Genetically Fused T4L Acts as a Shield in Covalent Enzyme Immobilisation Enhancing the Rescued Activity


Abstract
:
1. Introduction
2. Results
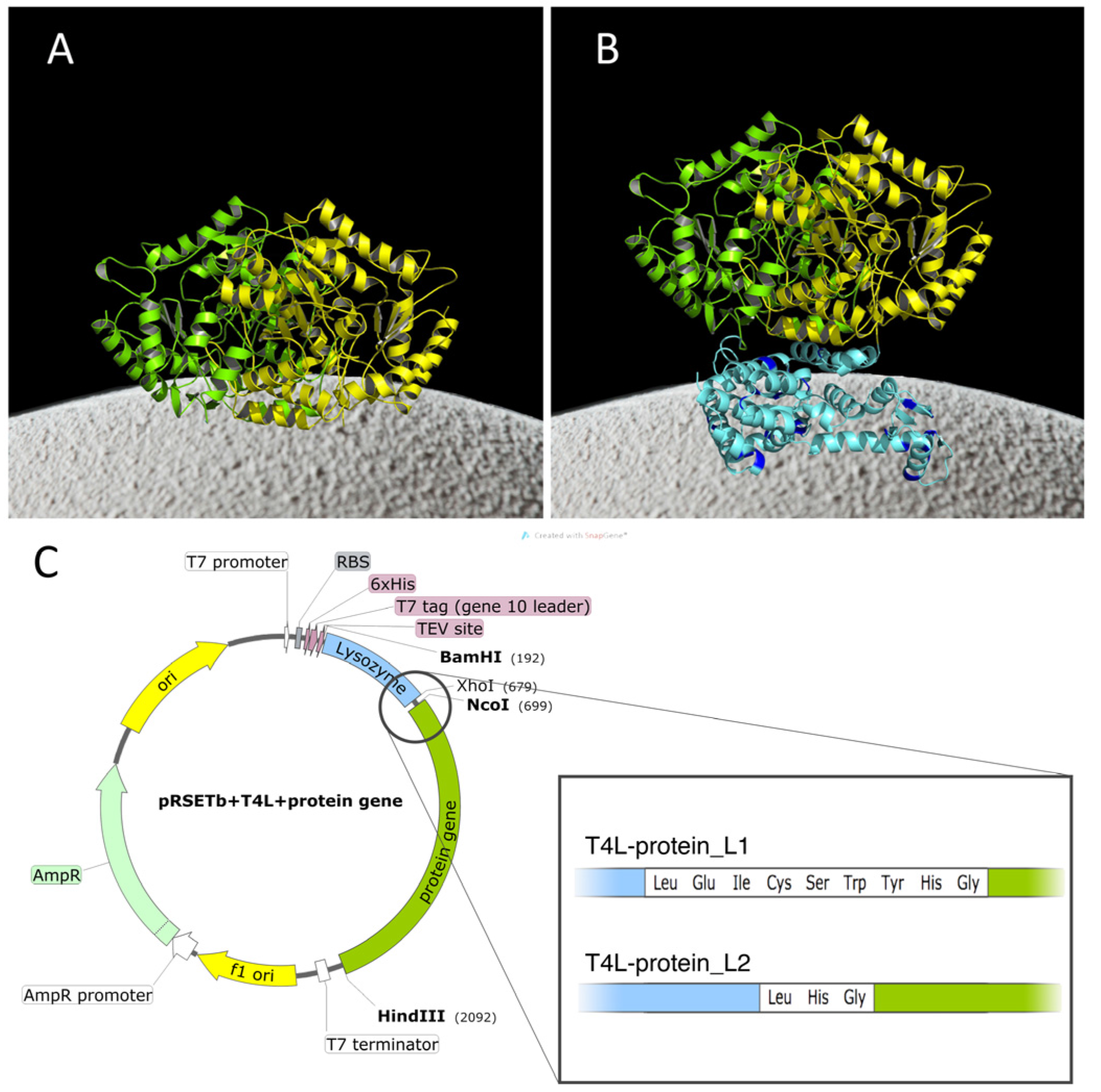
2.1. Construction of the Chimeras
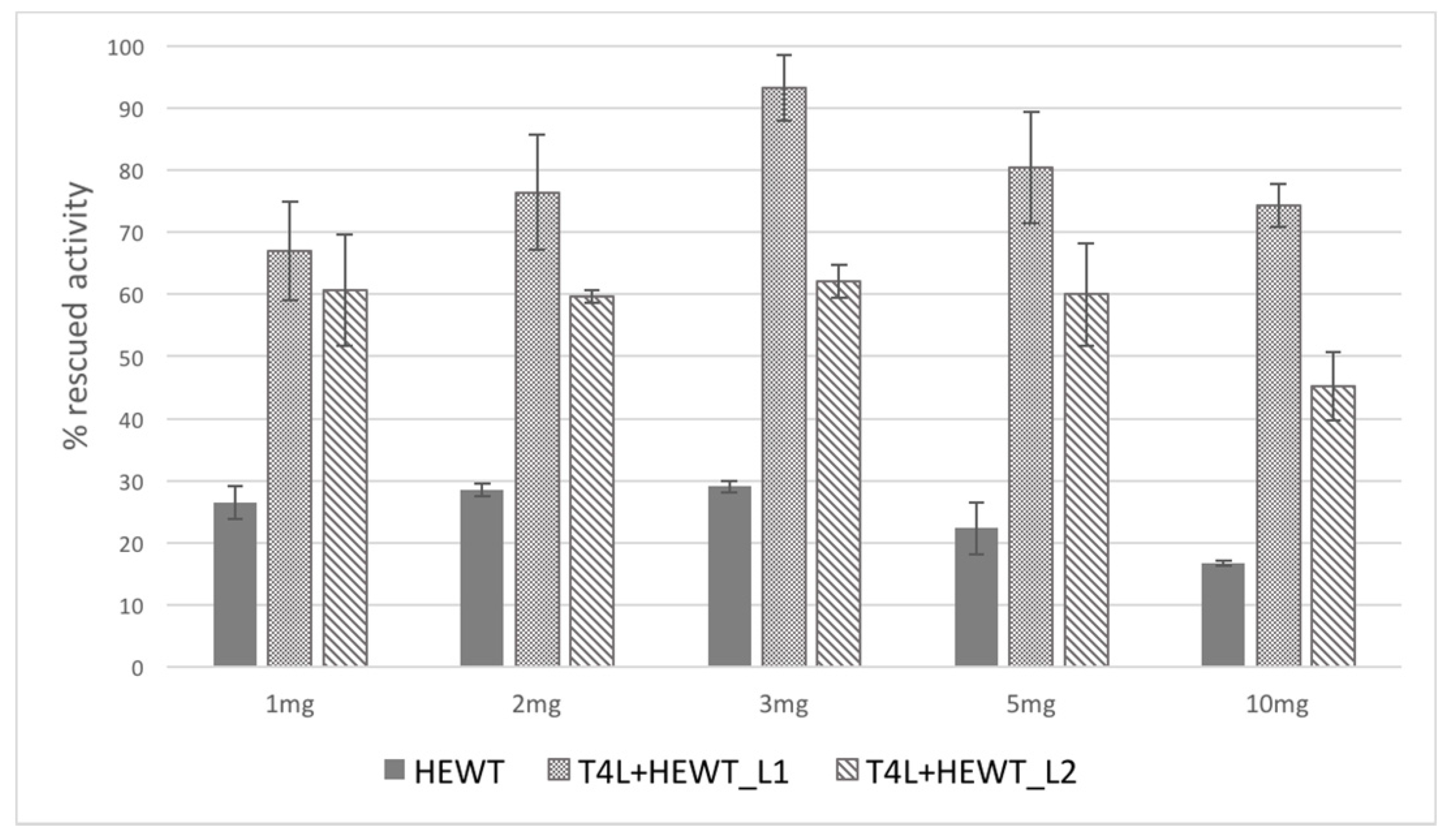
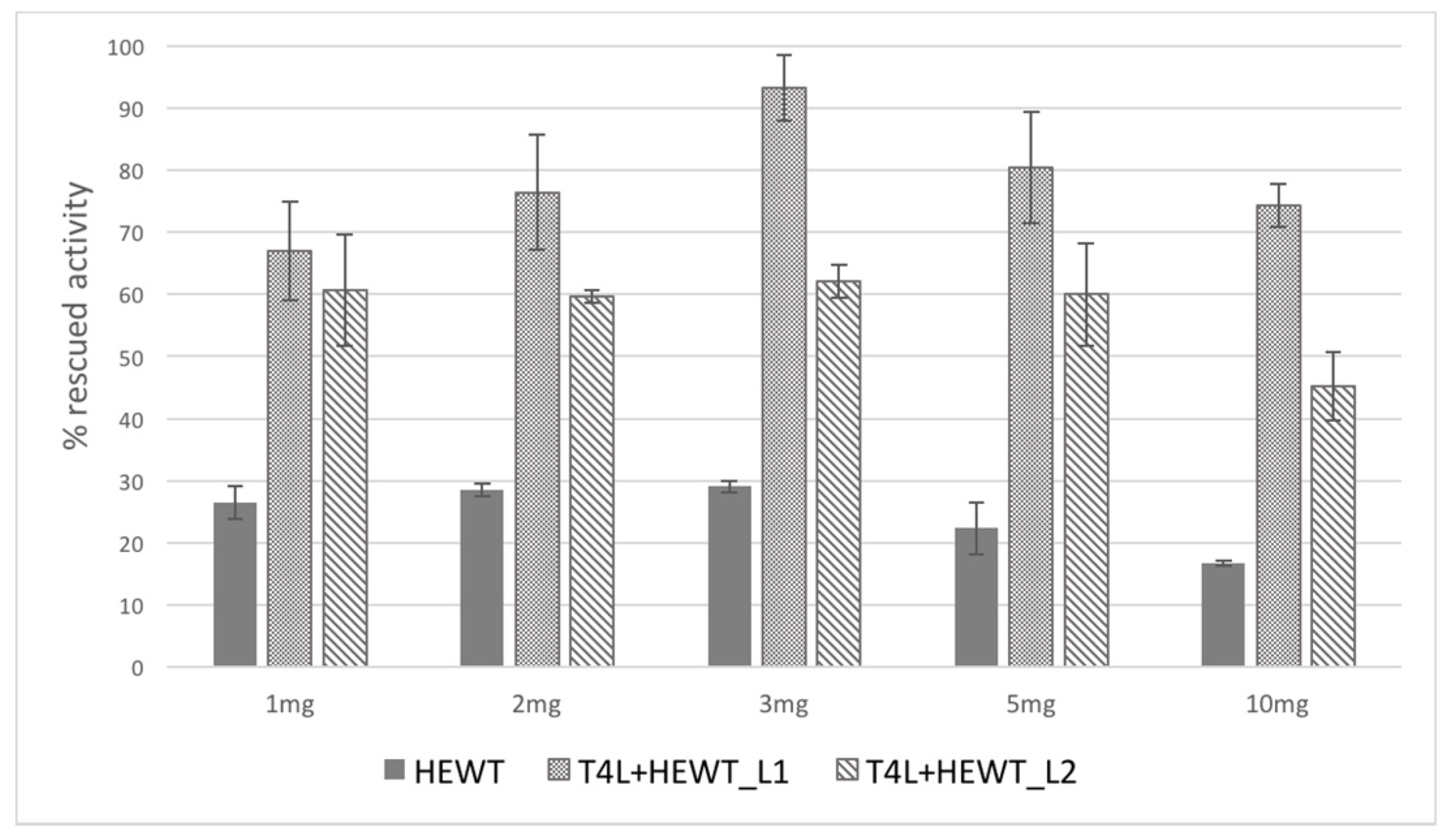
2.2. Halomonas elongata Aminotransferase
2.3. Bacillus subtilis Esterase
2.4. Horse Liver Alcohol Dehydrogenase
3. Discussion
4. Materials and Methods
4.1. T4L- HEWT, T4L-BS2m, and T4L-HLADH Constructs Generation
4.2. Expression, Purification, and Characterization of the HEWT, BS2m, HLADH, and T4L Proteins in E. coli
4.3. Immobilisation of HEWT, BS2m, and HLADH
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wenda, S.; Illner, S.; Mell, A.; Kragl, U. Industrial biotechnology—The future of green chemistry? Green Chem. 2011, 13, 3007. [Google Scholar] [CrossRef]
- Nestl, B.M.; Hammer, S.C.; Nebel, B.A.; Hauer, B. New generation of biocatalysts for organic synthesis. Angew. Chem. Int. Ed. 2014, 53, 3070–3095. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, U.; Cao, L.; Magner, E. Enzyme immobilisation: Fundamentals and application. Chem. Soc. Rev. 2013, 42, 6211–6212. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Galan, C.; Berenguer-Murcia, A.; Fernandez-Lafuente, R.; Rodrigues, R.C. Potential of different enzyme immobilization strategies to improve enzyme performance. Adv. Synth. Catal. 2011, 353, 2885–2904. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.S.; Lee, C.; Kim, J.; Kyung, D.; Kim, B.G.; Lee, Y.S. Covalent immobilization of ω-transaminase from Vibrio fluvialis JS17 on chitosan beads. Process Biochem. 2007, 42, 895–898. [Google Scholar] [CrossRef]
- Guisan, J.M. Immobilization of Enzymes and Cells IN Series Editor. Immobilization of Enzymes and Cells; Humana Press Inc.: New York, NY, USA, 2013; Volume 1051. [Google Scholar]
- Shuai, W.; Das, R.K.; Naghdi, M.; Brar, S.K.; Verma, M. A review on the important aspects of lipase immobilization on nanomaterials. Biotechnol. Appl. Biochem. 2017, 64, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Strategies for the one-step immobilization-purification of enzymes as industrial biocatalysts. Biotechnol. Adv. 2015, 33, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Christena, L.R.; Rajaram, Y.R.S. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Jeon, C.O. High-throughput recombinant protein expression in Escherichia coli: Current status and future perspectives. Open Biol. 2016, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, T.S.; Matt, R.; Weis, W.I.; Kobilka, B.K. Modified T4 Lysozyme Fusion Proteins Facilitate G Protein-Coupled Receptor Crystallogenesis. Structure 2014, 22, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Wiesbauer, J.; Bolivar, J.M.; Mueller, M.; Schiller, M.; Nidetzky, B. Oriented Immobilization of Enzymes Made Fit for Applied Biocatalysis: Non-Covalent Attachment to Anionic Supports using Zbasic2 Module. ChemCatChem 2011, 3, 1299–1303. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Nidetzky, B. Oriented and selective enzyme immobilization on functionalized silica carrier using the cationic binding module Z-basic2: Design of a heterogeneous D-amino acid oxidase catalyst on porous glass. Biotechnol. Bioeng. 2012, 109, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Fernandez-Lorente, G.; Cortes, E.; Garcia, J.L.; Fernandez-Lafuente, R.; Guisan, J.M. One-step purification, covalent immobilization, and additional stabilization of poly-his-tagged proteins using novel heterofunctional chelate-epoxy supports. Biotechnol. Bioeng. 2001, 76, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Cerioli, L.; Planchestainer, M.; Cassidy, J.; Tessaro, D.; Paradisi, F. Characterization of a novel amine transaminase from Halomonas elongata. J. Mol. Catal. B Enzym. 2015, 120, 141–150. [Google Scholar] [CrossRef]
- Planchestainer, M.; Contente, M.L.; Cassidy, J.; Molinari, F.; Tamborini, L.; Paradisi, F. Continuous flow biocatalysis: Production and in-line purification of amines by immobilised transaminase from Halomonas elongata. Green Chem 2017, 53, 3007–3048. [Google Scholar] [CrossRef]
- Contente, M.L.; Dall’Oglio, F.; Tamborini, L.; Molinari, F.; Paradisi, F. Highly Efficient Oxidation of Amines to Aldehydes with Flow-based Biocatalysis. ChemCatChem 2017, 9, 3843–3848. [Google Scholar] [CrossRef]
- Quaglia, D.; Irwin, J.A.; Paradisi, F. Horse liver alcohol dehydrogenase: New perspectives for an old enzyme. Mol. Biotechnol. 2012, 52, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Hackenschmidt, S.; Moldenhauer, E.J.; Behrens, G.A.; Gand, M.; Pavlidis, I.V.; Bornscheuer, U.T. Enhancement of promiscuous amidase activity of a Bacillus subtilis esterase by formation of a π-π Network. ChemCatChem 2014, 6, 1015–1020. [Google Scholar] [CrossRef]
- Zou, Y.; Weis, W.I.; Kobilka, B.K. N-Terminal T4 Lysozyme Fusion Facilitates Crystallization of a G Protein Coupled Receptor. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.S.; Mahoney, R.R. Enzyme thermostabilization by bovine serum albumin and other proteins: Evidence for hydrophobic interactions. Biotechnol. Appl. Biochem. 1995, 22, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Xing, Z.; Yang, J.; Jiang, W.; Zhang, G.; Tang, J.; Li, Q. Construction of an Immobilized Thermophilic Esterase on Epoxy Support for Poly(ε-caprolactone) Synthesis. Molecules 2016, 21, 796. [Google Scholar] [CrossRef] [PubMed]
- Sousa, H.A.; Rodrigues, C.; Klein, E.; Afonso, C.A.M.; Crespo, J.G. Immobilisation of pig liver esterase in hollow fibre membranes. Enzyme Microb. Technol. 2001, 29, 625–634. [Google Scholar] [CrossRef]
- Mei, Y.; Kumar, A.; Gross, R.A. Probing water-temperature relationships for Lipase-catalyzed lactone ring-opening polymerizations. Macromolecules 2002, 35, 5444–5448. [Google Scholar] [CrossRef]
- Pierre, S.J.; Thies, J.C.; Dureault, A.; Cameron, N.R.; Van Hest, J.C.M.; Carette, N.; Weberskirch, R. Covalent enzyme immobilization onto photopolymerized highly porous monoliths. Adv. Mater. 2006, 18, 1822–1826. [Google Scholar] [CrossRef]
- Quaglia, D.; Pori, M.; Galletti, P.; Emer, E.; Paradisi, F.; Giacomini, D. His-tagged Horse Liver Alcohol Dehydrogenase: Immobilization and application in the bio-based enantioselective synthesis of (S)-arylpropanols. Process Biochem. 2013, 48, 810–818. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S.; Benkovic, S.J. Relating protein motion to catalysis. Annu. Rev. Biochem. 2006, 75, 519–541. [Google Scholar] [CrossRef] [PubMed]
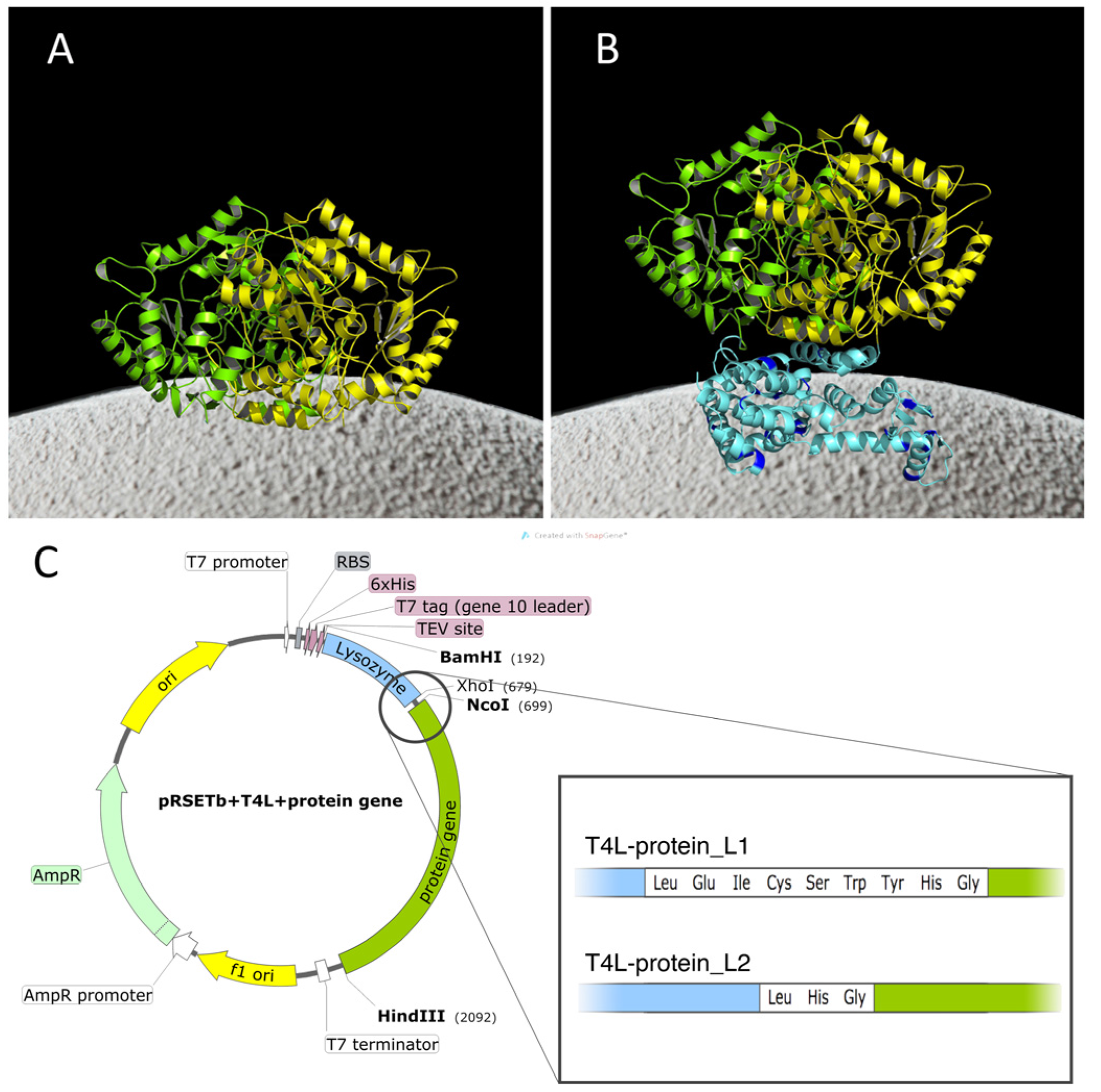

{kind=link}
{kind=link}
{kind=link}
| Free Enzyme | Immobilised Enzyme onto Sepabeads EC-EP/S (Pore ø 10–20 nm) | Immobilised Enzyme onto ReliZyme EP403/S (Pore ø 20–40 nm) | ||||
|---|---|---|---|---|---|---|
| Specific Activity (U/mg) | Expression (mg/L) | Specific Activity (U/g) | Rescued Activity (%) | Specific Activity (U/g) | Rescued Activity (%) | |
| HEWT | 4.51 ± 0.05 | 40 | 5.1 ± 0.8 | 22.3 | 5.8 ± 0.6 | 26.1 |
| T4L-HEWT_L1 | 1.37 ± 0.08 | 12 | 5.5 ± 0.5 | 80.4 | 6.0 ± 0.4 | 89.1 |
| T4L-HEWT_L2 | 0.50 ± 0.07 | 20 | 1.5 ± 0.3 | 59.8 | 2.1 ± 0.1 | 84.1 |
| Free Enzyme | Immobilised Enzyme onto Sepabeads EC-EP/S (Pore ø 10–20 nm) | Immobilised Enzyme onto ReliZyme EP403/S (Pore ø 20–40 nm) | ||||
|---|---|---|---|---|---|---|
| Specific Activity (U/mg) | Expression (mg/L) | Specific Activity (U/g) | Rescued Activity (%) | Specific Activity (U/g) | Rescued Activity (%) | |
| BS2m | 61 ± 4 | 30 | 1.3 ± 0.1 | 0.4 | 1.6 ± 0.3 | 0.5 |
| T4L-BS2m_L1 | 31 ± 3 | 15 | 5.5 ± 0.8 | 3.6 | 6.2 ± 0.8 | 3.9 |
| T4L-BS2m_L2 | 35 ± 2 | 13 | 2.9 ± 0.6 | 1.7 | 3.7 ± 0.4 | 2.1 |
| Free Enzyme | Immobilised Enzyme onto Sepabeads EC-EP/S (Pore ø 10–20 nm) | Immobilised Enzyme onto ReliZyme EP403/S (Pore ø 20–40 nm) | ||||
|---|---|---|---|---|---|---|
| Specific Activity (U/mg) | Expression (mg/L) | Specific Activity (U/g) | Rescued Activity (%) | Specific Activity (U/g) | Rescued Activity (%) | |
| HLADH | 2.6 ± 0.3 | 20 | 0.80 ± 0.08 | 30.8 | 1.11 ± 0.05 | 32.4 |
| T4L-HLADH_L1 | 0.18 ± 0.01 | >1 | 0.08 ± 0.01 | 48.1 | 0.07 ± 0.02 | 38.9 |
| T4L-HLADH_L2 | 0.15 ± 0.01 | >1 | 0.07 ± 0.02 | 45.7 | 0.06 ± 0.01 | 39.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Planchestainer, M.; Roura Padrosa, D.; Contente, M.L.; Paradisi, F. Genetically Fused T4L Acts as a Shield in Covalent Enzyme Immobilisation Enhancing the Rescued Activity. Catalysts 2018, 8, 40. https://doi.org/10.3390/catal8010040
Planchestainer M, Roura Padrosa D, Contente ML, Paradisi F. Genetically Fused T4L Acts as a Shield in Covalent Enzyme Immobilisation Enhancing the Rescued Activity. Catalysts. 2018; 8(1):40. https://doi.org/10.3390/catal8010040
Chicago/Turabian StylePlanchestainer, Matteo, David Roura Padrosa, Martina Letizia Contente, and Francesca Paradisi. 2018. "Genetically Fused T4L Acts as a Shield in Covalent Enzyme Immobilisation Enhancing the Rescued Activity" Catalysts 8, no. 1: 40. https://doi.org/10.3390/catal8010040
APA StylePlanchestainer, M., Roura Padrosa, D., Contente, M. L., & Paradisi, F. (2018). Genetically Fused T4L Acts as a Shield in Covalent Enzyme Immobilisation Enhancing the Rescued Activity. Catalysts, 8(1), 40. https://doi.org/10.3390/catal8010040