1. Introduction
It is undeniable that the biggest constraint for the wide use of hydrogen as a powerful and environmentally friendly fuel is the difficulty of its transport and storage. In this perspective, any effort to improve the transport and storage of the fuel becomes a great step forward to the use of the hydrogen fuel.
Alkaline fuel cells fed with hydrogen constitute one of the most environmentally friendly electrochemical devices for energy production developed to date. However, for these cells to become a viable alternative in fuel production, it is necessary to develop storage and transport in these cells even further, so that they meet practical and economic demands. Hydrogen production from water electrolysis at the purity levels required to feed fuel cells is still expensive [
1], not to mention that CO originating from the biomass reforming process causes electrode passivation [
2]. To overcome these limitations, one reasonable approach would be to generate hydrogen in a plant that produces electricity at low cost; i.e., a hydroelectric plant, and then transform it into a compound that is easier to transport and use in the PEM fuel cell located at the desired site. Sodium borohydride (NaBH
4) fits such a purpose well—it originates from the reaction between NaH and B(CH
3O)
3, the so-called Brown- Schlesinger reaction [
3].
NaBH
4 is a very stable white solid that melts at 400 °C at ordinary room pressure. It serves as a useful hydrogen vector with the additional bigger advantage of easy and safe transport and storage in comparison to the hydrogen gas. Once it reaches the site of electricity generation, NaBH
4 acts as a hydrogen source that feeds the fuel cell. Electrochemical NaBH
4 oxidation involves a series of chemical and electrochemical steps [
4], as follows:
or
These processes involve a slightly more complex mechanism than direct hydrogen gas electrooxidation. In both cases, H2 originates from preceding chemical steps followed by direct fuel electrooxidation (Steps (2) or (5)) or another electrochemical intermediate step that also produces H2 (Step (4)). Even so, these pathways are thermodynamically and kinetically more favorable than the electrooxidation of any organic compound. Despite these advantages, investigations into active electrode materials for electrochemical borohydride oxidation are not conclusive, and the development of more efficient, stable, and inexpensive electrocatalysts remains a matter of extreme urgency.
Several papers have reported on the use of borohydride in fuel cells. Studies have focused on the performance of Pt, other noble metals, and a few other metallic materials toward borohydride [
5,
6,
7,
8,
9]. Only a few papers have attempted to clarify the process of borohydride electrooxidation (BOR) on different electrode materials.
The last decade saw the publication of some interesting works.
Gyenge [
10] used cyclic voltammetry, potentiometry, amperometry, coulometry, and electrochemical impedance spectroscopy to study BOR on Pt and Au in NaOH solution. This author observed that catalytic borohydride hydrolysis, which produces H
2, as well as direct ion oxidation at more positive potentials, influenced the voltammetric behavior of borohydride on Pt. The author also found that thiourea inhibited hydrolysis effectively, and that only the direct pathway occurred in the presence of this compound. To develop electrode materials for borohydride-fed fuel cells, Liu et al. [
11] studied BOR on several electrocatalysts (Ni, Ni Raney, Pd, Pt, Cu, and Au). The open circuit potential depended on borohydride concentration and catalyst nature. The authors stated that BOR comprised a multi-step reaction with distinct pathways, and that the pathways depended on reaction conditions and catalyst nature. Martins et al. [
12] employed cyclic voltammetry to evaluate BOR on Pt in the presence and absence of thiourea. These authors confirmed that thiourea suppressed hydrolysis and found that the optimum thiourea/NaBH
4 concentration ratio was 0.6. Hydrolysis inhibition increased the coulombic efficiency of the fuel cell and drove eight electrons to the oxidation process. Concha and Chatenet [
13] investigated BOR on Pt, Ag, and Pt-Ag (alloy) nanoparticles by cyclic voltammetry in NaOH solution. To compare different electrocatalysts, these authors measured the kinetic parameters and estimated the number of electrons exchanged by the borohydride anion (faradaic efficiency), to find that BOR was faster on Pt/C than on Ag/C even though both materials afforded similar Tafel slope values. The authors suggested that Pt/C promoted borohydride hydrolysis followed by direct hydrogen oxidation or direct BOR. As for Ag/C, the authors suggested that direct but incomplete BOR occurred on this electrode material. BOR on Pt-Ag alloys provided non-consistent data. Simões, Baranton, and Countanceau [
14] verified how Pd, Au, and bimetallic Pd-Au nanoparticles supported on carbon, in different compositions and in alkaline medium, performed during BOR. Compared with Pt and Au, Pd
xAu
1−x/C catalysts presented lower open circuit potential. By means of voltammetric measurements using a rotating disc electrode and hydrogen evolution estimates, these authors proposed that BOR on Pd-based nanocatalysts took place via two pathways: at negative potentials, the process involved a BH
3OH
− intermediate that produced H
2, whereas direct BOR involving a six electron pathway occurred at higher overpotentials. Finkelstein et al. [
4] conducted a detailed electrochemical study of the BOR mechanism on Au and Pt surfaces by employing cyclic voltammetry using rotating disc and ring-disc electrode. These authors concluded that the electrode material determined the characteristics of the process; i.e., number of transferred electrons, heterogeneous charge transfer constant (k
b), and presence of coupling reactions and adsorbed intermediates. Au sped up the hydrolysis step and generated H
2 at the electrode surface. On the other hand, two main processes took place on Pt: at low potentials, a seven-electron process produced hydride on the Pt surface, and BH
3OH
− adsorption poisoned the electrode; at higher overpotentials, a five- or six-electron process occurred on partially oxidized Pt surface with adsorbed HO
− and BH
3OH
−. Liu, Yang, and Li [
15] evaluated BOR on Au and Ag electrodes supported on carbon. Reaction efficiency depended on the [HO
−]/[BH
4−] ratio in the solution. A reaction involving nearly eight electrons took place for [HO
−]/[BH
4−] ≥ 5. Otherwise, H
2 gas evolved and the fuel efficiency decreased. The authors suggested that the BH
3OH
− ion originating from the chemical step diminished process efficiency. Supported by DFT calculations and using quartz crystal microbalance (EQCM). Lam et al. [
16] investigated BOR on Pt under different experimental conditions, in the presence and absence of thiourea. The authors proposed that thiourea affected the electrode process, and that stable intermediates probably participated in the reaction. Santos et al. [
17] compared the performances of Pt-rare Earth binary alloys with Pt as anode towards the BOR, by employing cyclic voltammetry and chronoamperometry techniques in the range of temperature of 25–55 °C. The calculated transfer coefficient ranged from 0.75 to 0.81 for Pt, Pt-Ho < Pt-Sm, and Pt-Ce materials. The number of transferred electrons were from 1.4 to 2.0 for the Pt-RE (rare earth) materials while for Pt it was increased to the range 2.5–3.1. The kinetics parameters showed the best performance for the Pt-Ho alloy. The authors concluded that the Pt-RE alloys do not presented better performance than pure Pt for the BOR. Hou and co-workers [
18] published a paper that dealt with the performance of commercially available Pt
3Sn/C (40 wt %, E-TEK) as anode for borohydride electrooxidation reaction in alkaline medium. The authors compared the results of Pt
3Sn and Pt (30 wt %, De Nora) under the same experimental conditions and concluded on the crucial role of OH species in the electrode process. Even though they did not succeed determining
ik for the processes occurring at both surfaces due to uncommon Koutecky–Levich relationship, it was proposed a mechanism based on the experimental findings like as number of transferred electrons and dependence on the OH species concentration. Merino-Jimenez et al. [
19] evaluated the Pd-Ir (1:1) alloy covered by carbon microfibers and supported on a titanium plate as anode for the BOR. The authors calculated the number of transferred electrons in different concentration NaOH solutions and electrode potentials. The authors concluded that the Ir presented a beneficial effect enhancing the Pd catalytic activity towards the BOR. This enhancement was attributed to improvement of the BOR instead the hydrogen evolution reaction. Sljukic et al. [
20] reported the results obtained from the Pt-Dy alloys for the BOR. According the results, the number of transferred electrons was found to be about 2.5. Cyclic voltammetry essays presented higher values for the current densities for Pt
0.4Dy
0.6 electrode in comparison to the Pt
0.5Dy
0.5, for the whole range of temperatures studied (25–55 °C). The alloys showed performances comparable to the pure Pt at room temperature and higher coulombic efficiency at higher temperatures. Direct Boroydride Fuel Cell tests presented promising results for the material Pt
0.4Dy
0.6 employed as anode, where the power density at 25 °C was 298 mW cm
−1 and the current density was 595 mA cm
−2 at 0.5 V potential.
An interesting study was reported by Sljukic et al. [
21]. The authors presented the results obtained from the use of Pt/C and PtM/C (Pt
0.75M
0.25, M = Ni or Co) as anode for the BOR by using cyclic voltammetry and linear sweep with rotating disc electrode techniques. The results pointed to higher current density registered for the bimetallic Pt
0.75Co
0.25/C while Pt/C presented the poorer activity. The number of the transferred electrons calculated were 3.5; 6.6 and 4.5 for Pt/C; Pt
0.75Ni
0.25/C and Pt
0.75Co
0.25/C, respectively. The number of transferred electrons decreased with the increase of the NaBH
4 concentration, same effect observed for the temperature change. The authors concluded that the better activity of the bimetallic alloys was credited to alteration of the electronic condition of the Pt sites. In a rare study concerning to the BOR on intermetallic materials, Yi et al. [
22] synthesized and evaluated PtZn bimetallic catalysts supported on carbon and obtained the electrochemical characteristics for the reaction by employing cyclic voltammetry and chronoamperometry techniques. The authors studied three different compositions of the PtZn system and affirmed that the performances were better than Pt/C under the same experimental conditions towards the BH
4− oxidation reaction. Moreover, the authors pointed out the outstanding performance of the catalyst Pt
67Zn
33/C due to the Zn synergetic effect in the alloy. Briega-Martos et al. [
23] published the results obtained from the study of the BOR at Pt single crystal surfaces in NaOH solution by using stationary and rotating conditions. The results showed that the activity towards the BOR is structure sensitive. The (110) face presented the highest current densities at low potentials. At the limit of low potentials the accumulation of hydrolysis products of the borohydride was responsible to the increase of the current under stationary conditions since the products were not adsorbed on the surface and were easily removed under non-stationary diffusion condition. Recently, Grimer et al. [
24] performed a very complete study of the borohydride oxidation reaction at Pd/C by using cyclic voltammetry, rotating disc electrode, chronoamperometry coupled to NMR, and FTIR in situ techniques. According to the analysis of the RDE technique, the number of transferred electrons was 4 at 0.4 V vs. RHE, while at 0.8 V the number of transferred electrons increased to 8 at 0.8 V the reaction is considered complete and at 0.4 V 2 different pathways are possible: one consisting of the total oxidation to BO
2− and the production of two hydrogen molecules and through the second pathway occurred a four electron electrooxidation path resulting in BH
2− species. The authors proposed that at low potentials surface sites are blocked by Pd-BH
2OH species and for higher potentials this intermediate is completely oxidized via an eight electron process.
Electrocatalysis’ research group has published a series of papers on ordered intermetallic PtSn nanoparticles where it was found an outstanding performance of this material towards the hydrogen oxidation reaction (HOR). In this perspective, the aim of the present work was to investigate the kinetic and mechanism of the BOR on ordered intermetallic PtSn/C in alkaline medium by means of the rotating disc electrode and quasi steady-state techniques.
Despite the numerous works investigating BOR on noble metals, the state-of-the-art information presented above shows that the development of new electrocatalysts for this reaction is still incipient. However, a great deal of work is still demanded to improve and make feasible the use of borohydride as a safe energy vector and source of hydrogen for electrochemical devices. So, the present study aims to provide an alternative and highly efficient anode material for BOR in alkaline media.
2. Results and Discussion
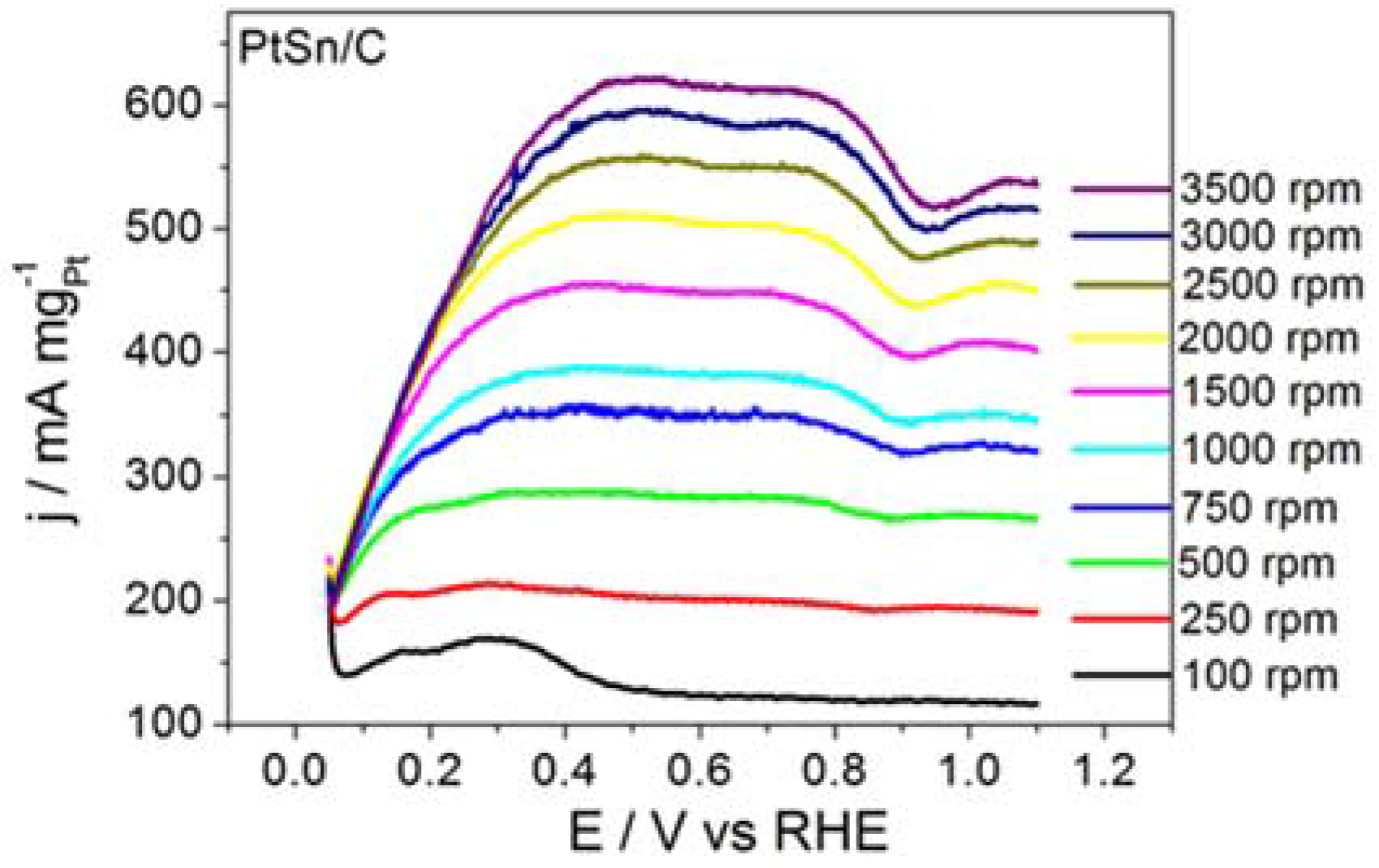
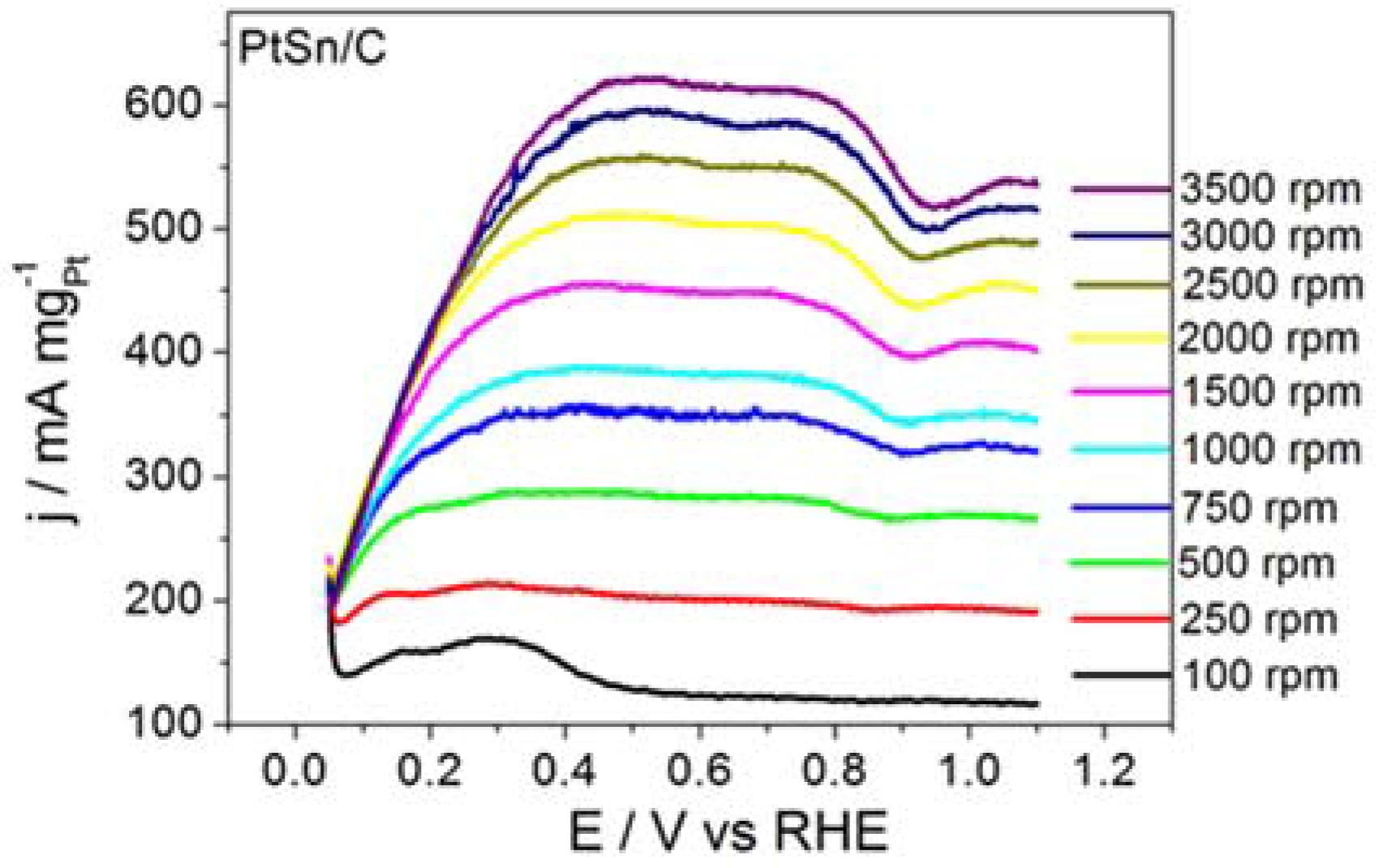
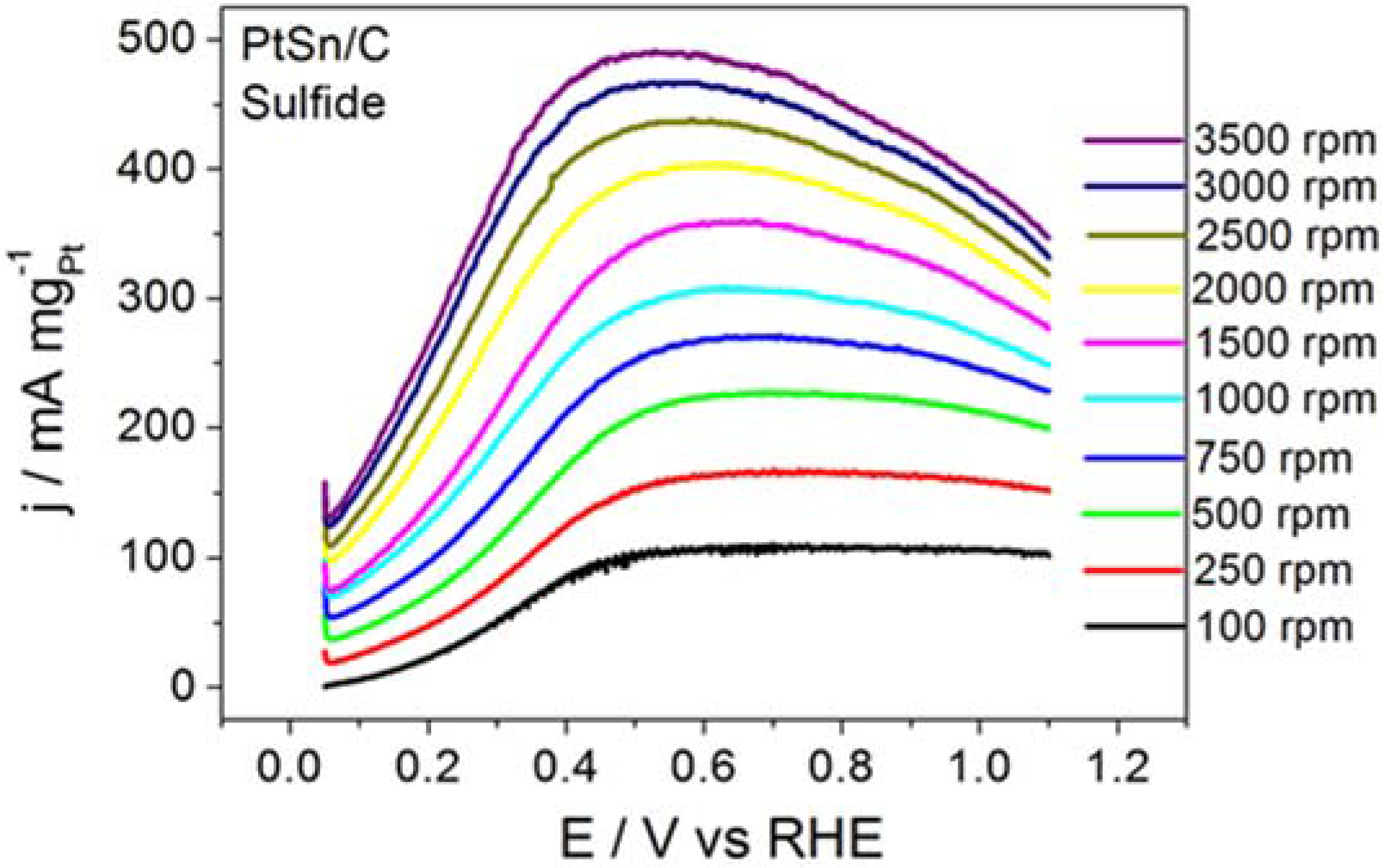
Figure 1 shows the RDE polarization curves obtained for BOR on ordered intermetallic PtSn/C electrodes in alkaline medium. BOR on PtSn/C generated a plateau-like profile that persisted all over the same rotation speed range. The shape of the polarization curve recorded for BOR on PtSn/C suggested that the adsorption and subsequent chemical and electrochemical steps improved along BOR on PtSn/C. Our previous paper had already discussed that adsorption site properties of the ordered intermetallic PtSn/C influenced the kinetics of hydrogen oxidation (HOR) positively [
25]. According to that study, in the intermetallic structure Sn atom exerts a pronounced electronic donor effect to Pt adsorption site that improves in a great deal its adsorption characteristics. Such as improvement is even responsible to the lower CO poisoning susceptibility of the ordered intermetallic PtSn surface in comparison to the Pt.
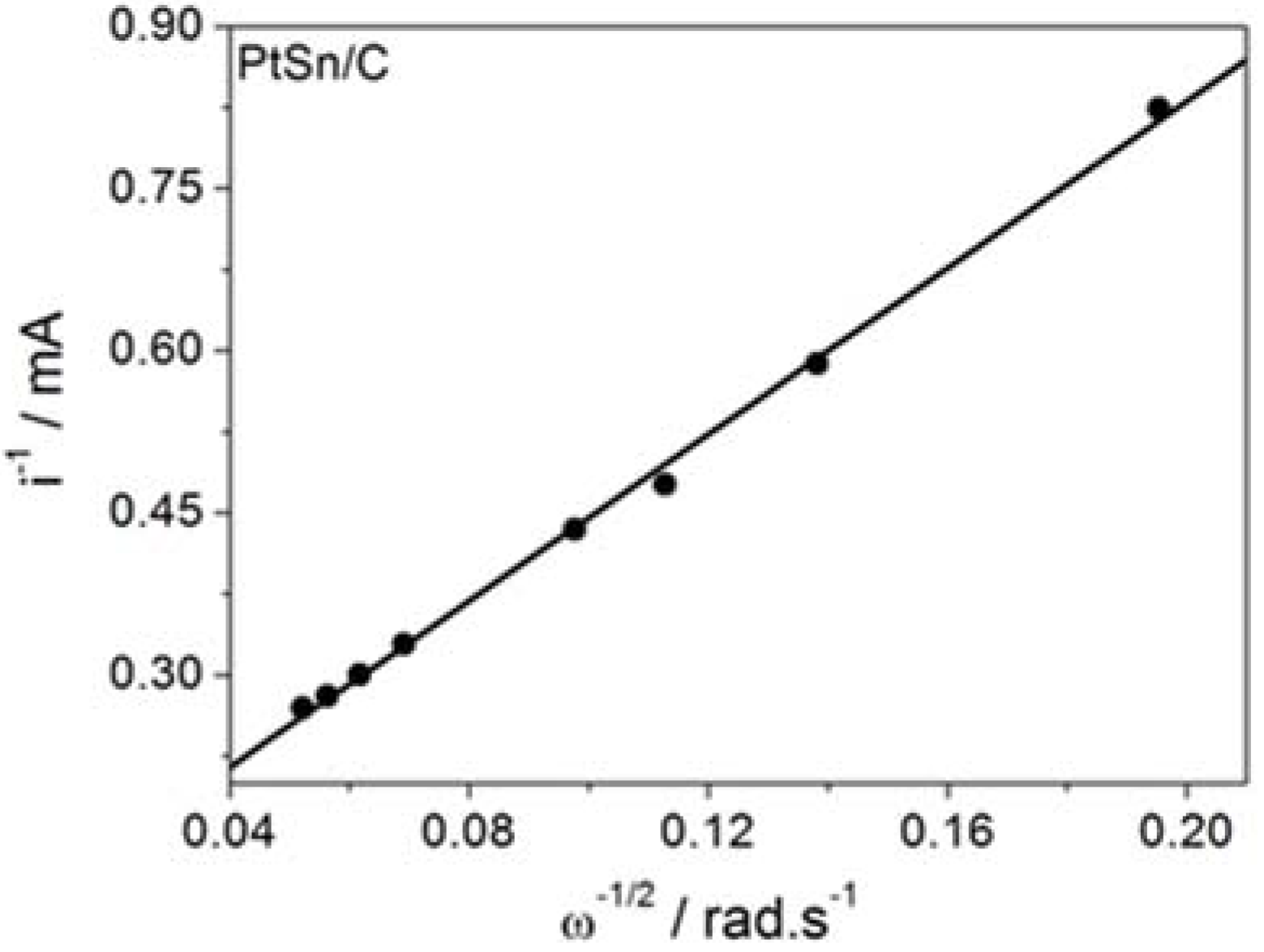
The kinetics parameters were calculated according to the Koutecky–Levich equation
where
ik is the kinetic current density,
id is the diffusion-limited density,
B = 0.620nFD
2/3υ
−1/6, and
co is the BH
4− concentration.
The data extracted from the curves displayed in
Figure 1 helped to build the Koutecky–Levich plots reproduced in
Figure 2 and to determine the kinetic data listed in
Table 1.
The PtSn/C material displayed remarkable kinetic performance. Considering the electrode surface geometric area, the
ik value for the BOR taking place on PtSn/C was 0.08 A cm
−2, which was significantly higher than the 0.06 A cm
−2 value reported by Concha and Chatenet [
13] for the reaction occurring on the surface of Pt/C nanoparticles under similar experimental conditions.
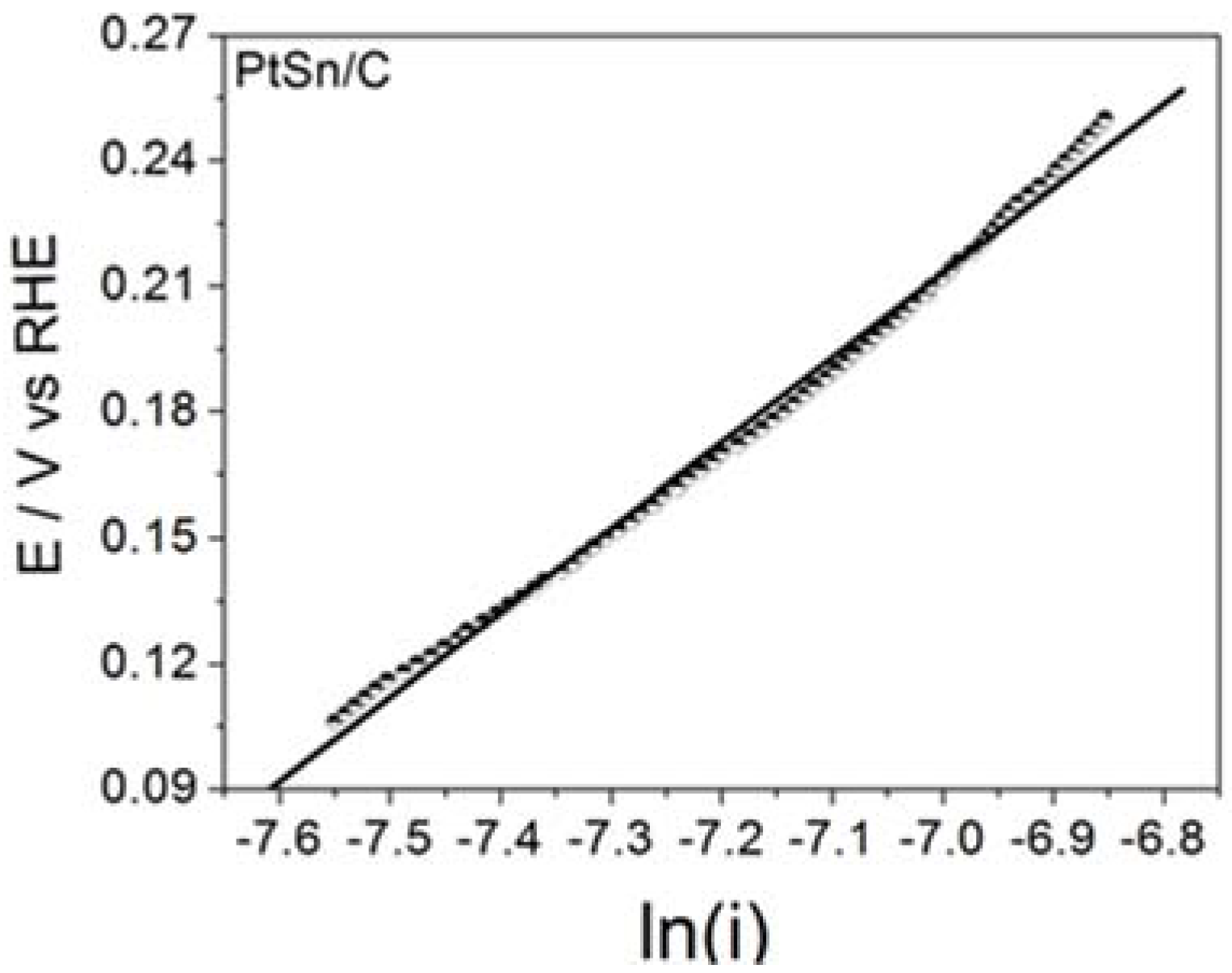
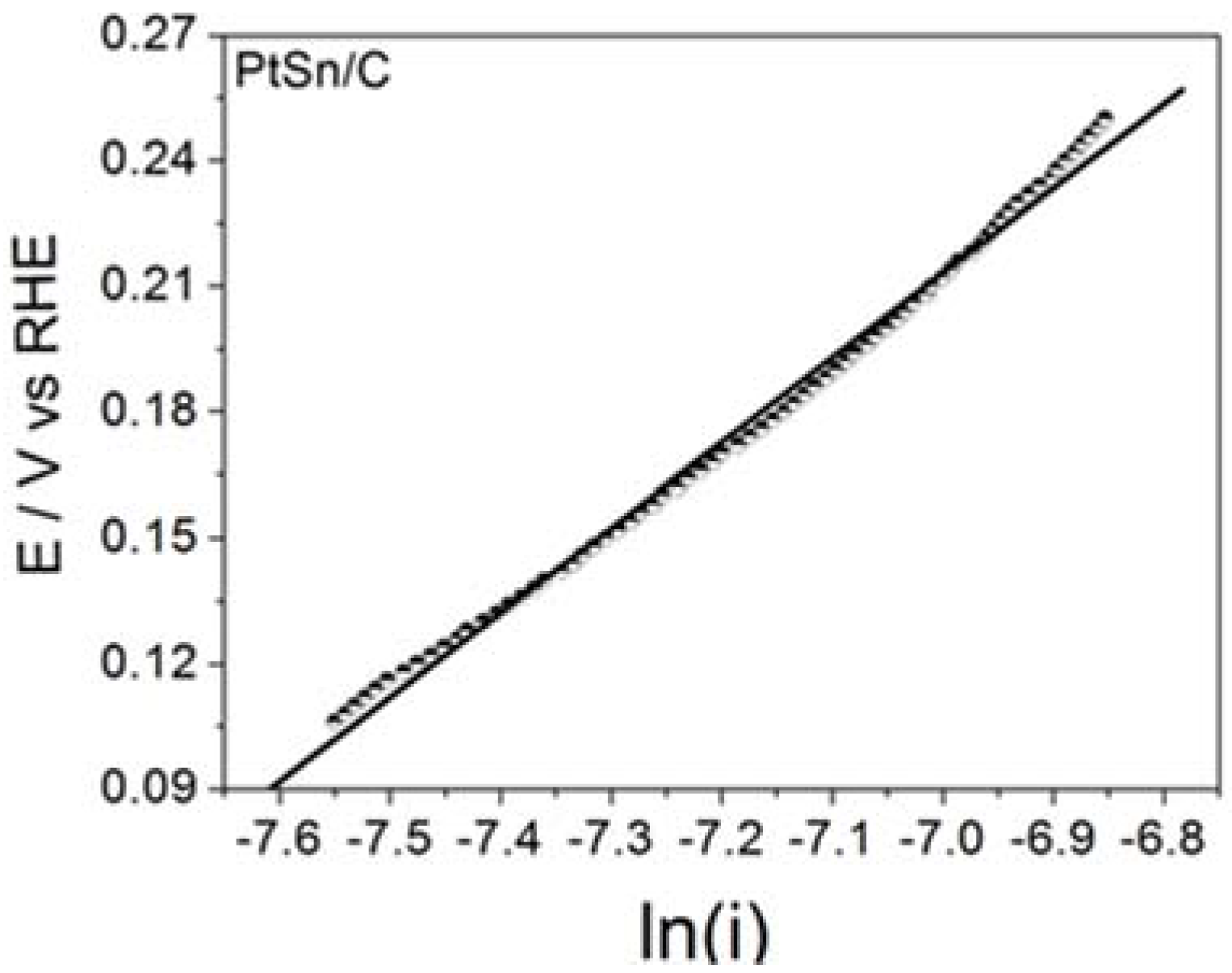
Figure 3 illustrates the steady state Tafel plots built from the data obtained for BOR on the assayed electrode material.
Table 2 summarizes the corresponding
b Tafel parameter (Tafel slope) and the calculated number of transferred electrons.
The surprisingly high
b values obtained for PtSn/C were noteworthy and may have stemmed from the active layer effect [
13], in which the hydrogen gas produced during the chemical (Reactions (1) and (3)) or electrochemical (reaction 4) steps may have been trapped in the porous surface of the electrode for further oxidation. Such a phenomenon could give rise to twice, three times, or even four times higher
b values than the ones predicted for bulk materials (usually 0.059 or 0.12 V dec
−1, depending on the rds) [
7]. The same effect may also have accounted for the number of transferred electrons. The values calculated for the reaction on the materials studied in this work and the values determined for Pt electrodes in the literature agreed well. Therefore, BOR on the ordered intermetallic nanostructured PtSn/C materials and on Pt/C nanoparticles most probably followed the same mechanism, and any improvement in BOR may have resulted from influence of the material properties. On the basis of this viewpoint and as already mentioned for HOR [
25], the enhanced adsorption properties of the Pt sites in PtSn/C may have stemmed from the electronic donor effect of Sn atoms [
26,
27]—this effect may also have greatly contributed to the electrocatalytic activity of PtSn/C toward BOR.
The number of electrons transferred in the reaction was determined by means of the equation [
13]
where
id is the diffusional limited current, n is the number of transferred electrons, C* is the BH
4− concentration in mol L
−1, ω is the electrode rotation speed, D is the BH
4− diffusion coefficient (3.5 × 10
−5 cm
2 s
−1 [
13]), and υ is the kinematic viscosity (1.01 × 10
−2 cm
2 s
−1 [
13]). Hou et al. [
18], in similar experiment on Pt
3Sn/C (E-TEK) found also 8 as the number of transferred electrons. Unfortunately, the authors did not succeed determining the kinetic current
jk.
According to various reports, the presence of thiourea enhances BOR on Pt surfaces because thiourea inhibits borohydride ion hydrolysis and improves hydrogen production and oxidation via faradaic processes [
10,
12,
16]. Hence, in this work, we performed BOR in the presence of thiourea as well as S
−2 to observe the role that these surface site blocking/modifier agents play in BOR kinetics. Is must be pointed out that while it is a rather solution effective agent, S
−2 ion usually acts as surface modifier [
28], so two different effects should be observed as an action of these agents.
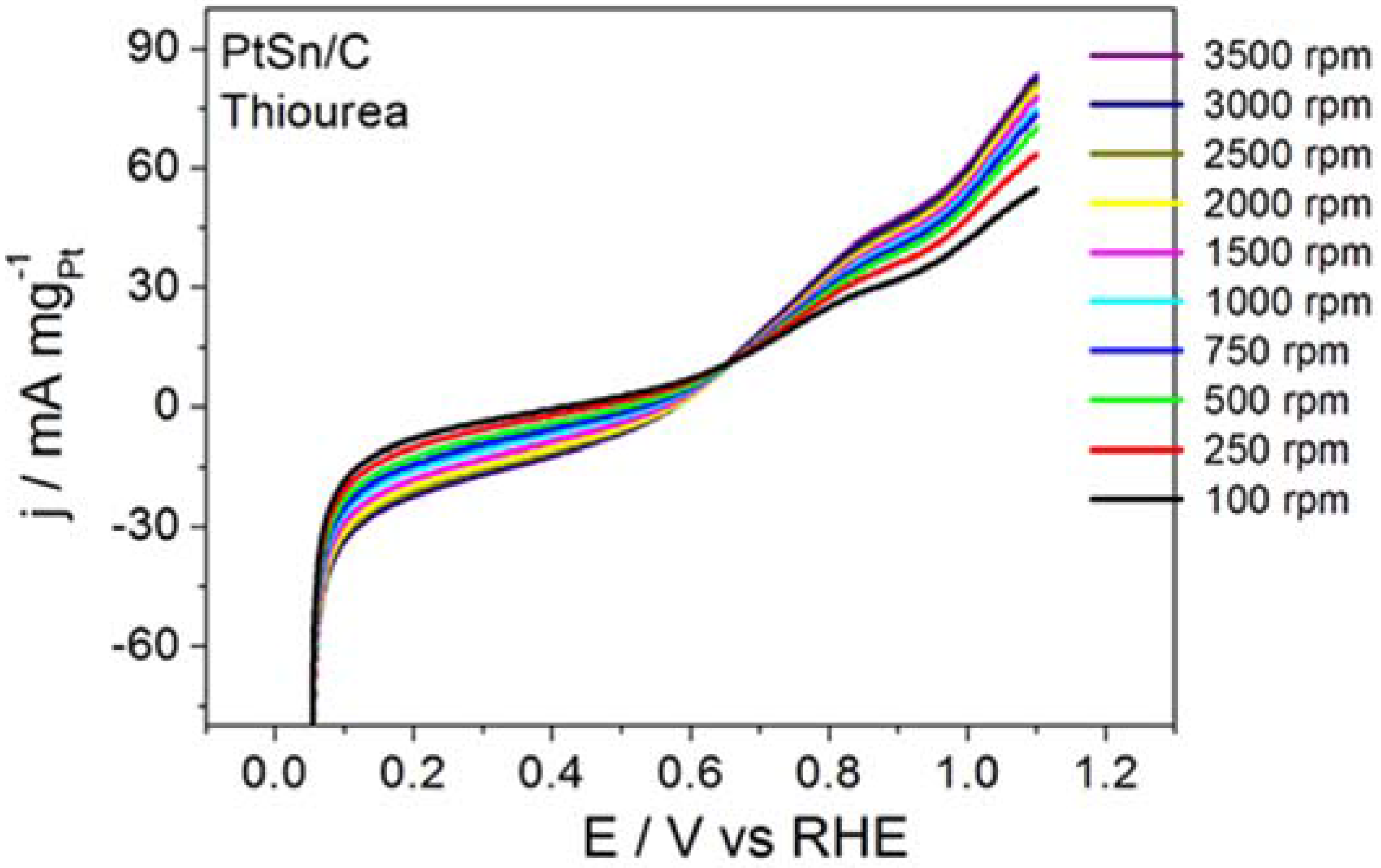
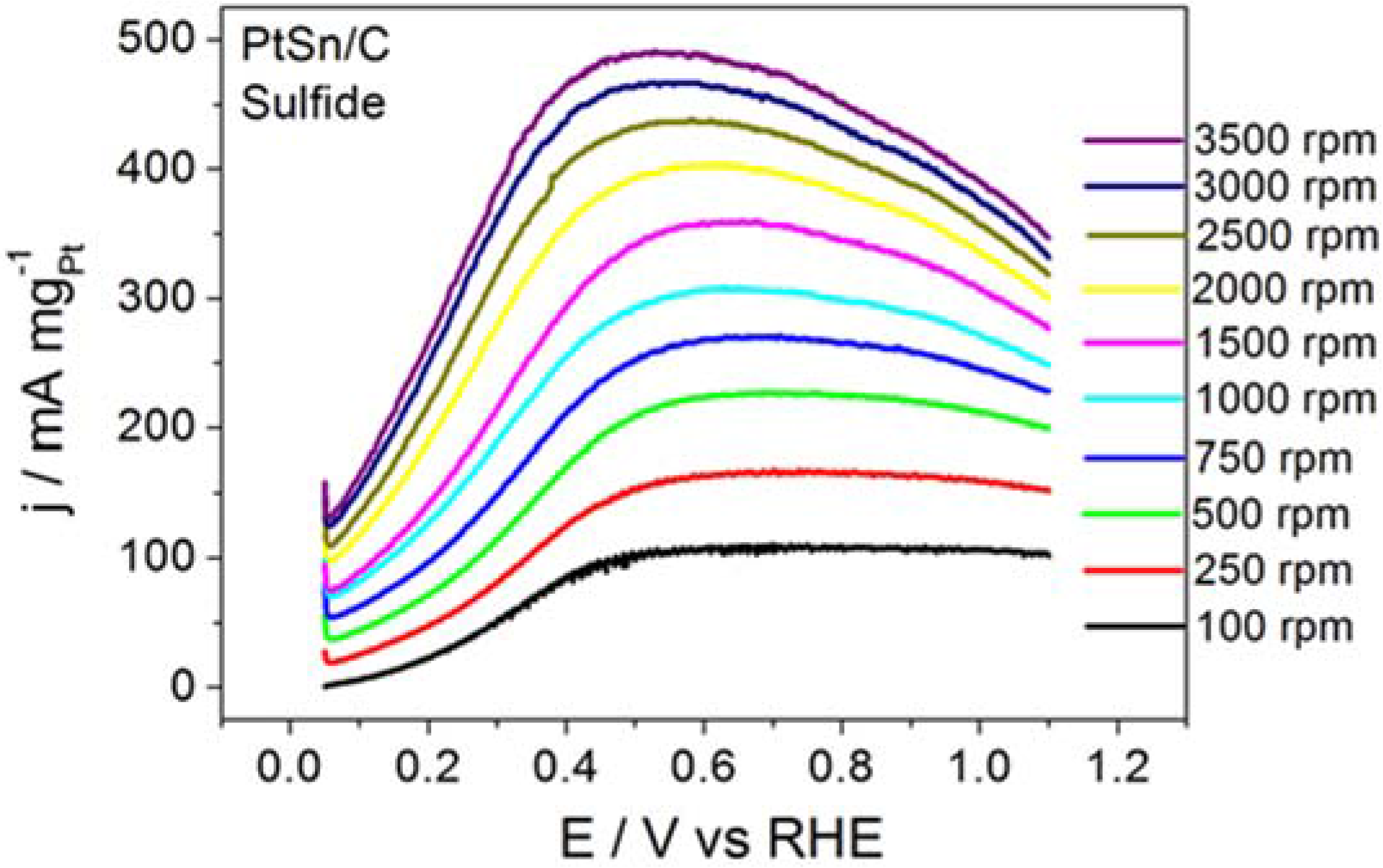
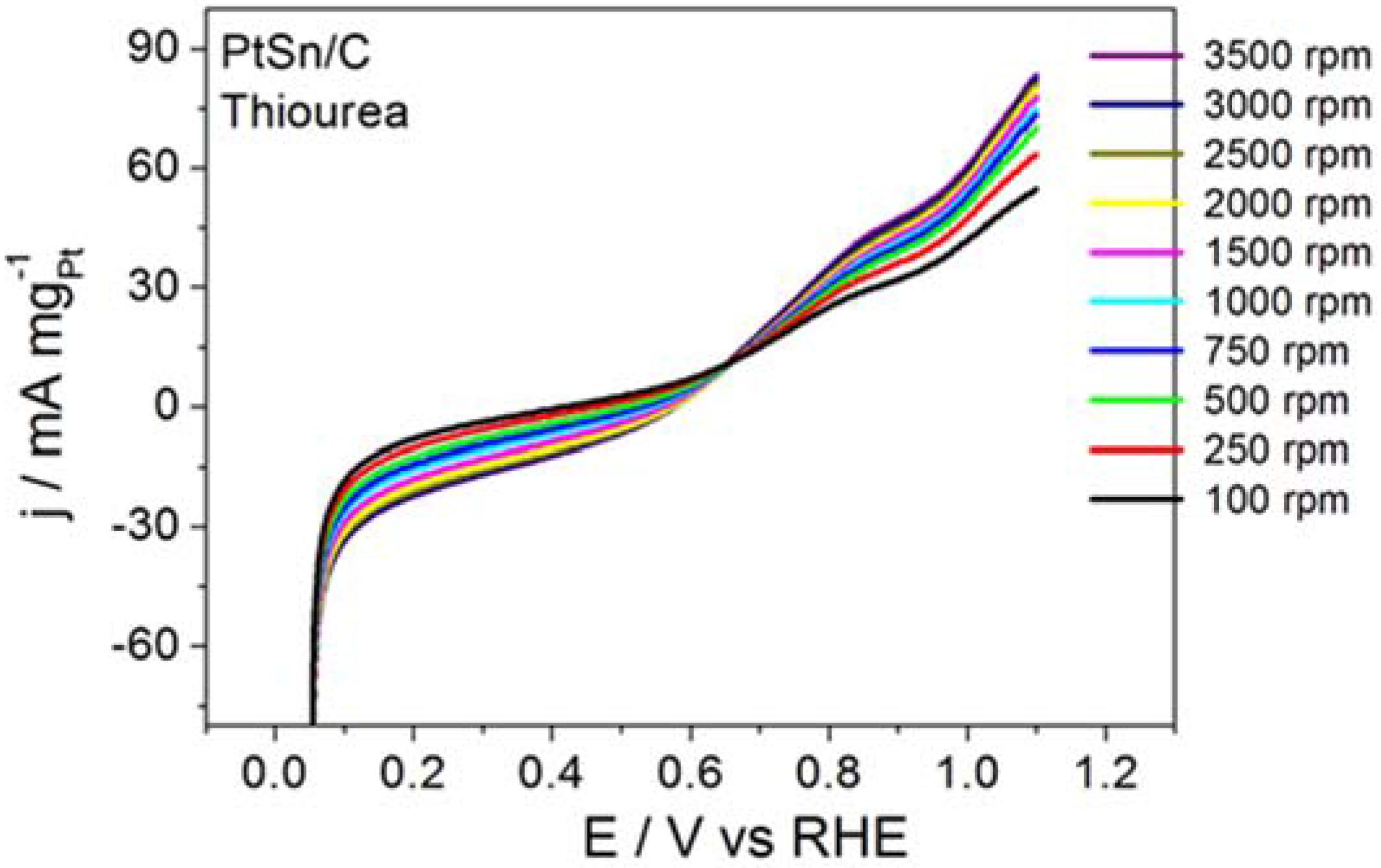
Figure 4 displays the RDE curves obtained for BOR on the PtSn/C material in the presence of thiourea and on S
2−-surface-modified PtSn/C.
Figure 4 also brings the curve recorded for BOR on a clean surface, for comparison purposes.
Thiourea acted intensely on BOR, to inhibit the electrode reaction, as corroborated by the ~0.65 V displacement in the onset potential toward more positive values and by the clearly inhibited electrode process. Although BOR on PtSn/C containing surface-adsorbed S2− did not experience such intense inhibition, the onset potential shifted toward slightly more positive potentials. The profile of the RDE curve also changed, suggesting that the reaction slowed down. The calculated kinetic parameters and the number of transferred electrons obtained from this study were jk = 39.7 mA mgPt−1 and n = 0.6 for the reaction that proceeded in the presence of thiourea, and jk = 2075 mA mgPt−1 and n = 6.4 for the reaction conducted on PtSn/C surface covered with adsorbed S2−. Together, the results reported herein demonstrated that the addition of thiourea or S2− did not enhance BOR on PtSn/C in alkaline medium. It is noteworthy that the addition of thiourea (hydrolysis step inhibitor) exerted a far more intense effect than the surface deposition of S2− (surface site modifier).
Considering the mechanisms earlier mentioned [
4] it is easily realized regarding Step (3) that differences both mechanisms involves an intermediate produced from the hydrolysis step, in solution, that gives rise to the electrochemically generated hydrogen molecule, which, on its turn, is electrooxidized at the electrode surface. Our experimental results showed an almost complete inhibition of the BOR when the reaction was carried out in the presence of thiourea that implies that the main reactant responsible for the H
2 production is being affect by the adsorption of thiourea molecules at the electrode surface. It should be pointed out that S
−2 ion is a surface modifier agent that does not inhibit the hydrogen reaction at the electrode surface [
28]. According to the exposed, it is reasonable to propose that the inhibition of the BOR was caused by the inhibition of Step (4), due to the inhibition of the adsorption of the BH
3HO
− specie that was, on its turn, competitively blocked by the adsorption of the thiourea molecules.
In this perspective, it is possible to propose that the BOR occurs on ordered intermetallic PtSn/C particles in alkaline solution through the mechanism represented by Steps 3–5 as earlier cited in this paper.
Moreover, the proposed mechanism agrees very well to the number of electrons transferred (
n = 8.0) displayed in
Table 2.
This outstanding performance of the PtSn/C material may be attributed to the unique electronic condition of the Pt adsorption surface site caused by the electron donor effect provided by the Sn atoms in the hexagonal geometric structure [
29].
3. Materials and Methods
Ordered intermetallic PtSn nanoparticles supported on carbon (Vulcan XC-72, Cabot) were synthesized via the polyol route proposed by Cable and Schaak [
30]. To this end, proper amounts of H
2Cl
6Pt·6H
2O (Merck, Hohenbrunn, Germany) and SnCl
2 (Merck, Rio de Janeiro, Brazil) were dissolved in tetraethylene glycol (Merck, Hohenbrunn, Germany) in the presence of carbon (CABOT, Billericay, MA, USA) previously treated in N
2 (White Martins, Bauru, Brazil) atmosphere at 400 °C for 4 h. Polyvinylpyrrolidone (PVP, Sigma-Aldrich, St. Louis, MO, USA) served as dispersing agent and stabilizer. Sodium borohydride (NaBH
4, Sigma-Aldrich, St. Louis, MO, USA) was employed as an auxiliary reducing agent. The material was deposited on a polished glassy carbon surface as a thin film obtained from a dispersion consisting of the supported nanoparticles, isopropyl alcohol (Sigma-Aldrich, St. Louis, MO, USA), Nafion 117
® (Sigma-Aldrich, St. Louis, MO, USA), and deionized water. The as-prepared surface was submitted to electrochemical studies. Characterization of the electrode material was performed by X-ray Diffraction, Electron Dispersive Spectroscopy, and Transmission Electron Microscopy (
Figures S1, S2 and Table S1) [
29].
The borohydride electrooxidation reaction (BOR) was studied by adding sodium borohydride (Sigma-Aldrich, St. Louis, MO, USA) to a solution of KOH (Merck, Darmstadt, HE, Germany) at 0.2 mol L
−1, which afforded a solution of BH
4− at 1 mmol L
−1. The as-prepared solution was deaerated by means of N
2 (White Martins, Bauru, SP, Brazil) bubbling for 15 min before each set of electrochemical measurements. A large surface Pt wire and Reversible Hydrogen Electrode (RHE) worked as the counter-electrode and reference electrode, respectively. Thiourea (Q.M., F. Maia Prod. Quim., Charqueada, Brazil) solution was used in a concentration of 1 mmol L
−1 and S
−2 was deposited on the PtSn/C particles by immersion in a 10 mmol L
−1 Na
2S.9H
2O (VETEC, Rio de Janeiro, Brazil) as previously described in details in our paper [
28]. During all sets of experiments, N
2 was passed over the solution. BOR was studied by the Rotating Disc Electrode (RDE) technique using the Linear Sweep Voltammetry (20 mV s
−1) setup. Several rotation speeds (between 100 and 3500 rpm) were investigated for each electrode potential ranging from 0.05 to 1.1 V.
Quasi steady-state data were also collected for BOR on the electrode material with the aid of Linear Sweep Voltammetry at a scan rate of 5 mV s−1. The selected rotation speed was 1500 rpm for all the steady-state measurements. Different rotation speeds were assayed, but no differences were observed.
The same experimental methodology depicted above was also employed for experiments conducted with addition of thiourea at 1 mmol L
−1 to the solution or after deposition of S
2− on the electrode. As often mentioned in the literature, thiourea inhibits BH
4− hydrolysis [
10,
12,
16], whereas S
2− blocks or modifies the electrode surface. Both play an important role in the hydrogen reaction mechanism [
28].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}