
A Simple and Efficient Process for Large Scale Glycerol Oligomerization by Microwave Irradiation

 ,
, 
Abstract
:1. Introduction
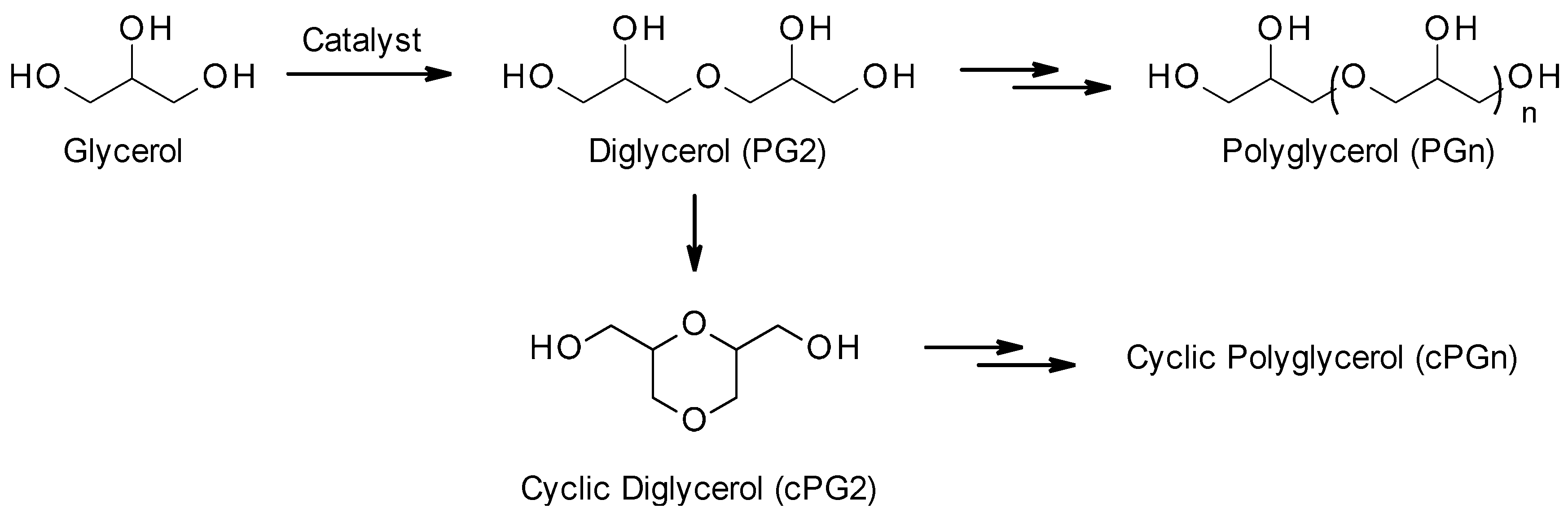
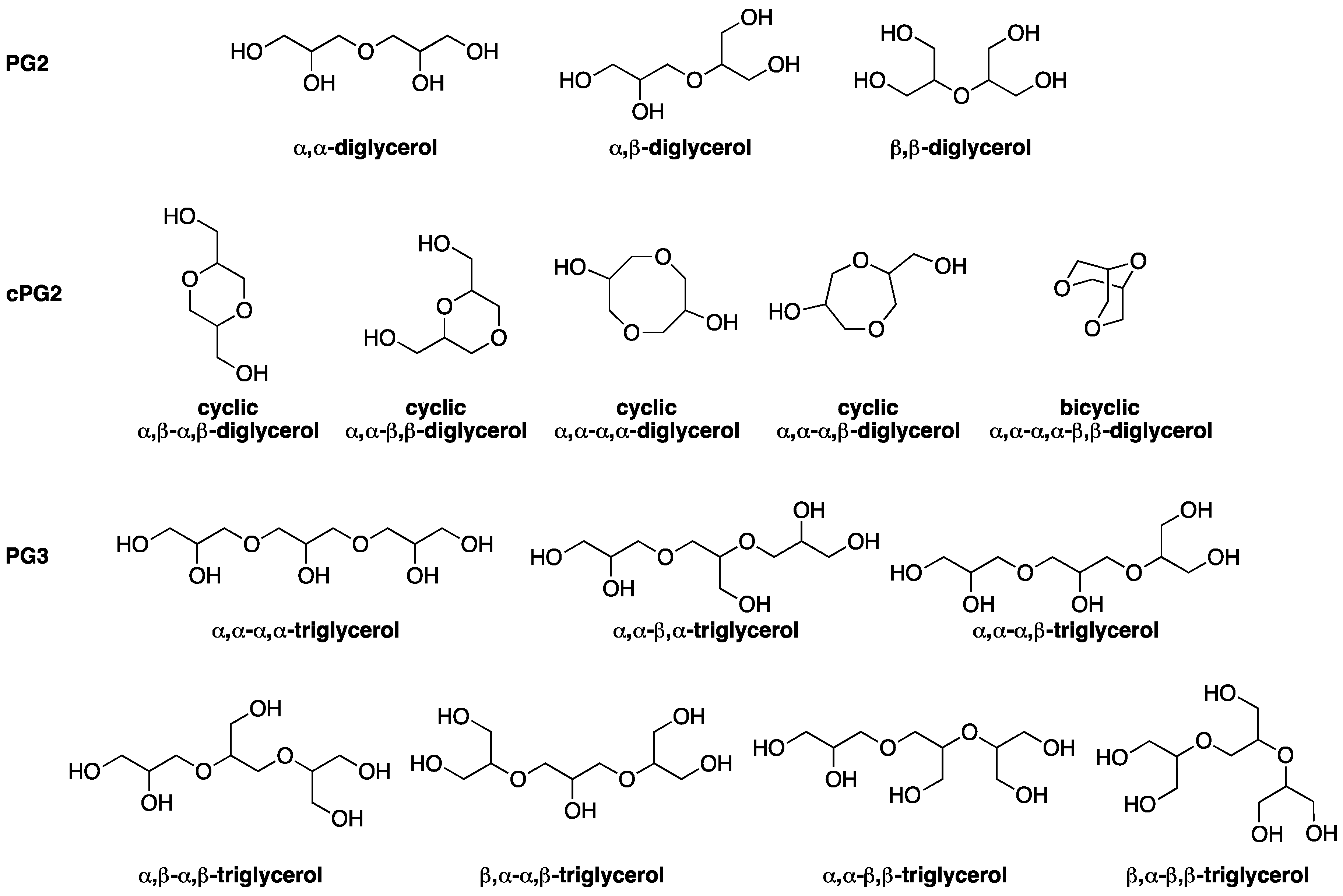
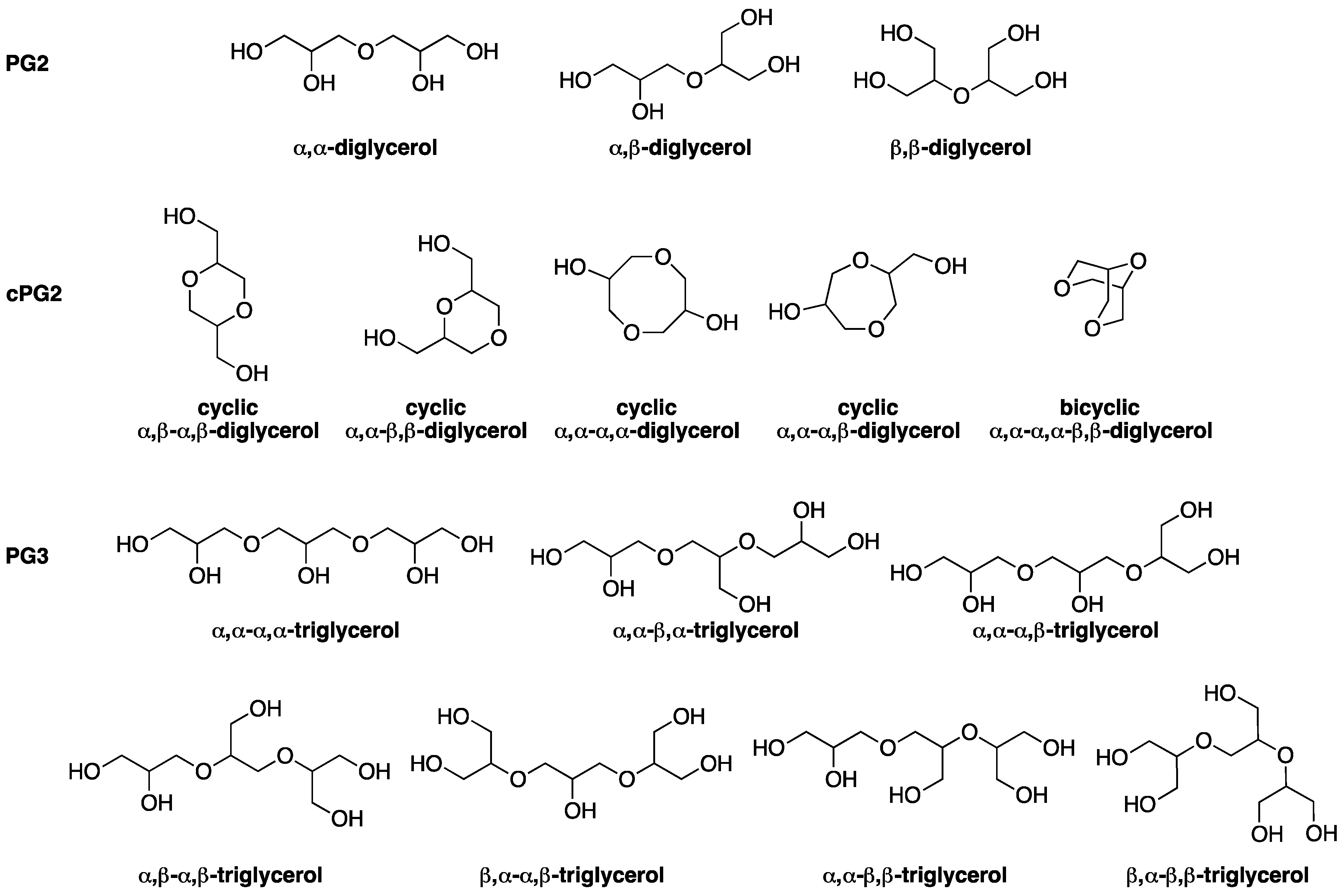
2. Results and Discussion
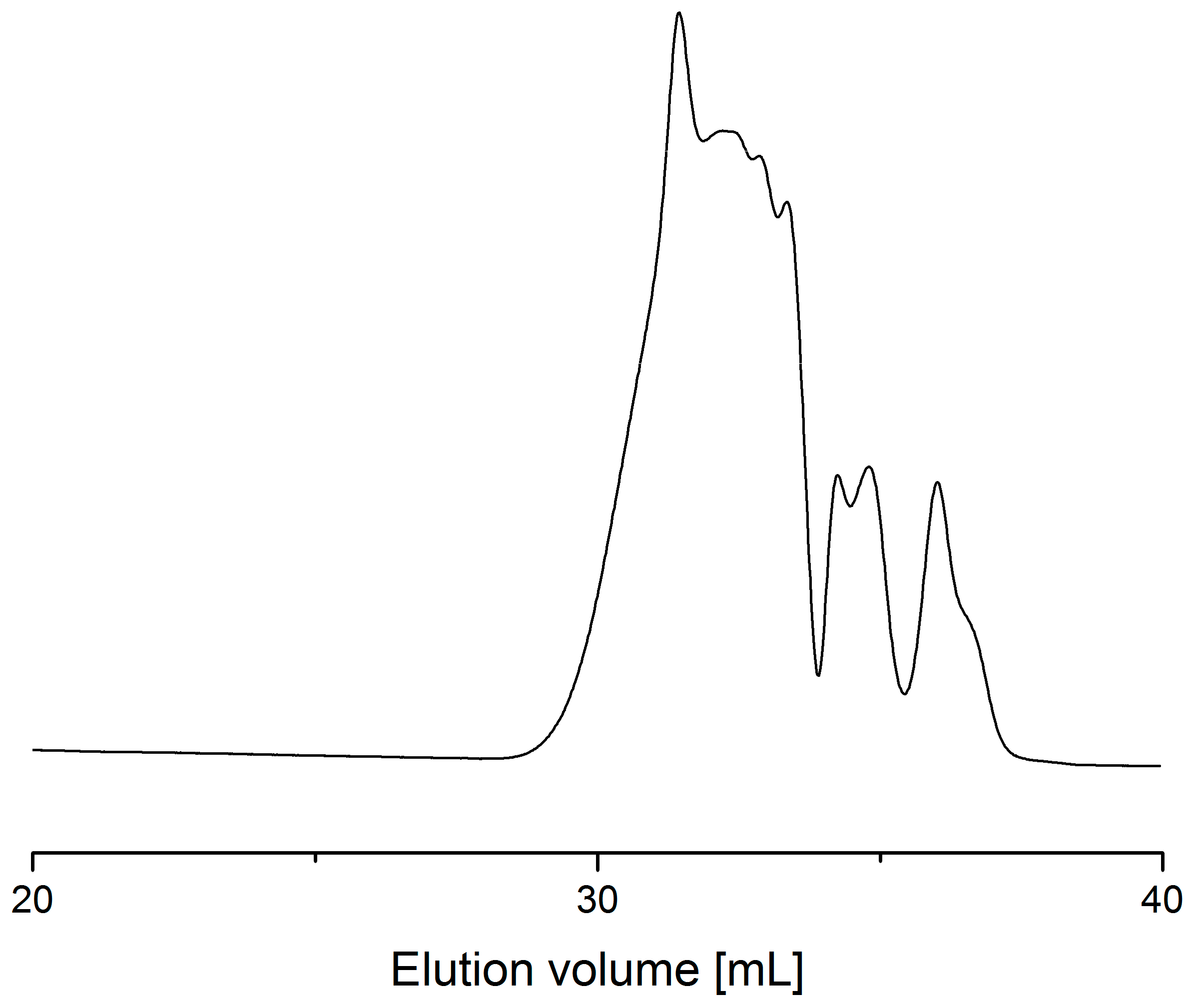
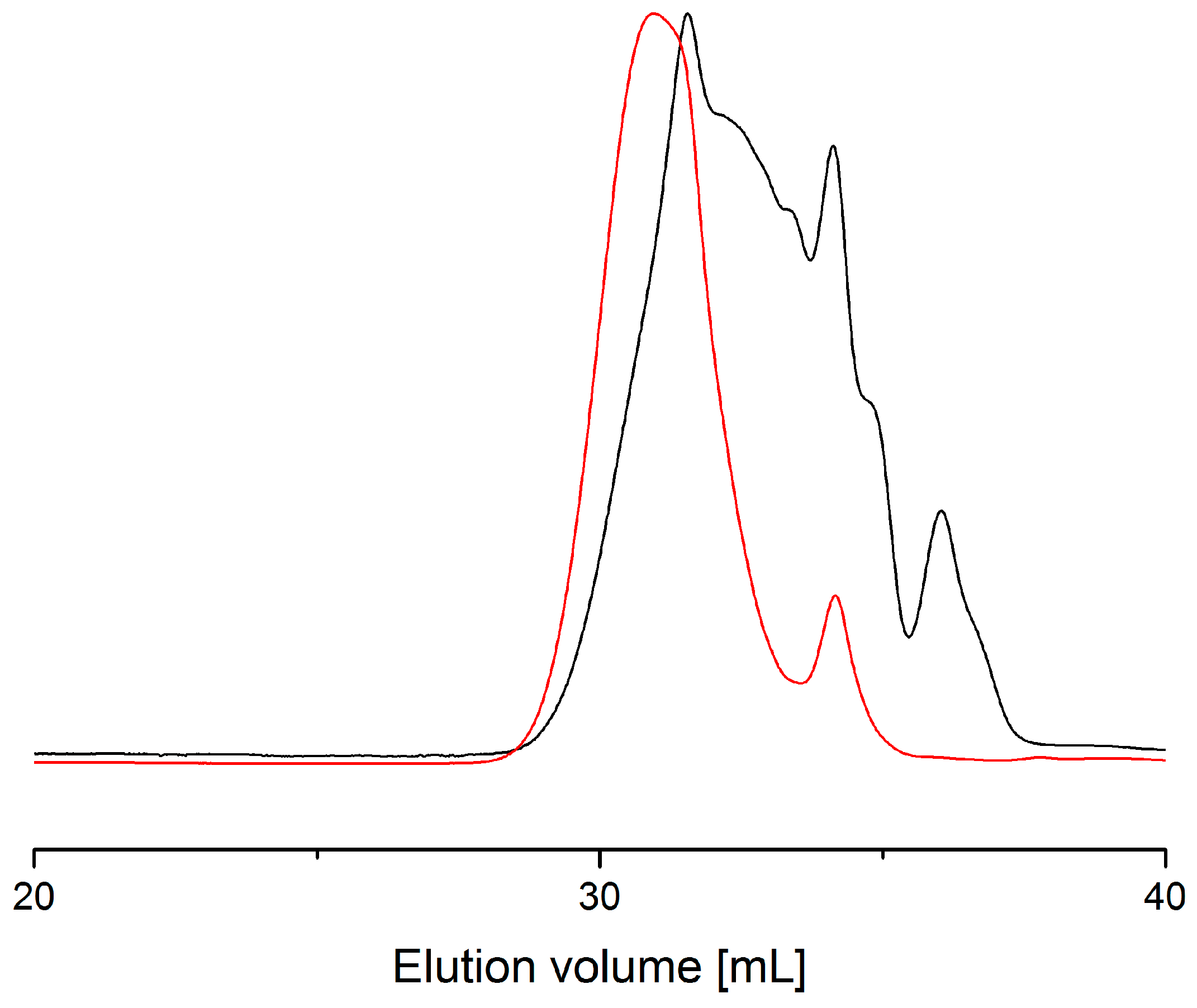
2.1. Preliminary Test with Monomode Reactor
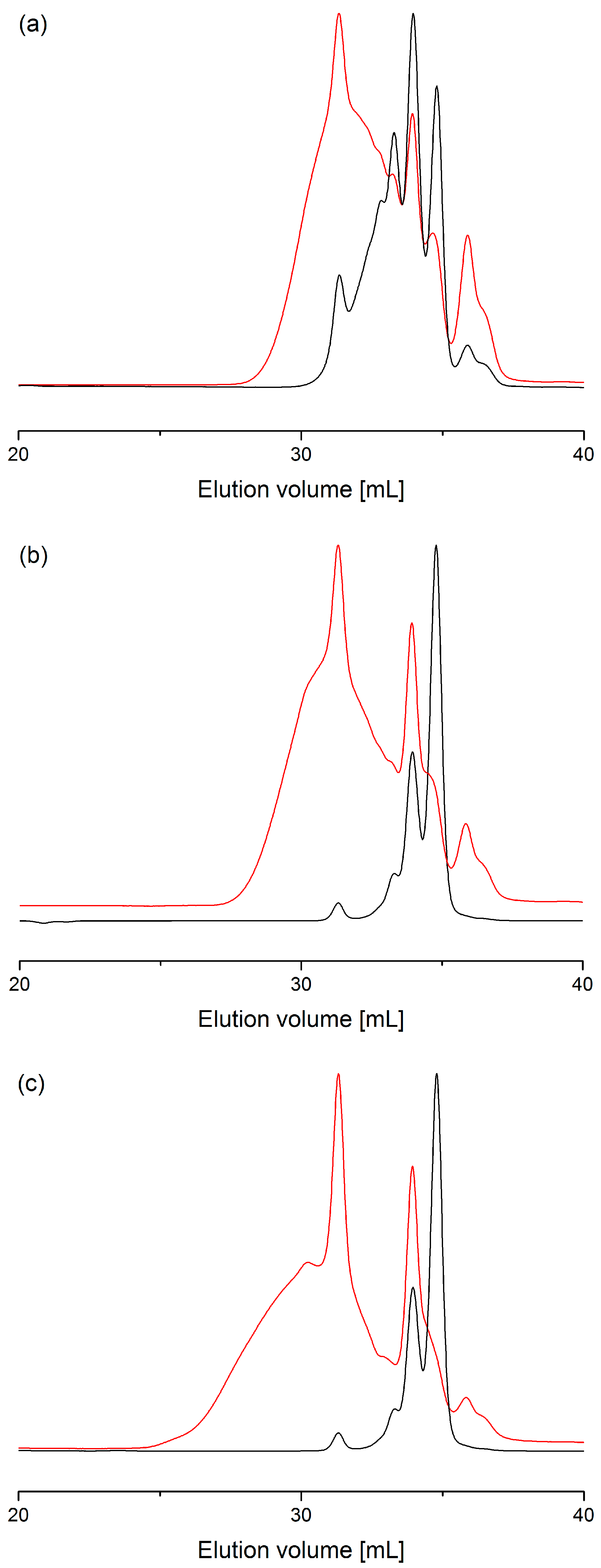
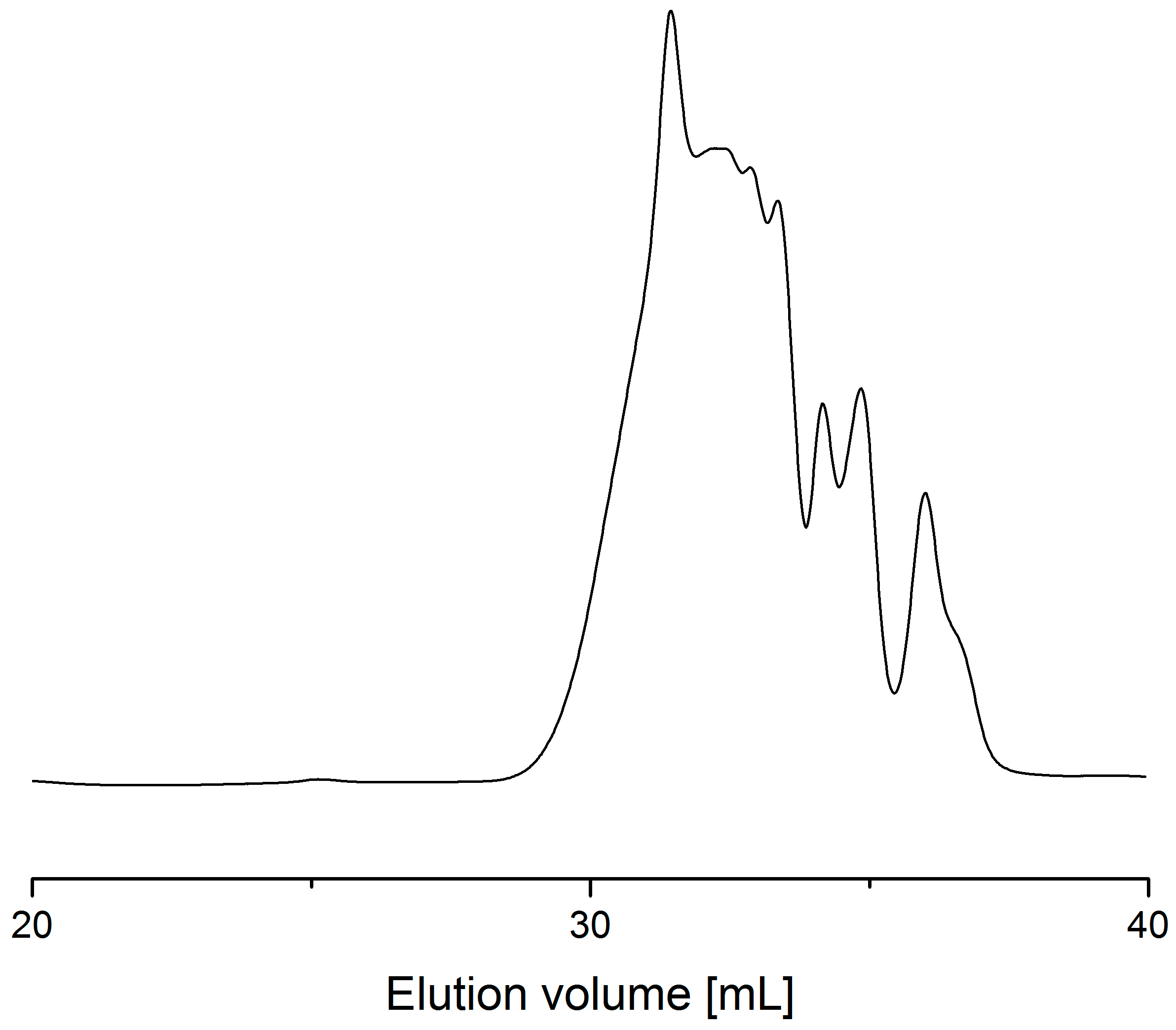
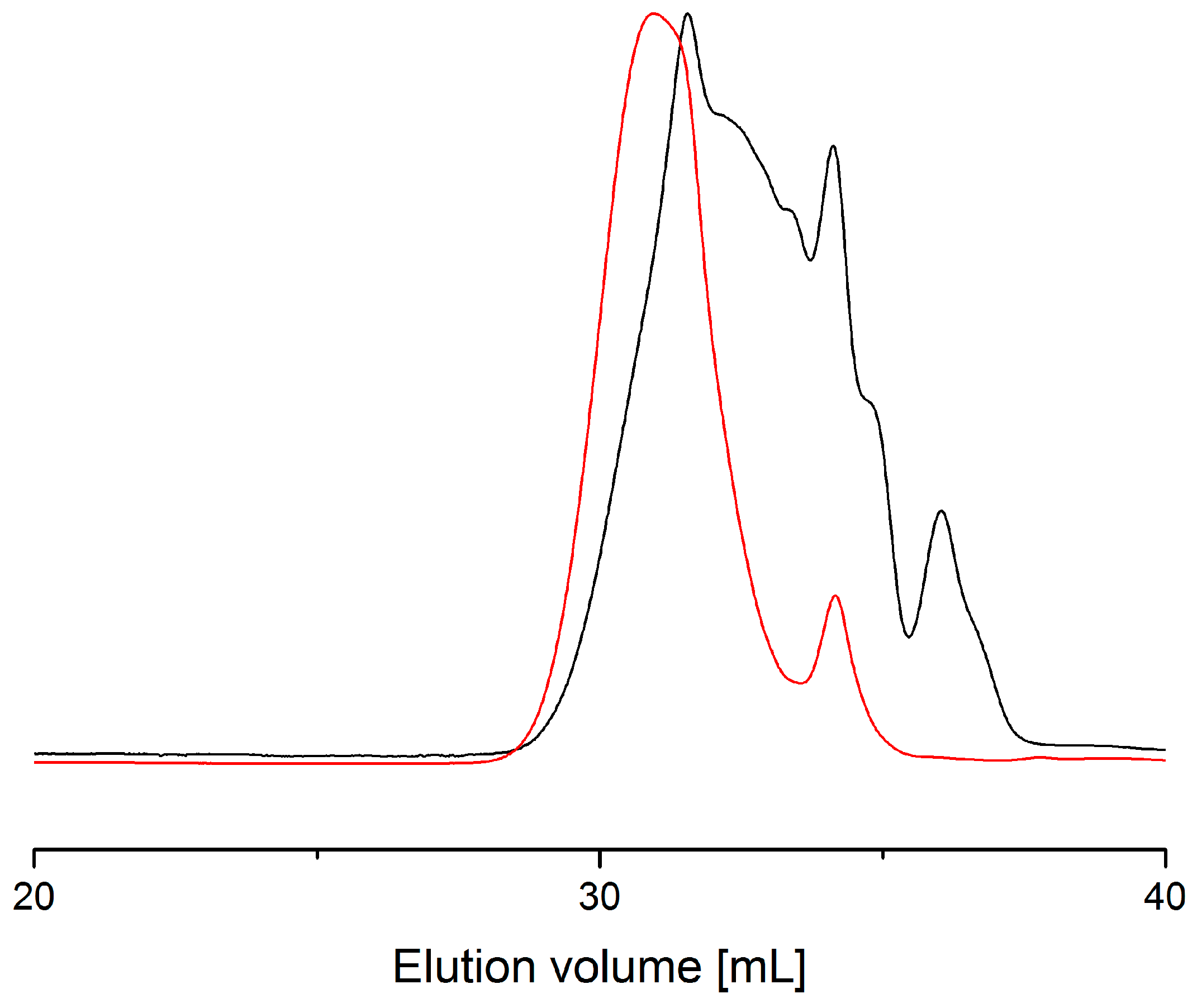
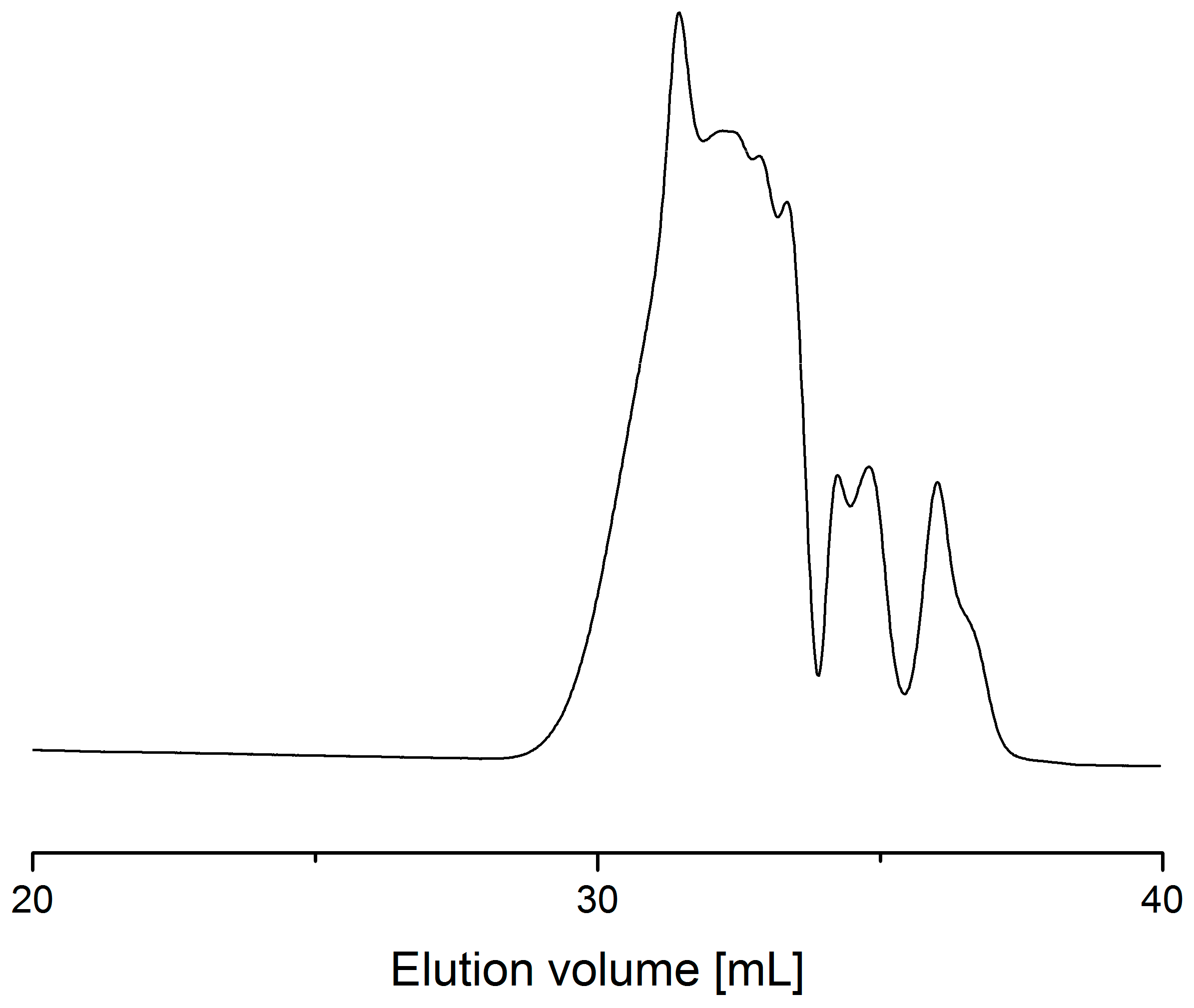
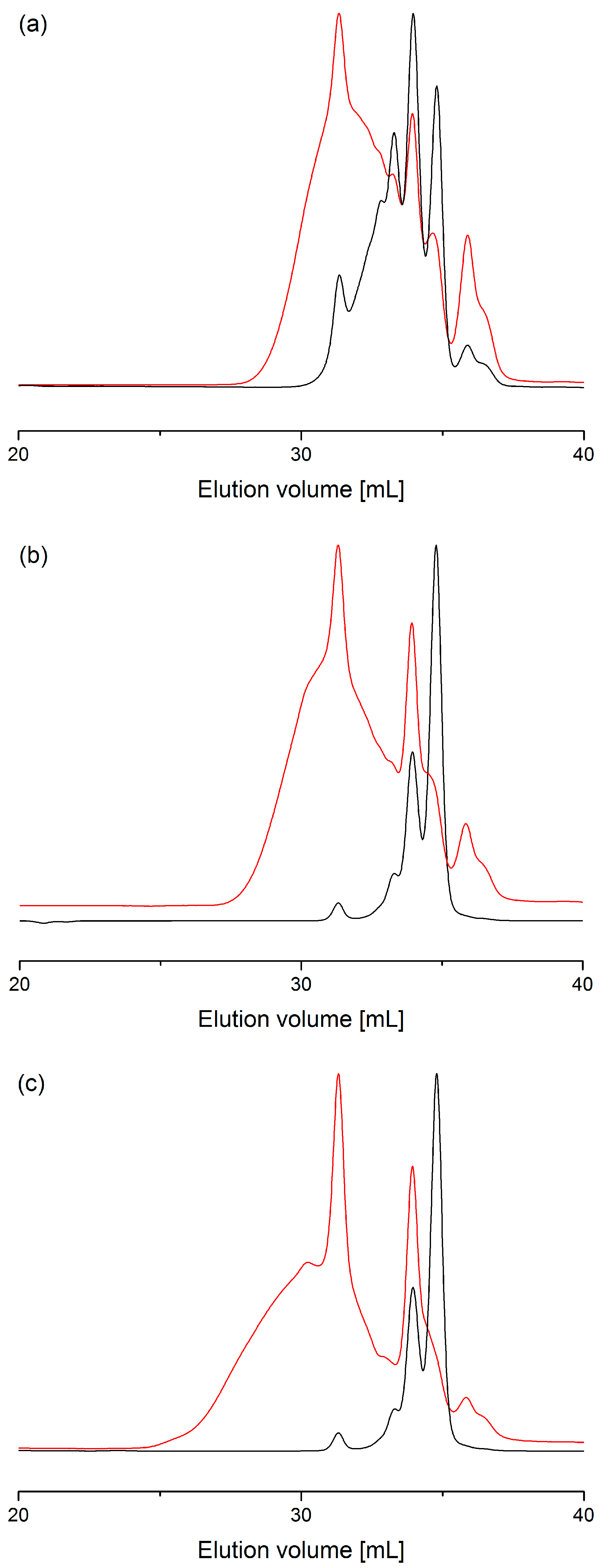
2.2. Process with Multimode Reactor
2.3. Process Optimization
3. Experimental Section
3.1. Oligomerization Process
3.1.1. Low Power Monomode Reaction (100–200 W)
3.1.2. High Power Multimode Reaction
3.1.3. Optimized High Power Multimode Process
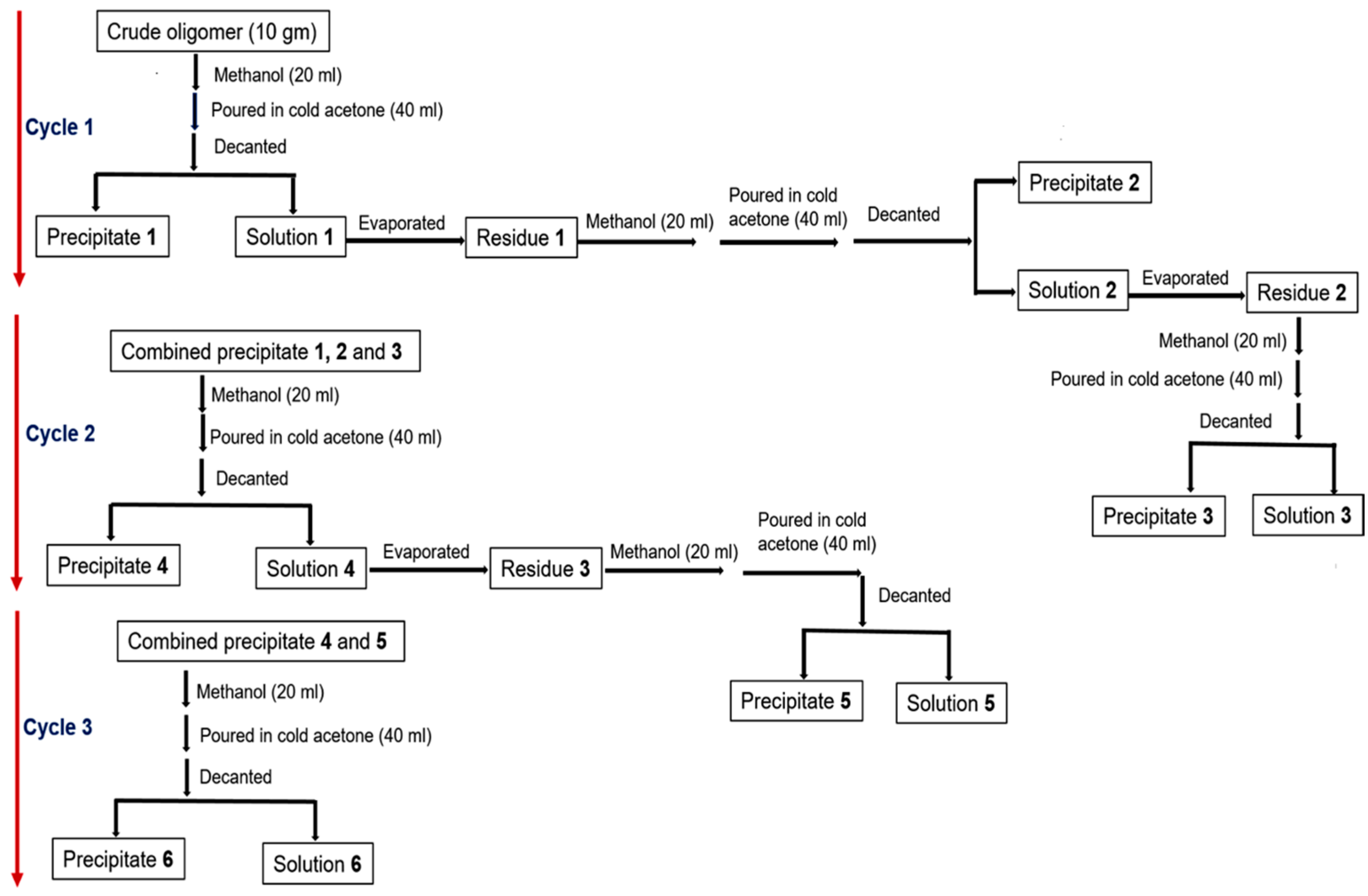
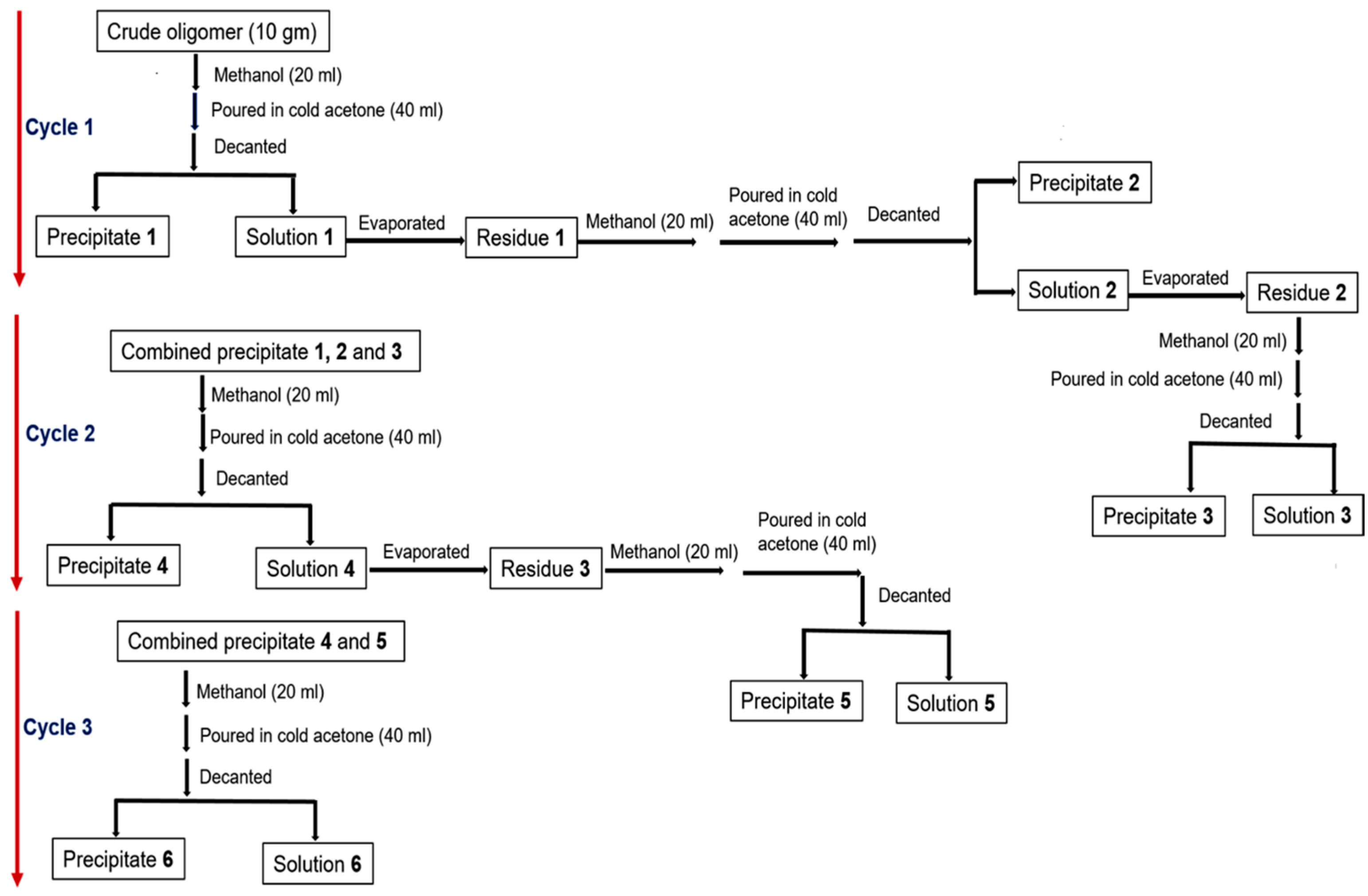
3.1.4. Purification
3.2. Analysis
3.2.1. Gas Chromatography (GC)
3.2.2. Gel Permeation Chromatography (GPC)
3.2.3. Mass Spectrometry
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Della Pina, C. From glycerol to value-added products. Angew. Chem. Int. Ed. 2007, 46, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.-H.; Beltramini, J.N.; Fan, Y.-X.; Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A. (Mitsubishi Chemical Corporation). Milk Beverage Containing Sucrose Fatty Acid Ester and Polyglycerol Fatty Acid Ester. World Patent 2004049813, 2004. [Google Scholar]
- Perrault, V. (L’Oréal). Cosmetic Composition, Useful in e.g. Spray Bottle, Roll on and Tubing, for Perfuming on Skin, Clothing and an Object, Comprises Aqueous Phase, Volatile Alcohol, Perfume, Non-Volatile Hydrocarbon Oil, and Polyglycerol Ester of Fatty Acid. Fr. Patent 2998783, 2014. [Google Scholar]
- Giuliani, G.; Benedusi, A.; Mascolo, A.; Marzani, B.; Bregaglio, G. (Giuliani S.P.A.). Polyglycerol-Azelaic Acid Polyesters for Cosmetic Applications. U.S. Patent 20150196480, 2015. [Google Scholar]
- Wilms, D.; Stiriba, S.-E.; Frey, H. Hyperbranched polyglycerols: From the controlled synthesis of biocompatible polyether polyols to multipurpose applications. Acc. Chem. Res. 2010, 43, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Richter, M. Oligomerization of glycerol—A critical review. Eur. J. Lipid Sci. Technol. 2011, 113, 100–117. [Google Scholar] [CrossRef]
- Sivaiah, M.V.; Robles-Manuel, S.; Valange, S.; Barrault, J. Recent developments in acid and base-catalyzed etherification of glycerol to polyglycerols. Catal. Today 2012, 198, 305–313. [Google Scholar] [CrossRef]
- Mingos, D.M.P.; Baghurst, D.R. Applications of microwave dielectric heating effects to synthetic problems in chemistry. Chem. Soc. Rev. 1991, 20, 1–47. [Google Scholar] [CrossRef]
- Perreux, L.; Loupy, A. A tentative rationalization of microwave effects in organic synthesis according to the reaction medium, and mechanistic considerations. Tetrahedron 2001, 57, 9199–9223. [Google Scholar] [CrossRef]
- Obermayer, D.; Gutmann, B.; Kappe, C.O. Microwave chemistry in silicon carbide reaction vials: Separating thermal from nonthermal effects. Angew. Chem. Int. Ed. 2009, 48, 8321–8324. [Google Scholar] [CrossRef] [PubMed]
- Mat Din, N.S.M.N.; Idris, Z.; Kian, Y.S.; Hassan, H.A. Preparation of polyglycerol from palm-biodiesel crude glycerin. J. Oil Palm Res. 2013, 25, 289–297. [Google Scholar]
- Bookong, P.; Ruchirawat, S.; Boonyarattanakalin, S. Optimization of microwave-assisted etherification of glycerol to polyglycerols by sodium carbonate as catalyst. Chem. Eng. J. 2015, 275, 253–261. [Google Scholar] [CrossRef]
- Clacens, J.-M.; Pouilloux, Y.; Barrault, J. Selective etherification of glycerol to polyglycerols over impregnated basic MCM-41 type mesoporous catalysts. Appl. Catal. A Gen. 2002, 227, 181–190. [Google Scholar] [CrossRef]
- Barrault, J.; Clacens, J.-M.; Pouilloux, Y. Selective oligomerization of glycerol over mesoporous catalysts. Top. Catal. 2004, 27, 137–142. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Meeldijk, J.D.; Kuipers, B.W.M.; Erné, B.H.; Weckhuysen, B.M. Glycerol etherification over highly active CaO-based materials: New mechanistic aspects and related colloidal particle formation. Chem. Eur. J. 2008, 14, 2016–2024. [Google Scholar] [CrossRef] [PubMed]
- Krisnandi, Y.K.; Eckelt, R.; Schneider, M.; Martin, A.; Richter, M. Glycerol upgrading over zeolithes by batch-reactor liquid-phase oligomerization: Heterogeneous versus homogeneous reaction. ChemSusChem 2008, 1, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Eckelt, R.; Krisnandi, Y.K.; Martin, A. Verfahren zur selektiven herstellung von linearem diglycerin. Chem. Ing. Tech. 2008, 80, 1573–1577. [Google Scholar] [CrossRef]
- Anuar, M.R.; Abdullah, A.Z.; Othman, M.R. Etherification of glycerol to polyglycerols over hydrotalcite catalyst prepared using a combustion method. Catal. Commun. 2013, 32, 67–70. [Google Scholar] [CrossRef]
- Katryniok, B.; Paul, S.; Capron, M.; Dumeignil, F. Towards the sustainable production of acrolein by glycerol dehydration. ChemSusChem 2009, 2, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Katryniok, B.; Paul, S.; Bellière-Baca, V.; Rey, P.; Dumeignil, F. Glycerol dehydration to acrolein in the context of new uses of glycerol. Green Chem. 2010, 12, 2079–2098. [Google Scholar] [CrossRef]
- Garti, N.; Aserin, A.; Zaidman, B. Polyglycerol esters: Optimization and techno-economic evaluation. J. Am. Oil Chem. Soc. 1981, 58, 878–883. [Google Scholar] [CrossRef]
- Cottin, K.; Clacens, J.-M.; Pouilloux, Y.; Barrault, J. Preparation of diglycerol and triglycerol by the direct polymerization of glycerol in the presence of the new solid catalysts. Oléagineux Corps Gras Lipides 1998, 5, 407–412. [Google Scholar]
- Charles, G.; Clacens, J.-M.; Pouilloux, Y.; Barrault, J. Preparation de diglycerol et triglycerol par polymerisation directe du glycerol en presence de catalyseurs solides. Oléagineux Corps Gras Lipides 2003, 10, 74–82. [Google Scholar] [CrossRef]
- Richter, M.; Krisnandi, Y.K.; Eckelt, R.; Martin, A. Homogeneous catalyzed batch reactor glycerol etherification by CsHCO3. Catal. Commun. 2008, 9, 2112–2116. [Google Scholar] [CrossRef]
- De Meulenaer, B.; Vanhoutte, B.; Huyghebaert, A. Development of chromatographic method fort he determination of degree of polymerisation of polyglycerols and polyglycerol fatty acid esters. Chromatographia 2000, 51, 44–52. [Google Scholar] [CrossRef]
- Cassel, S.; Debaig, C.; Benvegnu, T.; Chaimbault, P.; Lafosse, M.; Plusquellec, D.; Rollin, P. Original synthesis of linear, branched and cyclic oligoglycerol standards. Eur. J. Org. Chem. 2001, 2001, 875–896. [Google Scholar] [CrossRef]
- Iaych, K.; Dumarçay, S.; Fredon, E.; Gérardin, C.; Lemor, A.; Gérardin, P. Microwave-assisted synthesis of polyglycerol from glycerol carbonate. J. Appl. Polym. Sci. 2011, 120, 2354–2360. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Power (W) | Time [min] | MW a | MN b | D c | Evaporated Glycerol [Units] | Ratio d [%] |
|---|---|---|---|---|---|---|---|
| 1 | 300 | 30 | 887 | 358 | 2.5 | 2.8 | 3.6 |
| 2 | 600 | 10.5 | 1244 | 406 | 3.1 | 20.3 | 26.4 |
| 3 | 800 | 6.33 | 2833 | 479 | 5.9 | 29.1 | 37.9 |
| Entry | Time (min) | PG1 a [%] | cPG2 b [%] | PG2 [%] | cPG3 c [%] | PG3 [%] | PG4 [%] | PG5 [%] | PG6 [%] | PG7 [%] |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0 | 3.3 | 7.8 | 81.9 | 0.0 | 0.4 | 6.0 | 0.0 | 0.3 | 0.0 |
| 2 | 5 | 3.1 | 7.8 | 81.6 | 0.0 | 0.4 | 6.2 | 0.0 | 0.4 | 0.0 |
| 3 | 10 | 3.3 | 7.7 | 81.2 | 0.0 | 0.6 | 6.3 | 0.0 | 0.4 | 0.0 |
| 4 | 15 | 6.3 | 10.6 | 52.4 | 1.1 | 8.2 | 12.7 | 3.1 | 3.1 | 0.9 |
| 5 | 20 | 4.9 | 20.0 | 21.9 | 4.2 | 9.0 | 12.6 | 8.5 | 8.3 | 5.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, R.; Galy, N.; Singh, A.K.; Paulus, F.; Stöbener, D.; Schlesener, C.; Sharma, S.K.; Haag, R.; Len, C. A Simple and Efficient Process for Large Scale Glycerol Oligomerization by Microwave Irradiation. Catalysts 2017, 7, 123. https://doi.org/10.3390/catal7040123
Nguyen R, Galy N, Singh AK, Paulus F, Stöbener D, Schlesener C, Sharma SK, Haag R, Len C. A Simple and Efficient Process for Large Scale Glycerol Oligomerization by Microwave Irradiation. Catalysts. 2017; 7(4):123. https://doi.org/10.3390/catal7040123
Chicago/Turabian StyleNguyen, Rémi, Nicolas Galy, Abhishek K. Singh, Florian Paulus, Daniel Stöbener, Cathleen Schlesener, Sunil K. Sharma, Rainer Haag, and Christophe Len. 2017. "A Simple and Efficient Process for Large Scale Glycerol Oligomerization by Microwave Irradiation" Catalysts 7, no. 4: 123. https://doi.org/10.3390/catal7040123
APA StyleNguyen, R., Galy, N., Singh, A. K., Paulus, F., Stöbener, D., Schlesener, C., Sharma, S. K., Haag, R., & Len, C. (2017). A Simple and Efficient Process for Large Scale Glycerol Oligomerization by Microwave Irradiation. Catalysts, 7(4), 123. https://doi.org/10.3390/catal7040123