Theoretical Studies on the Direct Propylene Epoxidation Using Gold-Based Catalysts: A Mini-Review



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Background
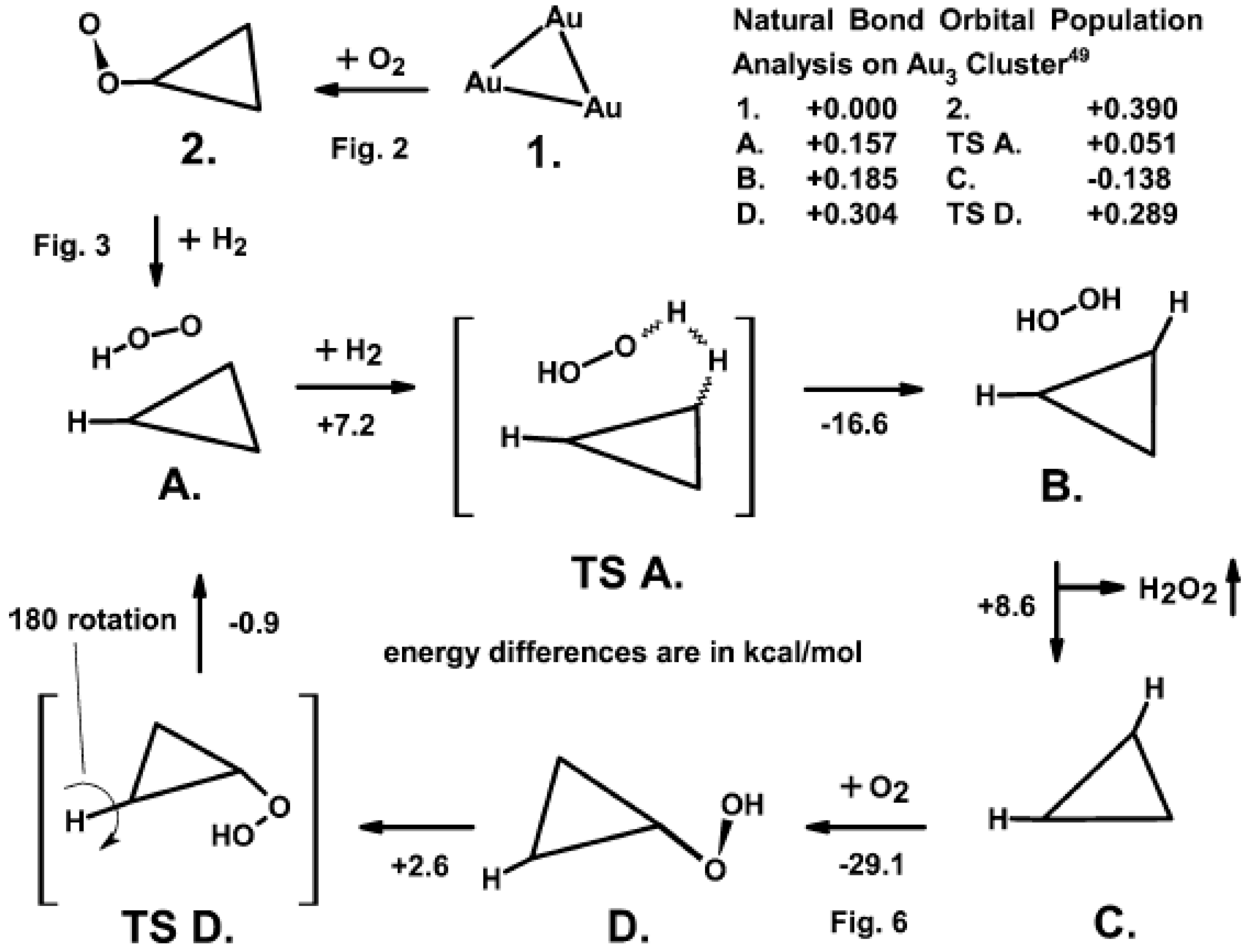
3. The Adsorption of O2 and H2 on Au
4. The Synthesis of H2O2 from O2 and H2
5. PO Reaction Mechanism
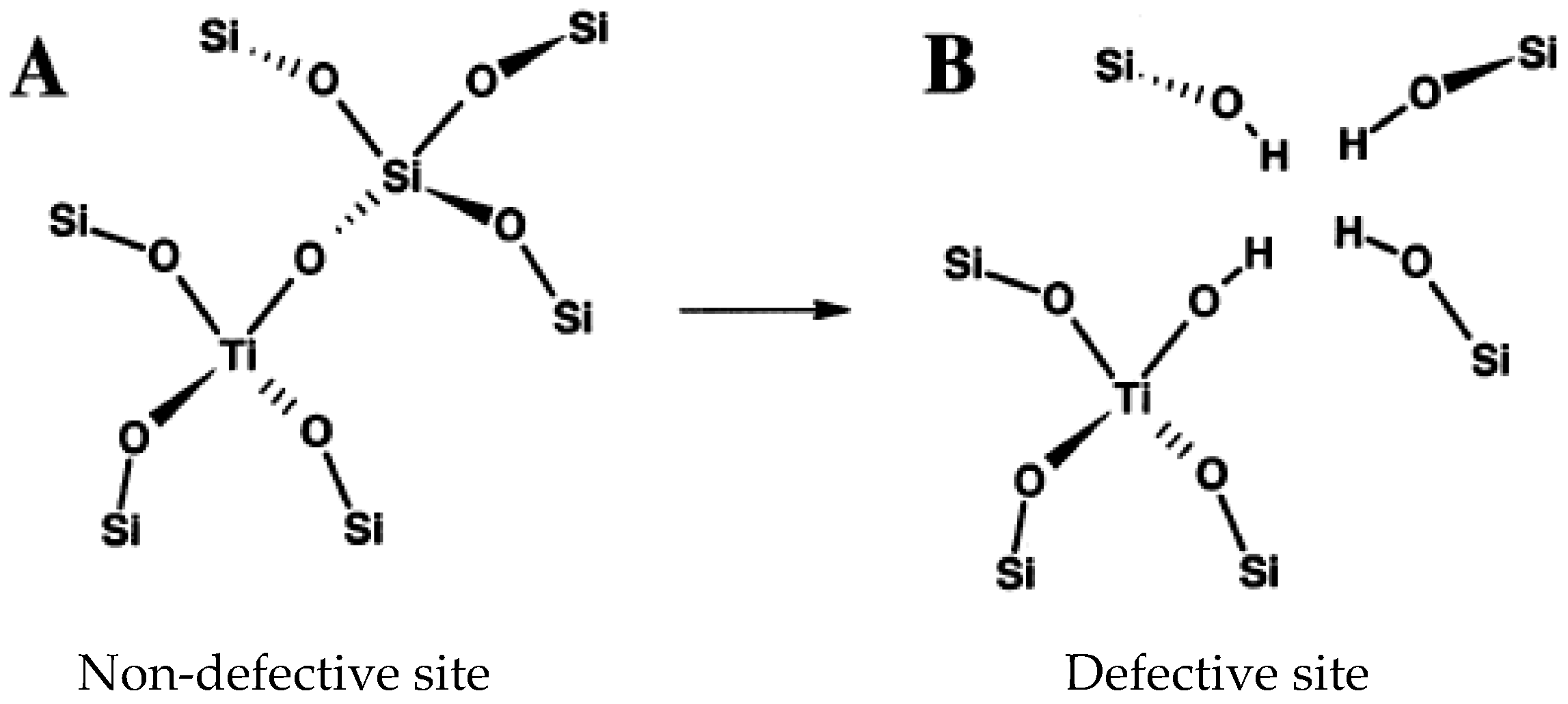
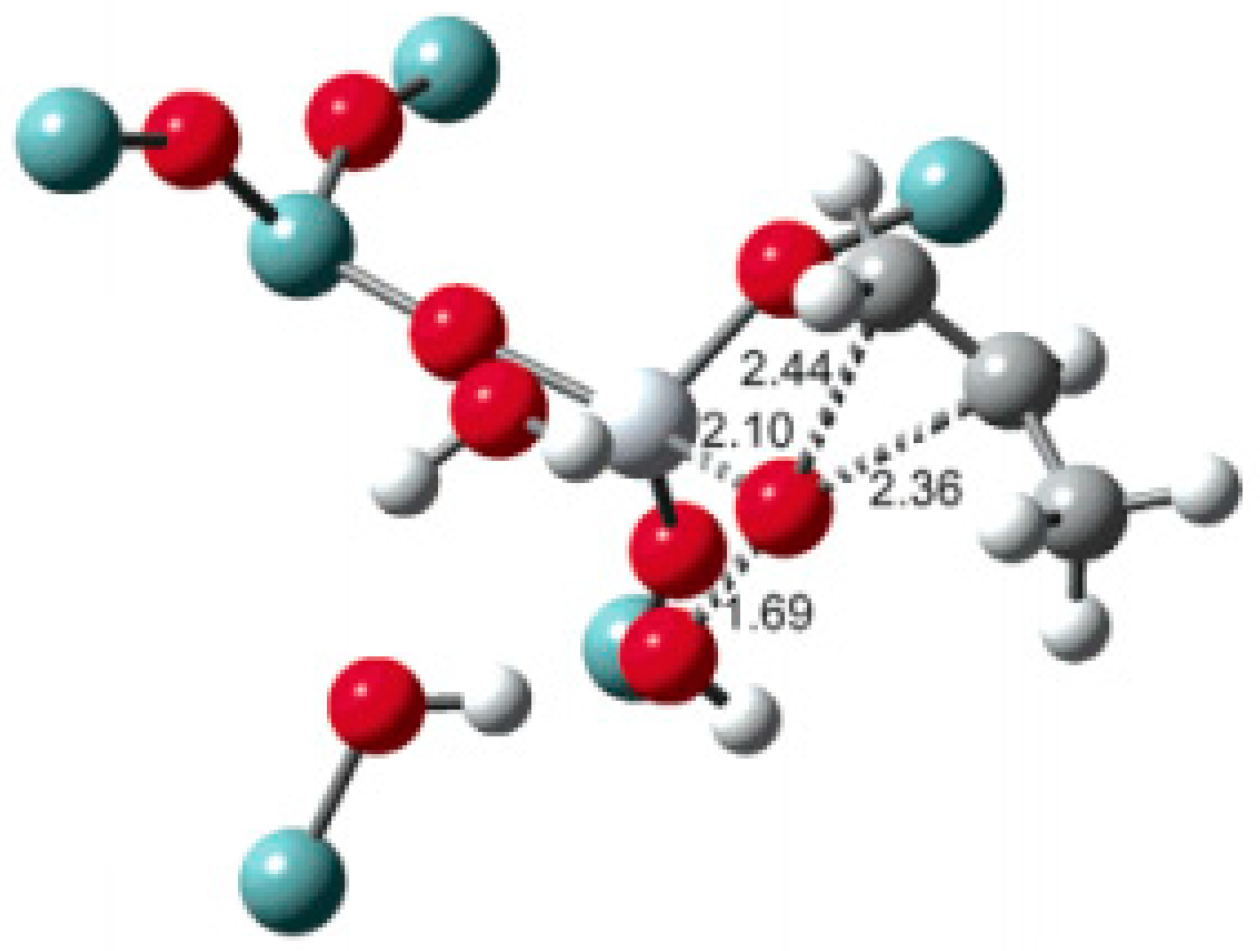
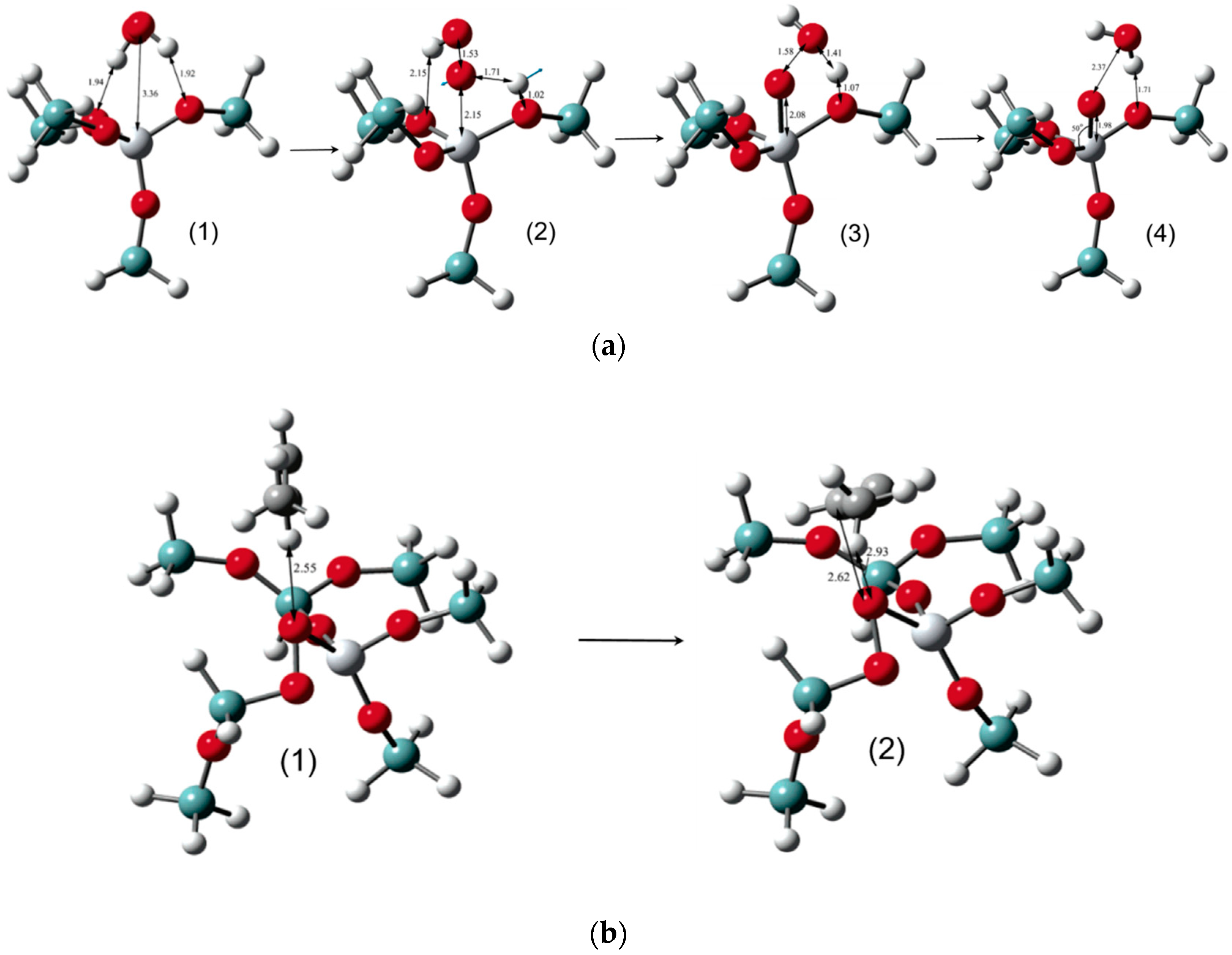
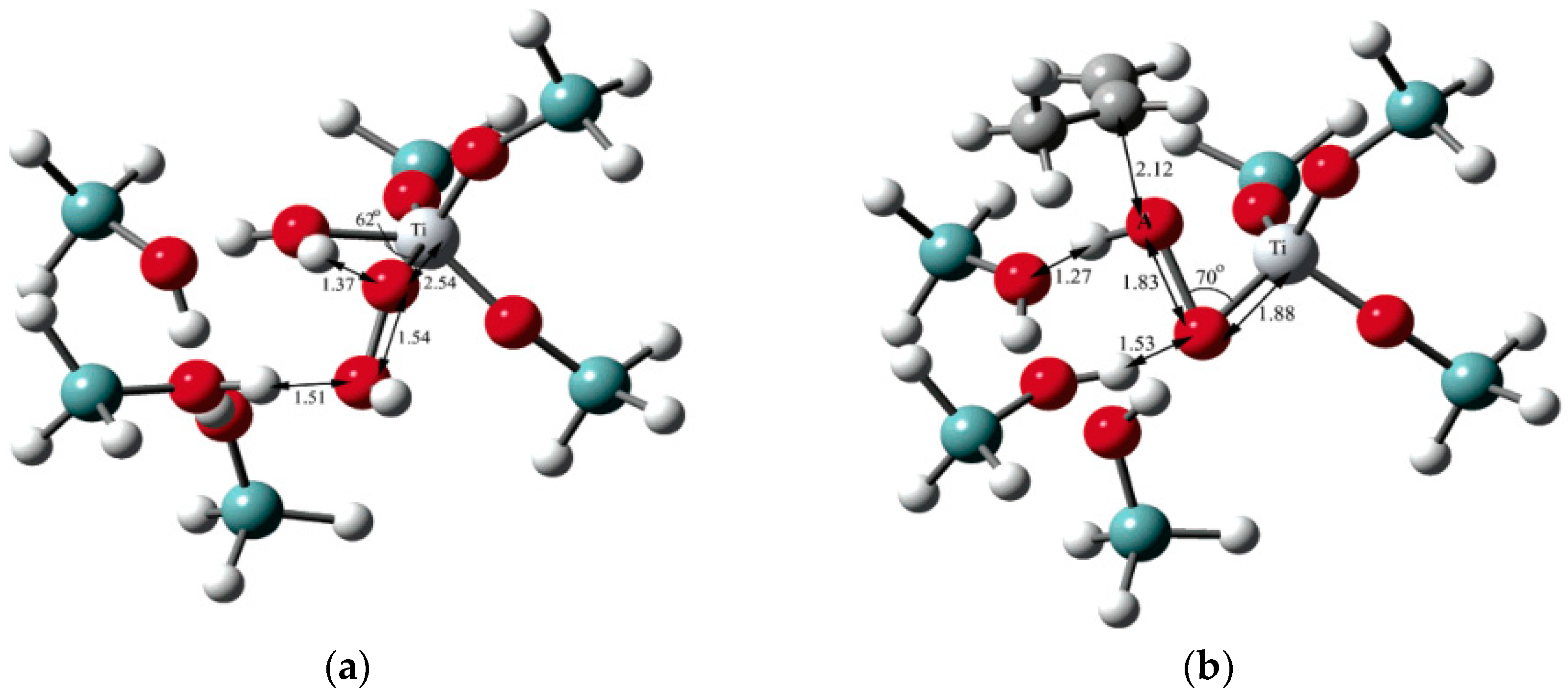
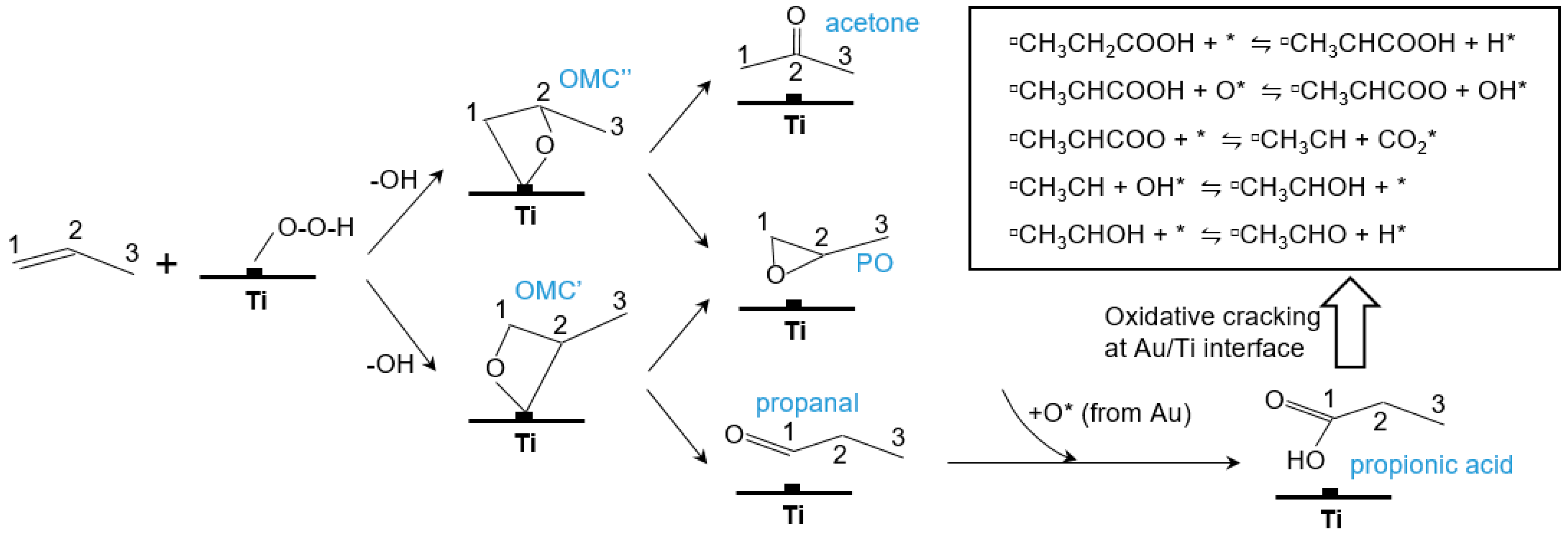
5.1. Reaction Mechanism on Titanosilicate
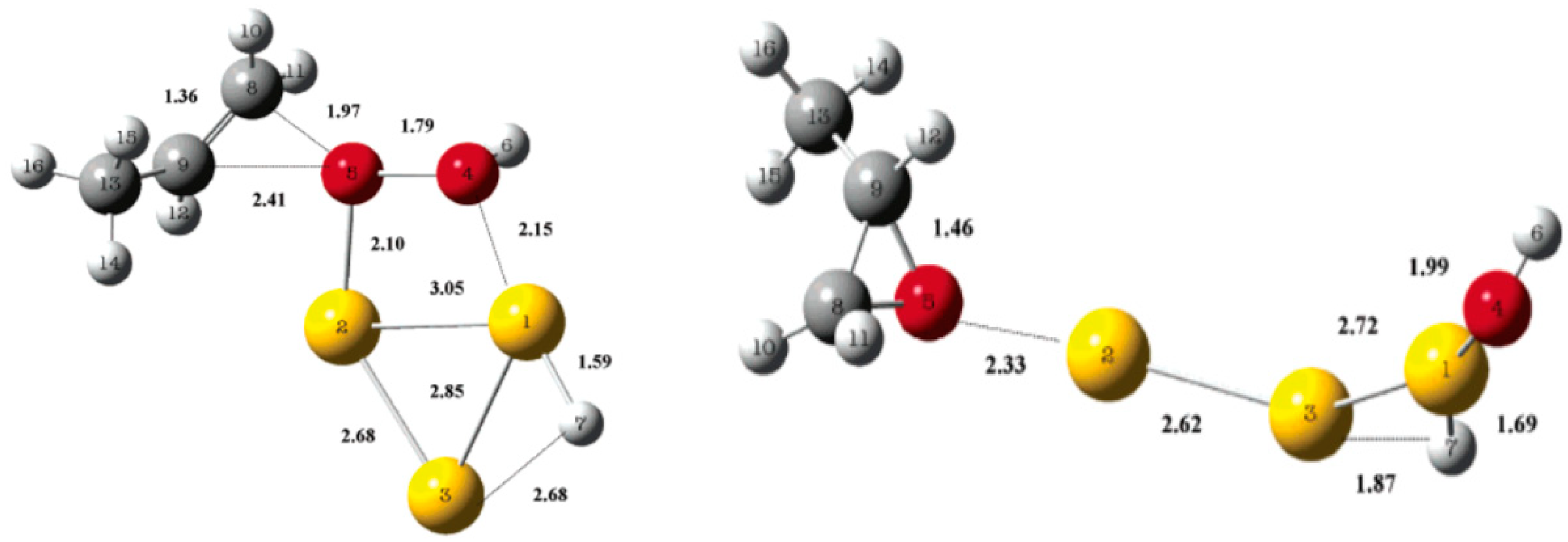
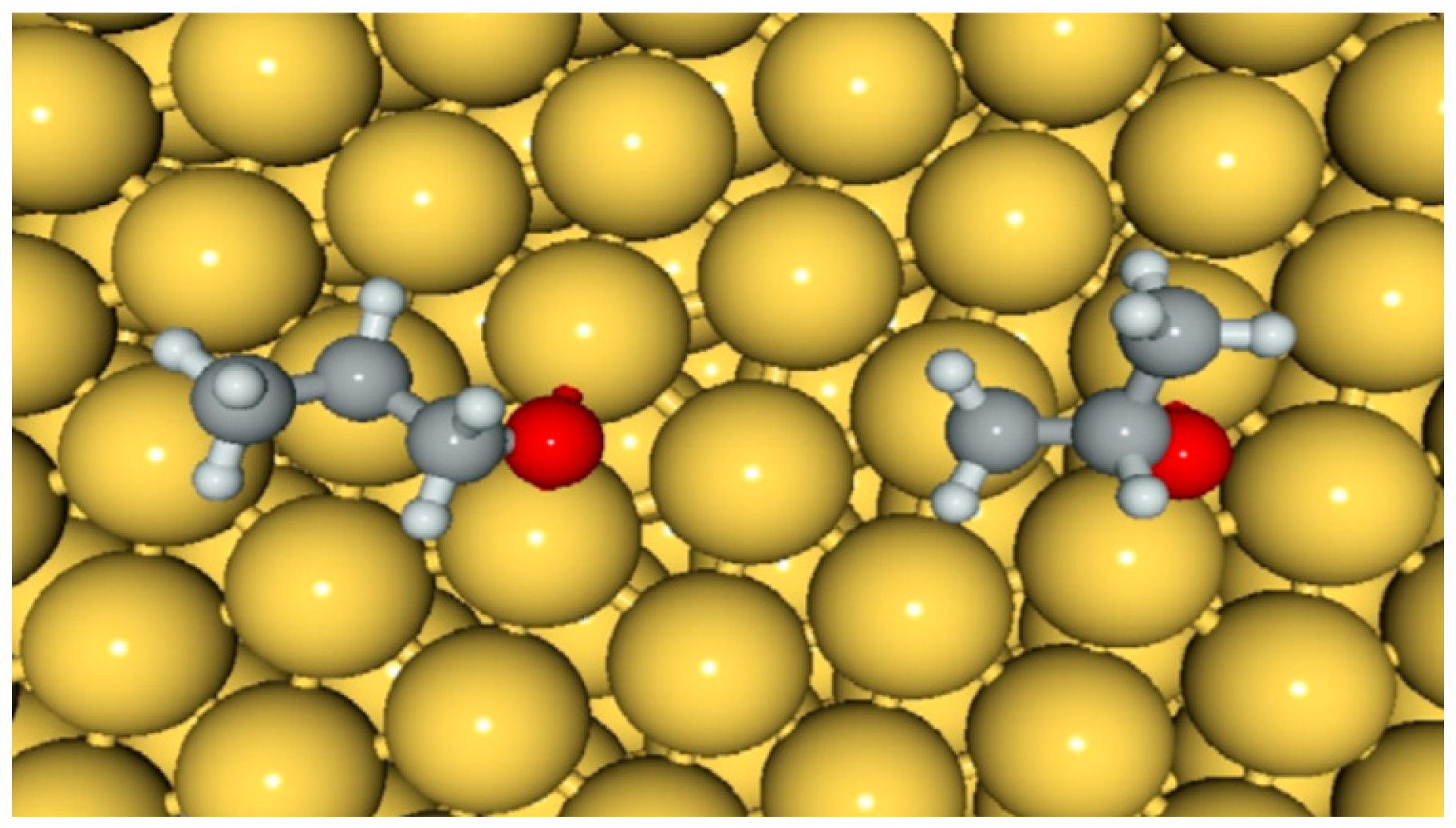
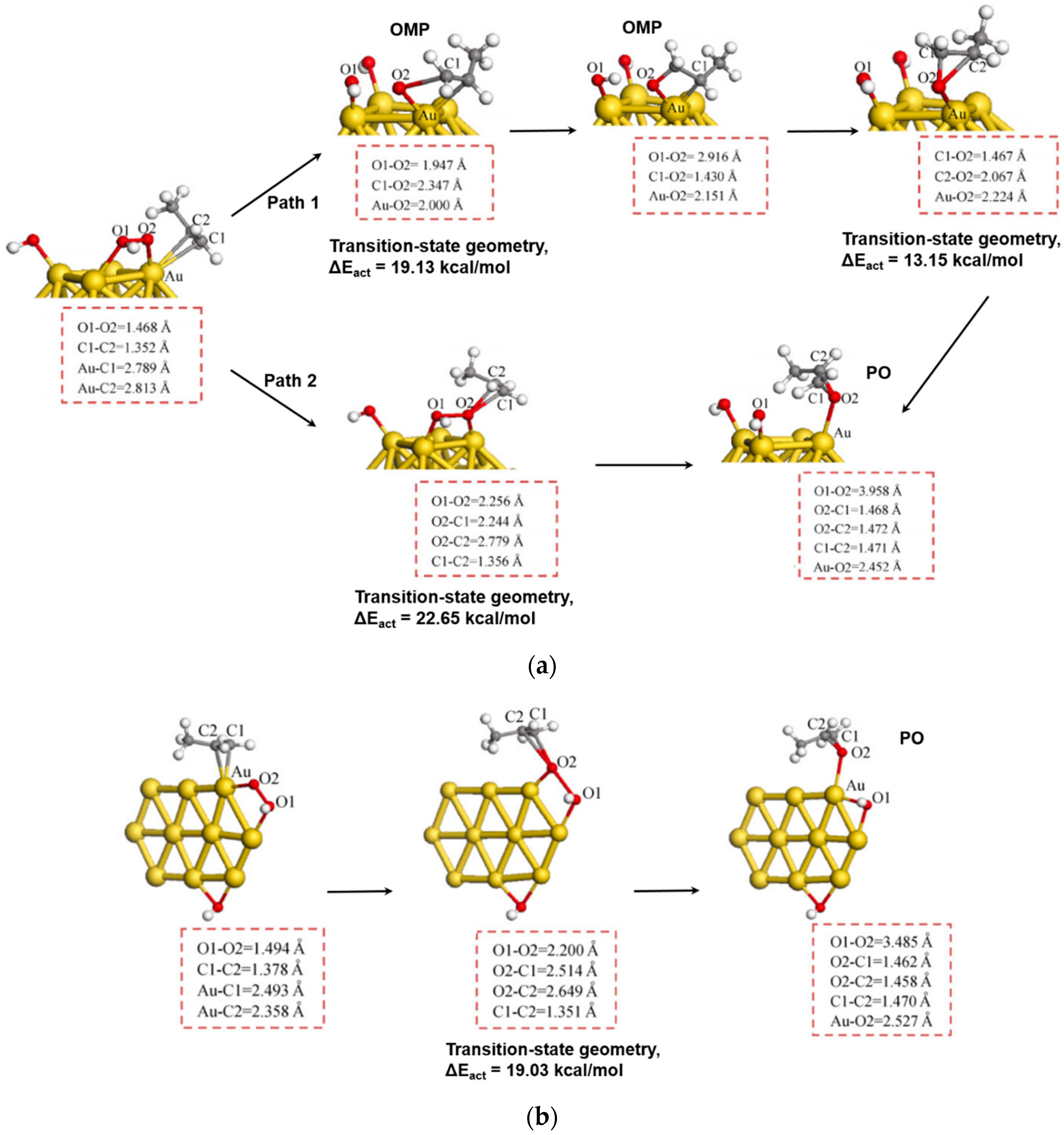
5.2. Reaction Mechanism on Gold
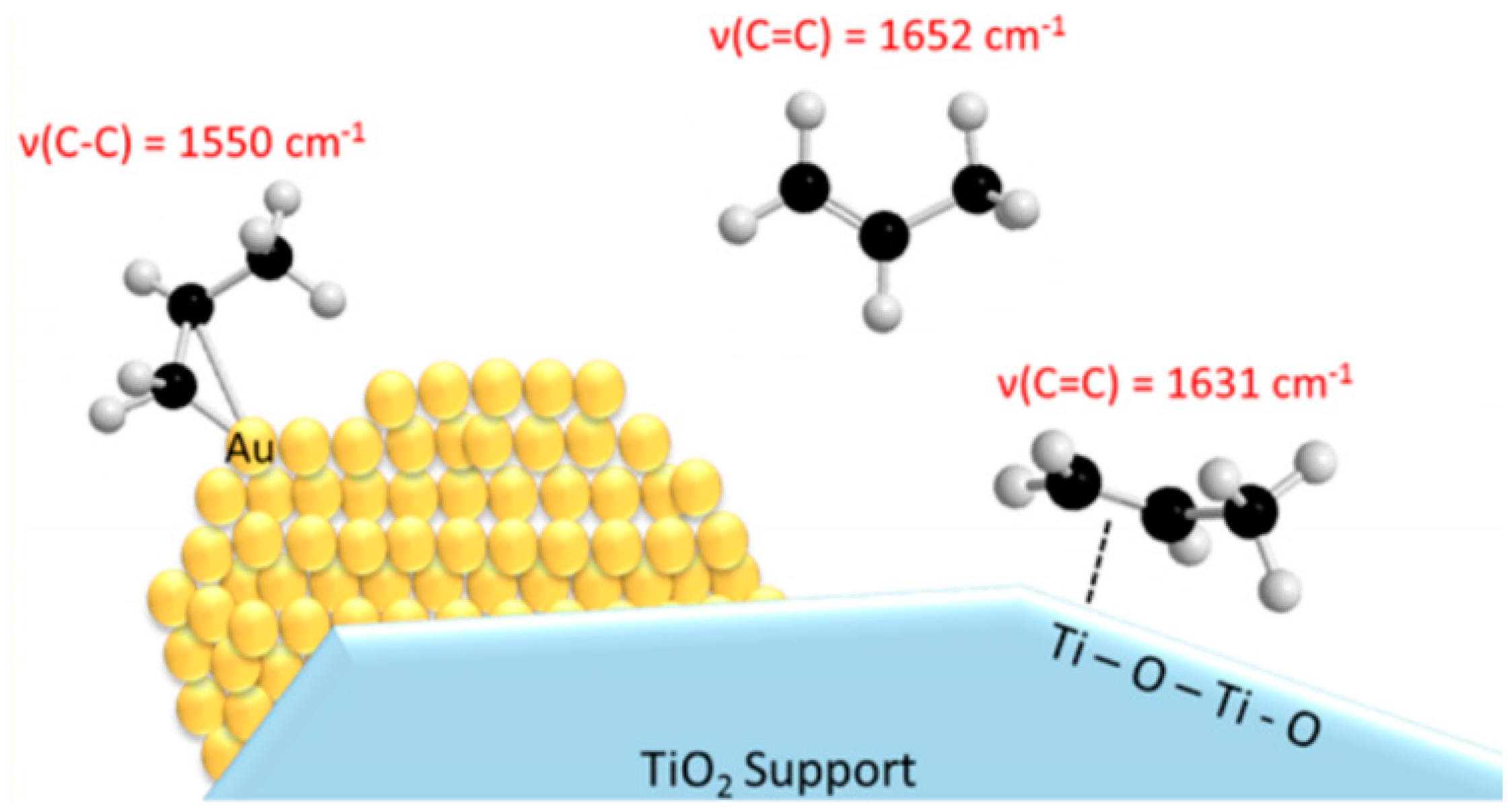
5.3. Reaction Mechanism on Au/Ti Interface Sites
6. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Shelef, M.; Graham, G.W. Why rhodium in automotive three-way catalysts? Catal. Rev. 1994, 36, 433–457. [Google Scholar] [CrossRef]
- Hoebink, J.H.B.J.; van Gemert, R.A.; van den Tillaart, J.A.A.; Marin, G.B. Competing reactions in three-way catalytic converters: Modelling of the NOx conversion maximum in the light-off curves under net oxidising conditions. Chem. Eng. Sci. 2000, 55, 1573–1581. [Google Scholar] [CrossRef]
- Angelici, R.J. Heterogeneous catalysis of the hydrodesulfurization of thiophenes in petroleum: An organometallic perspective of the mechanism. Acc. Chem. Res. 1988, 21, 387–394. [Google Scholar] [CrossRef]
- Barteau, M.A. New perspectives on direct heterogeneous olefin epoxidation. Top. Catal. 2003, 22, 3–8. [Google Scholar] [CrossRef]
- Hutchings, G.J. Promotion in heterogeneous catalysis: A topic requiring a new approach? Catal. Lett. 2001, 75, 1–12. [Google Scholar] [CrossRef]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Mavrikakis, M.; Stoltze, P.; Nørskov, J.K. Making gold less noble. Catal. Lett. 2000, 64, 101–106. [Google Scholar] [CrossRef]
- Yoon, B.; Häkkinen, H.; Landman, U.; Wörz, A.S.; Antonietti, J.-M.; Abbet, S.; Judai, K.; Heiz, U. Charging effects on bonding and catalyzed oxidation of CO on Au8 clusters on MgO. Science 2005, 307, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, T.A.; Makkee, M.; Moulijn, J.A.; Weckhuysen, B.M. The production of propene oxide: Catalytic processes and recent developments. Ind. Eng. Chem. Res. 2006, 45, 3447–3459. [Google Scholar] [CrossRef]
- Yap, N.; Andres, R.P.; Delgass, W.N. Reactivity and stability of Au in and on TS-1 for epoxidation of propylene with H2 and O2. J. Catal. 2004, 226, 156–170. [Google Scholar] [CrossRef]
- Huang, J.; Haruta, M. Gas-phase propene epoxidation over coinage metal catalysts. Res. Chem. Intermed. 2012, 38, 1–24. [Google Scholar] [CrossRef]
- Tullo, A. Dow, BASF to build propylene oxide. Chem. Eng. News 2004, 82, 15. [Google Scholar]
- Masatake, H.; Tetsuhiko, K.; Hiroshi, S.; Nobumasa, Y. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar]
- Hayashi, T.; Tanaka, K.; Haruta, M. Selective vapor-phase epoxidation of propylene over Au/TiO2 catalysts in the presence of oxygen and hydrogen. J. Catal. 1998, 178, 566–575. [Google Scholar] [CrossRef]
- Haruta, M. Spiers memorial lecture role of perimeter interfaces in catalysis by gold nanoparticles. Faraday Discuss. 2011, 152, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Daté, M. Advances in the catalysis of Au nanoparticles. Appl. Catal. A 2001, 222, 427–437. [Google Scholar] [CrossRef]
- Okumura, M.; Fujitani, T.; Huang, J.; Ishida, T. A career in catalysis: Masatake haruta. ACS Catal. 2015, 5, 4699–4707. [Google Scholar] [CrossRef]
- Huang, J.; Takei, T.; Akita, T.; Ohashi, H.; Haruta, M. Gold clusters supported on alkaline treated TS-1 for highly efficient propene epoxidation with O2 and H2. Appl. Catal. B 2010, 95, 430–438. [Google Scholar] [CrossRef]
- Sinha, A.K.; Sindhu, S.; Susumu, T.; Masatake, H. A three-dimensional mesoporous titanosilicate support for gold nanoparticles: Vapor-phase epoxidation of propene with high conversion. Angew. Chem. Int. Ed. 2004, 43, 1546–1548. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, B.; Bravo-Suárez, J.J.; Daté, M.; Tsubota, S.; Haruta, M. Trimethylamine as a gas-phase promoter: Highly efficient epoxidation of propylene over supported gold catalysts. Angew. Chem. Int. Ed. 2006, 45, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.; Lauterbach, J.; Delgass, W.N. Gas-phase epoxidation of propylene over small gold ensembles on TS-1. Appl. Catal. A 2005, 291, 188–198. [Google Scholar] [CrossRef]
- Taylor, B.; Lauterbach, J.; Delgass, W.N. The effect of mesoporous scale defects on the activity of Au/TS-1 for the epoxidation of propylene. Catal. Today 2007, 123, 50–58. [Google Scholar] [CrossRef]
- McEntee, M.; Tang, W.; Neurock, M.; Yates, J.T. Mechanistic insights into the catalytic oxidation of carboxylic acids on Au/TiO2: Partial oxidation of propionic and butyric acid to gold ketenylidene through unsaturated acids. ACS Catal. 2015, 5, 744–753. [Google Scholar] [CrossRef]
- Moskaleva, L.V. Theoretical mechanistic insights into propylene epoxidation on Au-based catalysts: Surface O versus OOH as oxidizing agents. Catal. Today 2016, 278, 45–55. [Google Scholar] [CrossRef]
- Stangland, E.E.; Stavens, K.B.; Andres, R.P.; Delgass, W.N. Characterization of gold–titania catalysts via oxidation of propylene to propylene oxide. J. Catal. 2000, 191, 332–347. [Google Scholar] [CrossRef]
- Uphade, B.S.; Akita, T.; Nakamura, T.; Haruta, M. Vapor-phase epoxidation of propene using H2 and O2 over Au/Ti–MCM-48. J. Catal. 2002, 209, 331–340. [Google Scholar] [CrossRef]
- Kapoor, M.P.; Sinha, A.K.; Seelan, S.; Inagaki, S.; Tsubota, S.; Yoshida, H.; Haruta, M. Hydrophobicity induced vapor-phase oxidation of propene over gold supported on titanium incorporated hybrid mesoporous silsesquioxane. Chem. Commun. 2002, 2902–2903. [Google Scholar] [CrossRef]
- Stangland, E.E.; Taylor, B.; Andres, R.P.; Delgass, W.N. Direct vapor phase propylene epoxidation over deposition-precipitation gold-titania catalysts in the presence of H2/O2: Effects of support, neutralizing agent, and pretreatment. J. Phys. Chem. B 2005, 109, 2321–2330. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, T.A.; Huizinga, B.J.; Makkee, M.; Moulijn, J.A. Direct epoxidation of propene using gold dispersed on TS-1 and other titanium-containing supports. Ind. Eng. Chem. Res. 1999, 38, 884–891. [Google Scholar] [CrossRef]
- Baker, T.A.; Xu, B.; Jensen, S.C.; Friend, C.M.; Kaxiras, E. Role of defects in propene adsorption and reaction on a partially O-covered Au(111) surface. Catal. Sci. Technol. 2011, 1, 1166–1174. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, Z.; Guo, Y.; Lu, G.; Zhao, Y.; Wang, H.; Hu, P. Significant enhancement of the selectivity of propylene epoxidation for propylene oxide: A molecular oxygen mechanism. Phys. Chem. Chem. Phys. 2017, 19, 25129–25139. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.H.; Delgass, W.N.; Thomson, K.T. Evidence of defect-promoted reactivity for epoxidation of propylene in titanosilicate (TS-1) catalysts: A dft study. J. Am. Chem. Soc. 2004, 126, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Corti, C.W.; Holliday, R.J.; Thompson, D.T. Developing new industrial applications for gold: Gold nanotechnology. Gold Bull. 2002, 35, 111–117. [Google Scholar] [CrossRef]
- Min, B.K.; Friend, C.M. Heterogeneous gold-based catalysis for green chemistry: Low-temperature CO oxidation and propene oxidation. Chem. Rev. 2007, 107, 2709–2724. [Google Scholar] [CrossRef] [PubMed]
- Mei, D.; Neurock, M.; Smith, C.M. Hydrogenation of acetylene–ethylene mixtures over Pd and Pd–Ag alloys: First-principles-based kinetic Monte Carlo simulations. J. Catal. 2009, 268, 181–195. [Google Scholar] [CrossRef]
- Mei, D.; Du, J.; Neurock, M. First-principles-based kinetic Monte Carlo simulation of nitric oxide reduction over platinum nanoparticles under lean-burn conditions. Ind. Eng. Chem. Res. 2010, 49, 10364–10373. [Google Scholar] [CrossRef]
- Hess, F.; Over, H. Rate-determining step or rate-determining configuration? The deacon reaction over RuO2(110) studied by DFT-based KMC simulations. ACS Catal. 2017, 7, 128–138. [Google Scholar] [CrossRef]
- Hess, F.; Sack, C.; Langsdorf, D.; Over, H. Probing the activity of different oxygen species in the CO oxidation over RuO2(110) by combining transient reflection–absorption infrared spectroscopy with kinetic Monte Carlo simulations. ACS Catal. 2017, 7, 8420–8428. [Google Scholar] [CrossRef]
- Salisbury, B.E.; Wallace, W.T.; Whetten, R.L. Low-temperature activation of molecular oxygen by gold clusters: A stoichiometric process correlated to electron affinity. Chem. Phys. 2000, 262, 131–141. [Google Scholar] [CrossRef]
- Okumura, M.; Kitagawa, Y.; Haruta, M.; Yamaguchi, K. DFT studies of interaction between O2 and Au clusters. The role of anionic surface Au atoms on Au clusters for catalyzed oxygenation. Chem. Phys. Lett. 2001, 346, 163–168. [Google Scholar] [CrossRef]
- Yoon, B.; Häkkinen, H.; Landman, U. Interaction of O2 with gold clusters: Molecular and dissociative adsorption. J. Phys. Chem. A 2003, 107, 4066–4071. [Google Scholar] [CrossRef]
- Mills, G.; Gordon, M.S.; Metiu, H. The adsorption of molecular oxygen on neutral and negative Aun clusters (n = 2–5). Chem. Phys. Lett. 2002, 359, 493–499. [Google Scholar] [CrossRef]
- Shi, H.X.; Sun, W.G.; Kuang, X.Y.; Lu, C.; Xia, X.X.; Chen, B.L.; Hermann, A. Probing the interactions of O2 with small gold cluster (n = 2–10, Q = 0, −1): A neutral chemisorbed complex Au5O2 cluster predicted. J. Phys. Chem. C 2017, 121, 24886–24893. [Google Scholar] [CrossRef]
- Xu, Y.; Mavrikakis, M. Adsorption and dissociation of O2 on gold surfaces: Effect of steps and strain. J. Phys. Chem. B 2003, 107, 9298–9307. [Google Scholar] [CrossRef]
- Lopez, N.; Nørskov, J.K. Catalytic CO oxidation by a gold nanoparticle: A density functional study. J. Am. Chem. Soc. 2002, 124, 11262–11263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gong, X.G. First-principles study of interaction of cluster Au32 with CO, H2, and O2. J. Chem. Phys. 2006, 125, 124703. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Horita, D.; Ono, Y.; Lyalin, A.; Maeda, S.; Taketsugu, T. Isomerization in gold clusters upon O2 adsorption. J. Phys. Chem. C 2017, 121, 2661–2668. [Google Scholar] [CrossRef]
- Roldan, A.; Ricart, J.M.; Illas, F.; Pacchioni, G. O2 adsorption and dissociation on neutral, positively and negatively charged Aun (n = 5–79) clusters. Phys. Chem. Chem. Phys. 2010, 12, 10723–10729. [Google Scholar] [CrossRef] [PubMed]
- Alberto, R.; Silvia, G.; Manel, R.J.; Francesc, I. Critical size for O2 dissociation by Au nanoparticles. ChemPhysChem 2009, 10, 348–351. [Google Scholar]
- Roldán, A.; Viñes, F.; Illas, F.; Ricart, J.M.; Neyman, K.M. Density functional studies of coinage metal nanoparticles: Scalability of their properties to bulk. Theor. Chem. Accounts 2008, 120, 565–573. [Google Scholar] [CrossRef]
- Boronat, M.; Corma, A. Oxygen activation on gold nanoparticles: Separating the influence of particle size, particle shape and support interaction. Dalton Trans. 2010, 39, 8538–8546. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Goodman, D.W. The structure of catalytically active gold on titania. Science 2004, 306, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Bond, G.C.; Thompson, D.T. Catalysis by gold. Catal. Rev. 1999, 41, 319–388. [Google Scholar] [CrossRef]
- Carrettin, S.; Concepción, P.; Corma, A.; Lopez Nieto, J.M.; Puntes, V.F. Nanocrystalline CeO2 increases the activity of Au for CO oxidation by two orders of magnitude. Angew. Chem. Int. Ed. 2004, 43, 2538–2540. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.; Carrettin, S.; Corma, A. Spectroscopic evidence for the supply of reactive oxygen during CO oxidation catalyzed by gold supported on nanocrystalline CeO2. J. Am. Chem. Soc. 2005, 127, 3286–3287. [Google Scholar] [CrossRef] [PubMed]
- Hernández, N.C.; Sanz, J.F.; Rodriguez, J.A. Unravelling the origin of the high-catalytic activity of supported Au: A density-functional theory-based interpretation. J. Am. Chem. Soc. 2006, 128, 15600–15601. [Google Scholar] [CrossRef] [PubMed]
- Kotobuki, M.; Leppelt, R.; Hansgen, D.A.; Widmann, D.; Behm, R.J. Reactive oxygen on a Au/TiO2 supported catalyst. J. Catal. 2009, 264, 67–76. [Google Scholar] [CrossRef]
- Florez, E.; Feria, L.; Viñes, F.; Rodriguez, J.A.; Illas, F. Effect of the support on the electronic structure of Au nanoparticles supported on transition metal carbides: Choice of the best substrate for Au activation. J. Phys. Chem. C 2009, 113, 19994–20001. [Google Scholar] [CrossRef]
- Schubert, M.M.; Hackenberg, S.; van Veen, A.C.; Muhler, M.; Plzak, V.; Behm, R.J. CO oxidation over supported gold catalysts—“Inert” and “active” support materials and their role for the oxygen supply during reaction. J. Catal. 2001, 197, 113–122. [Google Scholar] [CrossRef]
- Wang, J.G.; Hammer, B. Oxidation state of oxide supported nanometric gold. Top. Catal. 2007, 44, 49–56. [Google Scholar] [CrossRef]
- Laursen, S.; Linic, S. Strong chemical interactions between au and off-stoichiometric defects on TiO2 as a possible source of chemical activity of nanosized Au supported on the oxide. J. Phys. Chem. C 2009, 113, 6689–6693. [Google Scholar] [CrossRef]
- Greeley, J.; Nørskov, J.K. A general scheme for the estimation of oxygen binding energies on binary transition metal surface alloys. Surf. Sci. 2005, 592, 104–111. [Google Scholar] [CrossRef]
- Joshi, A.M.; Delgass, W.N.; Thomson, K.T. Analysis of O2 adsorption on binary−alloy clusters of gold: Energetics and correlations. J. Phys. Chem. B 2006, 110, 23373–23387. [Google Scholar] [CrossRef] [PubMed]
- Polynskaya, Y.G.; Pichugina, D.A.; Kuz’menko, N.E. Correlation between electronic properties and reactivity toward oxygen of tetrahedral gold–silver clusters. Comput. Theor. Chem. 2015, 1055, 61–67. [Google Scholar] [CrossRef]
- Feng, X.; Yang, J.; Duan, X.; Cao, Y.; Chen, B.; Chen, W.; Lin, D.; Qian, G.; Chen, D.; Yang, C.; et al. Enhanced catalytic performance for propene epoxidation with H2 and O2 over bimetallic Au–Ag/uncalcined titanium silicate-1 catalysts. ACS Catal. 2018, 7799–7808. [Google Scholar] [CrossRef]
- Varganov, S.A.; Olson, R.M.; Gordon, M.S.; Mills, G.; Metiu, H. A study of the reactions of molecular hydrogen with small gold clusters. J. Chem. Phys. 2004, 120, 5169–5175. [Google Scholar] [CrossRef] [PubMed]
- Okumura, M.; Kitagawa, Y.; Haruta, M.; Yamaguchi, K. The interaction of neutral and charged Au clusters with O2, CO and H2. Appl. Catal. A 2005, 291, 37–44. [Google Scholar] [CrossRef]
- Gao, M.; Lyalin, A.; Takagi, M.; Maeda, S.; Taketsugu, T. Reactivity of gold clusters in the regime of structural fluxionality. J. Phys. Chem. C 2015, 119, 11120–11130. [Google Scholar] [CrossRef]
- Barrio, L.; Liu, P.; Rodríguez, J.A.; Campos-Martín, J.M.; Fierro, J.L.G. A density functional theory study of the dissociation of H2 on gold clusters: Importance of fluxionality and ensemble effects. J. Chem. Phys. 2006, 125, 164715. [Google Scholar] [CrossRef] [PubMed]
- Barrio, L.; Liu, P.; Rodriguez, J.A.; Campos-Martin, J.M.; Fierro, J.L.G. Effects of hydrogen on the reactivity of O2 toward gold nanoparticles and surfaces. J. Phys. Chem. C 2007, 111, 19001–19008. [Google Scholar] [CrossRef]
- Hammer, B.; Norskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Jelínek, P.; Pérez, R.; Ortega, J.; Flores, F. Hydrogen dissociation over Au nanowires and the fractional conductance quantum. Phys. Rev. Lett. 2006, 96, 046803. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Cao, X.-M.; Gong, X.-Q.; Hu, P. A density functional theory study of hydrogen dissociation and diffusion at the perimeter sites of Au/TiO2. Phys. Chem. Chem. Phys. 2012, 14, 3741–3745. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kohyama, M.; Tanaka, S.; Takeda, S. A study on the mechanism for H2 dissociation on Au/TiO2 catalysts. J. Phys. Chem. C 2014, 118, 1611–1617. [Google Scholar] [CrossRef]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T. Low-temperature catalytic H2 oxidation over Au nanoparticle/TiO2 dual perimeter sites. Angew. Chem. Int. Ed. 2011, 50, 10186–10189. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, T.; Nakamura, I.; Akita, T.; Okumura, M.; Haruta, M. Hydrogen dissociation by gold clusters. Angew. Chem. Int. Ed. 2009, 48, 9515–9518. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Wang, L.; Li, G.; Guo, H. A review on research progress in the direct synthesis of hydrogen peroxide from hydrogen and oxygen: Noble-metal catalytic method, fuel-cell method and plasma method. Catal. Sci. Technol. 2016, 6, 1593–1610. [Google Scholar] [CrossRef]
- Solvay, Solvay’s Position and Strategy in Hydrogen Peroxide. London Investors Morning. Available online: http://www.solvay.com/EN/Investors/FinancialCalendarAndPresentations/Financial-Calendar/Calendar%20Documents/20100930_London-Morning.pdf (accessed on 30 September 2010).
- Ronald, H.; Achim, L. Applications of transition-metal catalysts to textile and wood-pulp bleaching. Angew. Chem. Int. Ed. 2006, 45, 206–222. [Google Scholar]
- Hess, H.T. Kirk-Othmer Encyclopedia of Chemical Engineering; Wiley: New York, NY, USA, 1995; p. 961. [Google Scholar]
- Pugge, J.W.a.L. Catalysis and Zeolites: Fundamentals and Applications; Springer: Berlin, Germany, 1996. [Google Scholar]
- Lane, B.S.; Burgess, K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003, 103, 2457–2474. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.H.; Delgass, W.N.; Thomson, K.T. Formation of hydrogen peroxide from H2 and O2 over a neutral gold trimer: A DFT study. J. Catal. 2004, 225, 69–77. [Google Scholar] [CrossRef]
- Kacprzak, K.A.; Akola, J.; Hakkinen, H. First-principles simulations of hydrogen peroxide formation catalyzed by small neutral gold clusters. Phys. Chem. Chem. Phys. 2009, 11, 6359–6364. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.M.; Delgass, W.N.; Thomson, K.T. Comparison of the catalytic activity of Au3, , Au5, and in the gas-phase reaction of H2 and O2 to form hydrogen peroxide: A density functional theory investigation. J. Phys. Chem. B 2005, 109, 22392–22406. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.C.; Nilekar, A.U.; Xu, Y.; Mavrikakis, M. Partial and complete reduction of O2 by hydrogen on transition metal surfaces. Surf. Sci. 2010, 604, 1565–1575. [Google Scholar] [CrossRef]
- Landon, P.; Collier, P.J.; Papworth, A.J.; Kiely, C.J.; Hutchings, G.J. Direct formation of hydrogen peroxide from H2/O2 using a gold catalyst. Chem. Commun. 2002, 2058–2059. [Google Scholar] [CrossRef]
- Joshi, A.M.; Delgass, W.N.; Thomson, K.T. Investigation of gold–silver, gold–copper, and gold–palladium dimers and trimers for hydrogen peroxide formation from H2 and O2. J. Phys. Chem. C 2007, 111, 7384–7395. [Google Scholar] [CrossRef]
- Ham, H.C.; Hwang, G.S.; Han, J.; Nam, S.W.; Lim, T.H. On the role of Pd ensembles in selective H2O2 formation on PdAu alloys. J. Phys. Chem. C 2009, 113, 12943–12945. [Google Scholar] [CrossRef]
- Ham, H.C.; Hwang, G.S.; Han, J.; Nam, S.W.; Lim, T.H. Geometric parameter effects on ensemble contributions to catalysis: H2O2 formation from H2 and O2 on AuPd alloys. A first principles study. J. Phys. Chem. C 2010, 114, 14922–14928. [Google Scholar] [CrossRef]
- Liu, P.; Norskov, J.K. Ligand and ensemble effects in adsorption on alloy surfaces. Phys. Chem. Chem. Phys. 2001, 3, 3814–3818. [Google Scholar] [CrossRef]
- Beletskaya, A.V.; Pichugina, D.A.; Shestakov, A.F.; Kuz’menko, N.E. Formation of H2O2 on Au20 and Au19Pd clusters: Understanding the structure effect on the atomic level. J. Phys. Chem. A 2013, 117, 6817–6826. [Google Scholar] [CrossRef] [PubMed]
- Staykov, A.; Kamachi, T.; Ishihara, T.; Yoshizawa, K. Theoretical study of the direct synthesis of H2O2 on Pd and Pd/Au surfaces. J. Phys. Chem. C 2008, 112, 19501–19505. [Google Scholar] [CrossRef]
- Li, J.; Staykov, A.; Ishihara, T.; Yoshizawa, K. Theoretical study of the decomposition and hydrogenation of H2O2 on Pd and Au@Pd surfaces: Understanding toward high selectivity of H2O2 synthesis. J. Phys. Chem. C 2011, 115, 7392–7398. [Google Scholar] [CrossRef]
- Li, J.; Ishihara, T.; Yoshizawa, K. Theoretical revisit of the direct synthesis of H2O2 on Pd and Au@Pd surfaces: A comprehensive mechanistic study. J. Phys. Chem. C 2011, 115, 25359–25367. [Google Scholar] [CrossRef]
- Todorovic, R.; Meyer, R.J. A comparative density functional theory study of the direct synthesis of H2O2 on Pd, Pt and Au surfaces. Catal. Today 2011, 160, 242–248. [Google Scholar] [CrossRef]
- Grabow, L.C.; Hvolbæk, B.; Falsig, H.; Nørskov, J.K. Search directions for direct H2O2 synthesis catalysts starting from Au12 nanoclusters. Top. Catal. 2012, 55, 336–344. [Google Scholar] [CrossRef]
- Uphade, B.S.; Yamada, Y.; Akita, T.; Nakamura, T.; Haruta, M. Synthesis and characterization of Ti-MCM-41 and vapor-phase epoxidation of propylene using H2 and O2 over Au/Ti-MCM-41. Appl. Catal. A 2001, 215, 137–148. [Google Scholar] [CrossRef]
- Taramasso, M.; Perego, G.; Notari, B. Preparation of Porous Crystalline Synthetic Material Comprised of Silicon and Titanium Oxides. U.S. Patent 4,410,501, 18 October 1983. [Google Scholar]
- Wenping, F.; Yaquan, W.; Guoqiang, W.; Yi, L.; Juan, X.; Hainan, S.; Teng, Z.; Shuhai, W.; Xiaoxue, W.; Pengxu, Y. Liquid phase propylene epoxidation with H2O2 on TS-1/SiO2 catalyst in a fixed-bed reactor: Experiments and deactivation kinetics. J. Chem. Technol. Biotechnol. 2015, 90, 1489–1496. [Google Scholar]
- Clerici, M.G.; Bellussi, G.; Romano, U. Synthesis of propylene oxide from propylene and hydrogen peroxide catalyzed by titanium silicalite. J. Catal. 1991, 129, 159–167. [Google Scholar] [CrossRef]
- Sivadinarayana, C.; Choudhary, T.V.; Daemen, L.L.; Eckert, J.; Goodman, D.W. The nature of the surface species formed on Au/TiO2 during the reaction of H2 and O2: An inelastic neutron scattering study. J. Am. Chem. Soc. 2004, 126, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, B.; Bravo-Suárez, J.J.; Mimura, N.; Lu, J.; Bando, K.K.; Tsubota, S.; Haruta, M. In situ UV−vis and EPR study on the formation of hydroperoxide species during direct gas phase propylene epoxidation over Au/Ti-SiO2 catalyst. J. Phys. Chem. B 2006, 110, 22995–22999. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.H.; Joshi, A.M.; Delgass, W.N.; Thomson, K.T. A quantum chemical study of comparison of various propylene epoxidation mechanisms using H2O2 and TS-1 catalyst. J. Phys. Chem. B 2006, 110, 14627–14639. [Google Scholar] [CrossRef] [PubMed]
- Bellussi, G.; Carati, A.; Clerici, M.G.; Maddinelli, G.; Millini, R. Reactions of titanium silicalite with protic molecules and hydrogen peroxide. J. Catal. 1992, 133, 220–230. [Google Scholar] [CrossRef]
- Sinclair, P.E.; Catlow, C.R.A. Quantum chemical study of the mechanism of partial oxidation reactivity in titanosilicate catalysts: Active site formation, oxygen transfer, and catalyst deactivation. J. Phys. Chem. B 1999, 103, 1084–1095. [Google Scholar] [CrossRef]
- Vayssilov, G.N.; van Santen, R.A. Catalytic activity of titanium silicalites—A DFT study. J. Catal. 1998, 175, 170–174. [Google Scholar] [CrossRef]
- Munakata, H.; Oumi, Y.; Miyamoto, A. A DFT study on peroxo-complex in titanosilicate catalyst: Hydrogen peroxide activation on titanosilicalite-1 catalyst and reaction mechanisms for catalytic olefin epoxidation and for hydroxylamine formation from ammonia. J. Phys. Chem. B 2001, 105, 3493–3501. [Google Scholar] [CrossRef]
- Nie, X.; Ji, X.; Chen, Y.; Guo, X.; Song, C. Mechanistic investigation of propylene epoxidation with H2O2 over TS-1: Active site formation, intermediate identification, and oxygen transfer pathway. Mol. Catal. 2017, 441, 150–167. [Google Scholar] [CrossRef]
- Panayotov, D.; McEntee, M.; Burrows, S.; Driscoll, D.; Tang, W.; Neurock, M.; Morris, J. Infrared studies of propene and propene oxide adsorption on nanoparticulate Au/TiO2. Surf. Sci. 2016, 652, 172–182. [Google Scholar] [CrossRef]
- Driscoll, D.M.; Tang, W.; Burrows, S.P.; Panayotov, D.A.; Neurock, M.; McEntee, M.; Morris, J.R. Binding sites, geometry, and energetics of propene at nanoparticulate Au/TiO2. J. Phys. Chem. C 2017, 121, 1683–1689. [Google Scholar] [CrossRef]
- Bocquet, M.-L.; Loffreda, D. Ethene epoxidation selectivity inhibited by twisted oxametallacycle: A DFT study on Ag surface-oxide. J. Am. Chem. Soc. 2005, 127, 17207–17215. [Google Scholar] [CrossRef] [PubMed]
- Mavrikakis, M.; Doren, D.J.; Barteau, M.A. Density functional theory calculations for simple oxametallacycles: Trends across the periodic table. J. Phys. Chem. B 1998, 102, 394–399. [Google Scholar] [CrossRef]
- Torres, D.; Lopez, N.; Illas, F.; Lambert, R.M. Why copper is intrinsically more selective than silver in alkene epoxidation: Ethylene oxidation on Cu(111) versus Ag(111). J. Am. Chem. Soc. 2005, 127, 10774–10775. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.S.; Mavrikakis, M.; Barteau, M.A.; Vohs, J.M. First synthesis, experimental and theoretical vibrational spectra of an oxametallacycle on a metal surface. J. Am. Chem. Soc. 1998, 120, 3196–3204. [Google Scholar] [CrossRef]
- Linic, S.; Barteau, M.A. Formation of a stable surface oxametallacycle that produces ethylene oxide. J. Am. Chem. Soc. 2002, 124, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Linic, S.; Medlin, J.W.; Barteau, M.A. Synthesis of oxametallacycles from 2-iodoethanol on Ag(111) and the structure dependence of their reactivity. Langmuir 2002, 18, 5197–5204. [Google Scholar] [CrossRef]
- Özbek, M.O.; van Santen, R.A. The mechanism of ethylene epoxidation catalysis. Catal. Lett. 2013, 143, 131–141. [Google Scholar] [CrossRef]
- Joshi, A.M.; Delgass, W.N.; Thomson, K.T. Partial oxidation of propylene to propylene oxide over a neutral gold trimer in the gas phase: A density functional theory study. J. Phys. Chem. B 2006, 110, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Roldan, A.; Torres, D.; Ricart, J.M.; Illas, F. On the effectiveness of partial oxidation of propylene by gold: A density functional theory study. J. Mol. Catal. A Chem. 2009, 306, 6–10. [Google Scholar] [CrossRef]
- Deng, X.; Min, B.K.; Liu, X.; Friend, C.M. Partial oxidation of propene on oxygen-covered Au(111). J. Phys. Chem. B 2006, 110, 15982–15987. [Google Scholar] [CrossRef] [PubMed]
- Cant, N.W.; Hall, W.K. Catalytic oxidation. Iv. Ethylene and propylene oxidation over gold. J. Phys. Chem. 1971, 75, 2914–2921. [Google Scholar] [CrossRef]
- Geenen, P.V.; Boss, H.J.; Pott, G.T. A study of the vapor-phase epoxidation of propylene and ethylene on silver and silver-gold alloy catalysts. J. Catal. 1982, 77, 499–510. [Google Scholar] [CrossRef]
- Mullen, G.M.; Zhang, L.; Evans, E.J.; Yan, T.; Henkelman, G.; Mullins, C.B. Oxygen and hydroxyl species induce multiple reaction pathways for the partial oxidation of allyl alcohol on gold. J. Am. Chem. Soc. 2014, 136, 6489–6498. [Google Scholar] [CrossRef] [PubMed]
- Mullen, G.M.; Zhang, L.; Evans, E.J.; Yan, T.; Henkelman, G.; Mullins, C.B. Control of selectivity in allylic alcohol oxidation on gold surfaces: The role of oxygen adatoms and hydroxyl species. Phys. Chem. Chem. Phys. 2015, 17, 4730–4738. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, M.; Iglesia, E. Catalytic epoxidation of propene with H2O-O2 reactants on Au/TiO2. Chem. Commun. 2009, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Molina, L.M.; Lopez, M.J.; Alonso, J.A.; Hammer, B.; Lee, B.; Seifert, S.; Winans, R.E.; Elam, J.W.; Pellin, M.J.; Vajda, S. Selective propene epoxidation on immobilized Au6–10 clusters: The effect of hydrogen and water on activity and selectivity. Angew. Chem. Int. Ed. 2009, 48, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Jiahui, H.; Tomoki, A.; Jérémy, F.; Tadahiro, F.; Takashi, T.; Masatake, H. Propene epoxidation with dioxygen catalyzed by gold clusters. Angew. Chem. Int. Ed. 2009, 48, 7862–7866. [Google Scholar]
- Bongiorno, A.; Landman, U. Water-enhanced catalysis of CO oxidation on free and supported gold nanoclusters. Phys. Rev. Lett. 2005, 95, 106102. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-R.; Wang, Y.-G.; Li, J. Theoretical investigations of the catalytic role of water in propene epoxidation on gold nanoclusters: A hydroperoxyl-mediated pathway. Nano Res. 2011, 4, 131–142. [Google Scholar] [CrossRef]
- Chang, C.-R.; Huang, Z.-Q.; Li, J. Hydrogenation of molecular oxygen to hydroperoxyl: An alternative pathway for O2 activation on nanogold catalysts. Nano Res. 2015, 8, 3737–3748. [Google Scholar] [CrossRef]
- Liu, J.C.; Tang, Y.; Chang, C.R.; Wang, Y.G.; Li, J. Mechanistic insights into propene epoxidation with O2–H2O mixture on Au7/α-Al2O3: A hydroproxyl pathway from ab initio molecular dynamics simulations. ACS Catal. 2016, 6, 2525–2535. [Google Scholar] [CrossRef]
- Joshi, A.M.; Delgass, W.N.; Thomson, K.T. Mechanistic implications of Aun/Ti-lattice proximity for propylene epoxidation. J. Phys. Chem. C 2007, 111, 7841–7844. [Google Scholar] [CrossRef]
- Ajo, H.M.; Bondzie, V.A.; Campbell, C.T. Propene adsorption on gold particles on TiO2(110). Catal. Lett. 2002, 78, 359–368. [Google Scholar] [CrossRef]
- Nijhuis, T.A.; Gardner, T.Q.; Weckhuysen, B.M. Modeling of kinetics and deactivation in the direct epoxidation of propene over gold–titania catalysts. J. Catal. 2005, 236, 153–163. [Google Scholar] [CrossRef]
- Lee, W.S.; Lai, L.C.; Cem Akatay, M.; Stach, E.A.; Ribeiro, F.H.; Delgass, W.N. Probing the gold active sites in Au/TS-1 for gas-phase epoxidation of propylene in the presence of hydrogen and oxygen. J. Catal. 2012, 296, 31–42. [Google Scholar] [CrossRef]
- Haruta, M. Size- and support-dependency in the catalysis of gold. Catal. Today 1997, 36, 153–166. [Google Scholar] [CrossRef]
- Nijhuis, T.A.; Visser, T.; Weckhuysen, B.M. The role of gold in gold–titania epoxidation catalysts. Angew. Chem. Int. Ed. 2005, 44, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.H.; Ji, J.; Lu, Z.; Lei, Y. Analysis of the propylene epoxidation mechanism on supported gold nanoparticles. Chem. Eng. Sci. 2017, 174, 229–237. [Google Scholar] [CrossRef]
- Lu, Z.; Piernavieja-Hermida, M.; Turner, C.H.; Wu, Z.; Lei, Y. Effects of TiO2 in low temperature propylene epoxidation using gold catalysts. J. Phys. Chem. C 2018, 122, 1688–1698. [Google Scholar] [CrossRef]
- Ji, J.; Lu, Z.; Lei, Y.; Turner, C.H. Mechanistic insights into the direct propylene epoxidation using Au nanoparticles dispersed on TiO2/SiO2. Chem. Eng. Sci. 2018, 191, 169–182. [Google Scholar] [CrossRef]
- Chen, J.; Halin, S.J.A.; Schouten, J.C.; Nijhuis, T.A. Kinetic study of propylene epoxidation with H2 and O2 over Au/Ti-SiO2 in the explosive regime. Faraday Discuss. 2011, 152, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.; Lauterbach, J.; Blau, G.E.; Delgass, W.N. Reaction kinetic analysis of the gas-phase epoxidation of propylene over Au/TS-1. J. Catal. 2006, 242, 142–152. [Google Scholar] [CrossRef]
- Zwijnenburg, A.; Makkee, M.; Moulijn, J.A. Increasing the low propene epoxidation product yield of gold/titania-based catalysts. Appl. Catal. A 2004, 270, 49–56. [Google Scholar] [CrossRef]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T. Localized partial oxidation of acetic acid at the dual perimeter sites of the Au/TiO2 catalyst—Formation of gold ketenylidene. J. Am. Chem. Soc. 2012, 134, 13569–13572. [Google Scholar] [CrossRef] [PubMed]
- Green, I.X.; Tang, W.; Neurock, M.; Yates, J.T. Mechanistic insights into the partial oxidation of acetic acid by O2 at the dual perimeter sites of a Au/TiO2 catalyst. Faraday Discuss. 2013, 162, 247–265. [Google Scholar] [CrossRef] [PubMed]
- McEntee, M.; Tang, W.; Neurock, M.; Yates, J.T. Selective catalytic oxidative-dehydrogenation of carboxylic acids—Acrylate and crotonate formation at the Au/TiO2 interface. J. Am. Chem. Soc. 2014, 136, 5116–5120. [Google Scholar] [CrossRef] [PubMed]
- Ntainjua, E.N.; Freakley, S.J.; Hutchings, G.J. Direct synthesis of hydrogen peroxide using ruthenium catalysts. Top. Catal. 2012, 55, 718–722. [Google Scholar] [CrossRef]
- Edwards, J.K.; Freakley, S.J.; Carley, A.F.; Kiely, C.J.; Hutchings, G.J. Strategies for designing supported gold–palladium bimetallic catalysts for the direct synthesis of hydrogen peroxide. Acc. Chem. Res. 2014, 47, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Gudarzi, D.; Ratchananusorn, W.; Turunen, I.; Heinonen, M.; Salmi, T. Promotional effects of au in Pd–Au bimetallic catalysts supported on activated carbon cloth (ACC) for direct synthesis of H2O2 from H2 and O2. Catal. Today 2015, 248, 58–68. [Google Scholar] [CrossRef]
- Yohei, N.; Tatsumi, I.; Yuiko, H.; Kotaro, K.; Kenji, K.; Hiroshige, M. Nanocolloidal Pd-Au as catalyst for the direct synthesis of hydrogen peroxide from H2 and O2. ChemSusChem 2008, 1, 619–621. [Google Scholar]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Haruta, M. Catalysis of gold nanoparticles deposited on metal oxides. CATTECH 2002, 6, 102–115. [Google Scholar] [CrossRef]
- Hashmi, A.S.; Hutchings, G.J. Gold catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, J.; Lu, Z.; Lei, Y.; Turner, C.H. Theoretical Studies on the Direct Propylene Epoxidation Using Gold-Based Catalysts: A Mini-Review. Catalysts 2018, 8, 421. https://doi.org/10.3390/catal8100421
Ji J, Lu Z, Lei Y, Turner CH. Theoretical Studies on the Direct Propylene Epoxidation Using Gold-Based Catalysts: A Mini-Review. Catalysts. 2018; 8(10):421. https://doi.org/10.3390/catal8100421
Chicago/Turabian StyleJi, Jingjing, Zheng Lu, Yu Lei, and C. Heath Turner. 2018. "Theoretical Studies on the Direct Propylene Epoxidation Using Gold-Based Catalysts: A Mini-Review" Catalysts 8, no. 10: 421. https://doi.org/10.3390/catal8100421
APA StyleJi, J., Lu, Z., Lei, Y., & Turner, C. H. (2018). Theoretical Studies on the Direct Propylene Epoxidation Using Gold-Based Catalysts: A Mini-Review. Catalysts, 8(10), 421. https://doi.org/10.3390/catal8100421