Steam Reforming of Bio-Compounds with Auto-Reduced Nickel Catalyst



Abstract
:1. Introduction
2. Results and Discussion
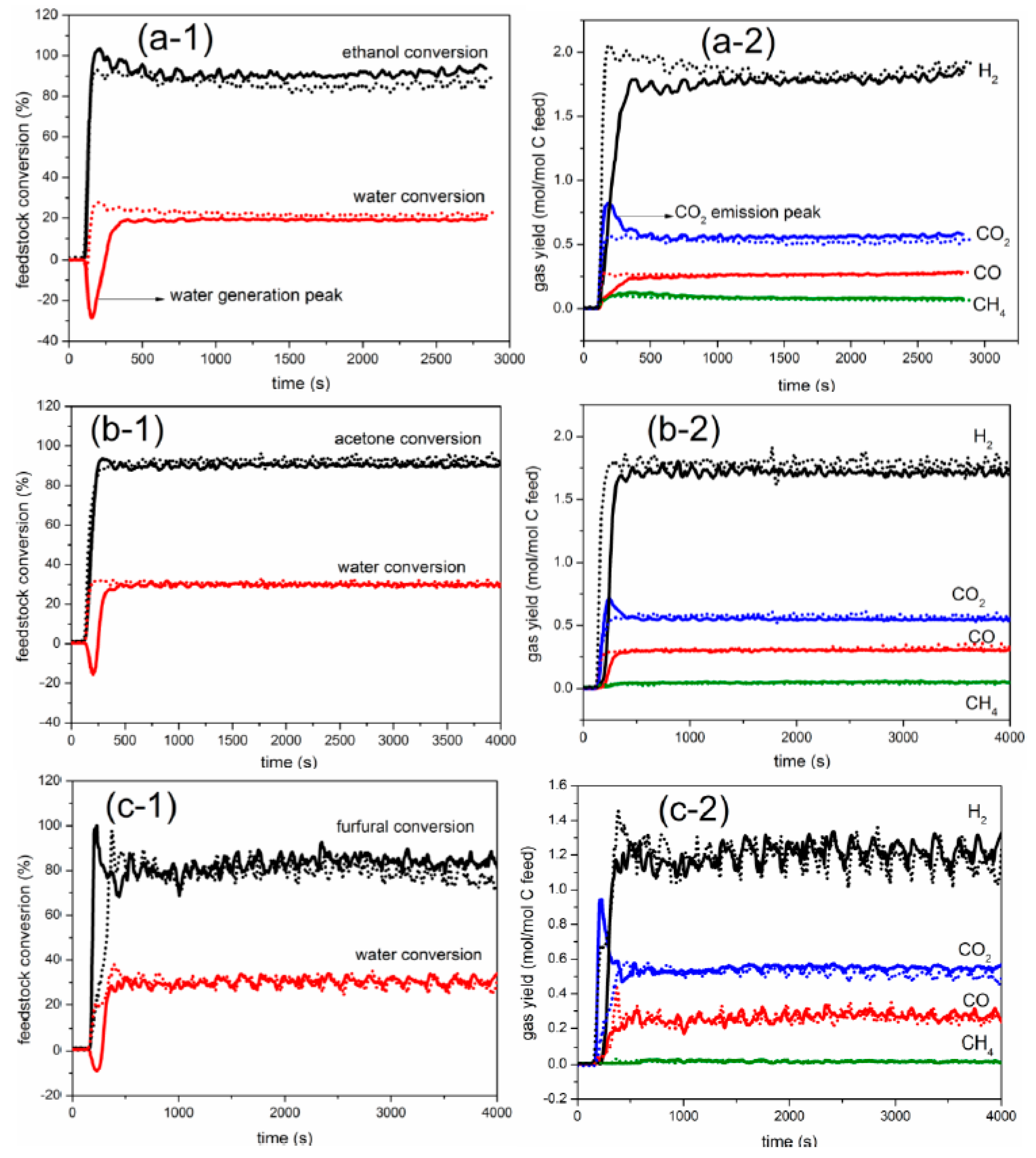
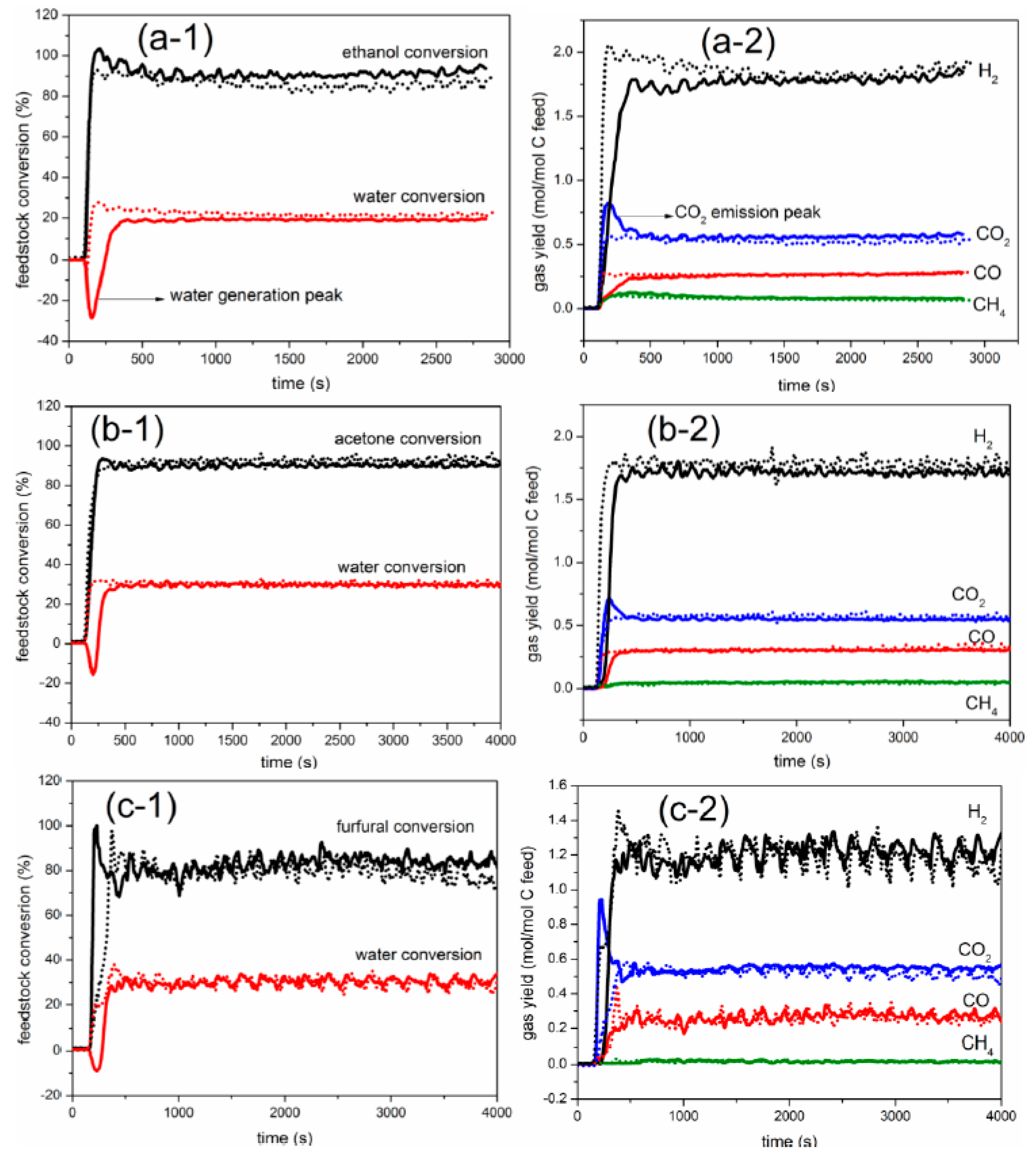
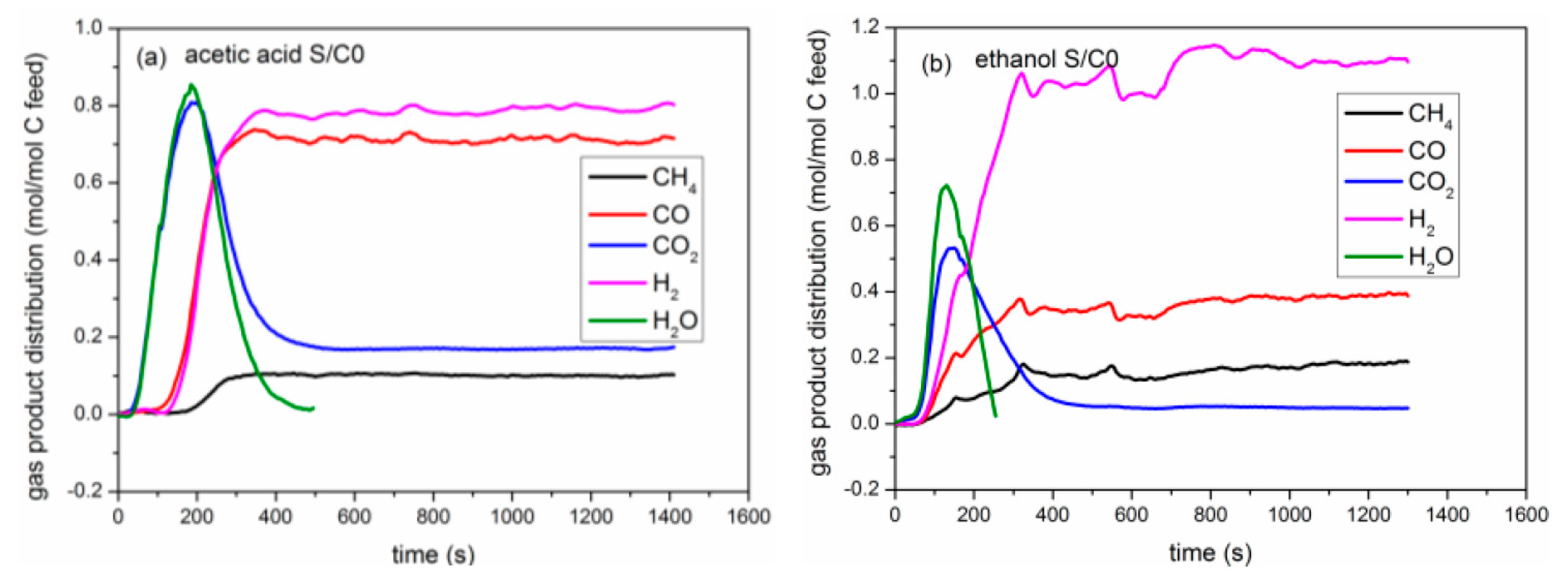
2.1. Comparison between Auto-Reduction and H2 Reduction
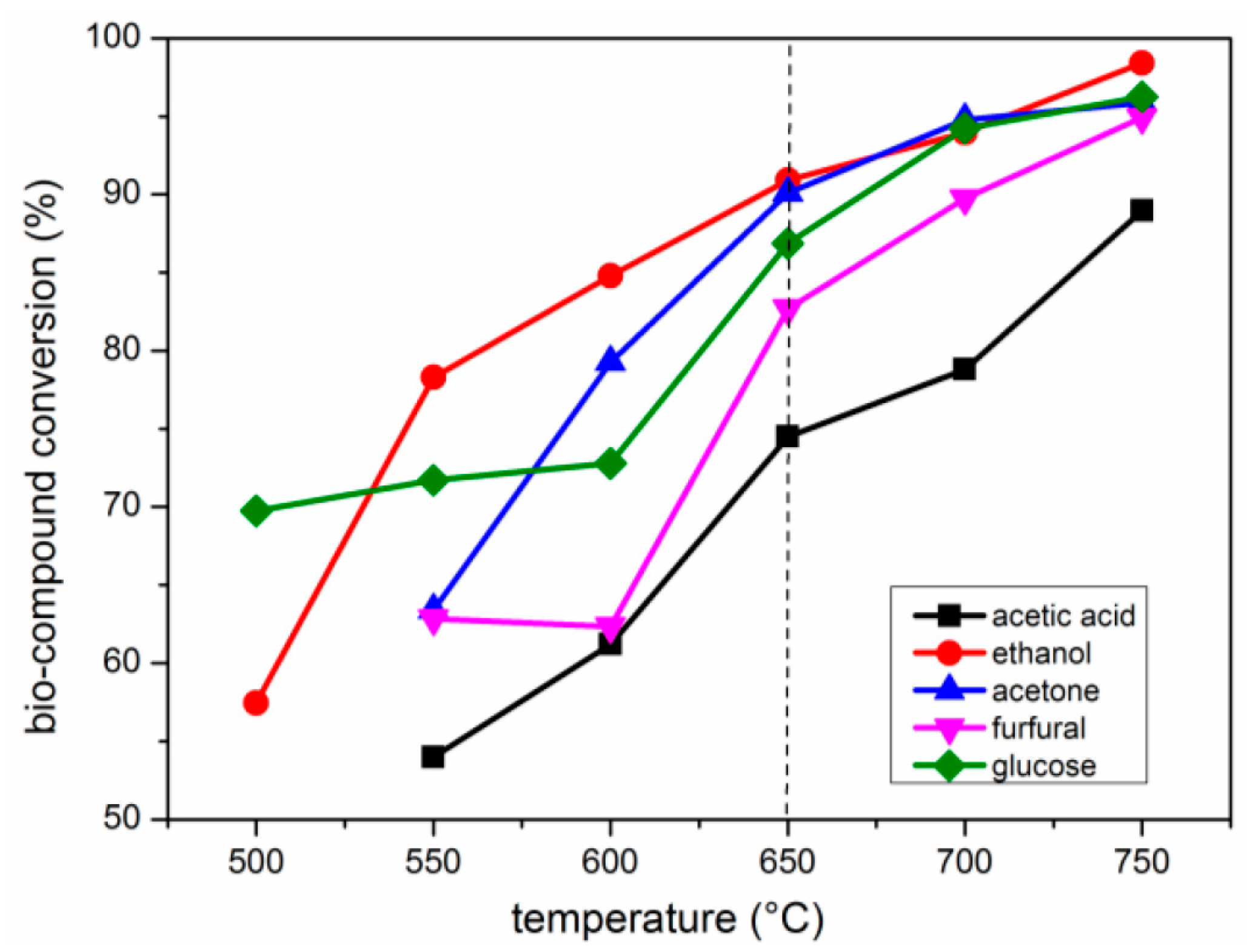
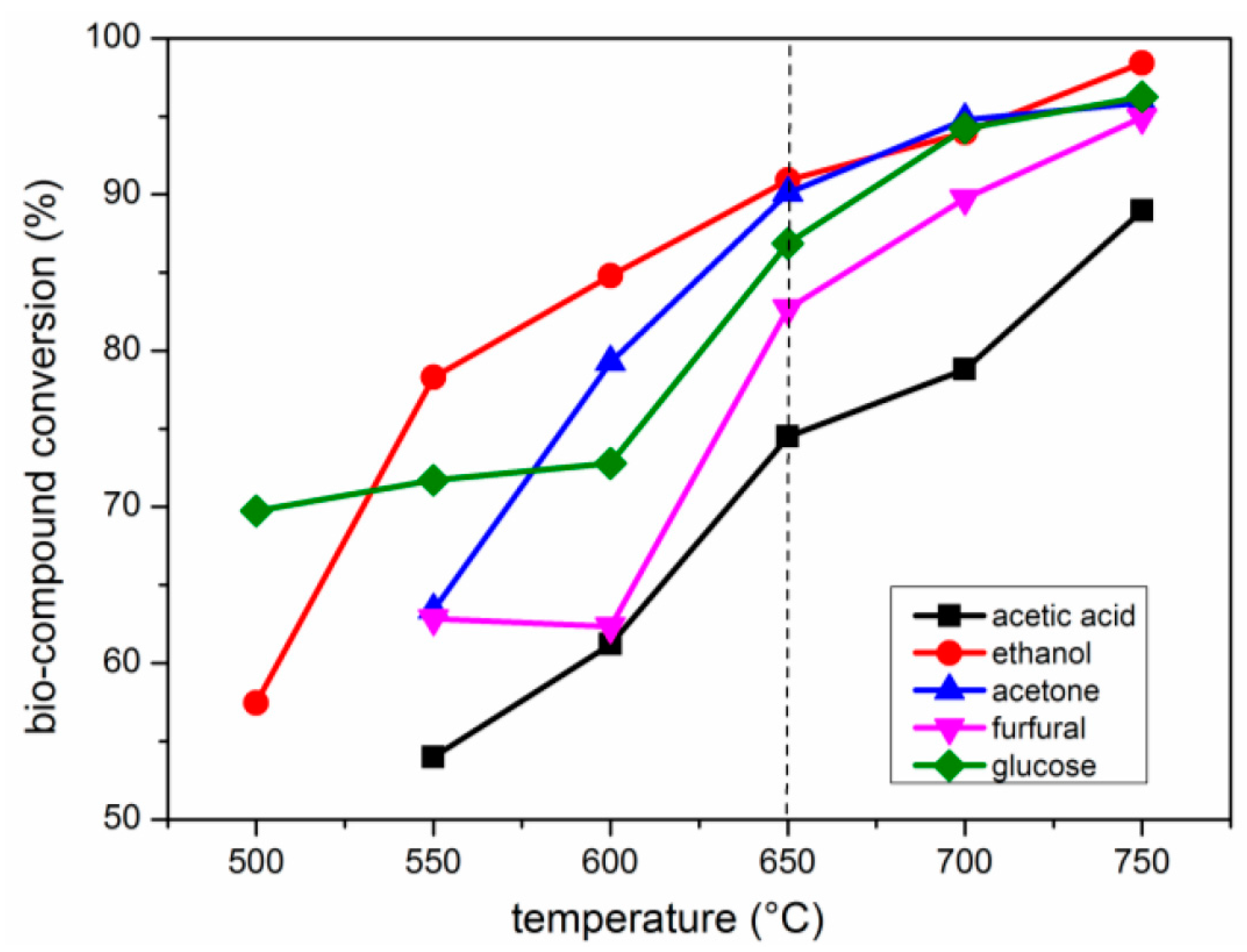
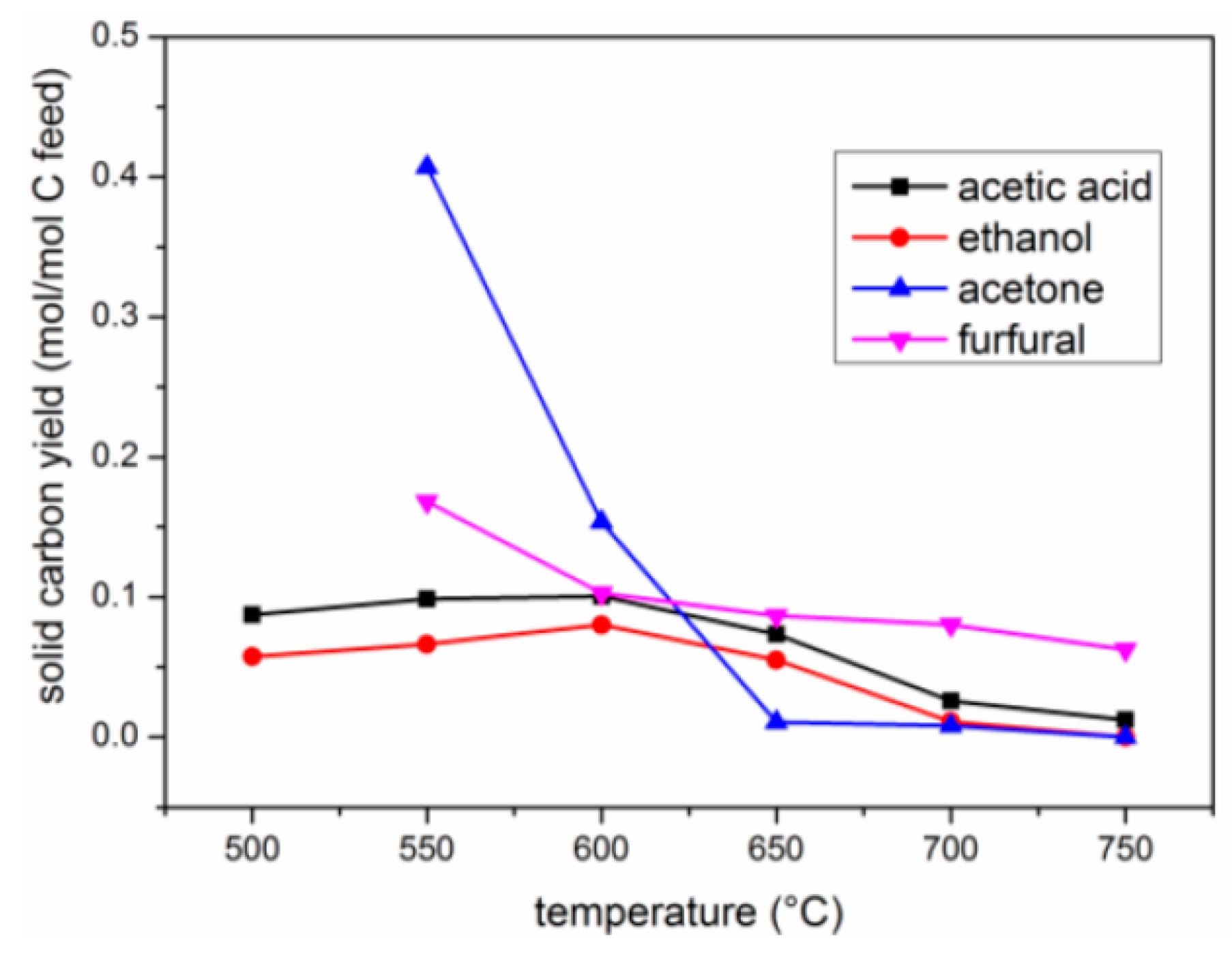
2.2. Effects of Temperature
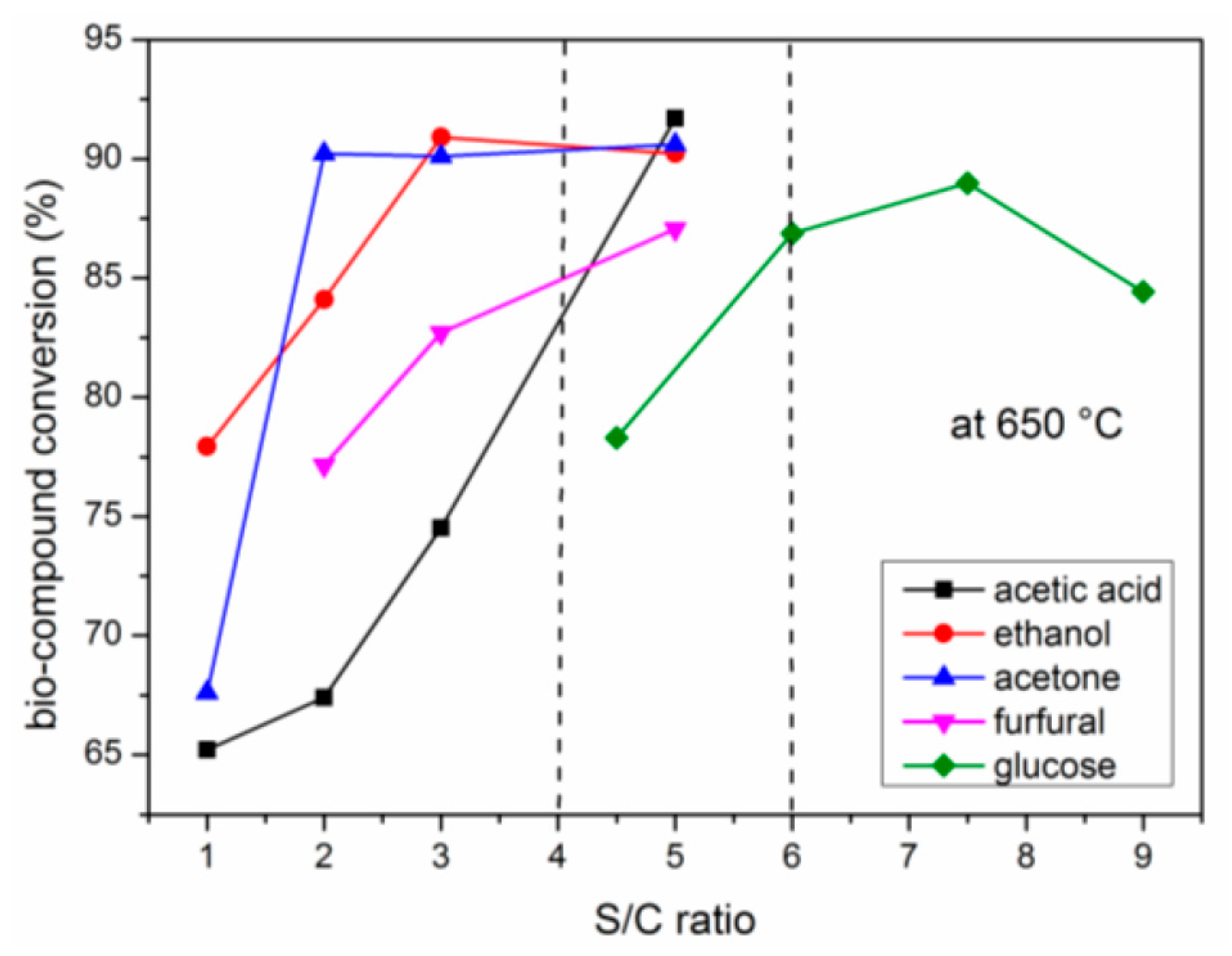
2.2.1. Carbon Conversion of Bio-Compounds to Gases
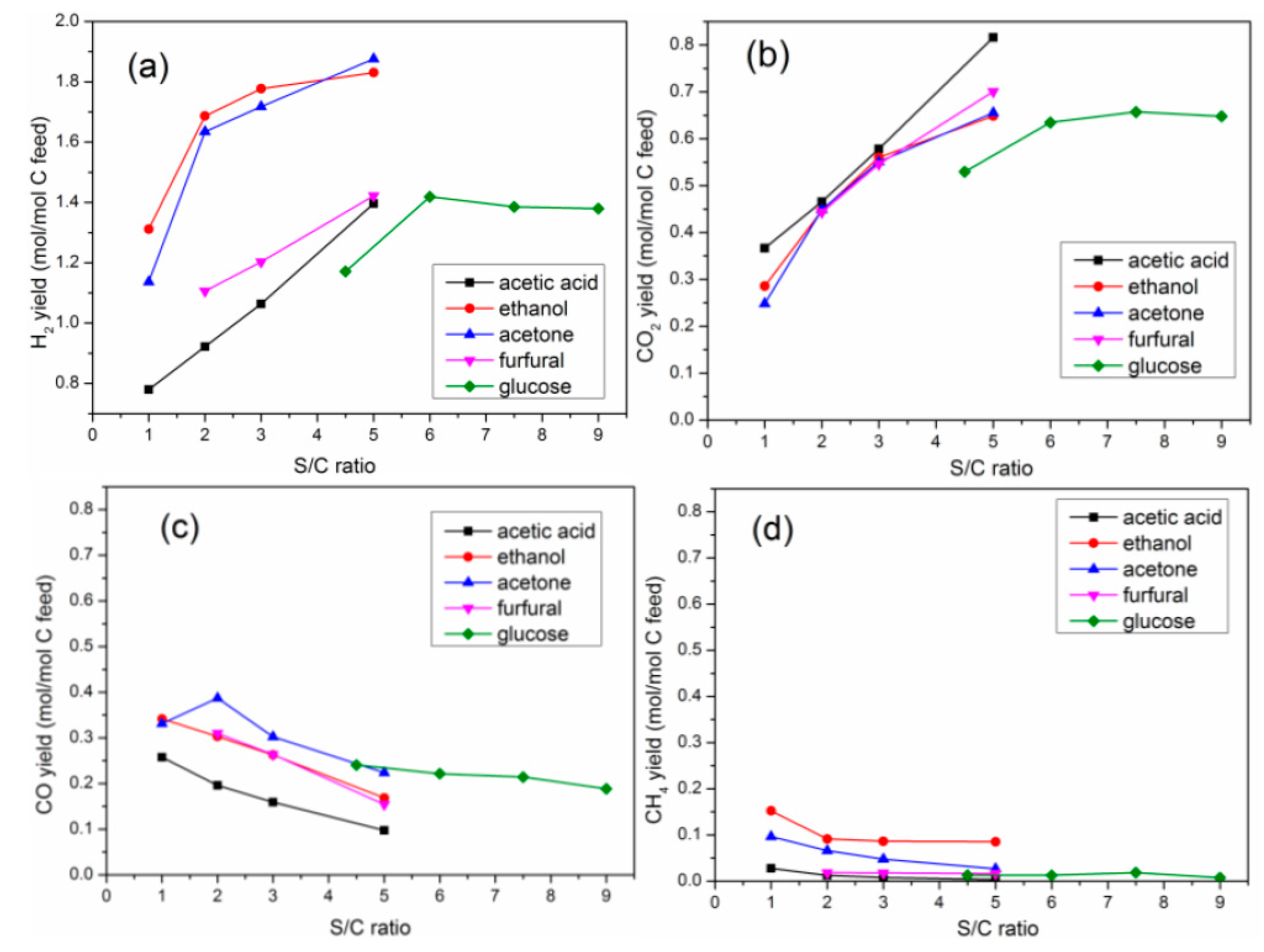
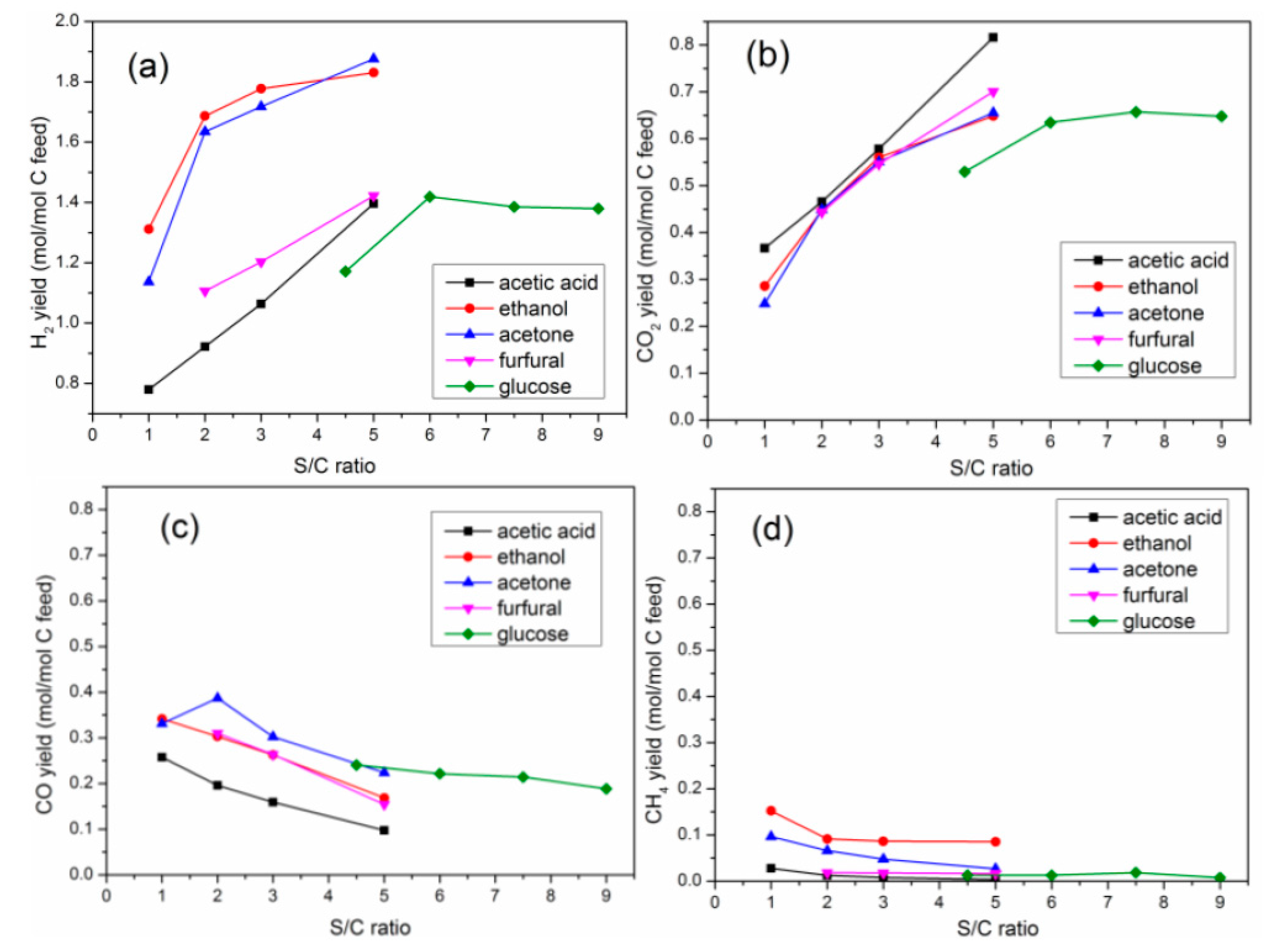
2.2.2. Yield of Gaseous Products
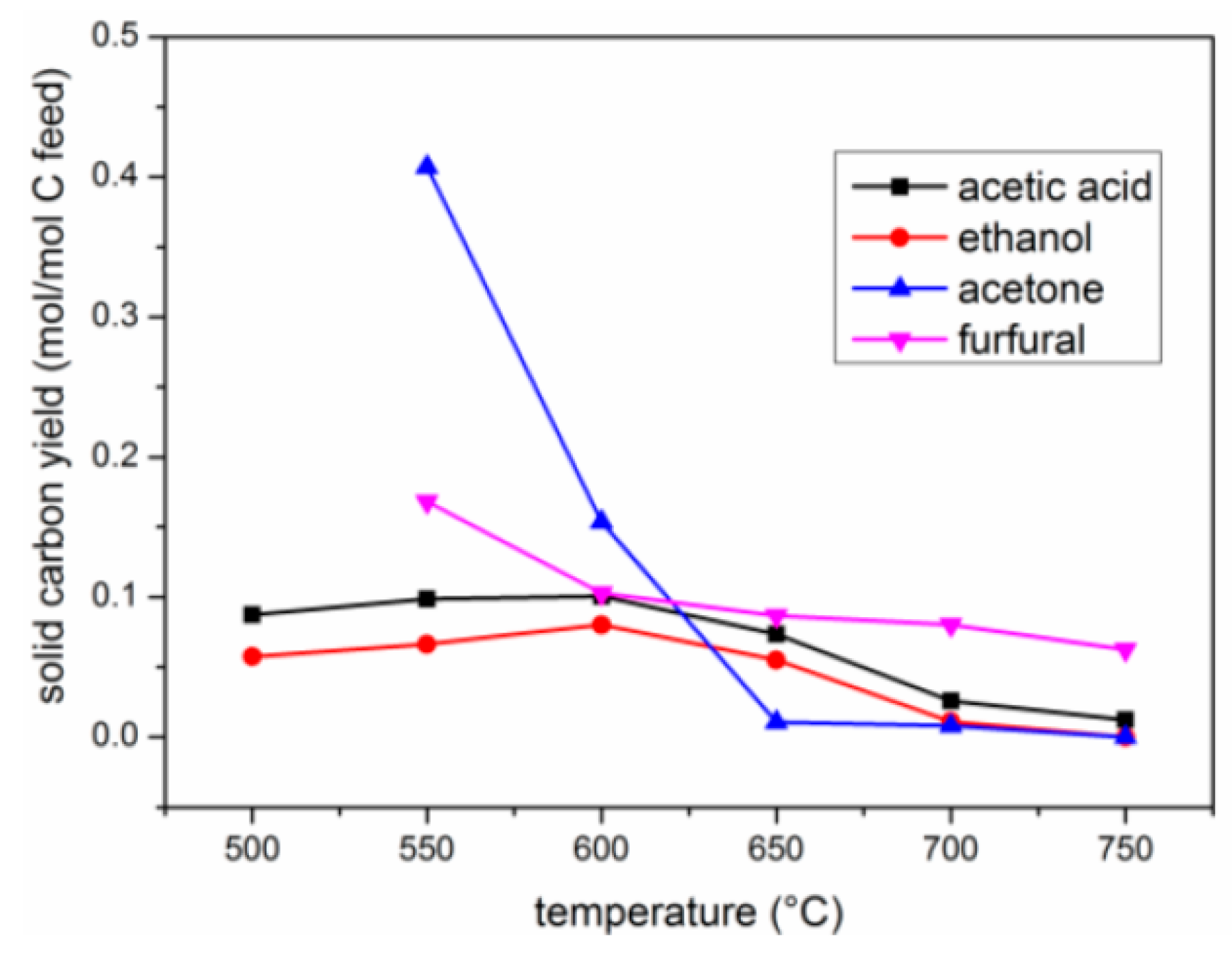
2.2.3. Yield of Carbon Deposits
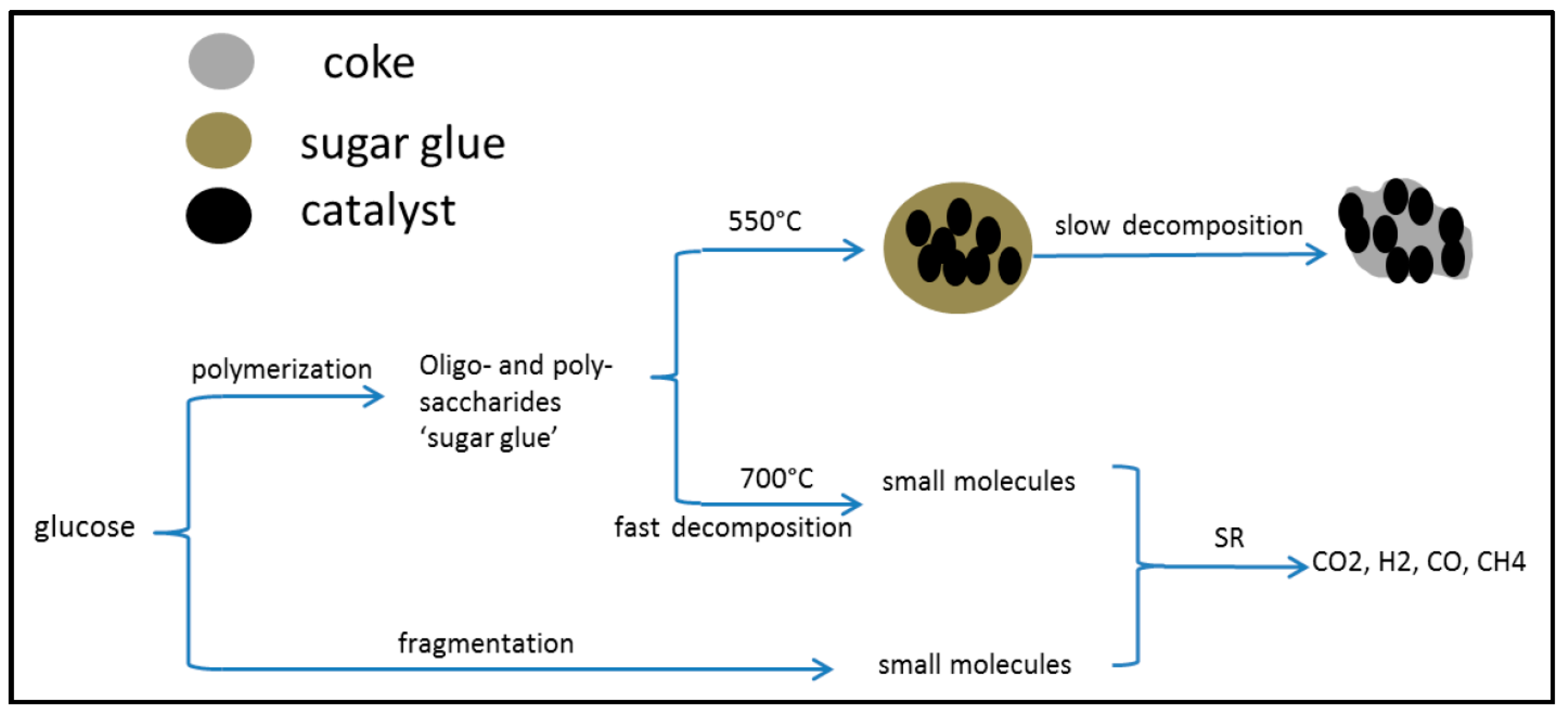
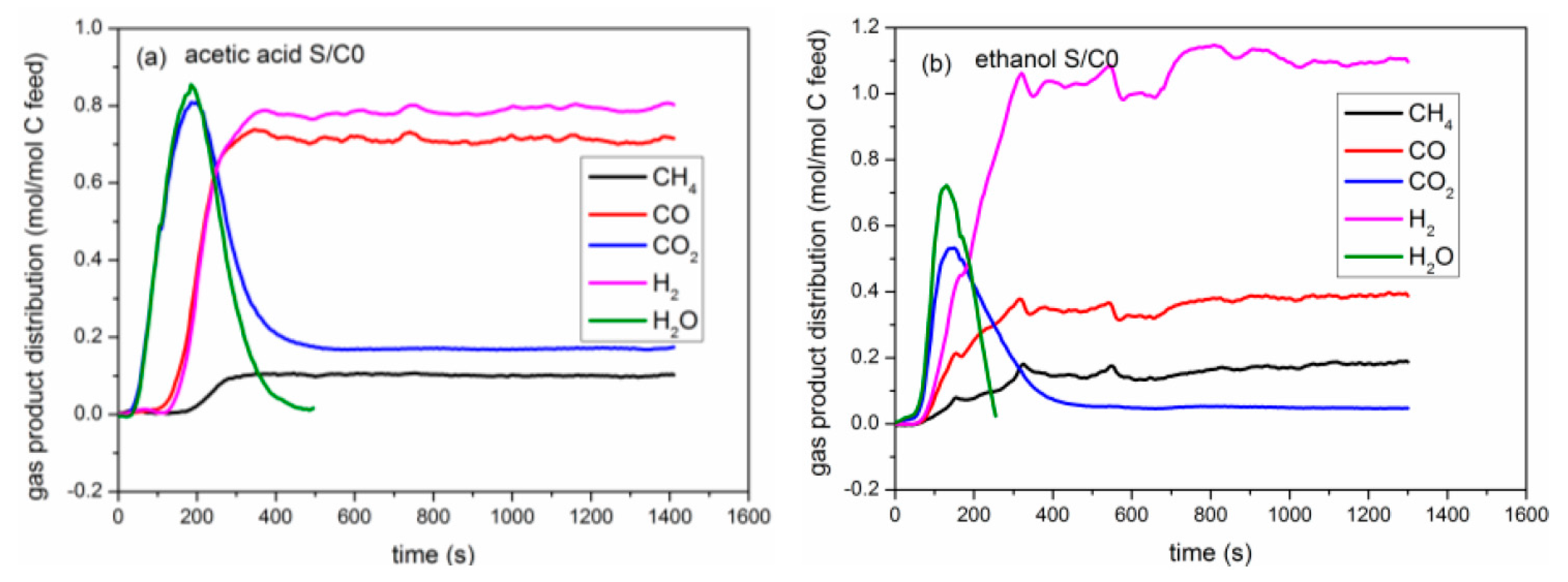
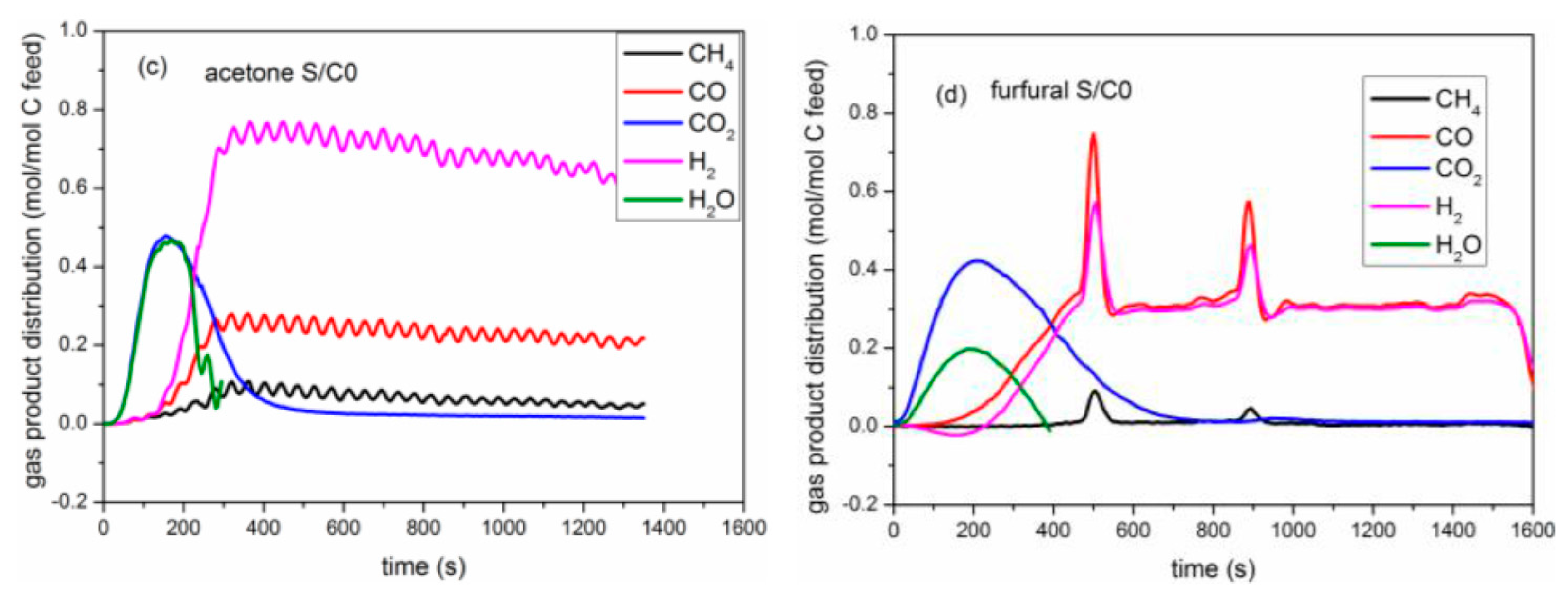
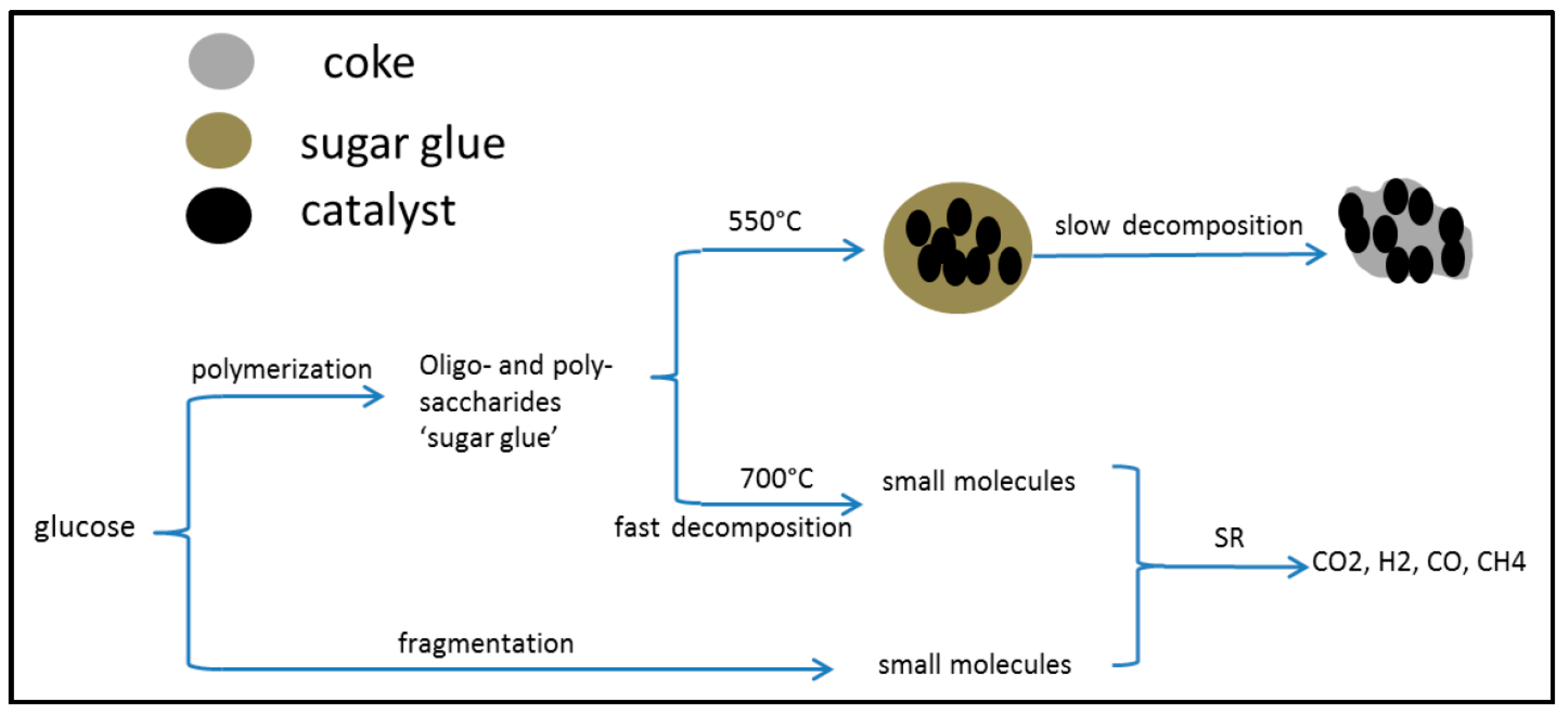
2.3. Catalytic Pyrolysis of Bio-Compounds (S/C = 0)
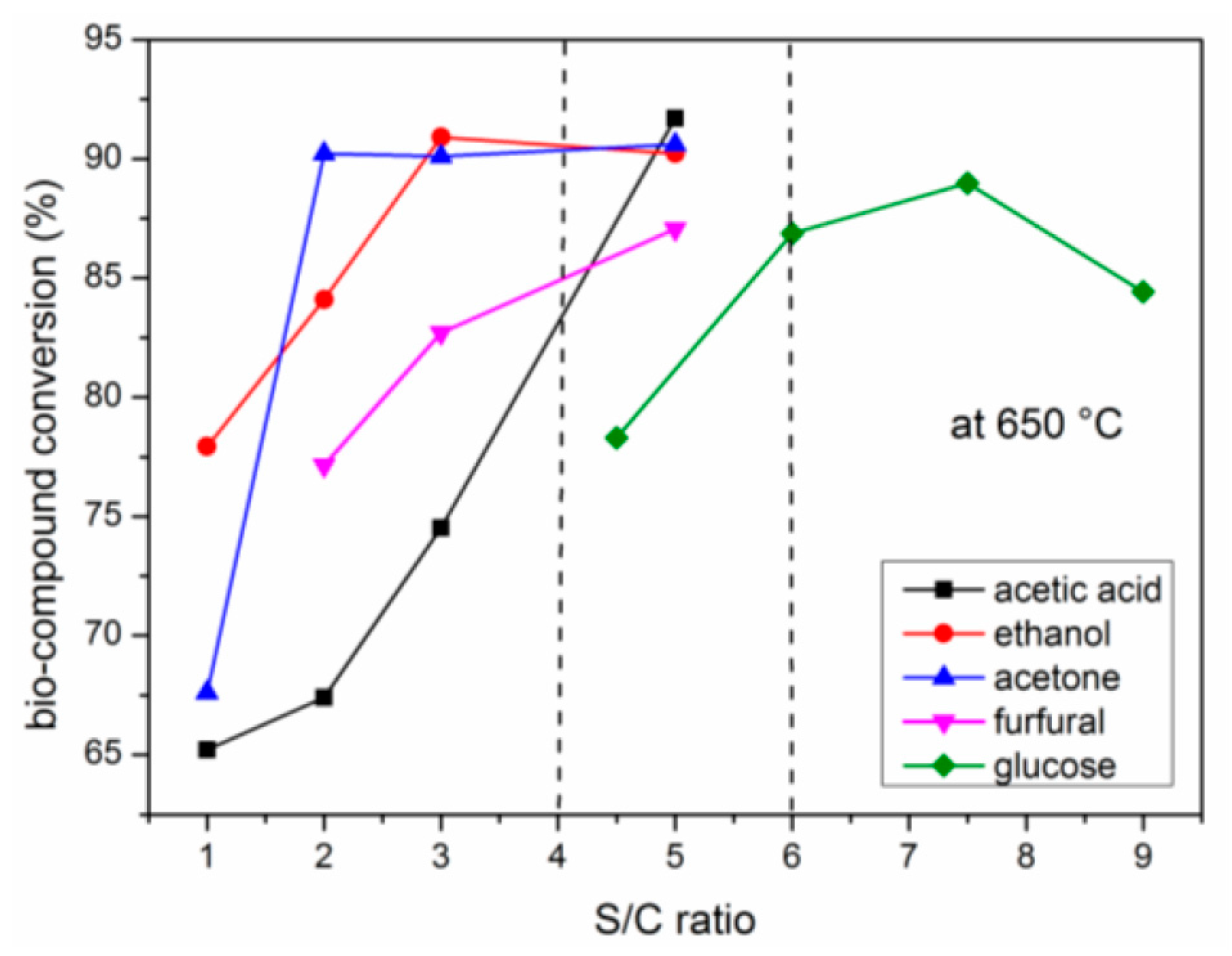
2.4. Effects of S/C
2.5. Micro-Morphology of Carbon Deposits
2.5.1. Acetic Acid, Ethanol, Acetone, and Furfural
2.5.2. Glucose
3. Materials and Methods
3.1. Materials
3.2. Reactor Set-Up and Operation Procedure
3.3. Elemental Balance and Definition of Process Outputs
3.4. Material Characterisation
3.5. Chemical Equilibrium Calculation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tanksale, A.; Beltramini, J.N.; Lu, G.M. A review of catalytic hydrogen production processes from biomass. Renew. Sustain. Energy Rev. 2010, 14, 166–182. [Google Scholar] [CrossRef]
- Cortright, R.D.; Davda, R.R.; Dumesic, J.A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. Nature 2002, 418, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Perez, M.; Chaala, A.; Pakdel, H.; Kretschmer, D.; Roy, C. Characterization of bio-oils in chemical families. Biomass Bioenergy 2007, 31, 222–242. [Google Scholar] [CrossRef]
- Wang, D.; Czernik, S.; Montane, D.; Mann, M.; Chornet, E. Biomass to hydrogen via fast pyrolysis and catalytic steam reforming of the pyrolysis oil or its fractions. Ind. Eng. Chem. Res. 1997, 36, 1507–1518. [Google Scholar] [CrossRef]
- Marquevich, M.; Czernik, S.; Chornet, E.; Montané, D. Hydrogen from biomass: Steam reforming of model compounds of fast-pyrolysis oil. Energy Fuels 1999, 13, 1160–1166. [Google Scholar] [CrossRef]
- Fatsikostas, A.N.; Verykios, X.E. Reaction network of steam reforming of ethanol over Ni-based catalysts. J. Catal. 2004, 225, 439–452. [Google Scholar] [CrossRef]
- Wang, D.; Montane, D.; Chornet, E. Catalytic steam reforming of biomass-derived oxygenates: Acetic acid and hydroxyacetaldehyde. Appl. Catal. A 1996, 143, 245–270. [Google Scholar] [CrossRef]
- Basagiannis, A.C.; Verykios, X.E. Reforming reactions of acetic acid on nickel catalysts over a wide temperature range. Appl. Catal. A 2006, 308, 182–193. [Google Scholar] [CrossRef]
- Ramos, M.C.; Navascués, A.I.; García, L.; Bilbao, R. Hydrogen production by catalytic steam reforming of acetol, a model compound of bio-oil. Ind. Eng. Chem. Res. 2007, 46, 2399–2406. [Google Scholar] [CrossRef]
- Hu, X.; Lu, G. Investigation of the steam reforming of a series of model compounds derived from bio-oil for hydrogen production. Appl. Catal. B 2009, 88, 376–385. [Google Scholar] [CrossRef]
- Graschinsky, C.; Giunta, P.; Amadeo, N.; Laborde, M. Thermodynamic analysis of hydrogen production by autothermal reforming of ethanol. Int. J. Hydrogen Energy 2012, 37, 10118–10124. [Google Scholar] [CrossRef]
- Subramani, V.; Song, C. Advances in catalysis and processes for hydrogen production from ethanol reforming. Catalysis 2007, 20, 65–106. [Google Scholar]
- Vagia, E.C.; Lemonidou, A.A. Thermodynamic analysis of hydrogen production via steam reforming of selected components of aqueous bio-oil fraction. Int. J. Hydrogen Energy 2007, 32, 212–223. [Google Scholar] [CrossRef]
- Sun, S.; Yan, W.; Sun, P.; Chen, J. Thermodynamic analysis of ethanol reforming for hydrogen production. Energy 2012, 44, 911–924. [Google Scholar] [CrossRef]
- Konsolakis, M.; Ioakimidis, Z.; Kraia, T.; Marnellos, G. Hydrogen production by ethanol steam reforming (ESR) over CeO2 supported transition metal (Fe, Co, Ni, Cu) catalysts: Insight into the structure-activity relationship. Catalysts 2016, 6, 39. [Google Scholar] [CrossRef]
- Cifuentes, B.; Figueredo, M.; Cobo, M. Response surface methodology and aspen plus integration for the simulation of the catalytic steam reforming of ethanol. Catalysts 2017, 7, 15. [Google Scholar] [CrossRef]
- Cifuentes, B.; Valero, M.; Conesa, J.; Cobo, M. Hydrogen production by steam reforming of ethanol on Rh-Pt catalysts: Influence of CeO2, ZrO2, and La2O3 as supports. Catalysts 2015, 5, 1872–1896. [Google Scholar] [CrossRef]
- Vaidya, P.D.; Rodrigues, A.E. Insight into steam reforming of ethanol to produce hydrogen for fuel cells. Chem. Eng. J. 2006, 117, 39–49. [Google Scholar] [CrossRef]
- Contreras, J.L.; Salmones, J.; Colin-Luna, J.A.; Nuno, L.; Quintana, B.; Cordova, I.; Zeifert, B.; Tapia, C.; Fuentes, G.A. Catalysts for H2 production using the ethanol steam reforming (a review). Int. J. Hydrogen Energy 2014, 39, 18835–18853. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, M.; Liang, T.; Yang, Z.; Yang, J.; Liu, S. Hydrogen generation from catalytic steam reforming of acetic acid by Ni/attapulgite catalysts. Catalysts 2016, 6, 172. [Google Scholar] [CrossRef]
- Adhikari, S.; Fernando, S.; Gwaltney, S.R.; Filip To, S.; Mark Bricka, R.; Steele, P.H.; Haryanto, A. A thermodynamic analysis of hydrogen production by steam reforming of glycerol. Int. J. Hydrogen Energy 2007, 32, 2875–2880. [Google Scholar] [CrossRef]
- Wu, C.; Liu, R. Carbon deposition behavior in steam reforming of bio-oil model compound for hydrogen production. Int. J. Hydrogen Energy 2010, 35, 7386–7398. [Google Scholar] [CrossRef]
- Khzouz, M.; Wood, J.; Pollet, B.; Bujalski, W. Characterization and activity test of commercial Ni/Al2O3, Cu/ZnO/Al2O3 and prepared Ni–Cu/Al2O3 catalysts for hydrogen production from methane and methanol fuels. Int. J. Hydrogen Energy 2013, 38, 1664–1675. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Wang, Y. Significantly improved catalytic performance of Ni-based MgO catalyst in steam reforming of phenol by inducing mesostructure. Catalysts 2015, 5, 1721–1736. [Google Scholar] [CrossRef]
- Hu, X.; Lu, G.X. Investigation of the effects of molecular structure on oxygenated hydrocarbon steam re-forming. Energy Fuels 2009, 23, 926–933. [Google Scholar] [CrossRef]
- Remon, J.; Broust, F.; Volle, G.; Garcia, L.; Arauzo, J. Hydrogen production from pine and poplar bio-oils by catalytic steam reforming. Influence of the bio-oil composition on the process. Int. J. Hydrogen Energy 2015, 40, 5593–5608. [Google Scholar] [CrossRef]
- Dupont, V.; Ross, A.; Knight, E.; Hanley, I.; Twigg, M. Production of hydrogen by unmixed steam reforming of methane. Chem. Eng. Sci. 2008, 63, 2966–2979. [Google Scholar] [CrossRef]
- Pimenidou, P.; Rickett, G.; Dupont, V.; Twigg, M. Chemical looping reforming of waste cooking oil in packed bed reactor. Bioresour. Technol. 2010, 101, 6389–6397. [Google Scholar] [CrossRef] [PubMed]
- Giannakeas, N.; Lea-Langton, A.; Dupont, V.; Twigg, M.V. Hydrogen from scrap tyre oil via steam reforming and chemical looping in a packed bed reactor. Appl. Catal. B 2012, 126, 249–257. [Google Scholar] [CrossRef]
- Zafar, Q.; Mattisson, T.; Gevert, B. Integrated hydrogen and power production with CO2 capture using chemical-looping reforming redox reactivity of particles of CuO, Mn2O3, NiO, and Fe2O3 using SiO2 as a support. Ind. Eng. Chem. Res. 2005, 44, 3485–3496. [Google Scholar] [CrossRef]
- Lyon, R.K.; Cole, J.A. Unmixed combustion: An alternative to fire. Combust. Flame 2000, 121, 249–261. [Google Scholar] [CrossRef]
- Mendiara, T.; Johansen, J.M.; Utrilla, R.; Geraldo, P.; Jensen, A.D.; Glarborg, P. Evaluation of different oxygen carriers for biomass tar reforming (I): Carbon deposition in experiments with toluene. Fuel 2011, 90, 1049–1060. [Google Scholar] [CrossRef]
- Adanez, J.; Abad, A.; Garcia-Labiano, F.; Gayan, P.; de Diego, L.F. Progress in chemical-looping combustion and reforming technologies. Prog. Energy Combust. Sci. 2012, 38, 215–282. [Google Scholar] [CrossRef]
- Dou, B.; Song, Y.; Wang, C.; Chen, H.; Yang, M.; Xu, Y. Hydrogen production by enhanced-sorption chemical looping steam reforming of glycerol in moving-bed reactors. Appl. Energy 2014, 130, 342–349. [Google Scholar] [CrossRef]
- Pimenidou, P.; Rickett, G.; Dupont, V.; Twigg, M.V. High purity H2 by sorption-enhanced chemical looping reforming of waste cooking oil in a packed bed reactor. Bioresour. Technol. 2010, 101, 9279–9286. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Dupont, V.; Twigg, M.V. Direct reduction of nickel catalyst with model bio-compounds. Appl. Catal. B 2017, 200, 121–132. [Google Scholar] [CrossRef]
- Tribalis, A.; Panagiotou, G.; Bourikas, K.; Sygellou, L.; Kennou, S.; Ladas, S.; Lycourghiotis, A.; Kordulis, C. Ni catalysts supported on modified alumina for diesel steam reforming. Catalysts 2016, 6, 11. [Google Scholar] [CrossRef]
- Goodman, D.R. Catalyst Handbook, 2nd ed.; Twigg, M.V., Ed.; Wolfe Publishing Ltd.: London, UK, 1996; pp. 161–174. [Google Scholar]
- Sehested, J. Four challenges for nickel steam-reforming catalysts. Catal. Today 2006, 111, 103–110. [Google Scholar] [CrossRef]
- Lea-Langton, A.; Zin, R.M.; Dupont, V.; Twigg, M.V. Biomass pyrolysis oils for hydrogen production using chemical looping reforming. Int. J. Hydrogen Energy 2012, 37, 2037–2043. [Google Scholar] [CrossRef]
- Dupont, V.; Ross, A.B.; Hanley, I.; Twigg, M.V. Unmixed steam reforming of methane and sunflower oil: A single-reactor process for H2-rich gas. Int. J. Hydrogen Energy 2007, 32, 67–79. [Google Scholar] [CrossRef]
- Feng, C.; Dupont, V. Nickel catalyst auto-reduction during steam reforming of bio-oil model compound acetic acid. Int. J. Hydrogen Energy 2013, 38, 15160–15172. [Google Scholar]
- Cheng, F. Bio-Compounds as Reducing Agents of Reforming Catalyst and Their Subsequent Steam Reforming Performance. Ph.D. Thesis, University of Leeds, Leeds, UK, 2014. [Google Scholar]
- Kato, K.; Takahashi, N. Pyrolysis of cellulose part II. Thermogravimetric analyses and determination of carbonyl and carboxyl groups in pyrocellulose. Agric. Biol. Chem. 1967, 31, 519–524. [Google Scholar]
- Wu, C.; Liu, R. Hydrogen production from steam reforming of m-cresol, a model compound derived from bio-oil: Green process evaluation based on liquid condensate recycling. Energy Fuels 2010, 24, 5139–5147. [Google Scholar] [CrossRef]
- Fagerson, I.S. Thermal degradation of carbohydrates; a review. J. Agric. Food Chem. 1969, 17, 747–750. [Google Scholar] [CrossRef]
- Sugisawa, H.; Edo, H. The thermal degradation of sugars I. Thermal polymerization of glucose. J. Food Sci. 1966, 31, 561–565. [Google Scholar] [CrossRef]
- Örsi, F. Kinetic studies on the thermal decomposition of glucose and fructose. J. Therm. Anal. 1973, 5, 329–335. [Google Scholar] [CrossRef]
- Gardner, R.A. The kinetics of silica reduction in hydrogen. J. Solid State Chem. 1974, 9, 336–344. [Google Scholar] [CrossRef]
- Vagia, E.C.; Lemonidou, A.A. Investigations on the properties of ceria–zirconia-supported Ni and Rh catalysts and their performance in acetic acid steam reforming. J. Catal. 2010, 269, 388–396. [Google Scholar] [CrossRef]
- Rossetti, I.; Lasso, J.; Nichele, V.; Signoretto, M.; Finocchio, E.; Ramis, G.; Di Michele, A. Silica and zirconia supported catalysts for the low-temperature ethanol steam reforming. Appl. Catal. B 2014, 150–151, 257–267. [Google Scholar] [CrossRef]
- Rossetti, I.; Gallo, A.; Dal Santo, V.; Bianchi, C.L.; Nichele, V.; Signoretto, M.; Finocchio, E.; Ramis, G.; Di Michele, A. Nickel catalysts supported over TiO2, SiO2 and ZrO2 for the steam reforming of glycerol. ChemCatChem 2013, 5, 294–306. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bio-Compound | Stoichiometric 1 | Equilibrium 2 | Experiment | H2 Yield Efficiency (%) |
|---|---|---|---|---|
| ethanol | 3 | 2.58 | 1.78 | 68.99 |
| acetone | 2.7 | 2.26 | 1.72 | 76.11 |
| glucose | 2 | 1.85 | 1.42 | 76.76 |
| acetic acid | 2 | 1.73 | 1.06 | 61.27 |
| furfural | 2 | 1.67 | 1.20 | 71.86 |
| Bio-Compounds | H2 | CO | CO2 | CH4 | Solid C |
|---|---|---|---|---|---|
| acetic acid | 0.79 | 0.71 | 0.17 | 0.1 | 0.02 |
| ethanol | 1.08 | 0.37 | 0.05 | 0.16 | 0.42 |
| acetone | 0.68 | 0.23 | 0.02 | 0.06 | 0.69 |
| furfural | 0.31 | 0.31 | 0.01 | 0.01 | 0.67 |
| Amount of Carbon Deposits in Descending Order | Furfural | Acetic Acid | Ethanol | Acetone |
|---|---|---|---|---|
| Density of carbon filaments | dense | dense | less dense | less dense |
| Diameter of carbon filaments | big | small | big | small |
| Sites | Al | O | Ni | C |
|---|---|---|---|---|
| A | 27 | 0 | 42 | 31 |
| B | 51 | 5 | 3 | 41 |
| C | 0 | 6 | 0 | 94 |
| D | 14 | 17 | 59 | 10 |
| E | 28 | 18 | 49 | 5 |
| F | 57 | 41 | 0 | 2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, F.; Dupont, V. Steam Reforming of Bio-Compounds with Auto-Reduced Nickel Catalyst. Catalysts 2017, 7, 114. https://doi.org/10.3390/catal7040114
Cheng F, Dupont V. Steam Reforming of Bio-Compounds with Auto-Reduced Nickel Catalyst. Catalysts. 2017; 7(4):114. https://doi.org/10.3390/catal7040114
Chicago/Turabian StyleCheng, Feng, and Valerie Dupont. 2017. "Steam Reforming of Bio-Compounds with Auto-Reduced Nickel Catalyst" Catalysts 7, no. 4: 114. https://doi.org/10.3390/catal7040114
APA StyleCheng, F., & Dupont, V. (2017). Steam Reforming of Bio-Compounds with Auto-Reduced Nickel Catalyst. Catalysts, 7(4), 114. https://doi.org/10.3390/catal7040114