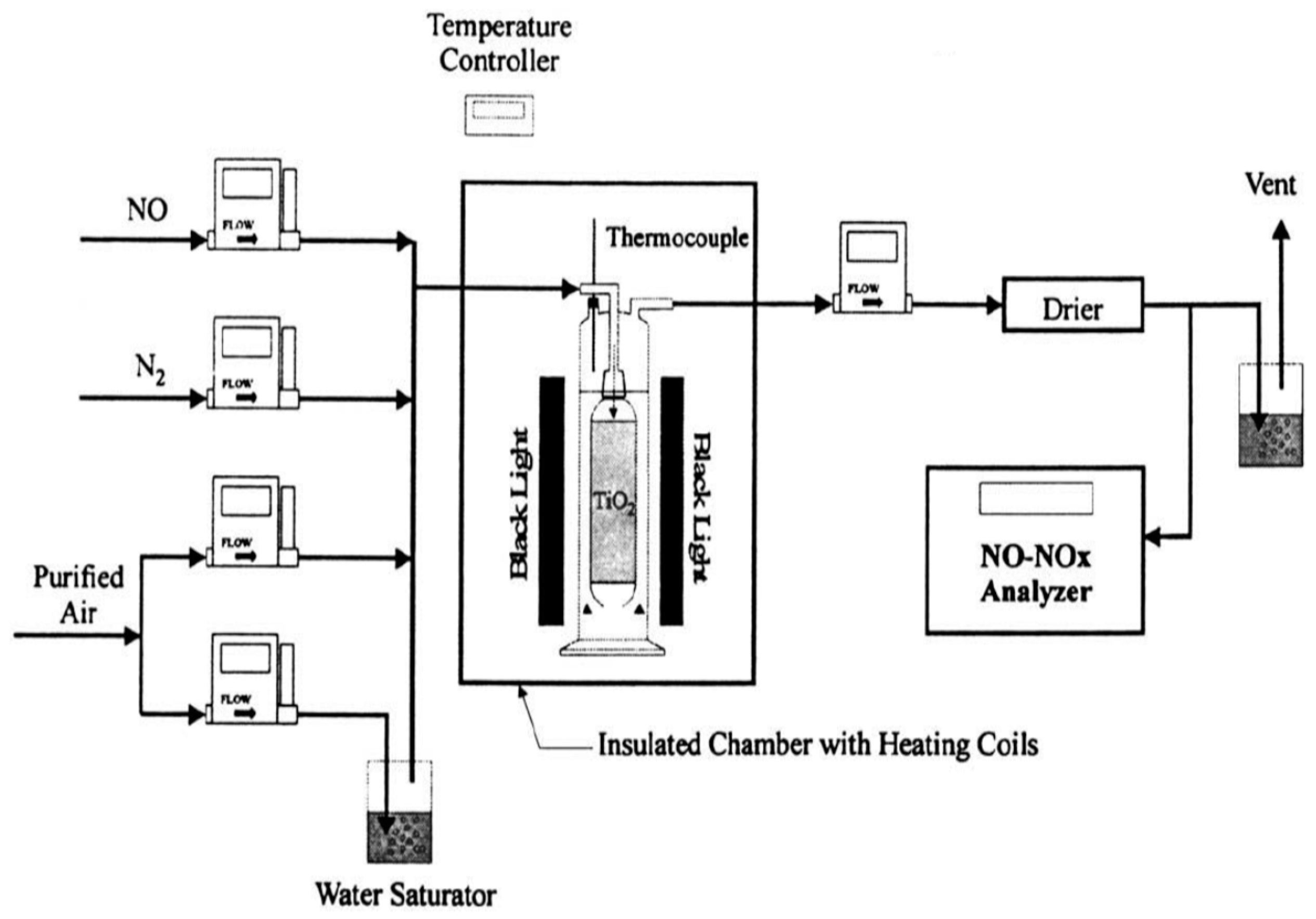{kind=link}
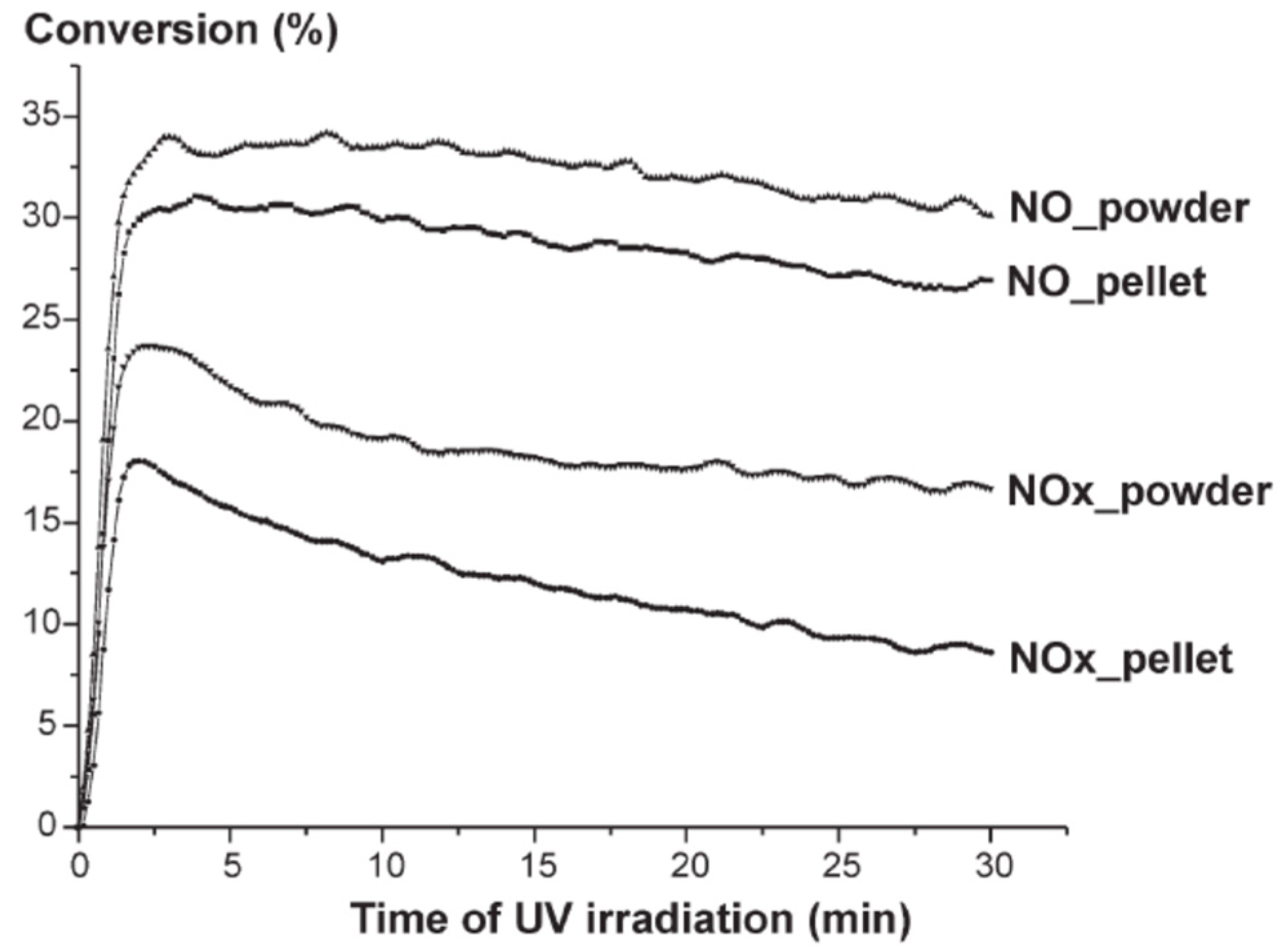{kind=link}
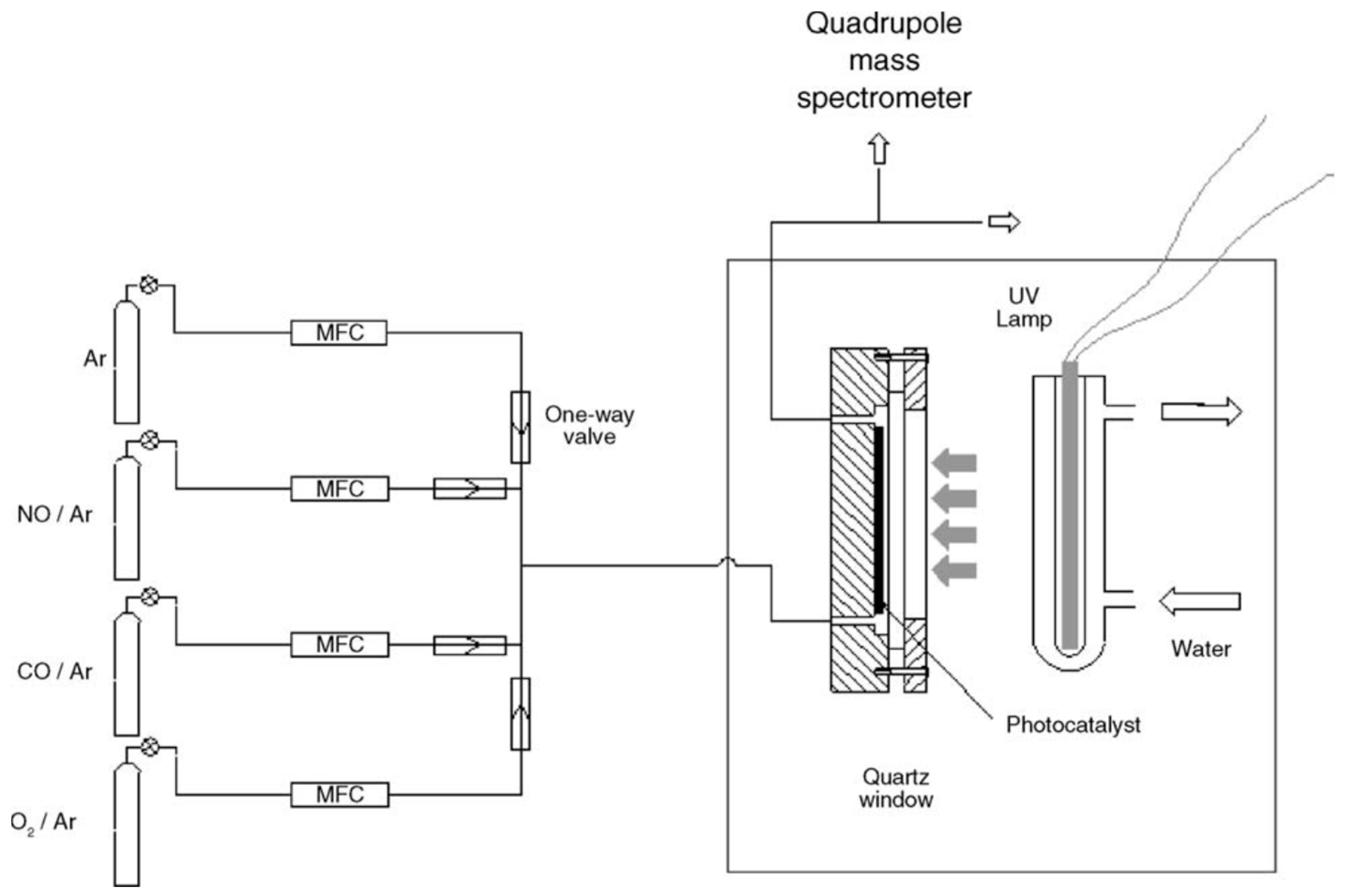{kind=link}
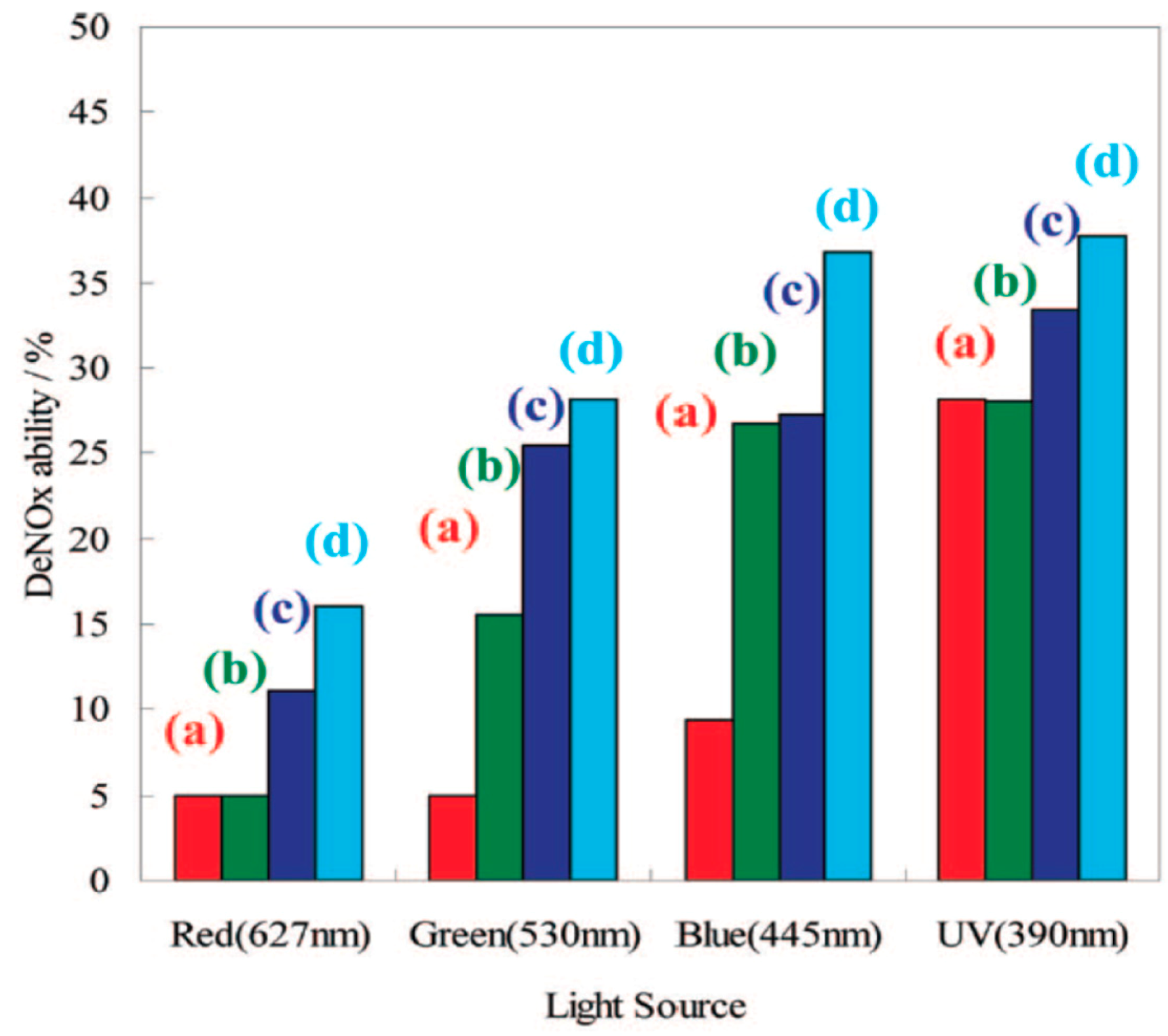{kind=link}
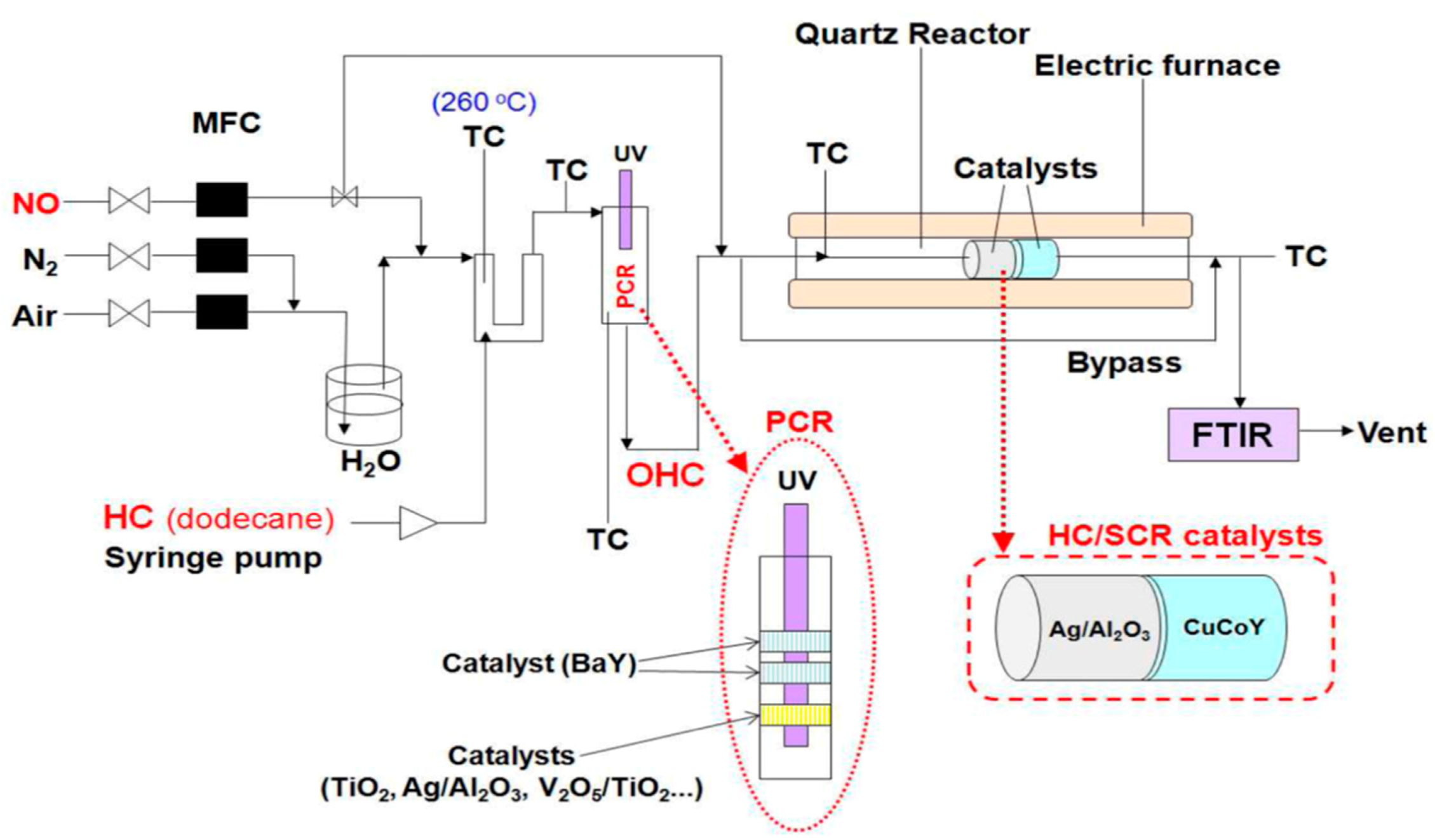{kind=link}
{kind=link}
{kind=link}
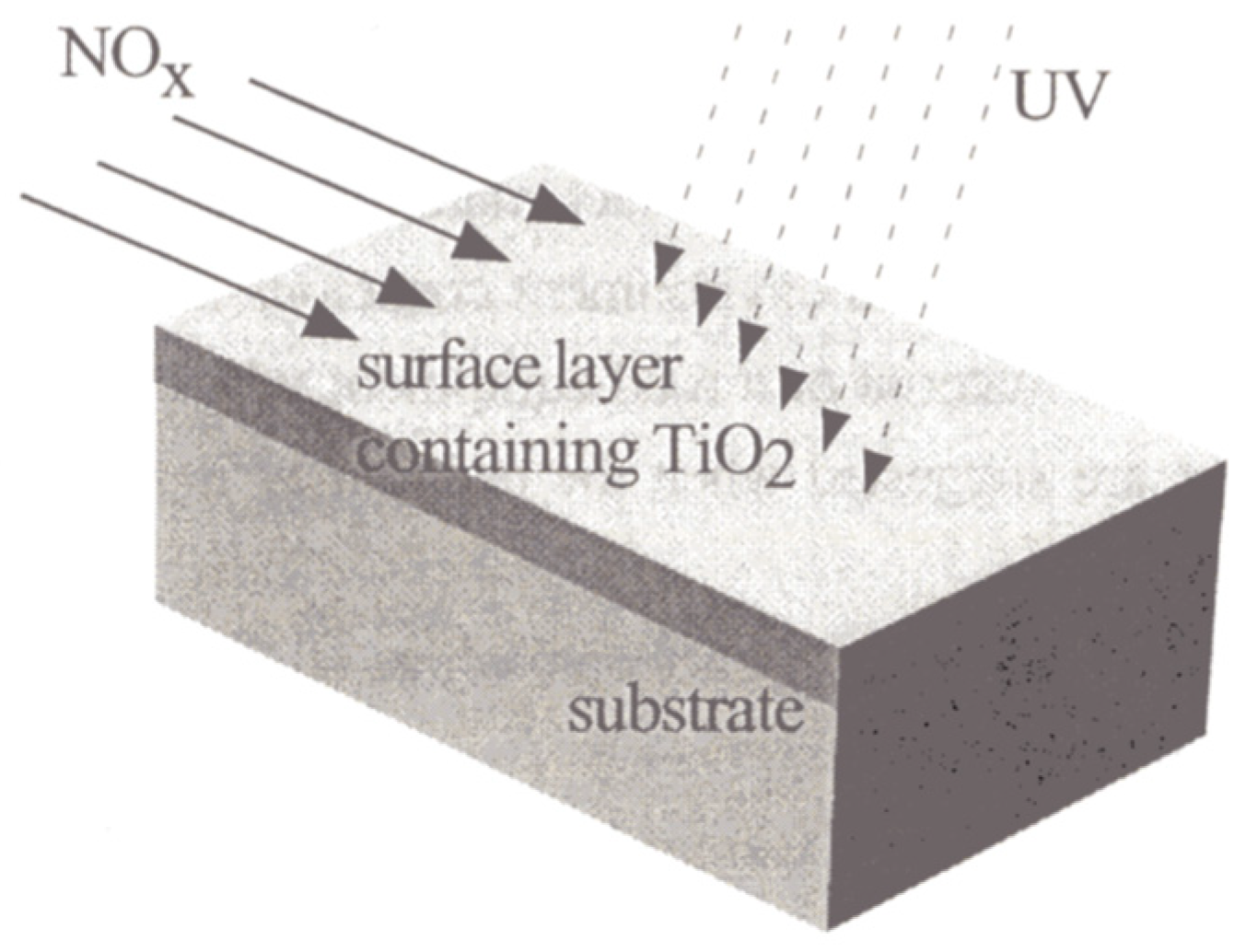{kind=link}
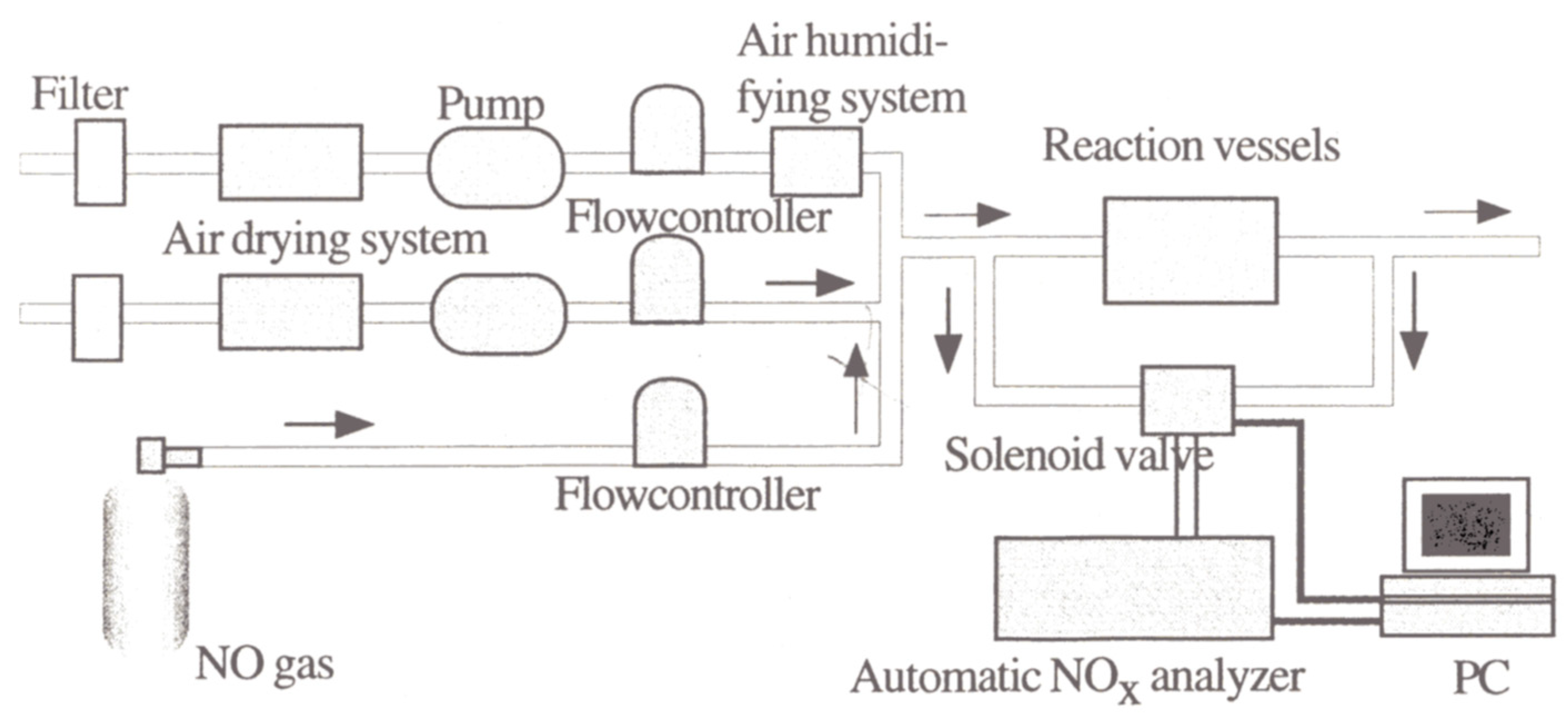{kind=link}
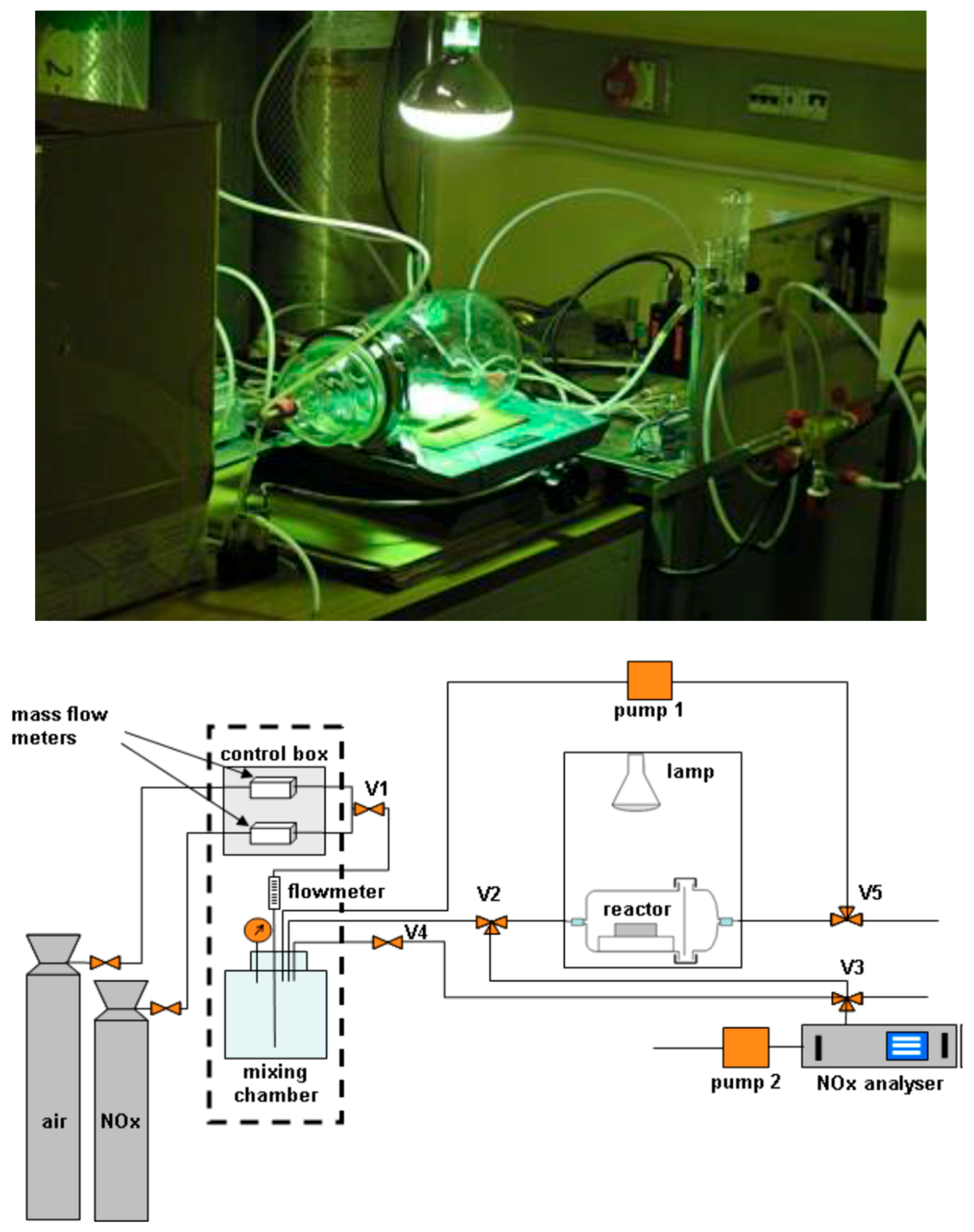{kind=link}
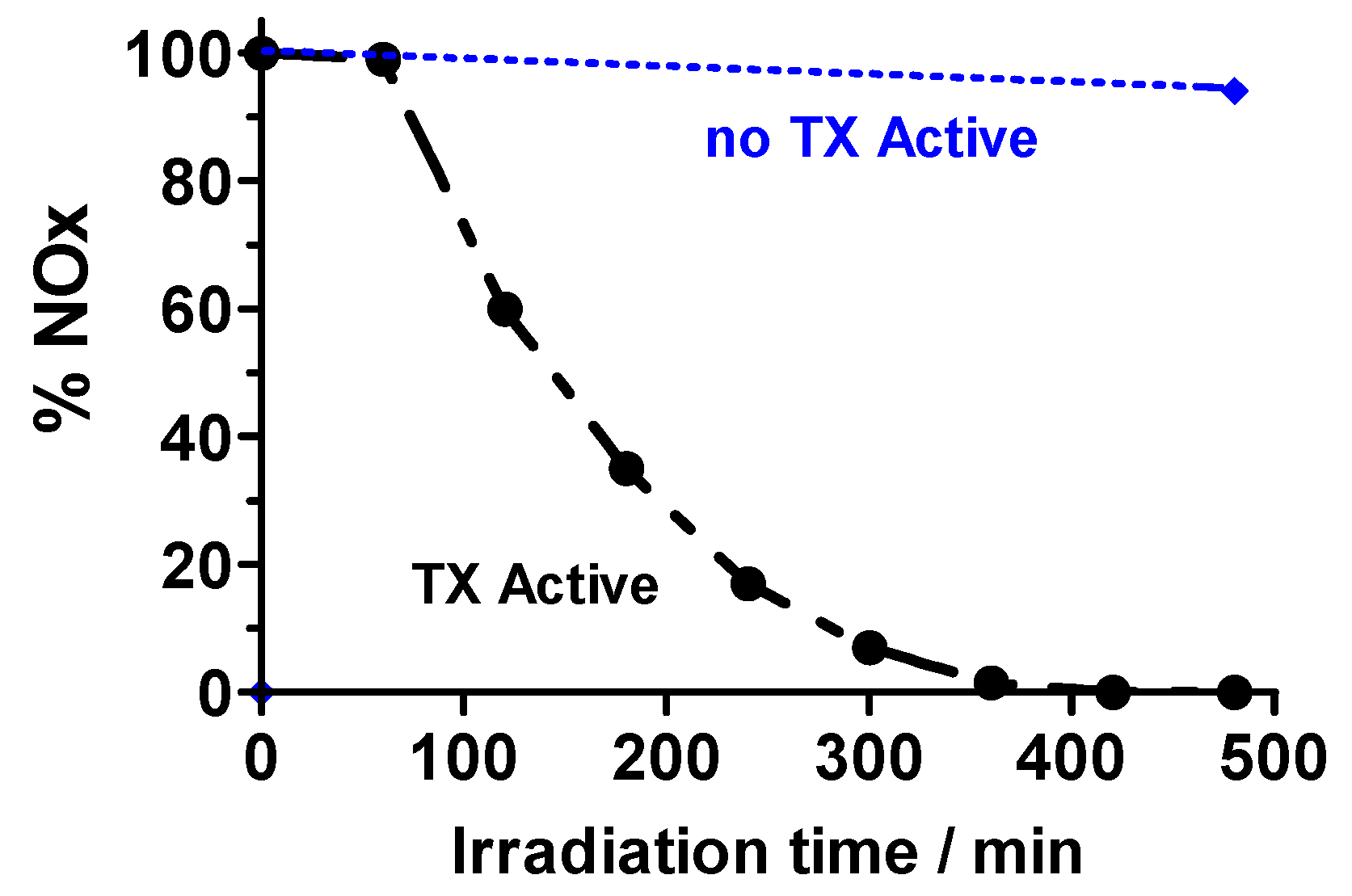{kind=link}
{kind=link}
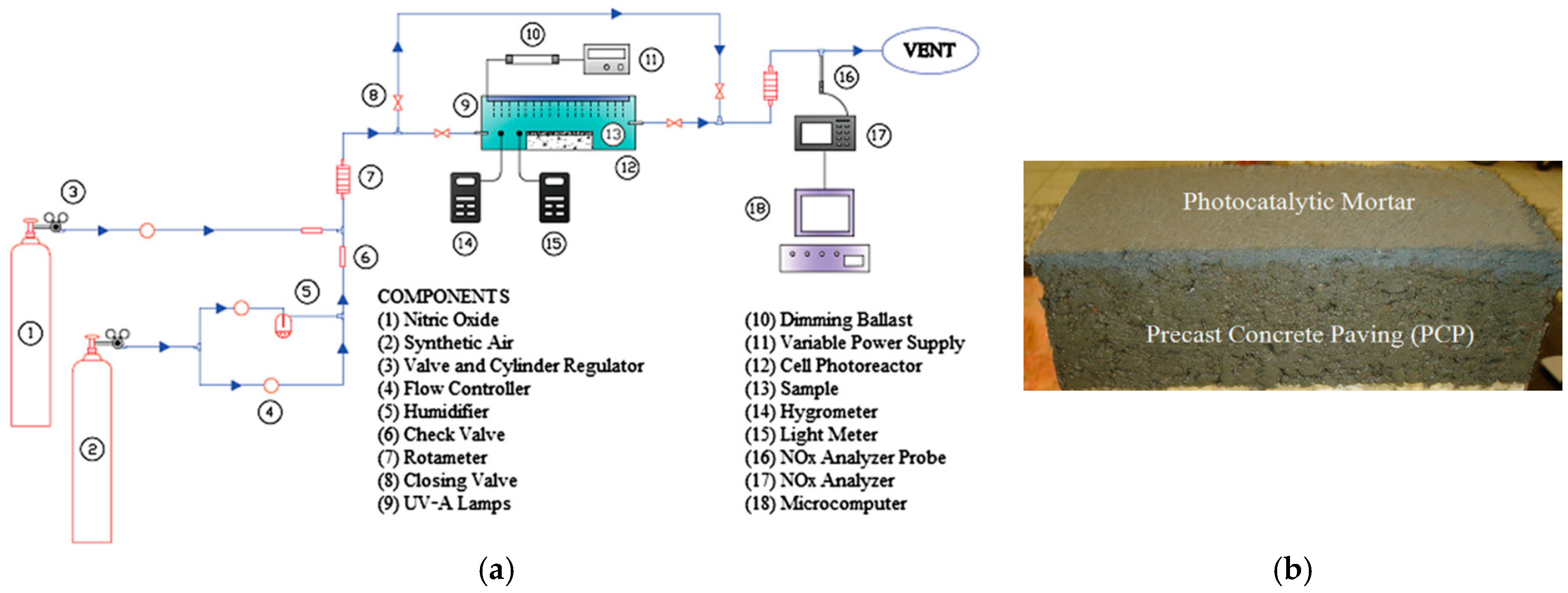{kind=link}
{kind=link}
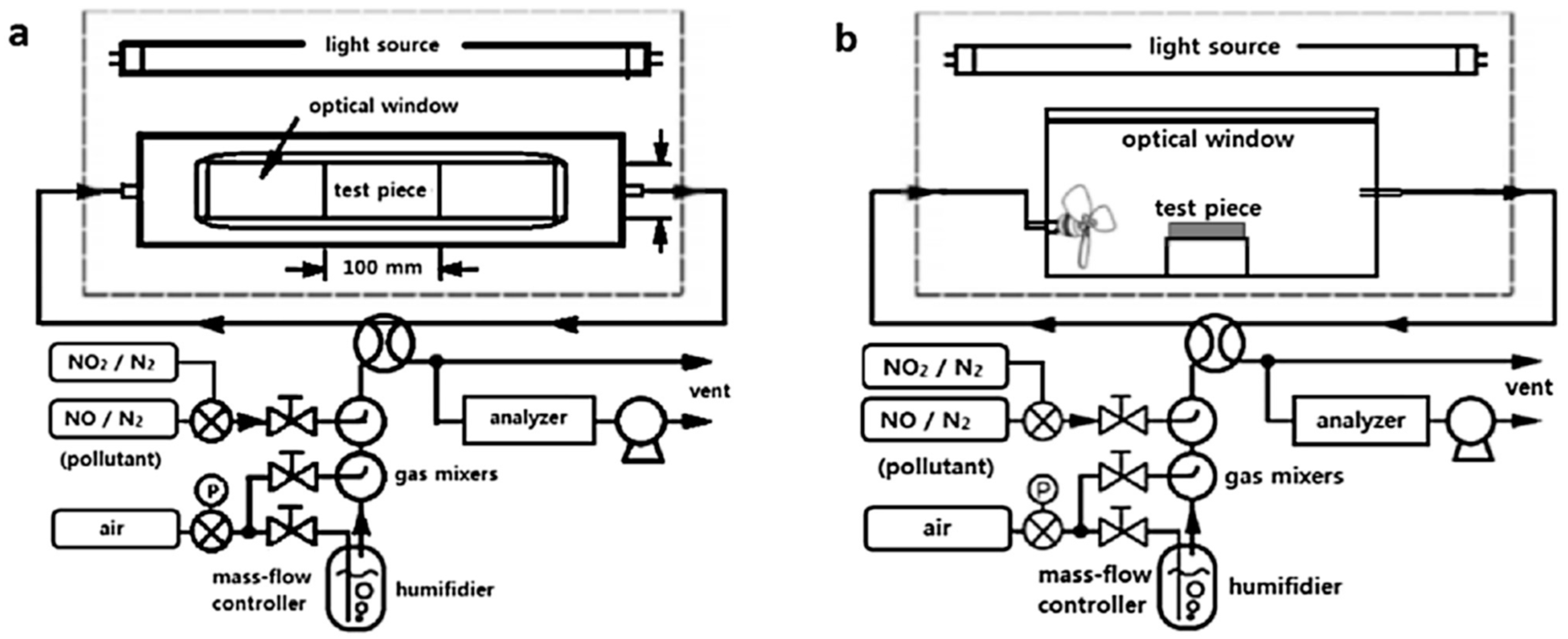{kind=link}
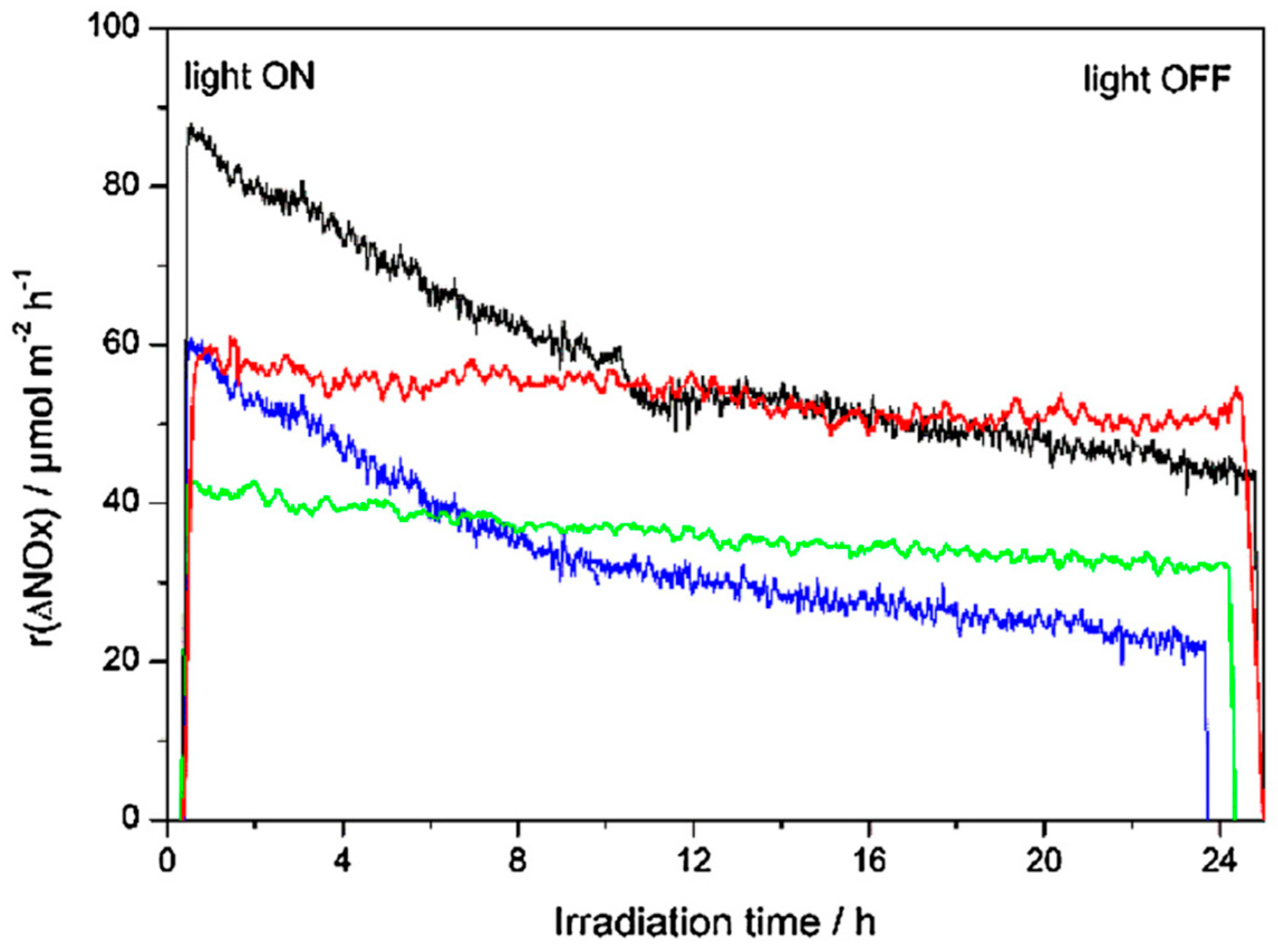{kind=link}
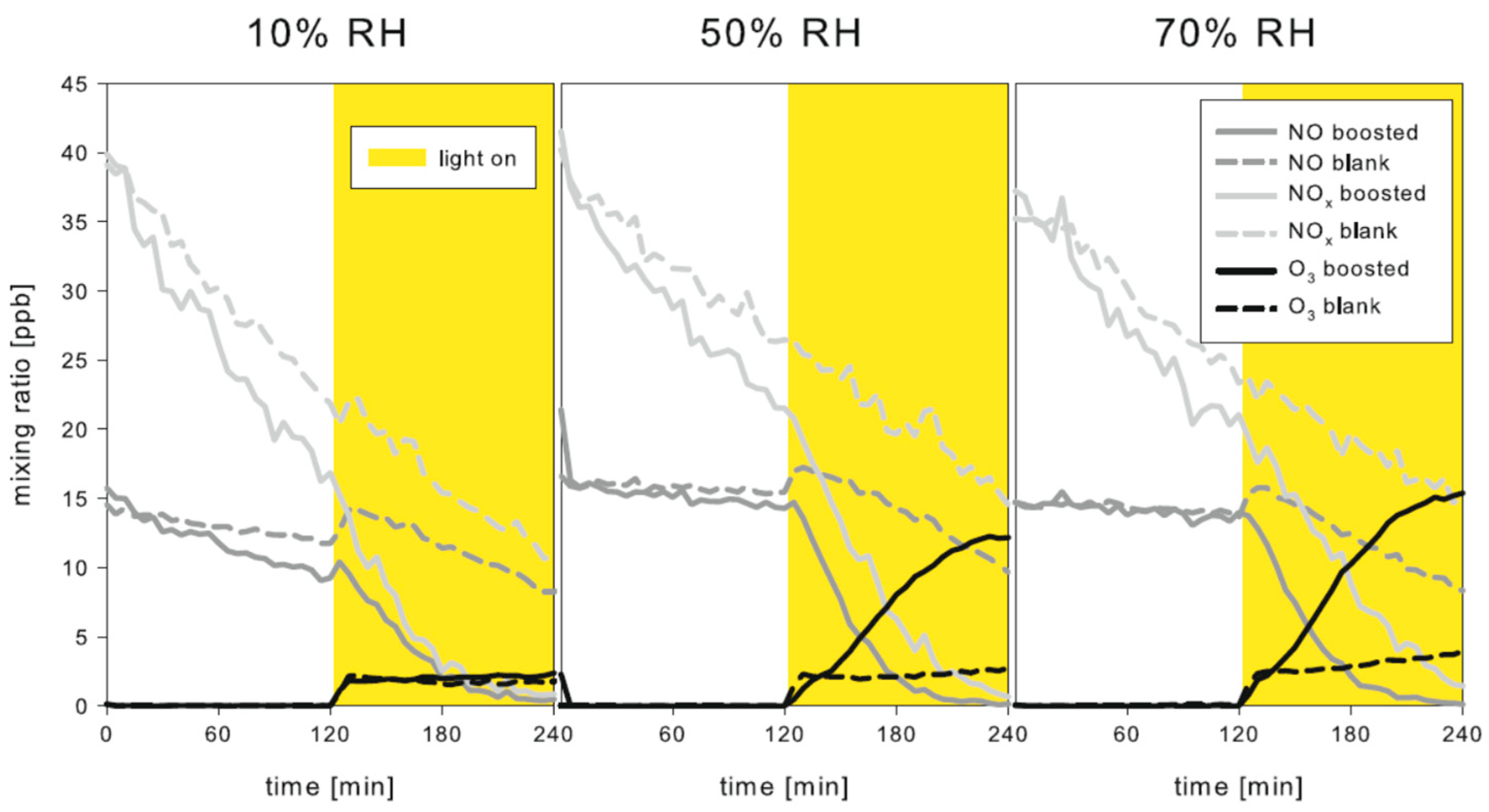{kind=link}
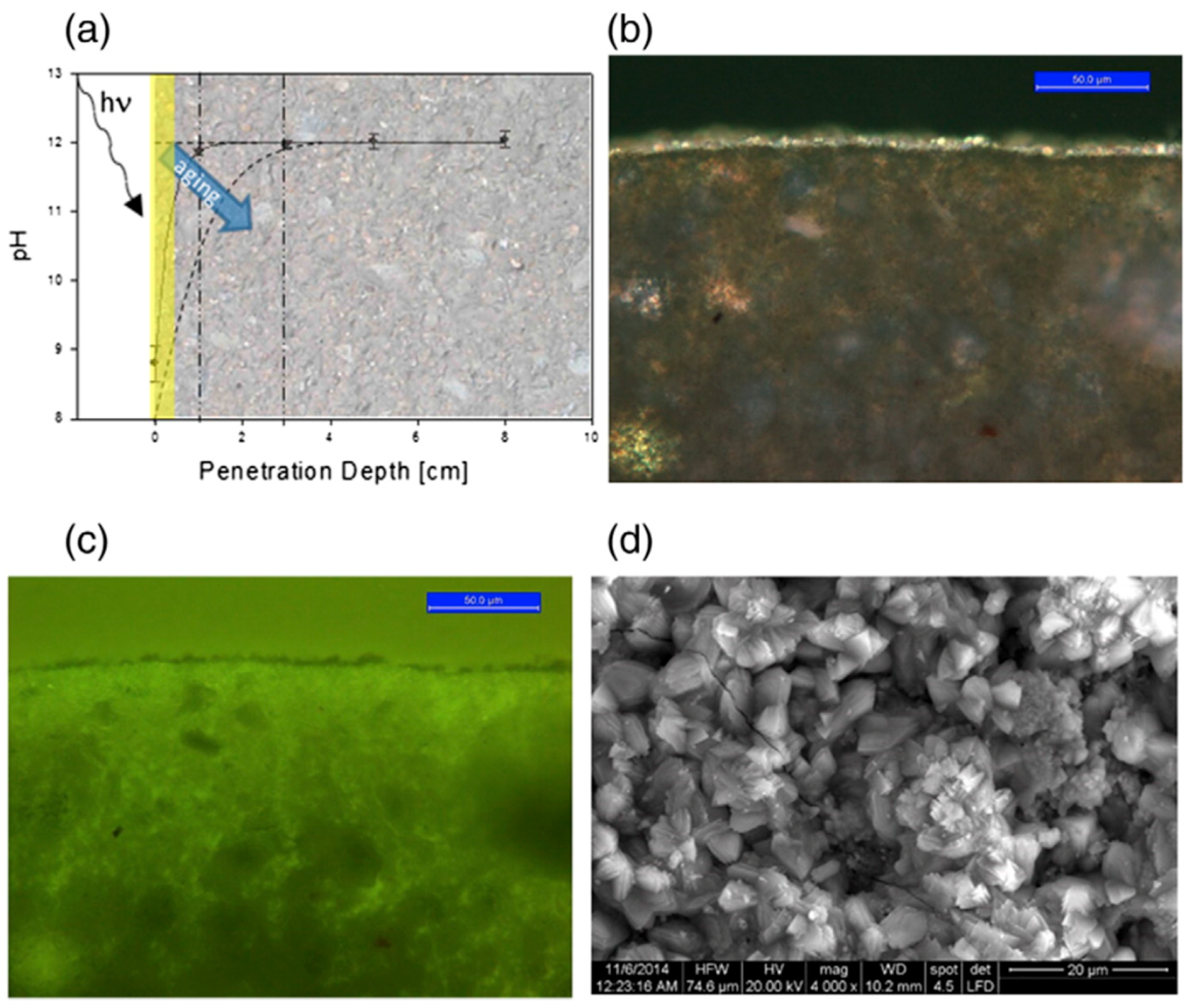{kind=link}
{kind=link}
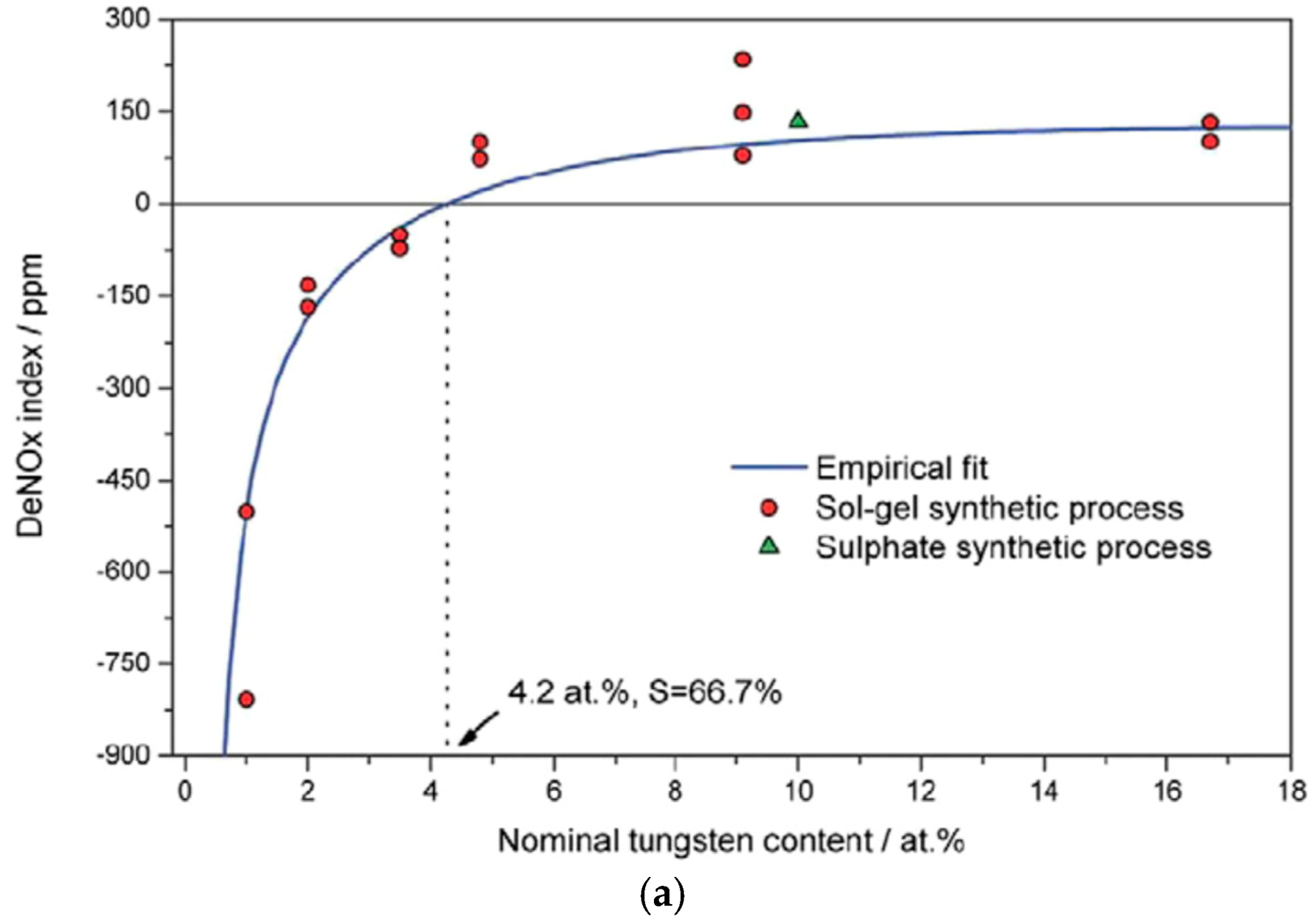{kind=link}
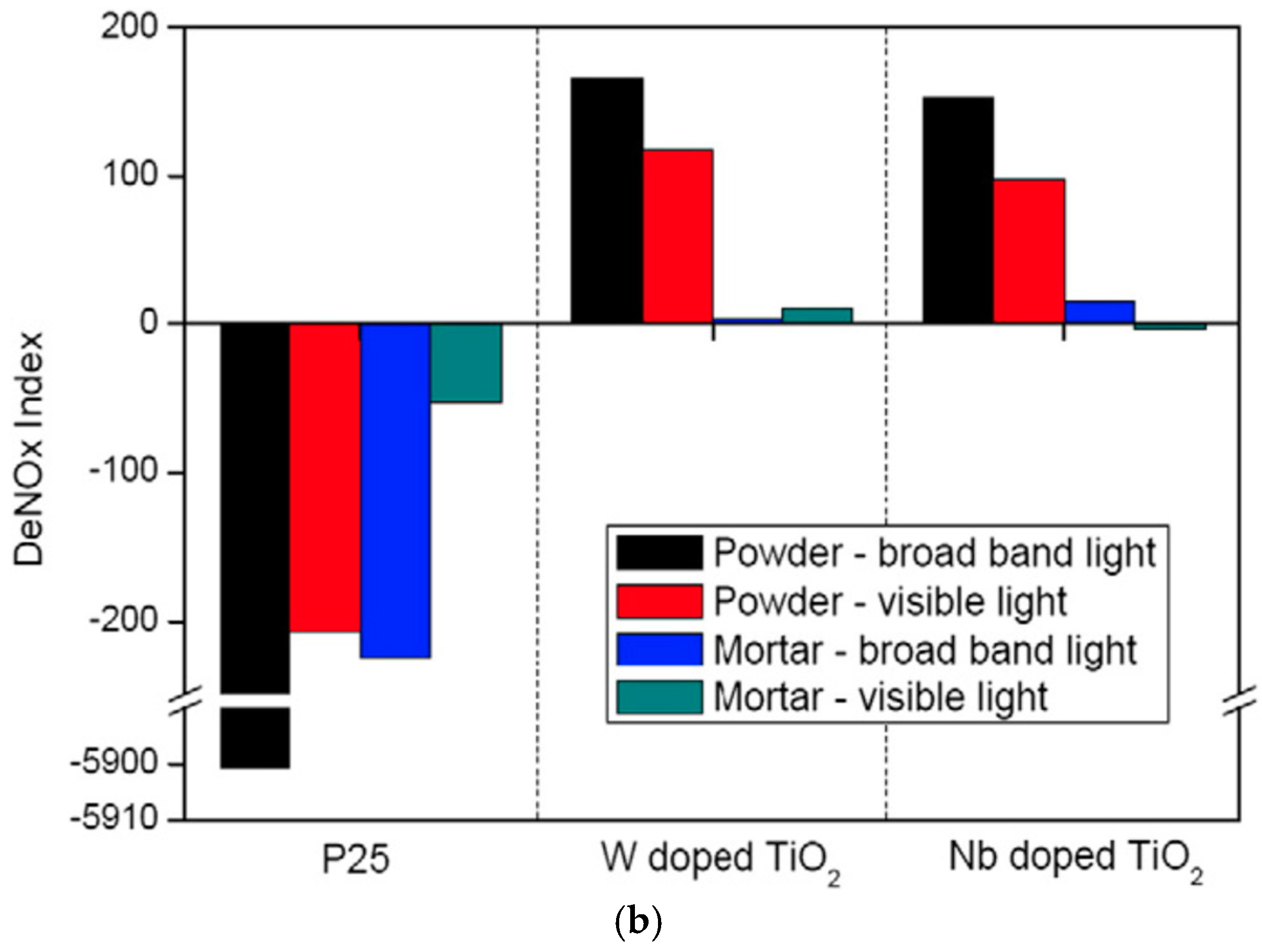{kind=link}
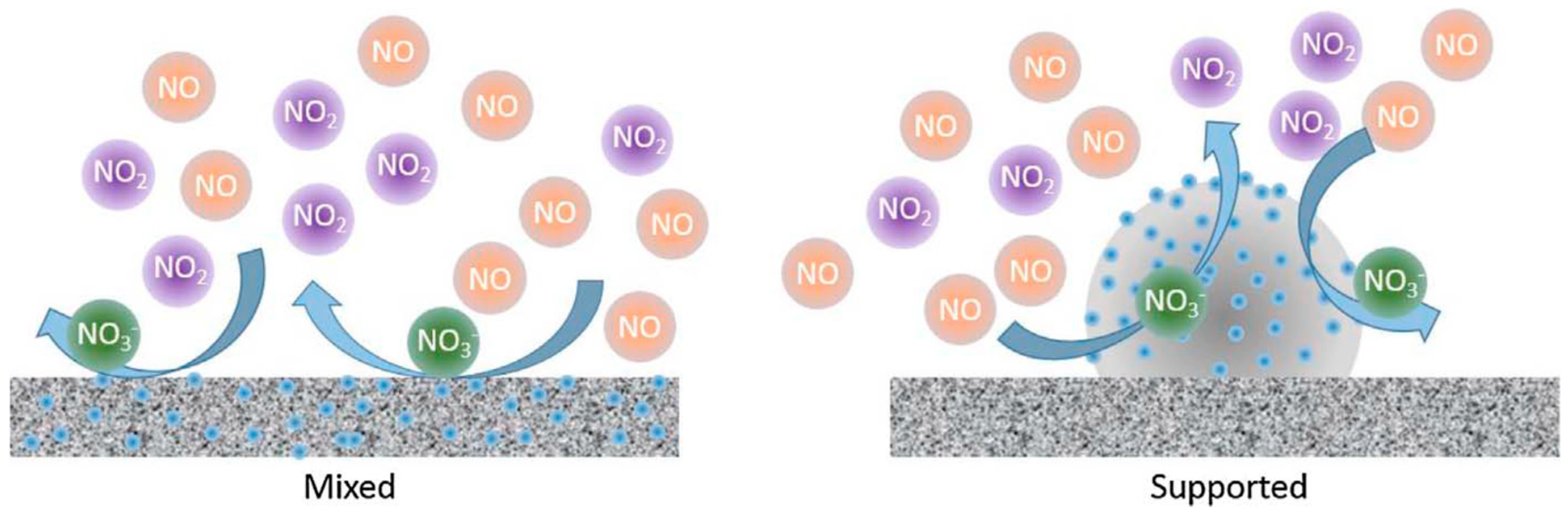{kind=link}
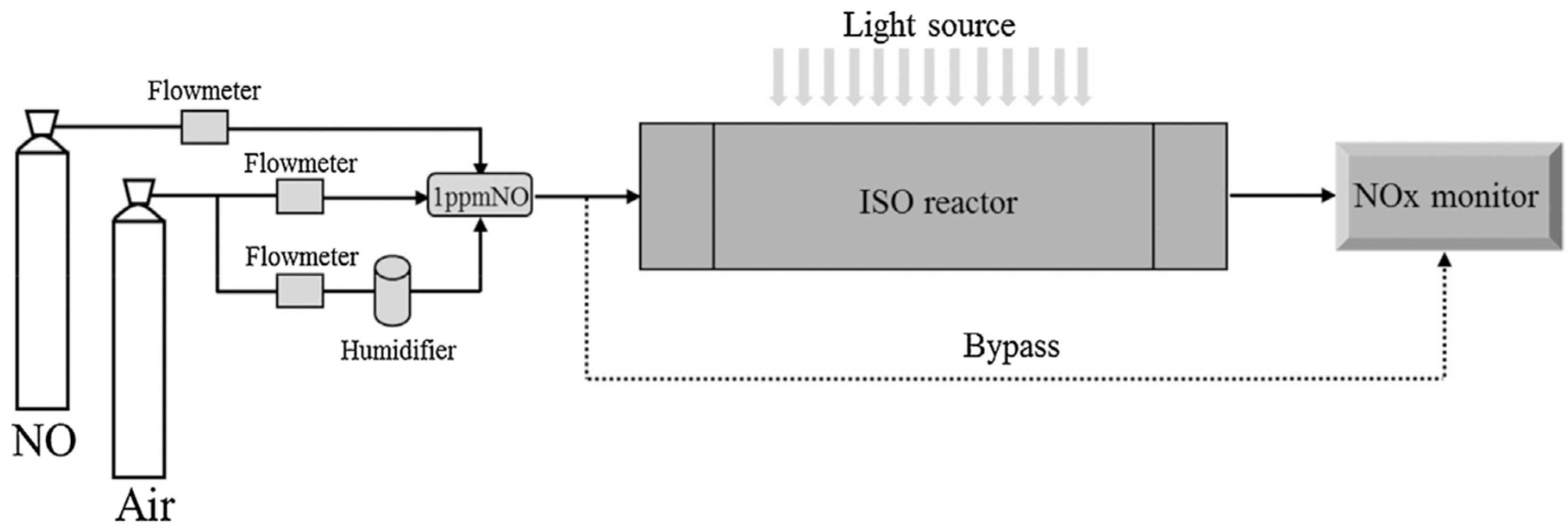{kind=link}
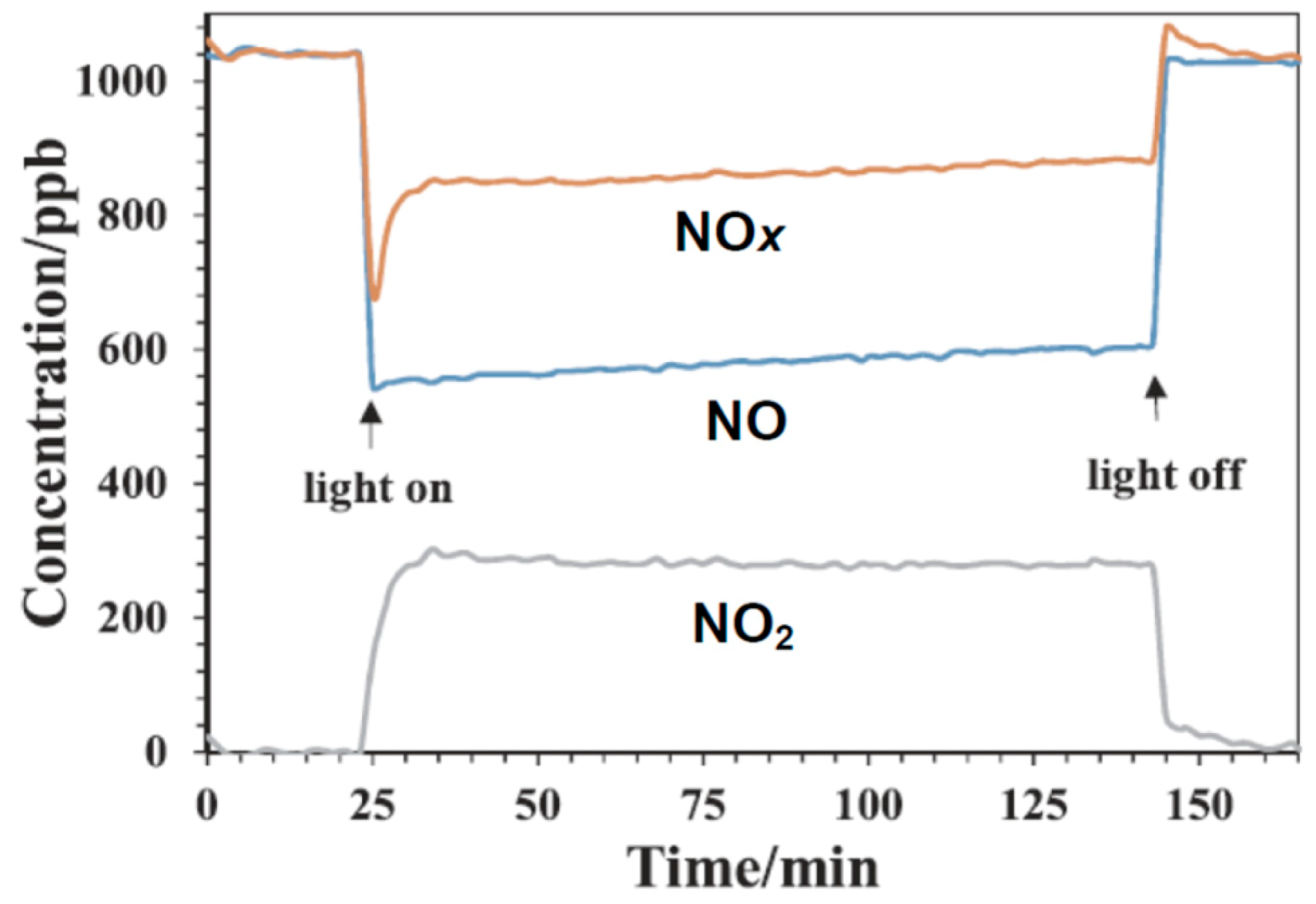{kind=link}
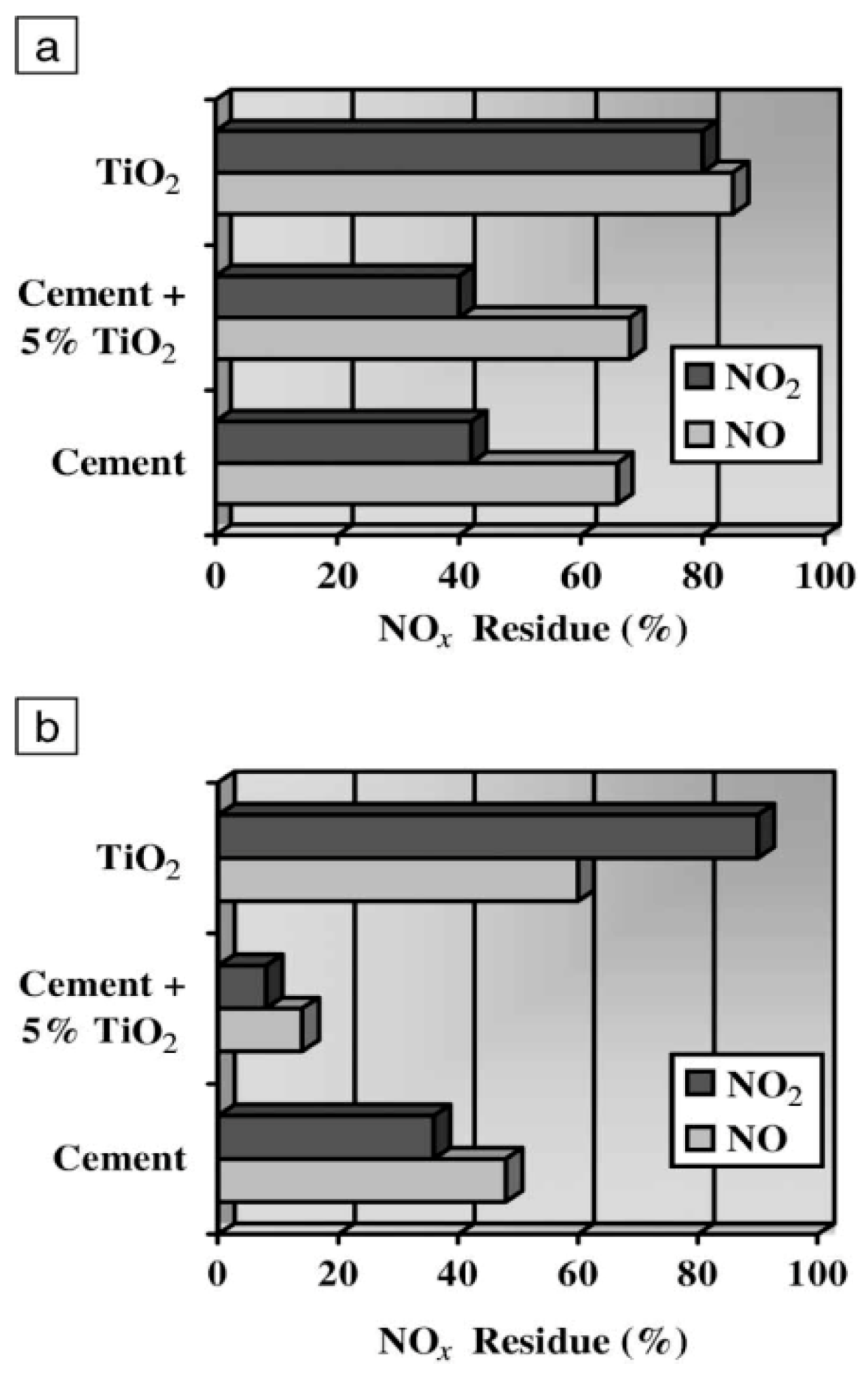{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
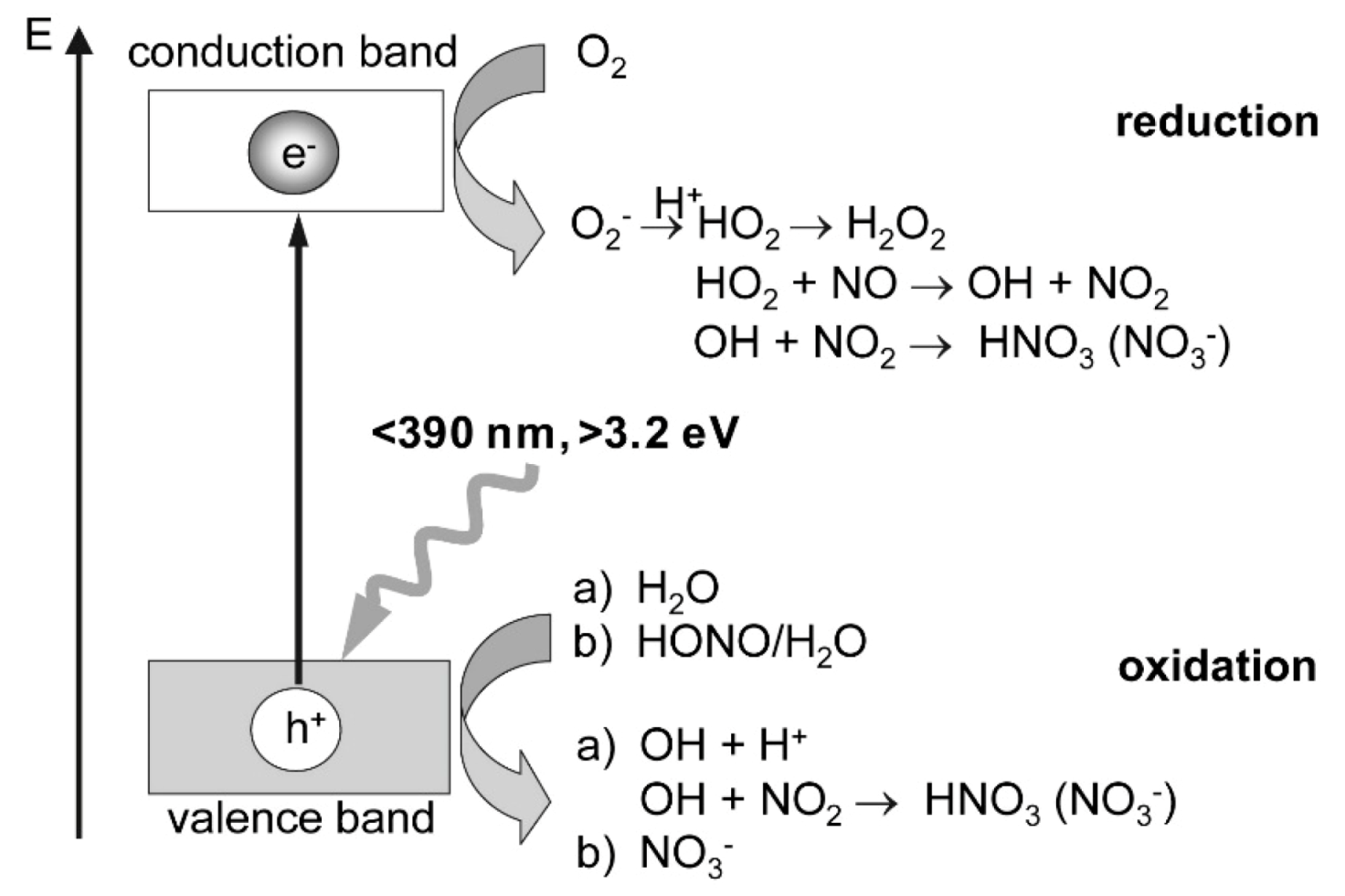{kind=link}
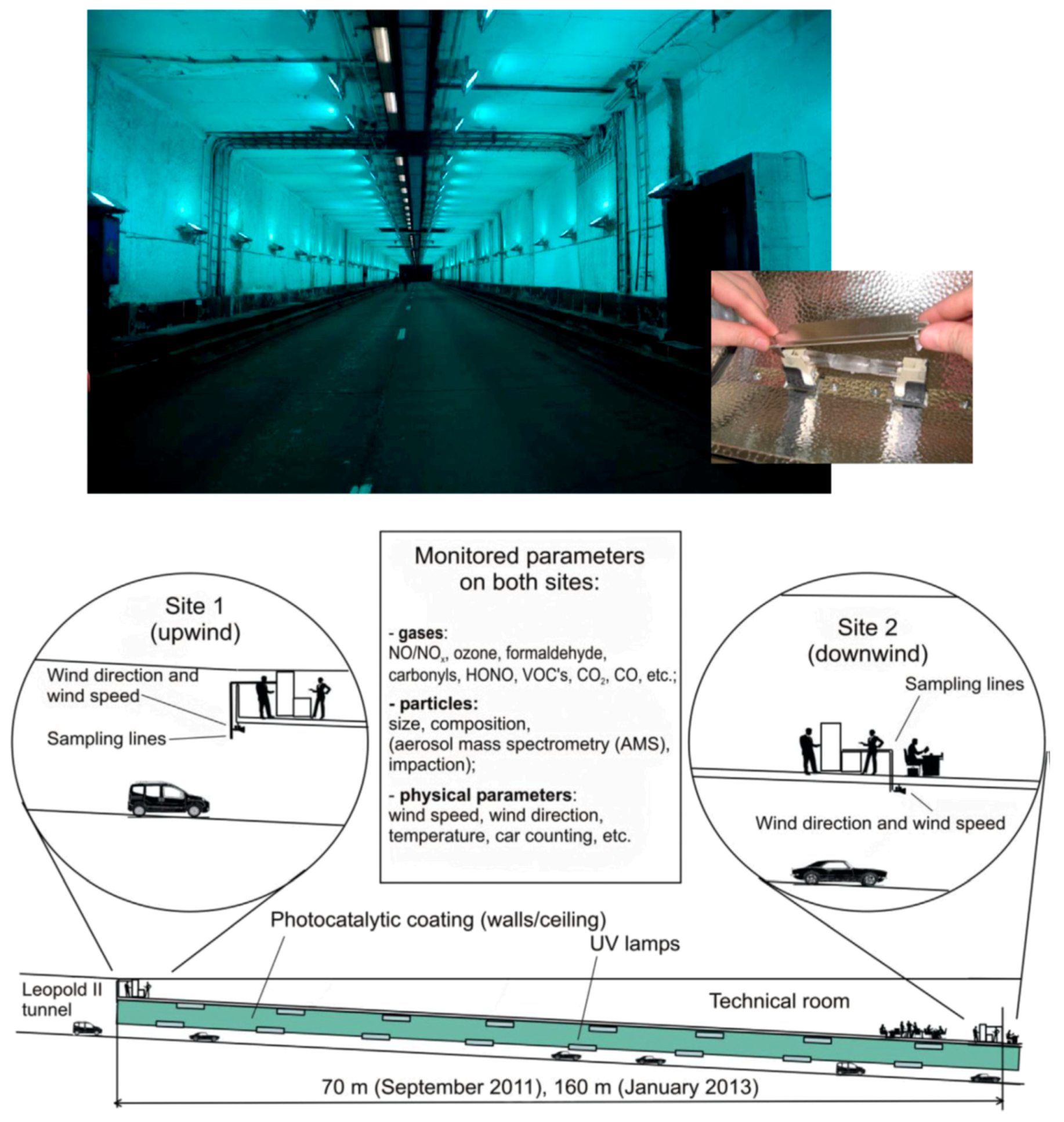{kind=link}
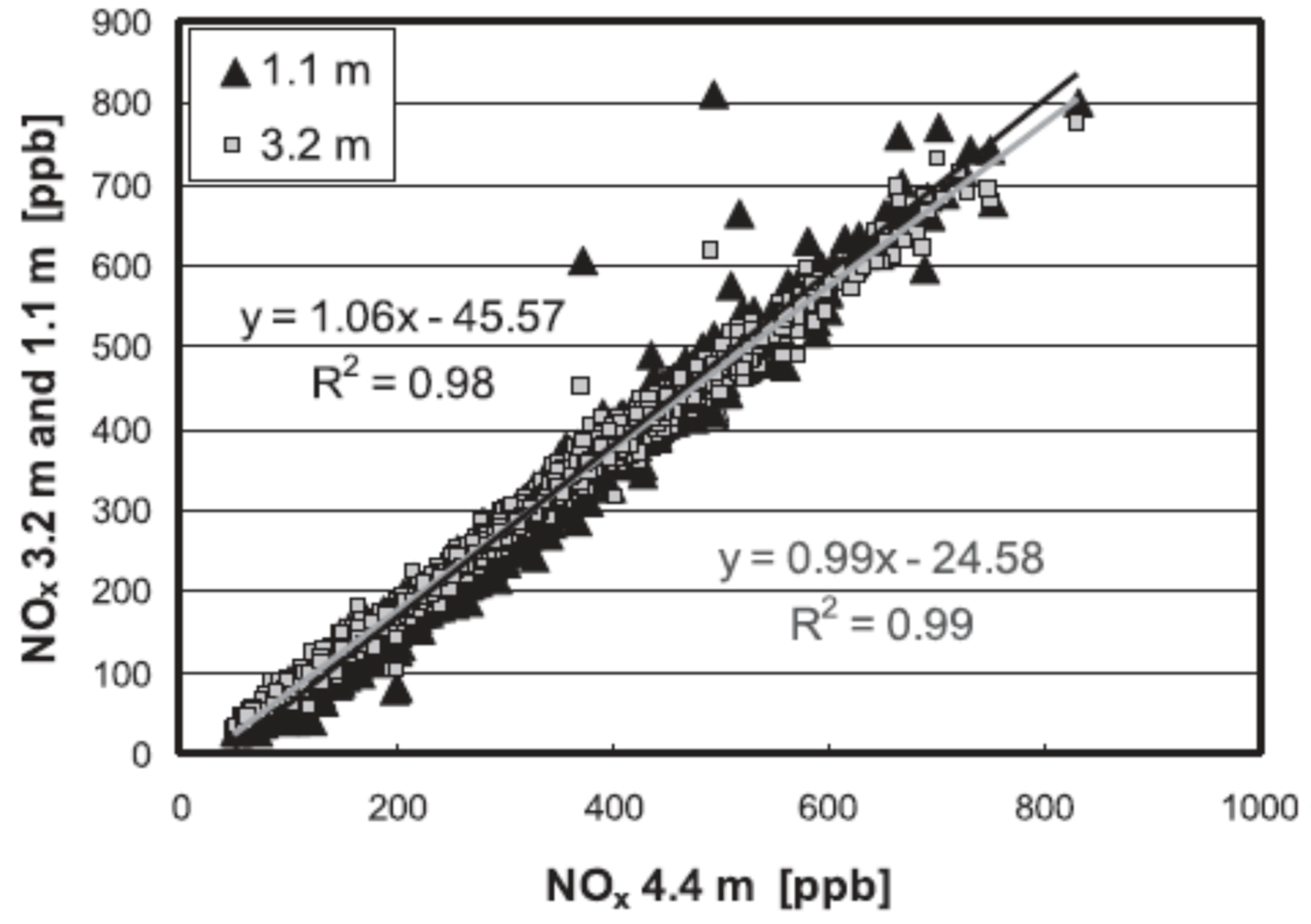{kind=link}
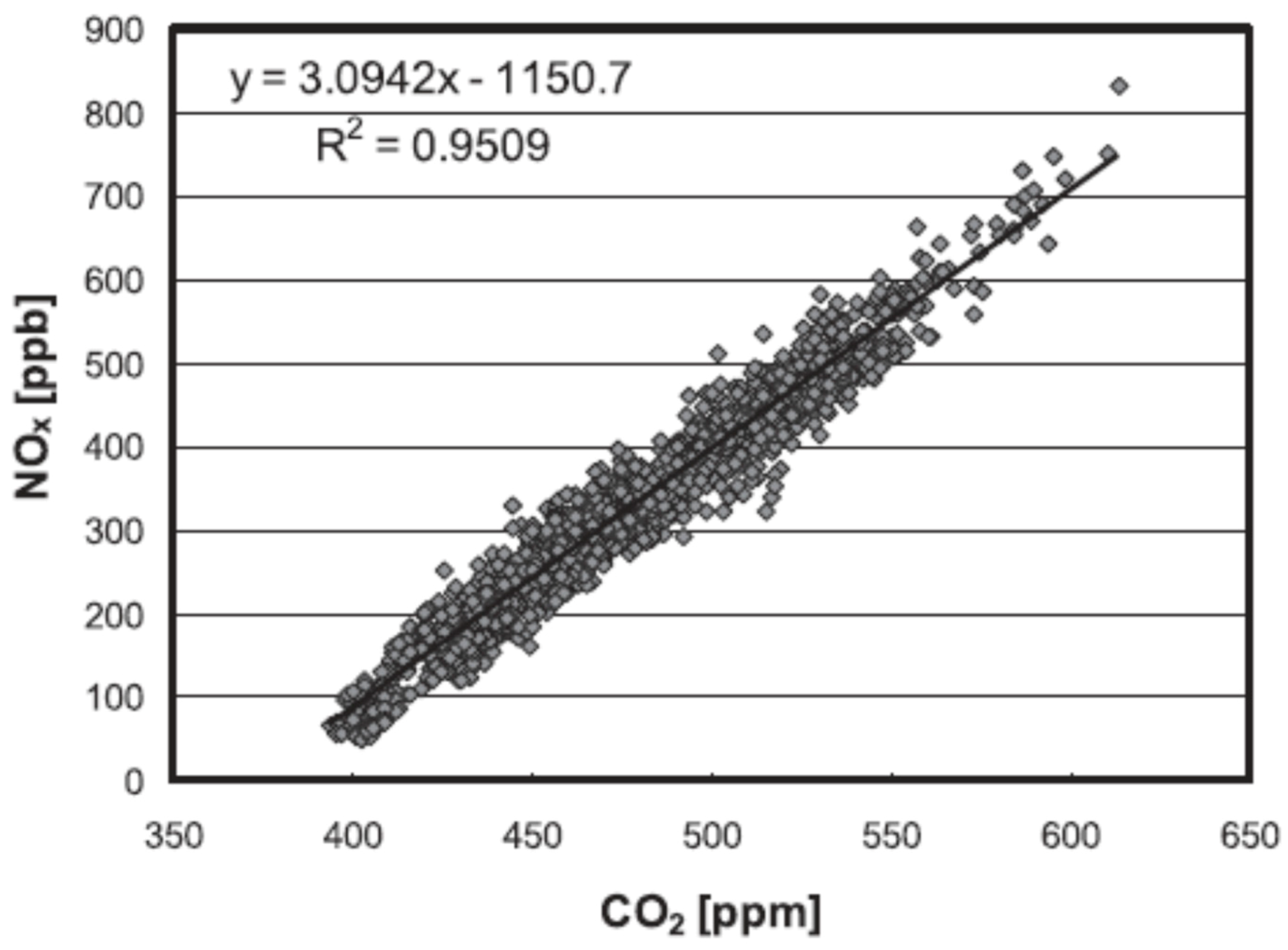{kind=link}
{kind=link}
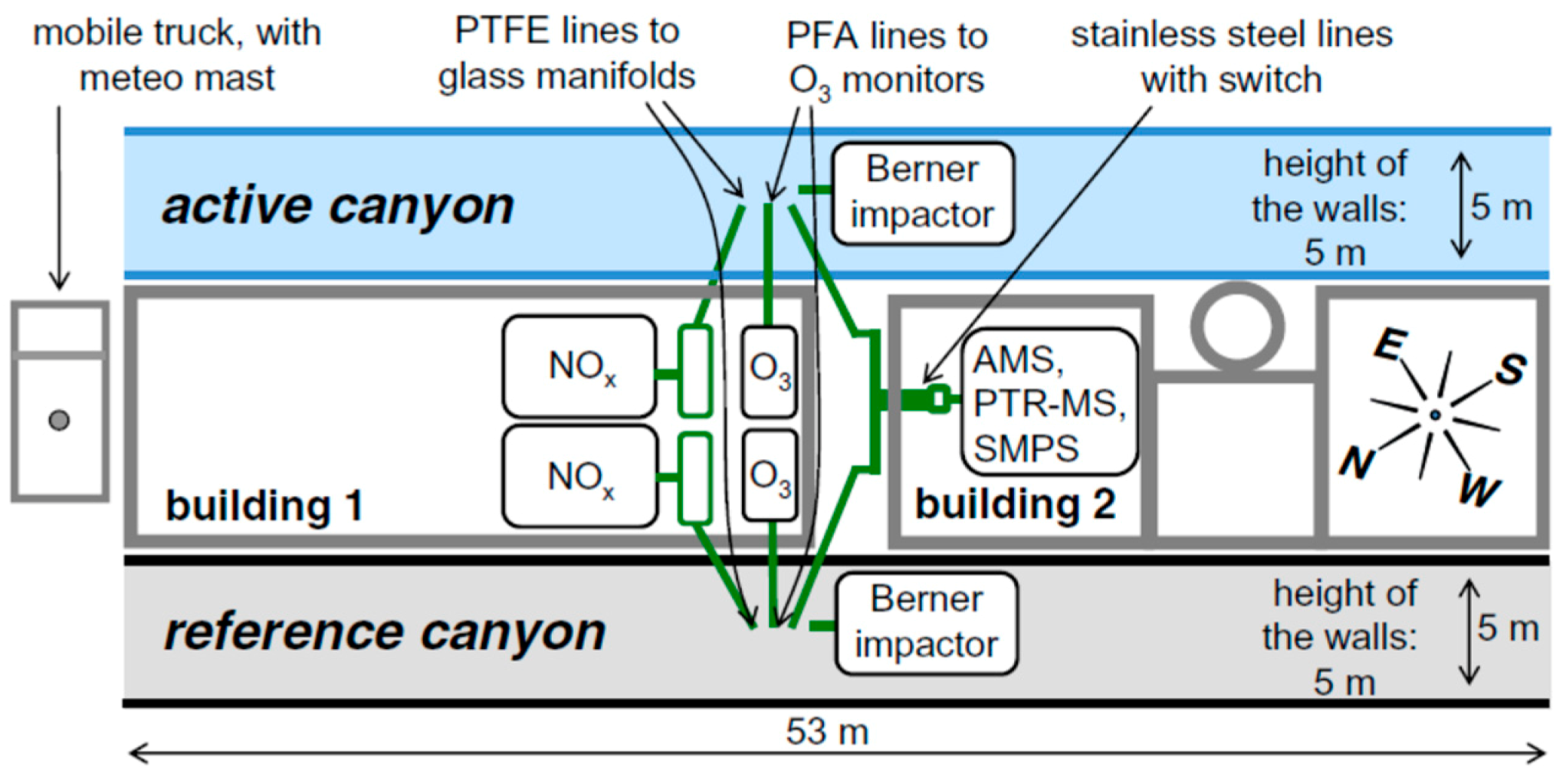{kind=link}
{kind=link}
{kind=link}
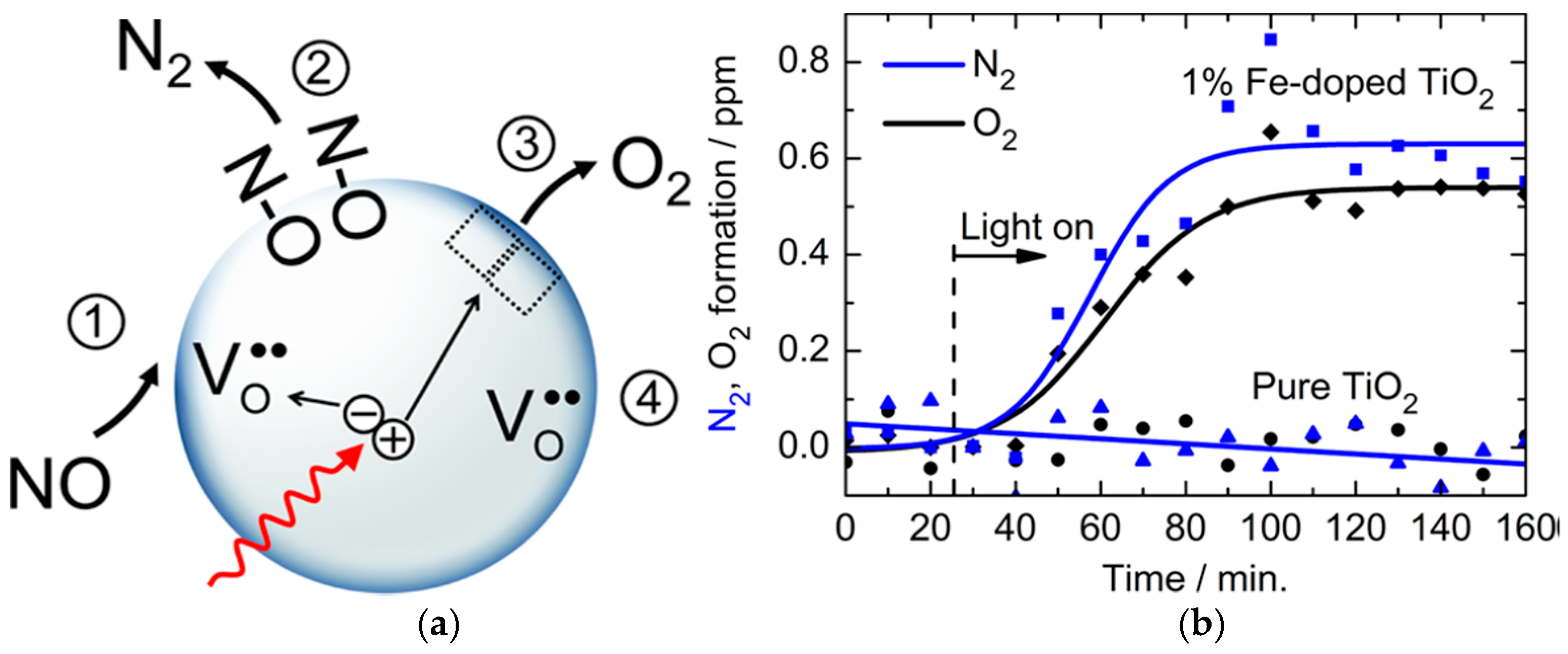{kind=link}
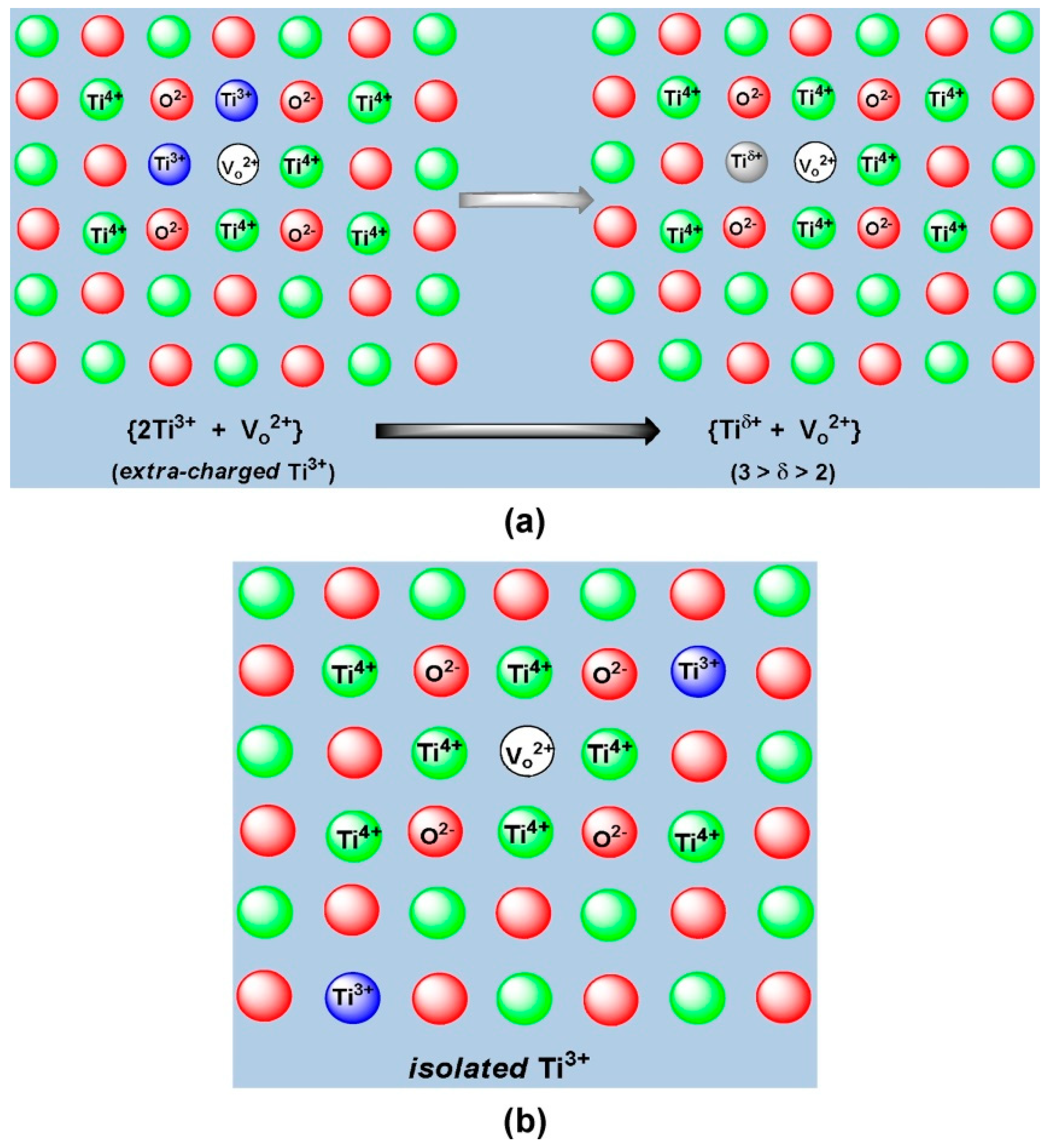{kind=link}
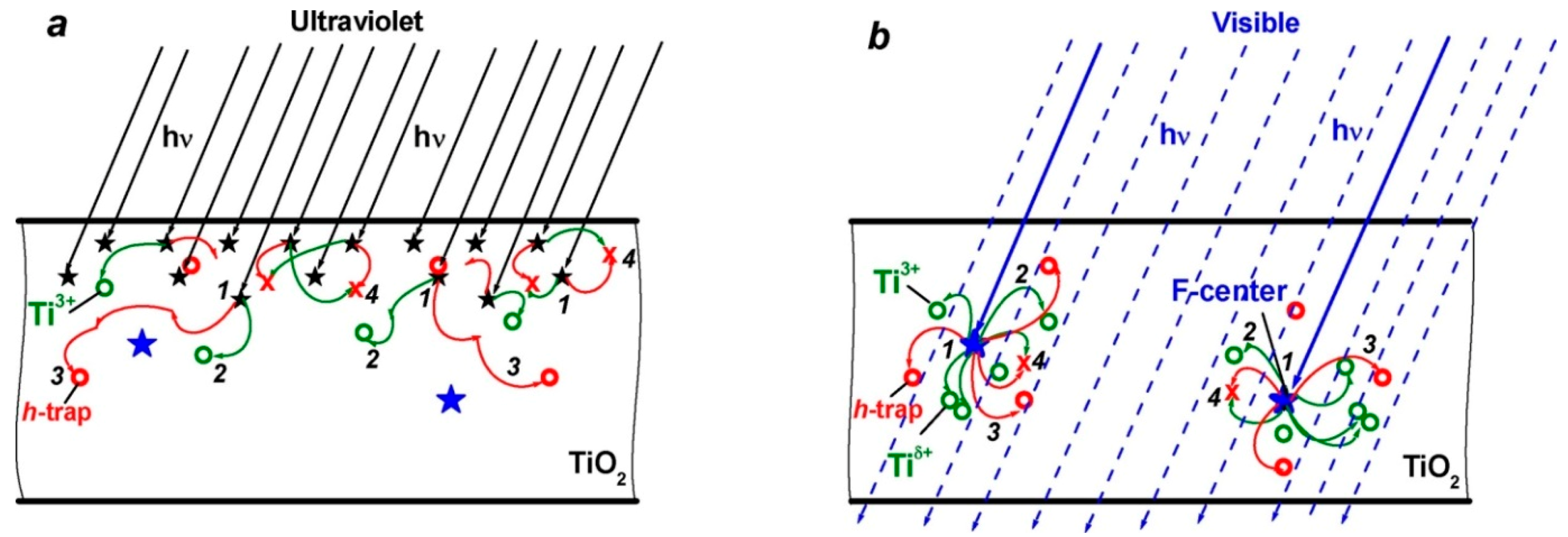{kind=link}