Synthesis and Characterization of Cationic Tetramethyl Tantalum(V) Complex

 ,
,  and
and
Abstract
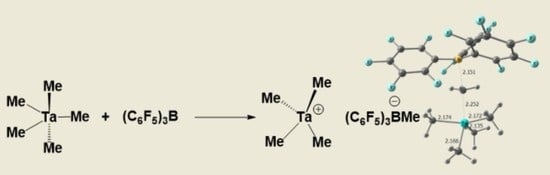
1. Introduction
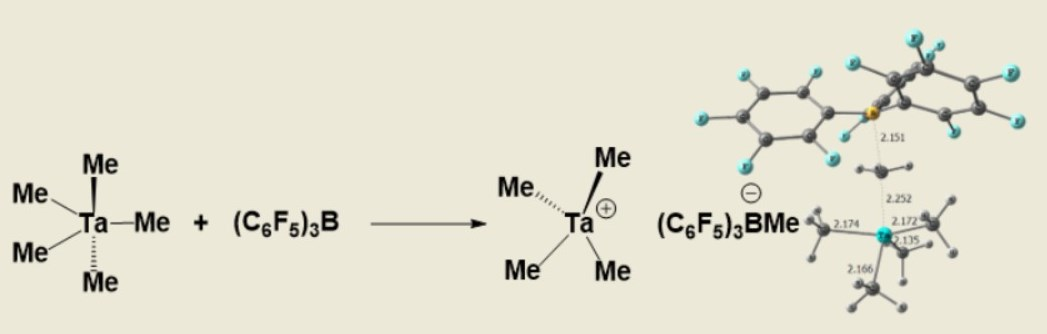
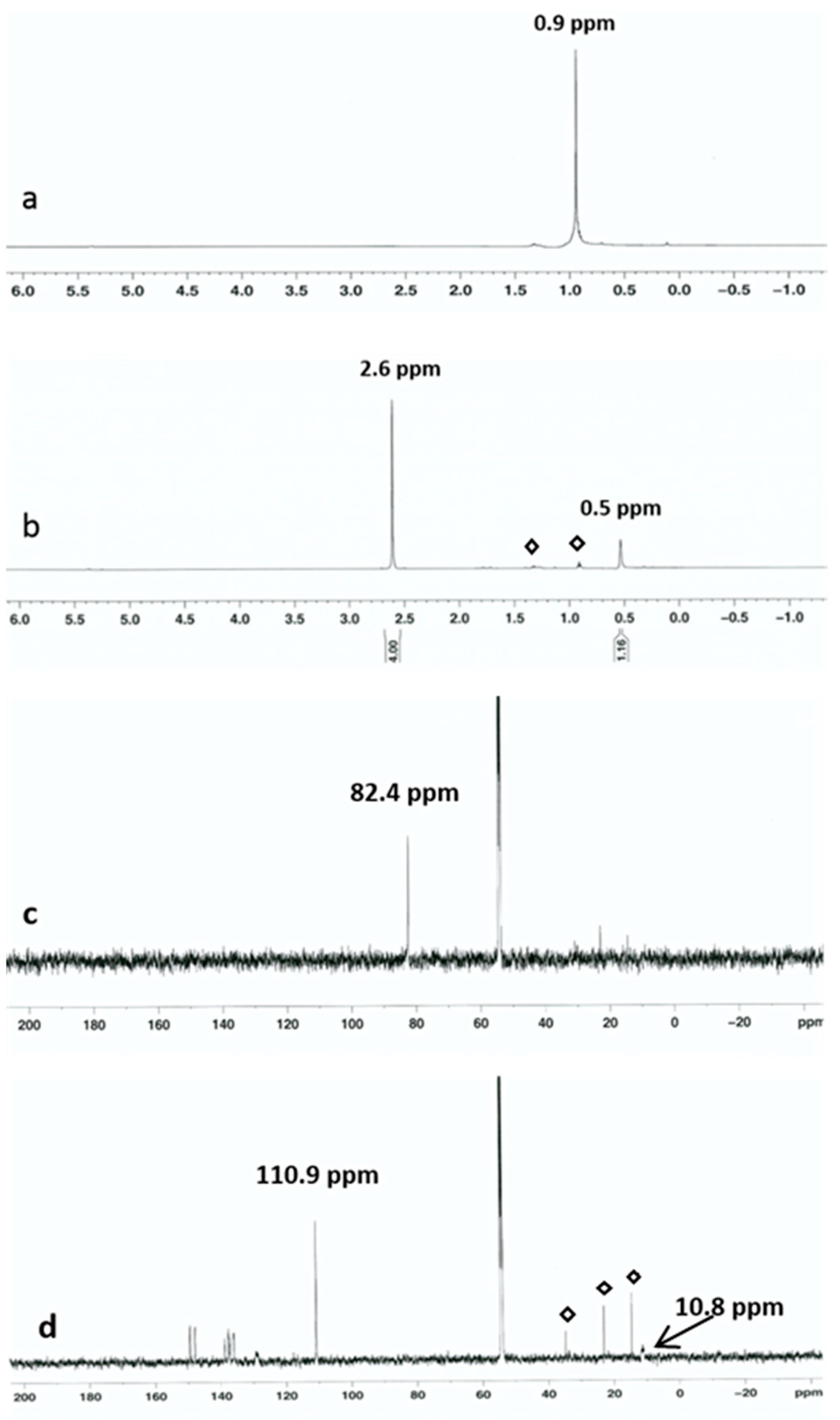
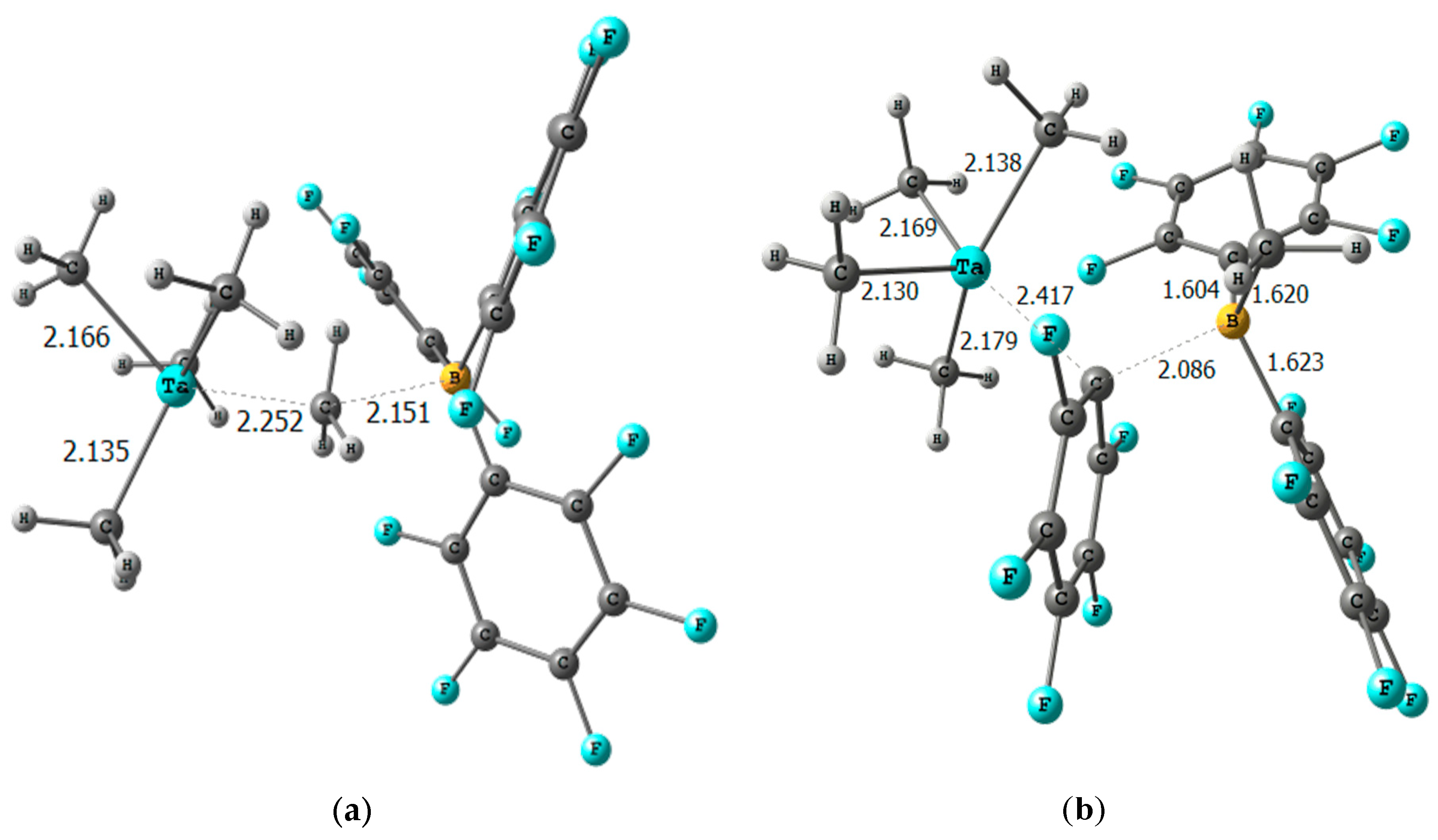
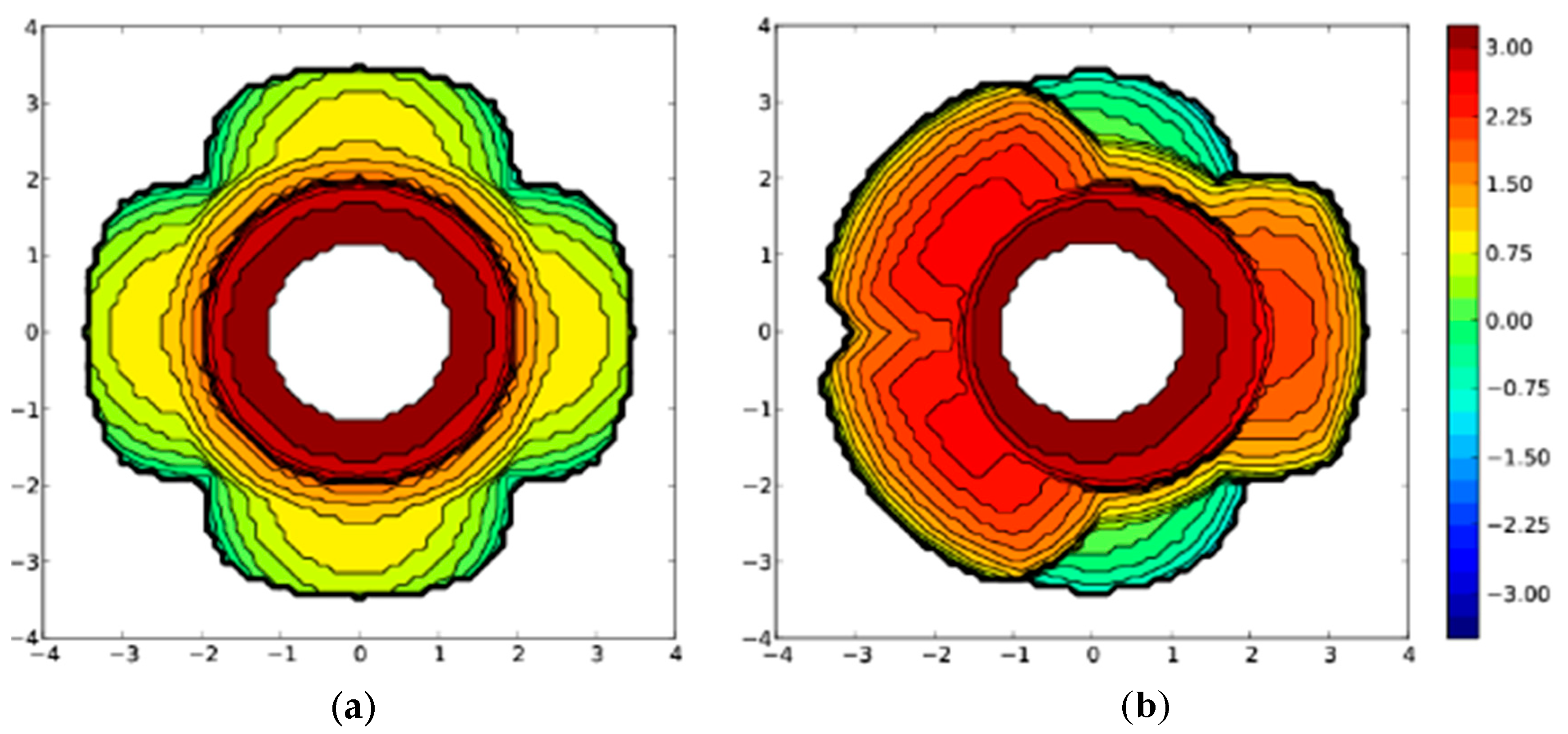
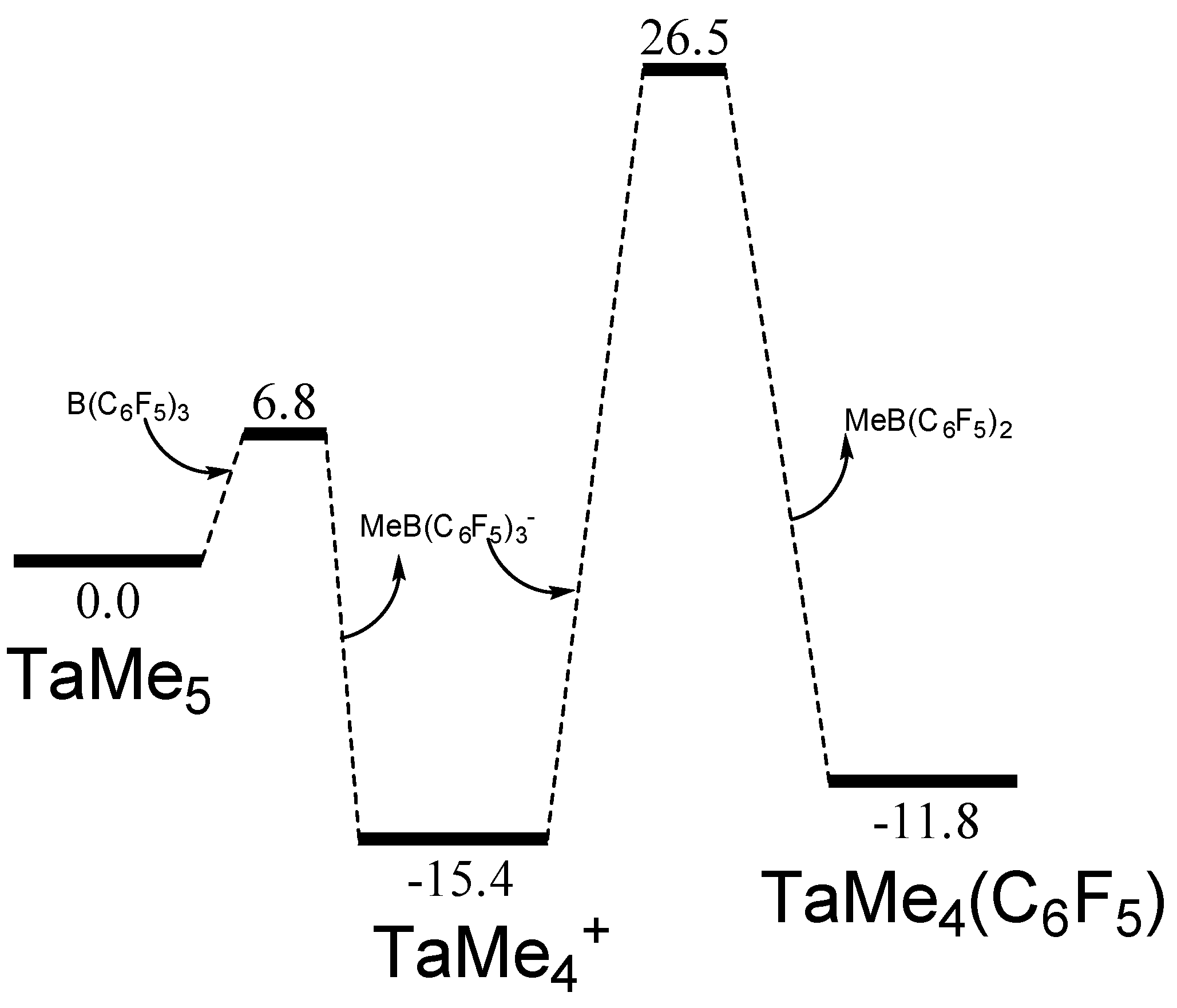
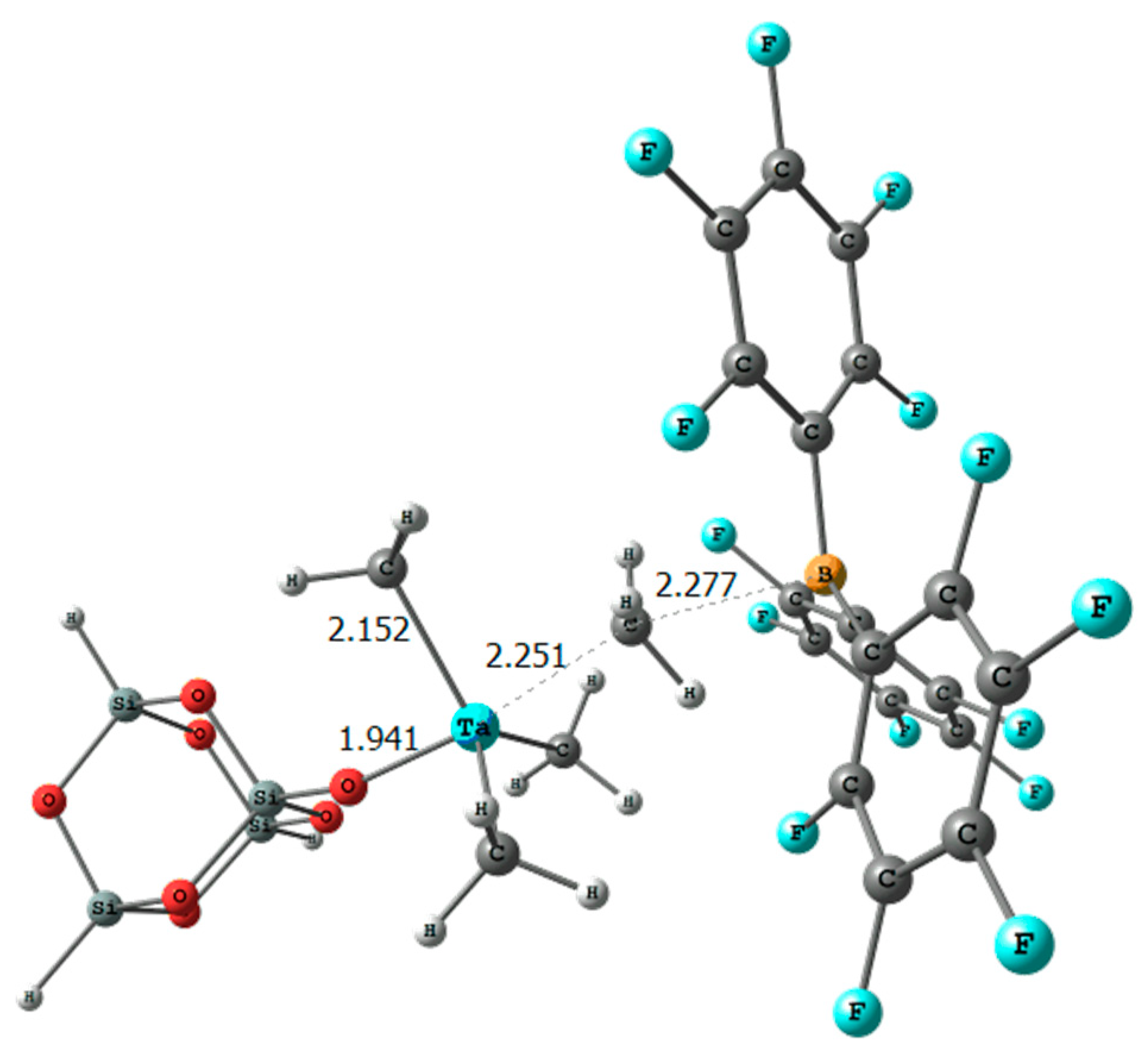
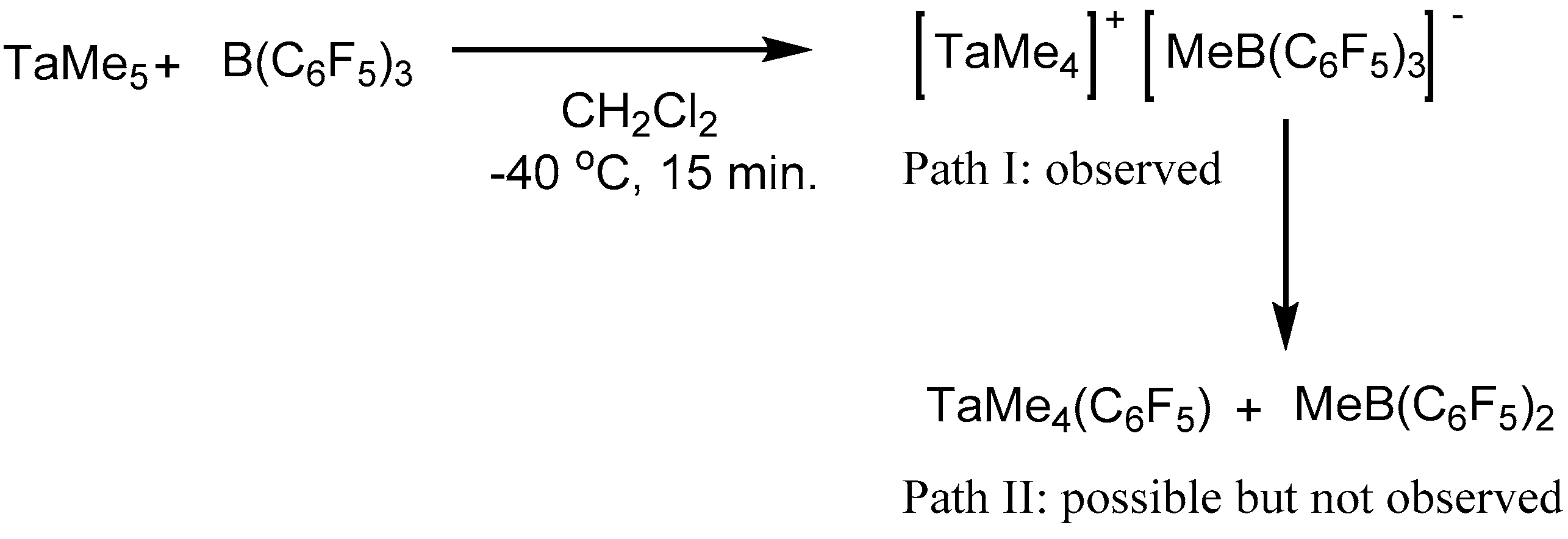
2. Results
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Schrock, R.R. Alkylidene Complexes of Niobium and Tantalum. Acc. Chem. Res. 1979, 12, 98–104. [Google Scholar] [CrossRef]
- Schrock, R.R. First Isolable Transition Metal Methylene Complex and Analogs. Characterization, Mode of Decomposition, and some Simple Reactions. J. Am. Chem. Soc. 1975, 97, 6577–6578. [Google Scholar] [CrossRef]
- Roux, E.L.; Chabanas, M.; Baudouin, A.; de-Mallmann, A.; Copéret, C.; Quadrelli, E.A.; Thivolle-Cazat, J.; Basset, J.-M.; Lukens, W.; Lesage, A.; et al. Detailed Structural Investigation of the Grafting of [Ta(=CHtBu)(CH2tBu)3] and [Cp*TaMe4] on Silica Partially Dehydroxylated at 700 °C and the Activity of the Grafted Complexes toward Alkane Metathesis. J. Am. Chem. Soc. 2004, 126, 13391–13399. [Google Scholar] [CrossRef] [PubMed]
- Freundlich, J.S.; Schrock, R.R.; Davis, W.M. Alkyl and Alkylidene Complexes of Tantalum That Contain a Triethylsilyl-Substituted Triamido–Amine Ligand. Organometallics 1996, 15, 2777–2783. [Google Scholar] [CrossRef]
- Schrock, R.R.; Meakin, P. Pentamethyl Complexes of Niobium and Tantalum. J. Am. Chem. Soc. 1974, 96, 5288–5290. [Google Scholar] [CrossRef]
- Schrock, R.R. Preparation and Characterization of M(CH3)5 (M = Nb or Ta) and Ta(CH2C6H5)5 and Evidence for Decomposition by α-Hydrogen Atom Abstraction. J. Organomet. Chem. 1976, 122, 209–225. [Google Scholar] [CrossRef]
- Albright, T.A.; Tang, H. The Structure of Pentamethyltantalum. Angew. Chem. Int. Ed. 1992, 31, 1462–1464. [Google Scholar] [CrossRef]
- Pulham, C.; Haaland, A.; Hammel, A.; Rypdal, K.; Verne, H.P.; Volden, H.V. Perfluorotriethylamine: An Amine with Unusual Structure and Reactivity. Angew. Chem. Int. Ed. 1992, 31, 1464–1467. [Google Scholar]
- Chen, Y.; Abou-hamad, E.; Hamieh, A.; Hamzaoui, B.; Emsley, L.; Basset, J.-M. Alkane Metathesis with the Tantalum Methylidene [(≡SiO)Ta(=CH2)Me2]/[(≡SiO)2Ta(=CH2)Me] Generated from Well-Defined Surface Organometallic Complex [(≡SiO)TaVMe4]. J. Am. Chem. Soc. 2015, 137, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ould-Chikh, S.; Abou-Hamad, E.; Callens, E.; Mohandas, J.C.; Khalid, S.; Basset, J.-M. Facile and Efficient Synthesis of the Surface Tantalum Hydride (≡SiO)2TaIIIH and Tris-Siloxy Tantalum (≡SiO)3TaIII Starting from Novel Tantalum Surface Species (≡SiO)TaMe4 and (≡SiO)2TaMe3. Organometallics 2014, 33, 1205–1211. [Google Scholar] [CrossRef]
- Schowner, R.; Frey, W.; Buchmeiser, M.R. Cationic Tungsten-Oxo-Alkylidene-N-Heterocyclic Carbene Complexes: Highly Active Olefin Metathesis Catalysts. J. Am. Chem. Soc. 2015, 137, 6188–6191. [Google Scholar] [CrossRef] [PubMed]
- Pucino, M.; Mougel, V.; Schowner, R.; Fedorov, A.; Buchmeiser, M.R.; Copéret, C. Cationic Silica-Supported N-Heterocyclic Carbene Tungsten Oxo Alkylidene Sites: Highly Active and Stable Catalysts for Olefin Metathesis. Angew. Chem. Int. Ed. 2016, 55, 4300–4302. [Google Scholar] [CrossRef] [PubMed]
- Dey, R.; Samantaray, M.K.; Poater, A.; Hamieh, A.; Kavitake, S.; Abou-Hamad, E.; Callens, E.; Emwas, A.-H.; Cavallo, L.; Basset, J.-M. Synthesis and Characterization of a Homogeneous and Silica Supported Homoleptic Cationic Tungsten(VI) Methyl Complex: Application in Olefin Metathesis. Chem. Commun. 2016, 52, 11270–11273. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Nieves, J.; Royo, P.; Mosquera, M.E.G. Synthesis of the Cation Complex [TaCp*Me3]+ and a Comparison of Its Reactivity with That of [TaCp*Me4]. Organometallics 2006, 25, 2331–2336. [Google Scholar] [CrossRef]
- Sanchez-Nieves, J.; Royo, P. Synthesis of Neutral and Cationic Monocyclopentadienyl Tantalum Alkoxo Complexes and Polymerization of Methyl Methacrylate. Organometallics 2007, 26, 2880–2884. [Google Scholar] [CrossRef]
- Mariott, W.R.; Gustafson, L.O.; Chen, E.Y.-X. Activation of Tantalocene(V) Alkyl and Alkylidene Complexes with Strong Organo Lewis Acids and Application to Polymerization Catalysis. Organometallics 2006, 25, 3721–3729. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Johnson, S.A.; Rettig, S.J. Synthesis and Structure of the Tantalum Trimethyl Complex [P2N2]TaMe3 and Its Conversion to the Tantalum Methylidene Species [P2N2]TaCH2(Me) ([P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh). Organometallics 1999, 18, 4059–4067. [Google Scholar] [CrossRef]
- Guérin, F.; Stephan, D.W. Synthesis and Structure of the Dicationic Bisborate Adduct. Angew. Chem. Int. Ed. 2000, 39, 1298–1300. [Google Scholar] [CrossRef]
- Anderson, L.L.; Schmidt, J.A.R.; Arnold, J.; Bergman, R.G. Neutral and Cationic Alkyl Tantalum Imido Complexes: Synthesis and Migratory Insertion Reactions. Organometallics 2006, 25, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.L.; Arnold, J.; Bergman, R.G. Catalytic Hydroamination of Alkynes and Norbornene with Neutral and Cationic Tantalum Imido Complexes. Org. Lett. 2004, 6, 2519–2522. [Google Scholar] [CrossRef] [PubMed]
- Tomson, N.C.; Arnold, J.; Bergman, R.G. Synthesis and Reactivity of Cationic Niobium and Tantalum Methyl Complexes Supported by Imido and β-diketiminato Ligands. Dalton Trans. 2011, 40, 7718–7729. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.G.; Salata, M.R.; Marks, T.J. B(C6F5)3- vs. Al(C6F5)3-Derived Metallocenium Ion Pairs. Structural, Thermochemical, and Structural Dynamic Divergences. J. Am. Chem. Soc. 2005, 127, 10898–10909. [Google Scholar] [CrossRef] [PubMed]
- Mathis, D.; Couzijn, E.P.A.; Chen, P. Structure, Dynamics, and Polymerization Activity of Zirconocenium Ion Pairs Generated with Boron-C6F5 Compounds and Al2R6. Organometallics 2011, 30, 3834–3843. [Google Scholar] [CrossRef]
- Alonso-Moreno, C.; Lancaster, S.J.; Wright, J.A.; Hughes, D.L.; Zuccaccia, C.; Correa, A.; Macchioni, A.; Cavallo, L.; Bochmann, M. Ligand Mobility and Solution Structures of the Metallocenium Ion Pairs [Me2C(Cp)(fluorenyl)MCH2SiMe3+···X−] (M = Zr, Hf; X = MeB(C6F5)3, B(C6F5)4). Organometallics 2008, 27, 5474–5487. [Google Scholar] [CrossRef]
- Spitzmesser, S.K.; Gibson, V.C. Dialkylaluminium Complexes Derived from 1,8-diphenyl-3,6-dimethylcarbazole: a New Sterically Hindered Monodentate Ligand System. J. Organomet. Chem. 2003, 673, 95–101. [Google Scholar] [CrossRef]
- Gillis, D.J.; Tudoret, M.-J.; Baird, M.C. Novel Arene Complexes of Titanium(IV), Zirconium(IV), and Hafnium(IV). J. Am. Chem. Soc. 1993, 115, 2543–2545. [Google Scholar] [CrossRef]
- Preparation of [TaMe4+ B(C6F5)3Me-](2): A cold solution (−40 °C) of B(C6F5)3 (125 mg, 0.25 mmol) in dichloromethane was added drop wise to the cold (−40 °C) solution of tantalumpentamethyl (50 mg, 0.2 mmol) in dichloromethane. The mixture was stirred for another 15 minutes. Colour of the solution intensified to dark yellowish which indicated the formation of the ionic complex. 1H-NMR(600 MHz) δ (ppm) 0.5(s, 3H, BCH3), 2.6(s, 12H, TaCH3). 13C-NMR(150 MHz) δ (ppm) 10.8(s, 1C, BCH3), 110.9 (s, 4C, TaCH3). (Caution! This 8e- compound is highly unstable and decomposes into black tantalum power while drying.).
- All the DFT calculations were performed with the Gaussian09, using the PBE0 functional, corrected by the D3 Grimme pairwise scheme, using the SVP basis set except for the metals the standard SDD. Single-point energy calculations in solution (SMD) were performed with the M06 functional and the Def2TZVPP basis set for main group atoms and again the same SDD pseudopotential for the metals.
- Martin, R.L.; Hay, P.J.; Pratt, L.R. Hydrolysis of Ferric Ion in Water and Conformational Equilibrium. J. Phys. Chem. A 1998, 102, 3565–3573. [Google Scholar] [CrossRef]
- Poater, A.; Pump, E.; Vummaleti, S.V.C.; Cavallo, L. The Right Computational Recipe for Olefin Metathesis with Ru-Based Catalysts: The Whole Mechanism of Ring-Closing Olefin Metathesis. J. Chem. Theory Comput. 2014, 10, 4442–4448. [Google Scholar] [CrossRef] [PubMed]
- Pump, E.; Slugovc, C.; Cavallo, L.; Poater, A. Mechanism of the Ru−allenylidene to Ru−Indenylidene Rearrangement in Ruthenium Precatalysts for Olefin Metathesis. Organometallics 2015, 34, 3107–3111. [Google Scholar] [CrossRef]
- Manzini, S.; Poater, A.; Nelson, D.J.; Cavallo, L.; Nolan, S.P. How Phenyl Makes a Difference: Mechanistic Insights into the Ruthenium(II)-Catalysed Isomerisation of Allylic Alcohols. Chem. Sci. 2014, 5, 180–188. [Google Scholar] [CrossRef]
- Manzini, S.; Poater, A.; Nelson, D.J.; Cavallo, L.; Slawin, A.M.Z.; Nolan, S.P. Insights into the Decomposition of Olefin Metathesis Precatalysts. Angew. Chem. Int. Ed. 2014, 53, 8995–8999. [Google Scholar] [CrossRef] [PubMed]
- Richmond, C.J.; Matheu, R.; Poater, A.; Falivene, L.; Benet-Buchholz, J.; Sala, X.; Cavallo, L.; Llobet, A. Supramolecular Water Oxidation with Ru–bda-based Catalysts. Chem. Eur. J. 2014, 20, 17282–17286. [Google Scholar] [CrossRef] [PubMed]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef]
- Poater, A.; Cosenza, B.; Correa, A.; Giudice, S.; Ragone, F.; Scarano, V.; Cavallo, L. SambVca: A Web Application for the Calculation of Buried Volumes of N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2009, 2009, 1759–1766. [Google Scholar] [CrossRef]
- Poater, A.; Ragone, F.; Giudice, S.; Costabile, C.; Dorta, R.; Nolan, S.P.; Cavallo, L. Thermodynamics of N-Heterocyclic Carbene Dimerization: The Balance of Sterics and Electronics. Organometallics 2008, 27, 2679–2681. [Google Scholar] [CrossRef]
- Poater, A.; Falivene, L.; Urbina-Blanco, C.A.; Manzini, S.; Nolan, S.P.; Cavallo, L. How does the Addition of Steric Hindrance to a Typical N-Heterocyclic Carbene Ligand Affect Catalytic Activity in Olefin Metathesis? Dalton Trans. 2013, 42, 7433–7439. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
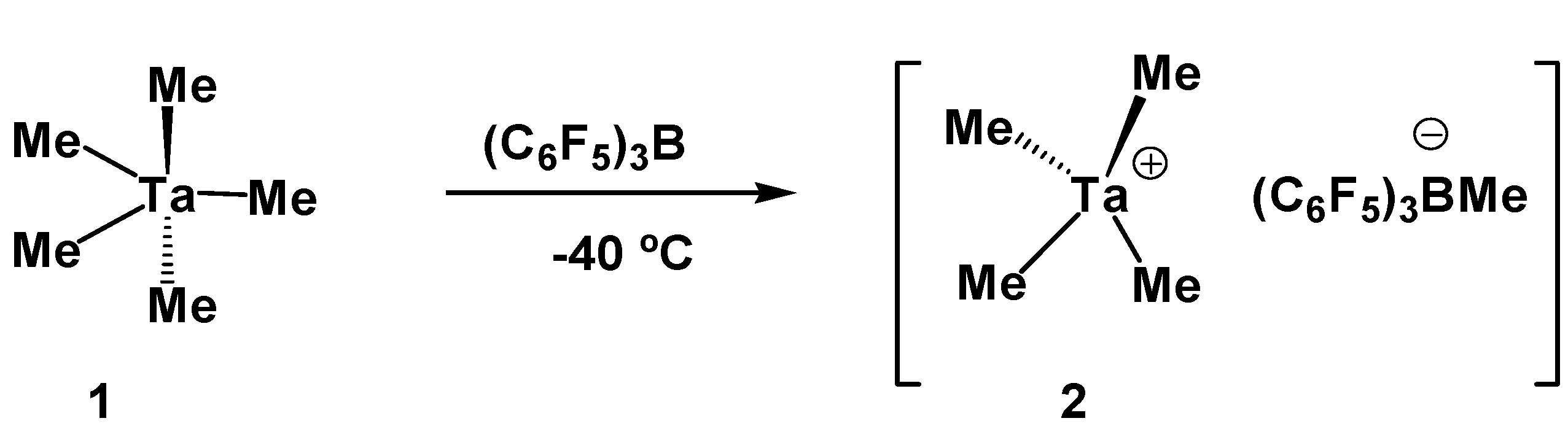{kind=link}
{kind=link}
| Catalyst | (a) | (b) | (c) | (d) |
|---|---|---|---|---|
| TaMe5 | 6.8 | −15.4 | 44.1 | −5.8 |
| WMe6 | 9.1 | −13.8 | 18.8 | 3.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dey, R.; Mohandas, J.C.; Samantaray, M.K.; Hamieh, A.; Kavitake, S.; Chen, Y.; Abou-Hamad, E.; Cavallo, L.; Poater, A.; Basset, J.-M. Synthesis and Characterization of Cationic Tetramethyl Tantalum(V) Complex. Catalysts 2018, 8, 507. https://doi.org/10.3390/catal8110507
Dey R, Mohandas JC, Samantaray MK, Hamieh A, Kavitake S, Chen Y, Abou-Hamad E, Cavallo L, Poater A, Basset J-M. Synthesis and Characterization of Cationic Tetramethyl Tantalum(V) Complex. Catalysts. 2018; 8(11):507. https://doi.org/10.3390/catal8110507
Chicago/Turabian StyleDey, Raju, Janet C. Mohandas, Manoja K. Samantaray, Ali Hamieh, Santosh Kavitake, Yin Chen, Edy Abou-Hamad, Luigi Cavallo, Albert Poater, and Jean-Marie Basset. 2018. "Synthesis and Characterization of Cationic Tetramethyl Tantalum(V) Complex" Catalysts 8, no. 11: 507. https://doi.org/10.3390/catal8110507
APA StyleDey, R., Mohandas, J. C., Samantaray, M. K., Hamieh, A., Kavitake, S., Chen, Y., Abou-Hamad, E., Cavallo, L., Poater, A., & Basset, J.-M. (2018). Synthesis and Characterization of Cationic Tetramethyl Tantalum(V) Complex. Catalysts, 8(11), 507. https://doi.org/10.3390/catal8110507