
Upgrading Lignocellulosic Biomasses: Hydrogenolysis of Platform Derived Molecules Promoted by Heterogeneous Pd-Fe Catalysts





Abstract
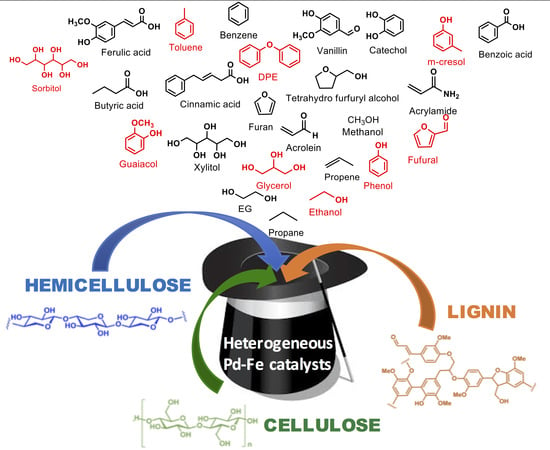
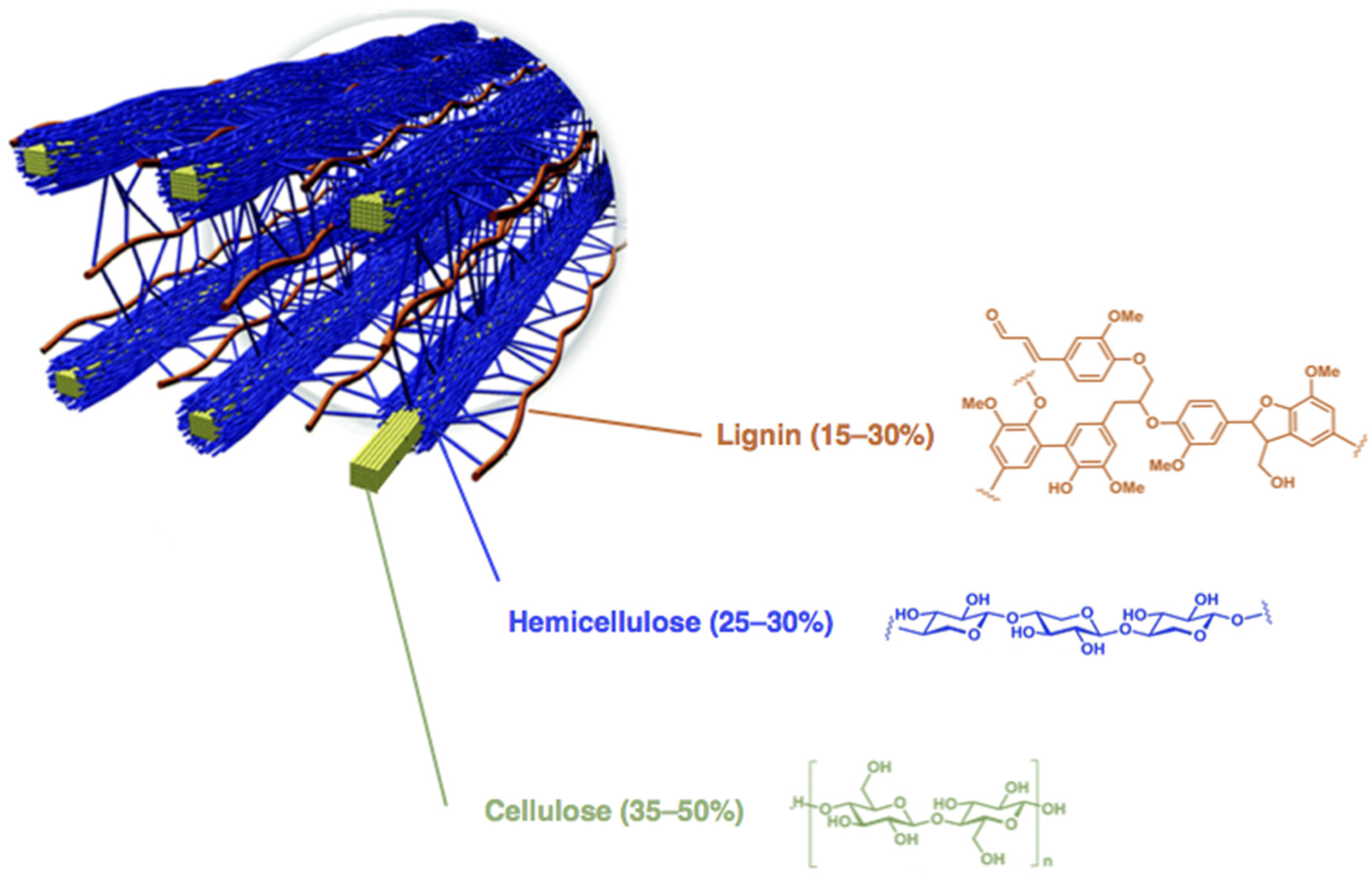
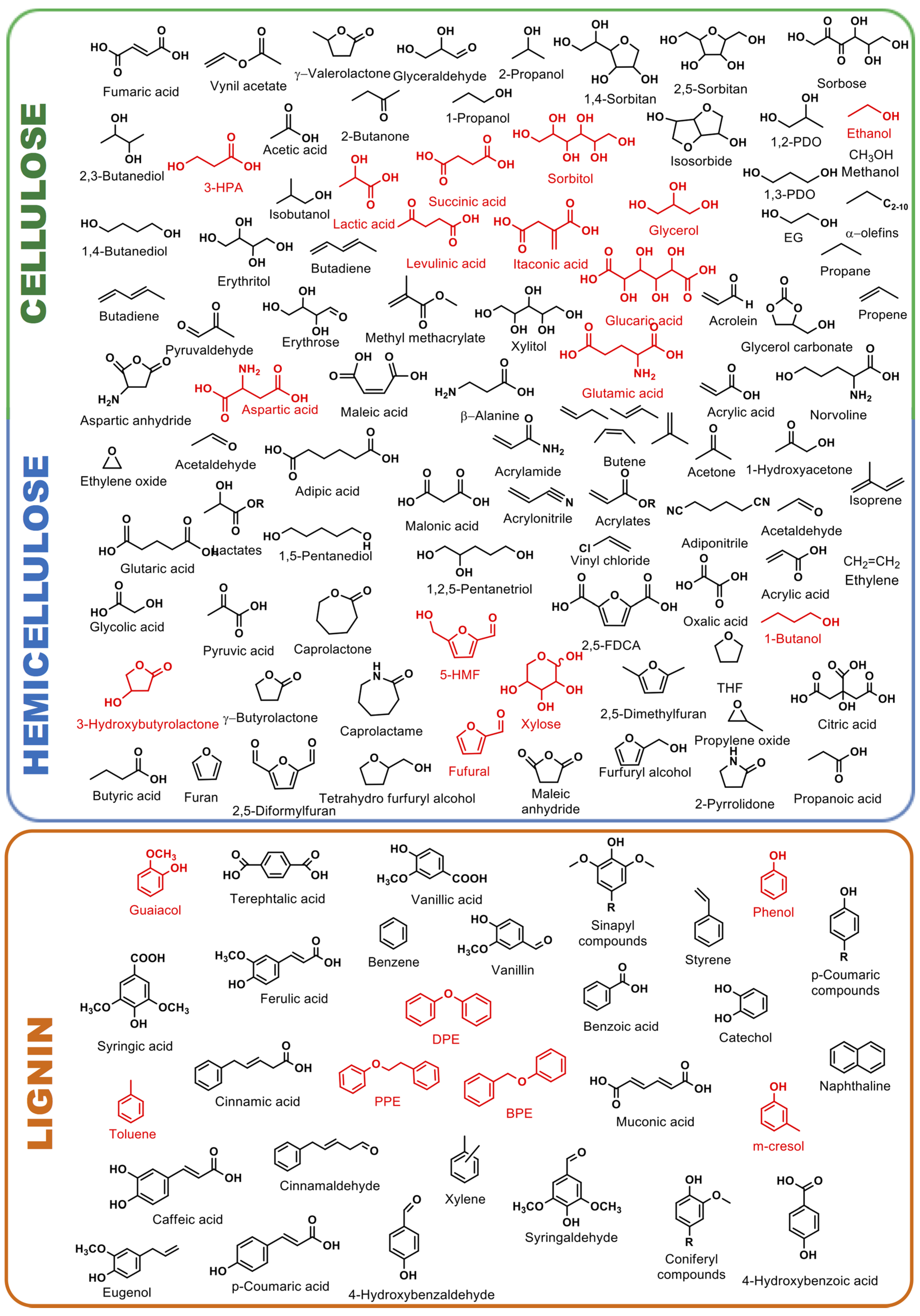
:
1. Introduction
2. Synthesis and Characterization of Pd/Fe Catalysts
2.1. Preparation Methods
2.1.1. Incipient Wetness Impregnation—IWi
2.1.2. Deposition-Precipitation—DP
2.1.3. Co-Precipitation—CP
2.2. Physico-Chemical Characterization
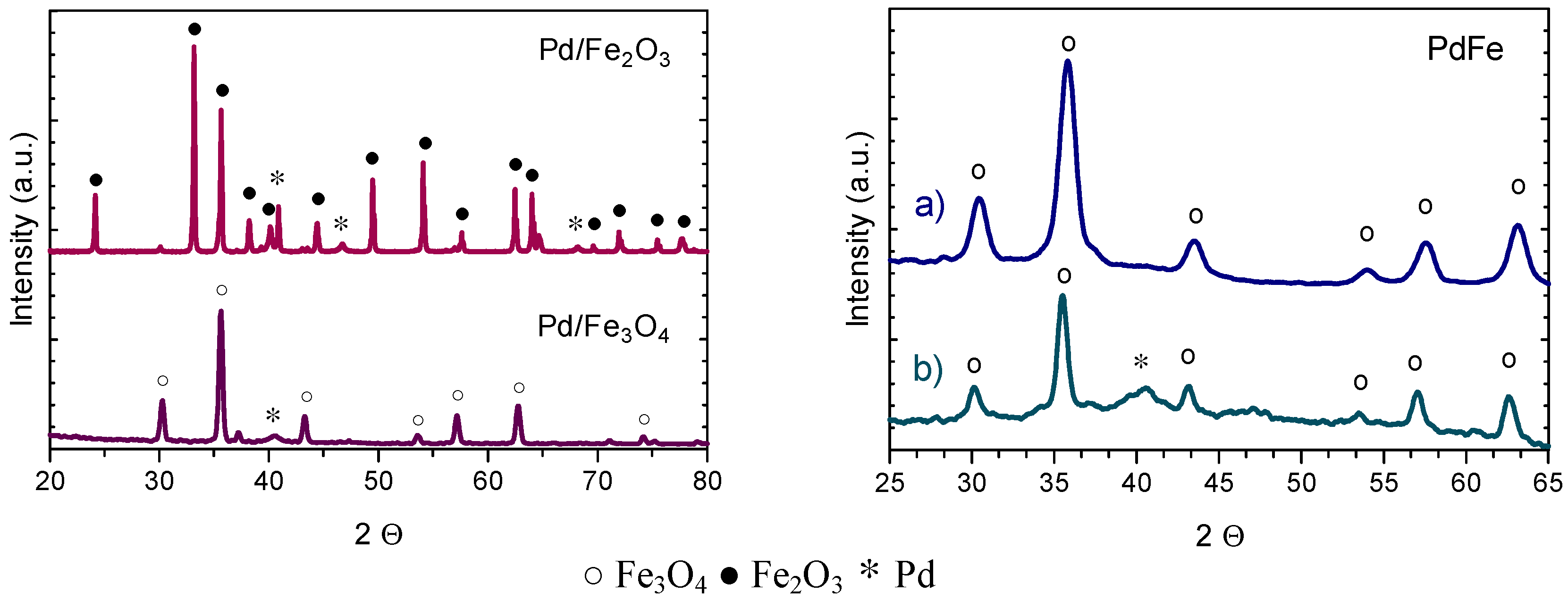
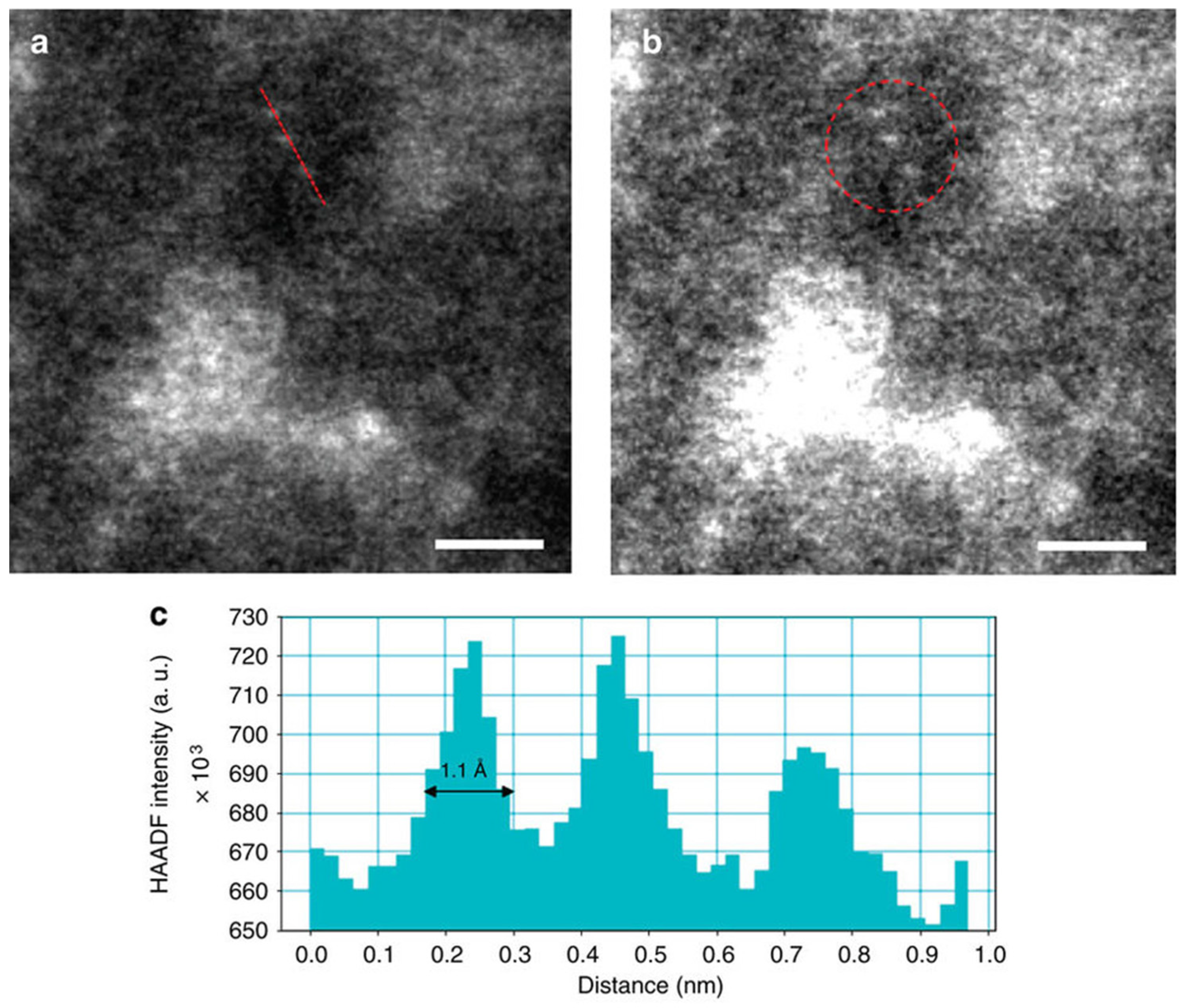
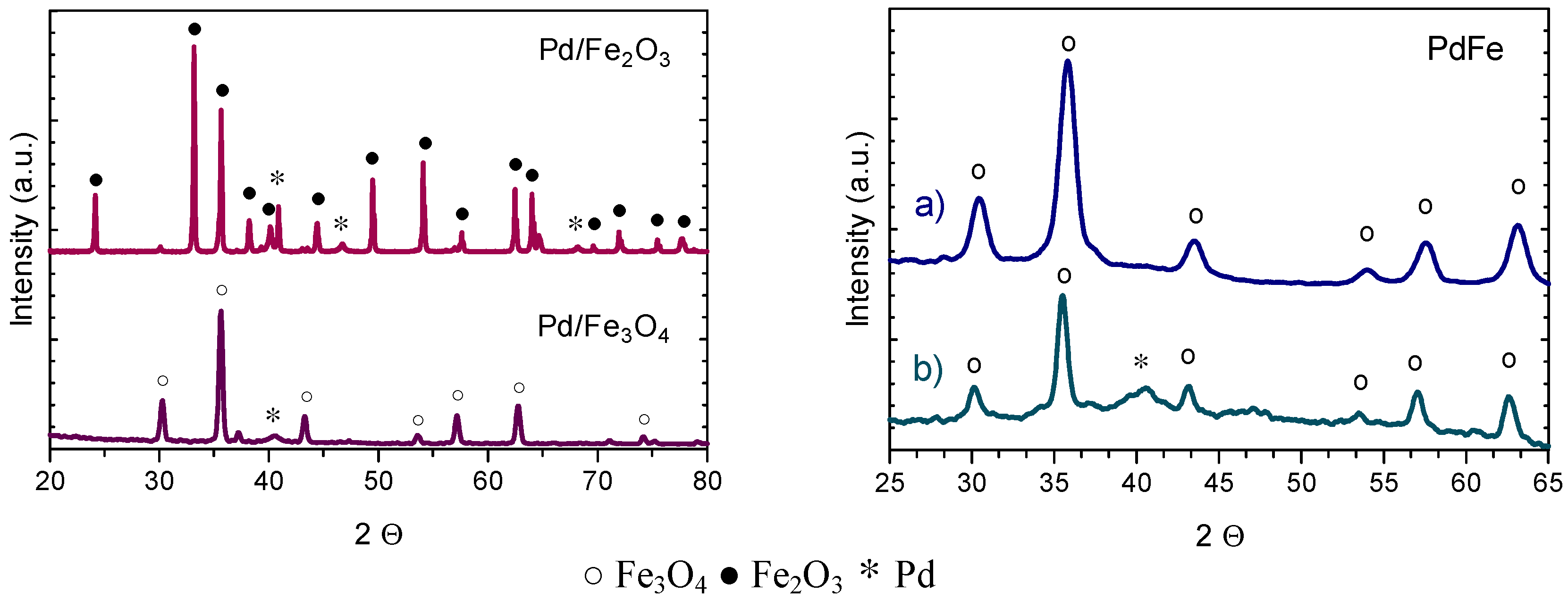
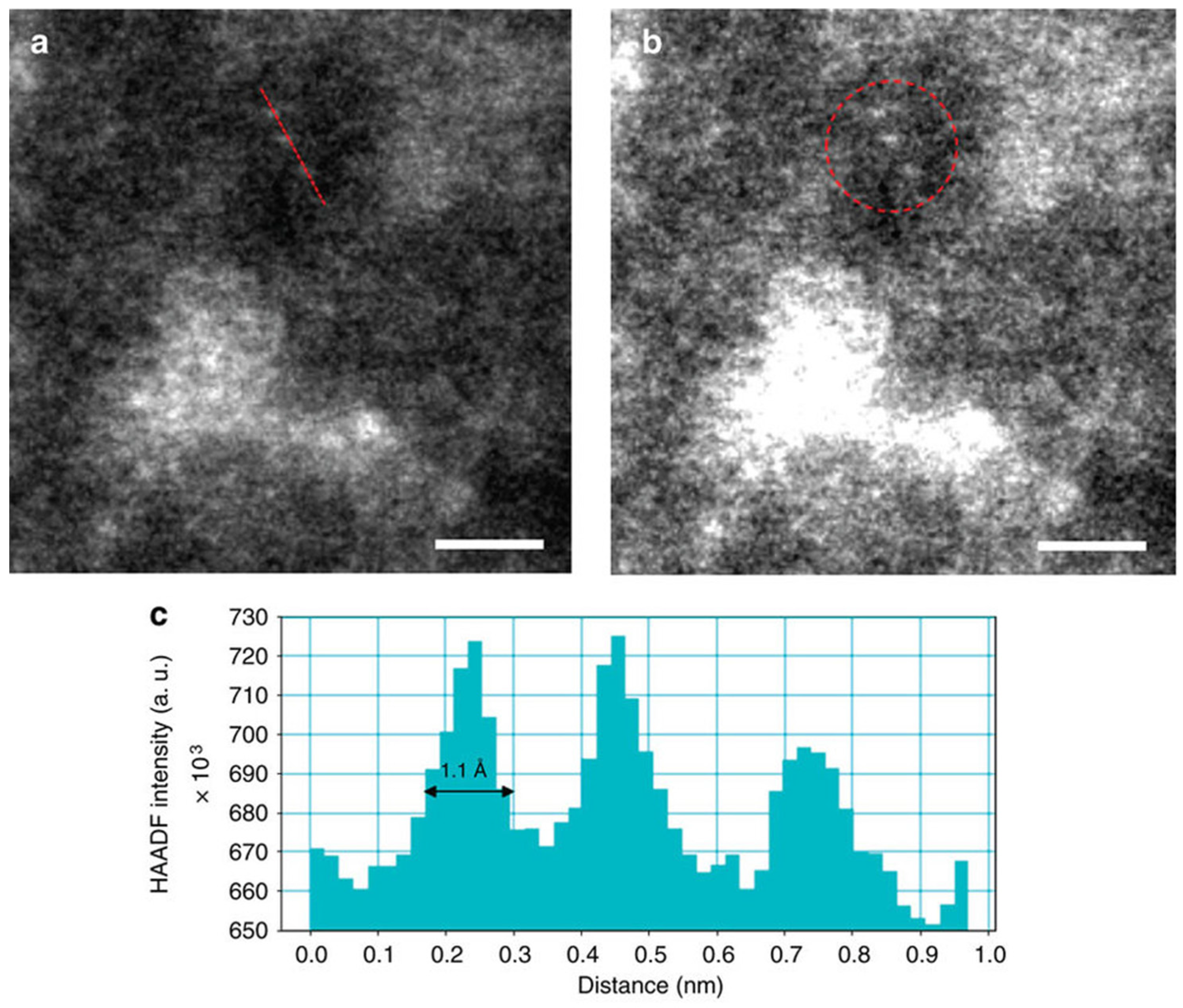
2.2.1. X-ray Diffraction (XRD) and Transmission Electron Microscopy (TEM)
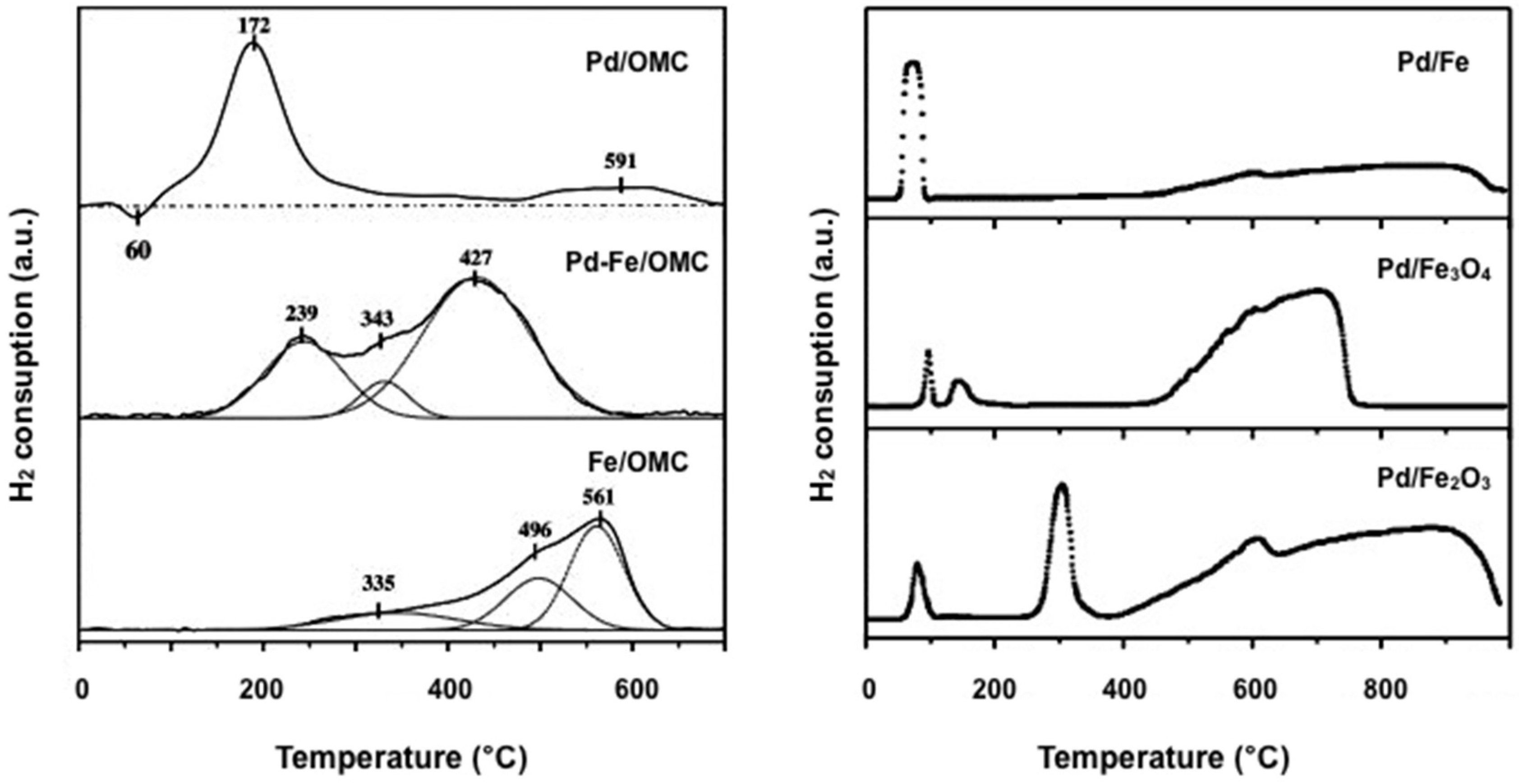
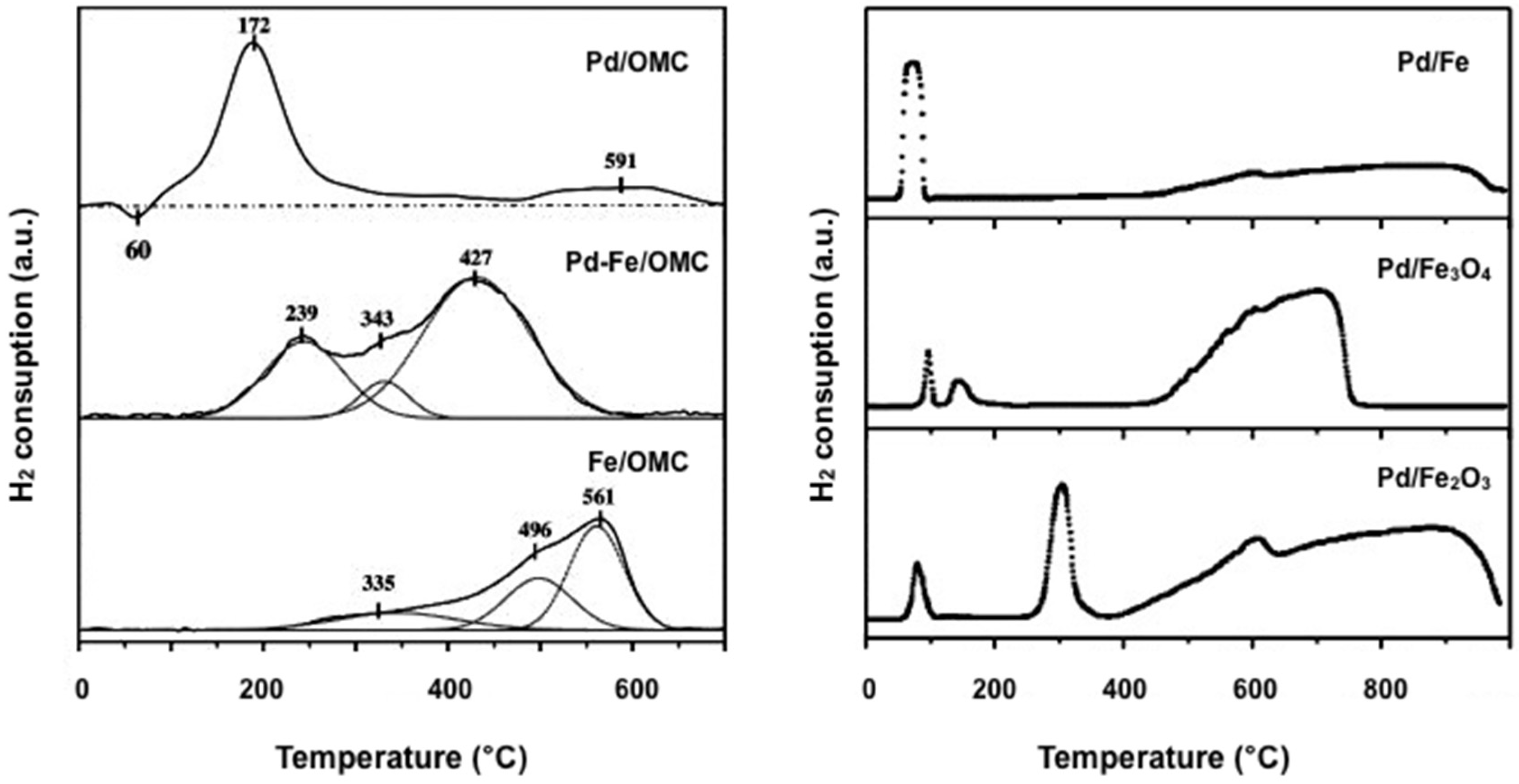
2.2.2. Hydrogen Temperature-Programmed Reduction (H2-TPR)
2.2.3. X-ray Photoelectron Spectroscopy (XPS)
2.2.4. Extended X-ray Absorption Fine Structure (EXAFS)
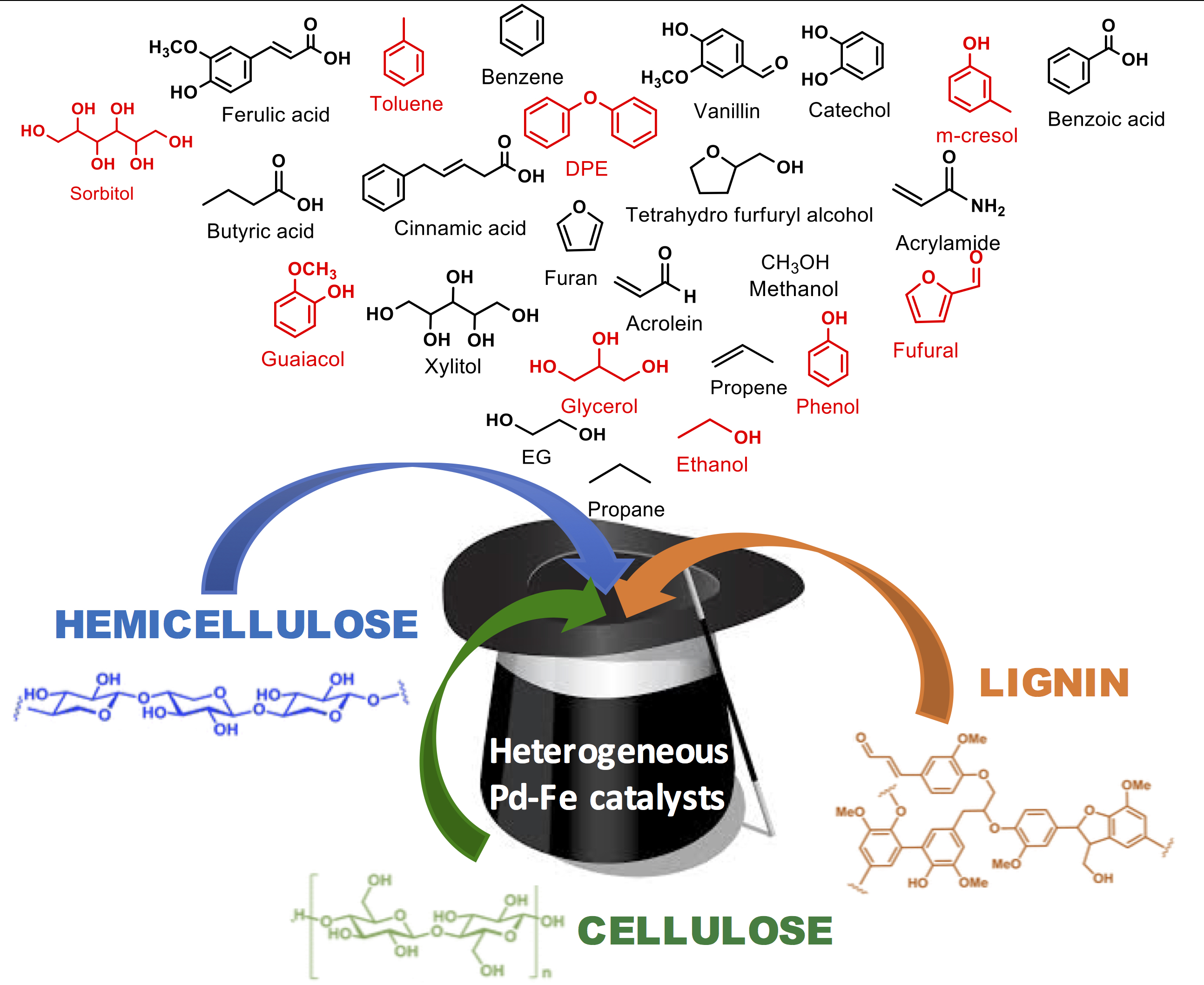
3. Hydrogenolysis Reactions of Biomass-Derived Platform Molecules Promoted by Heterogeneous Pd-Fe Catalysts
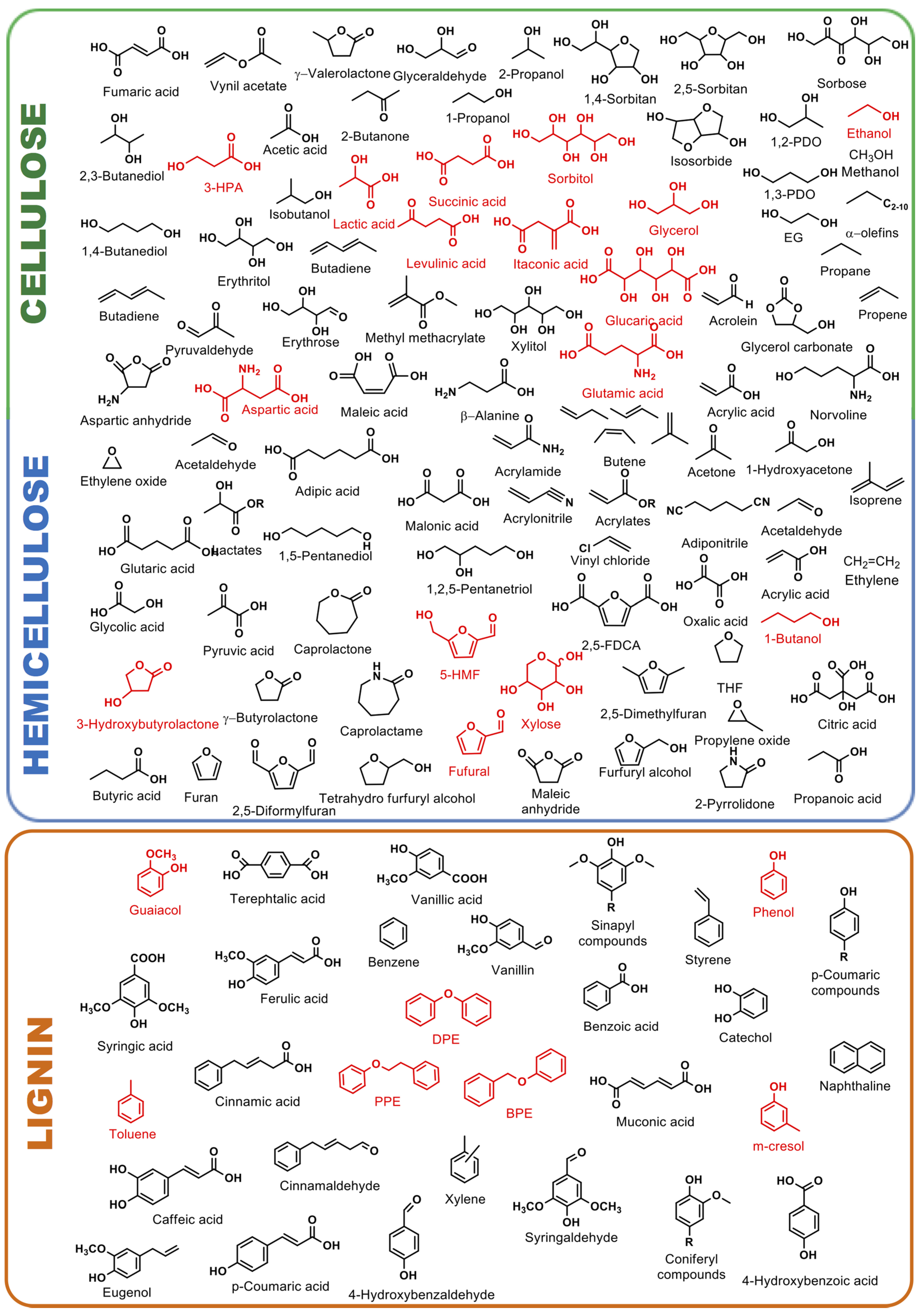
3.1. Valorization of Cellulose and Hemicellulose Derived Molecules
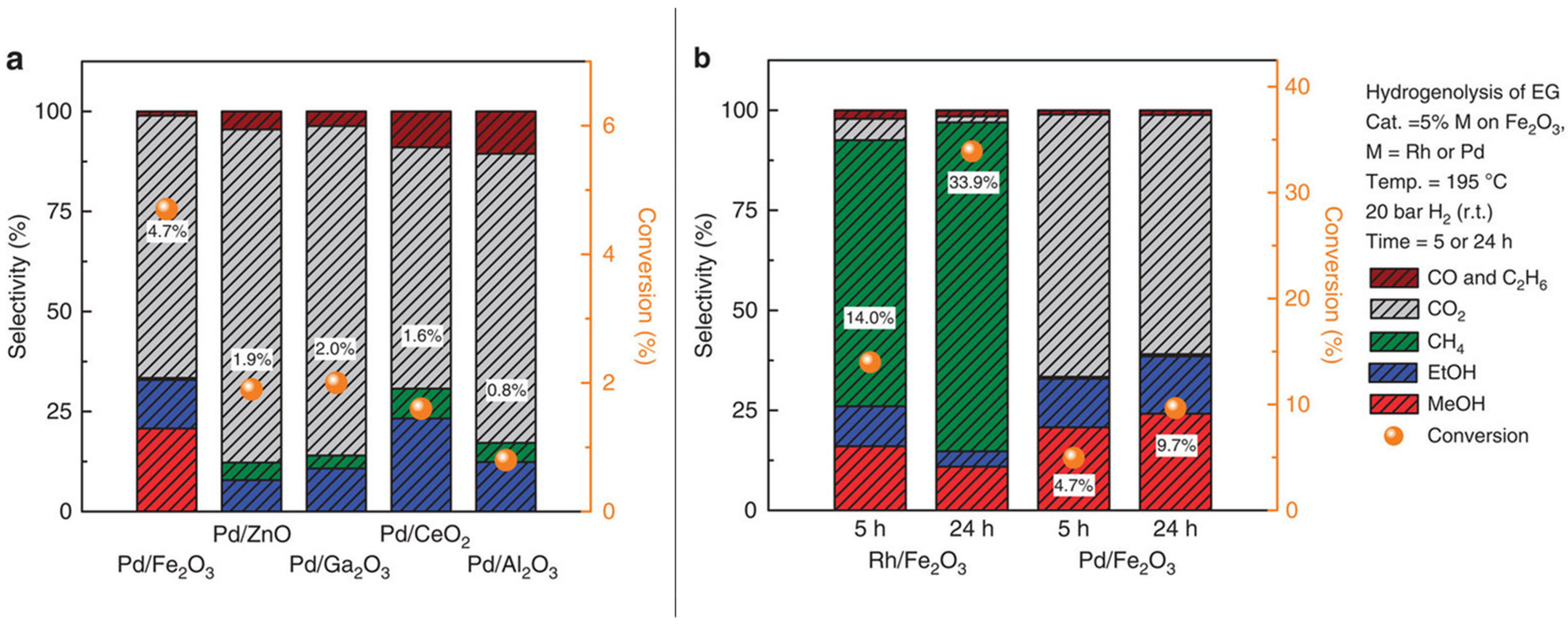
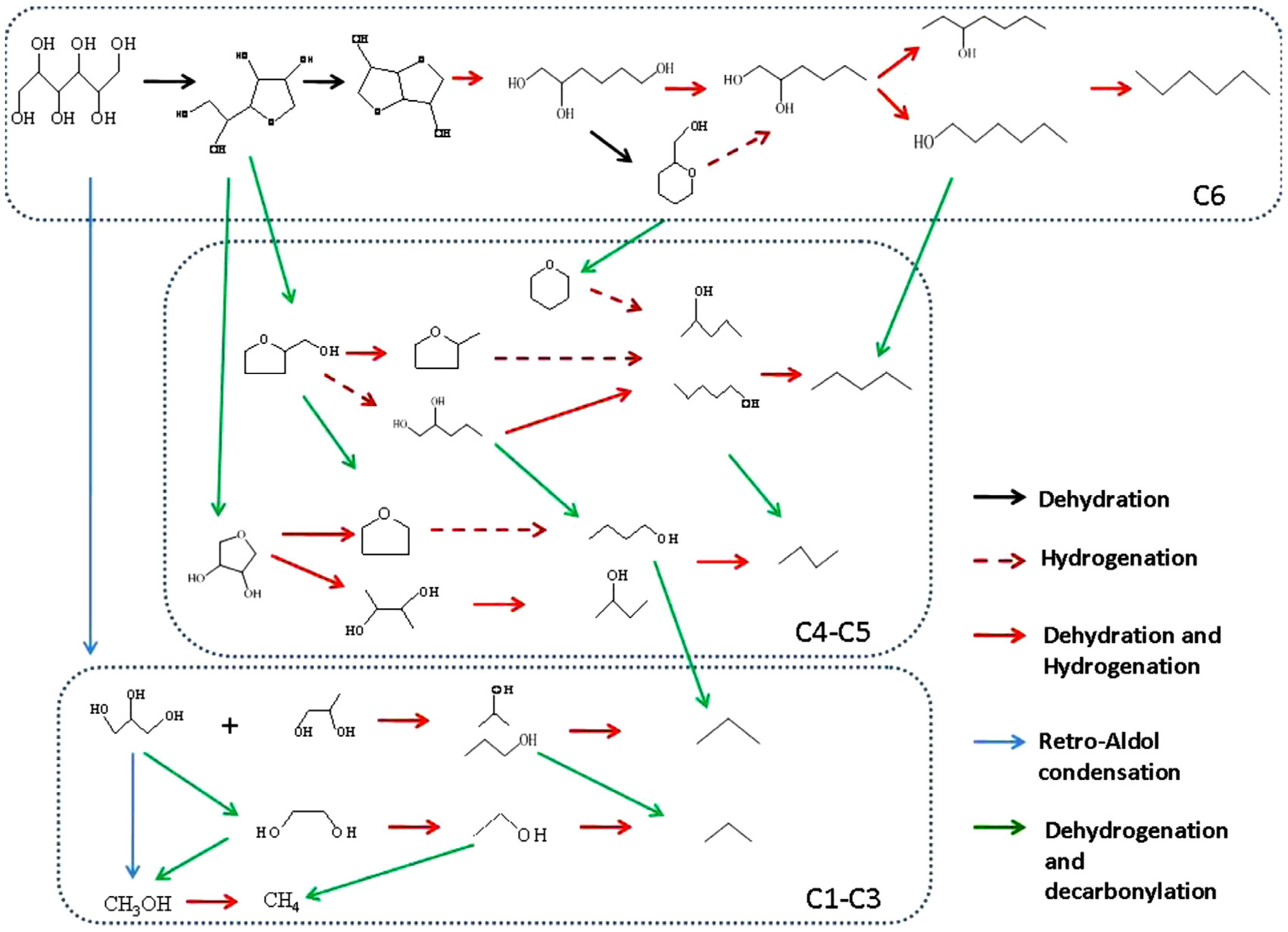
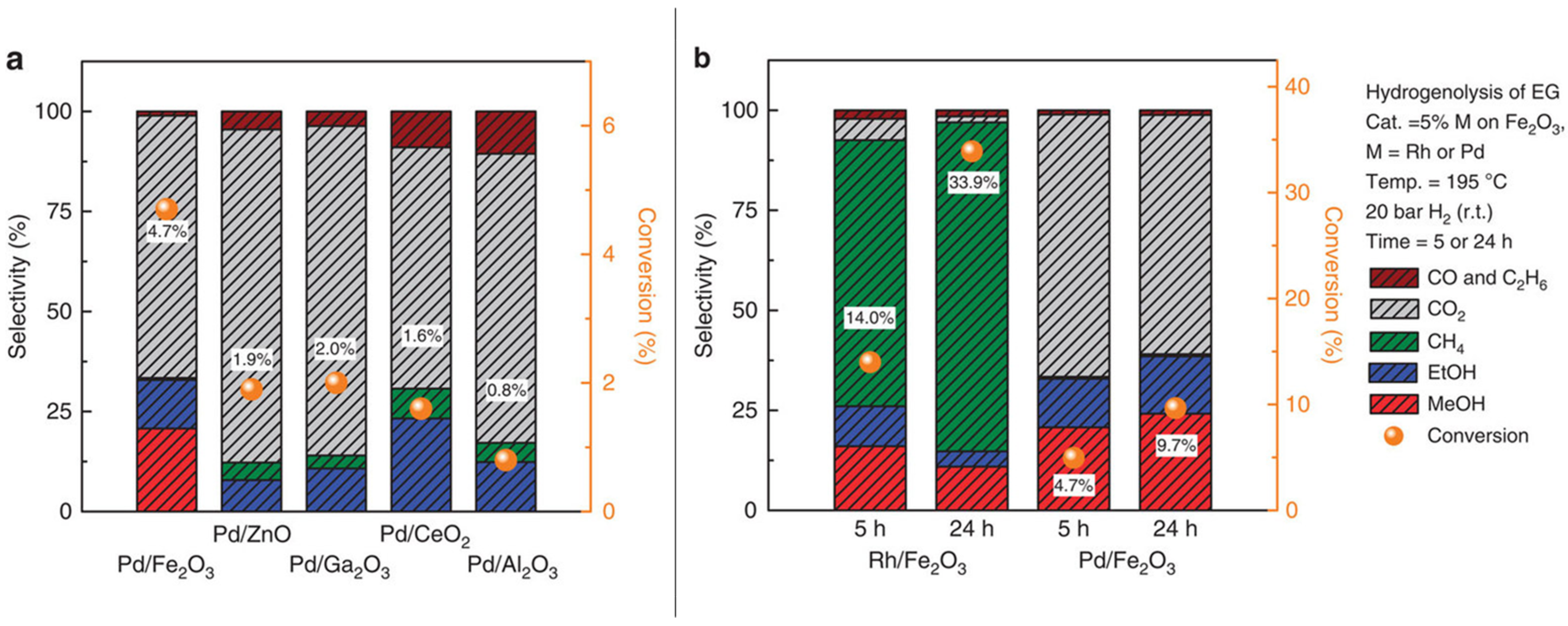
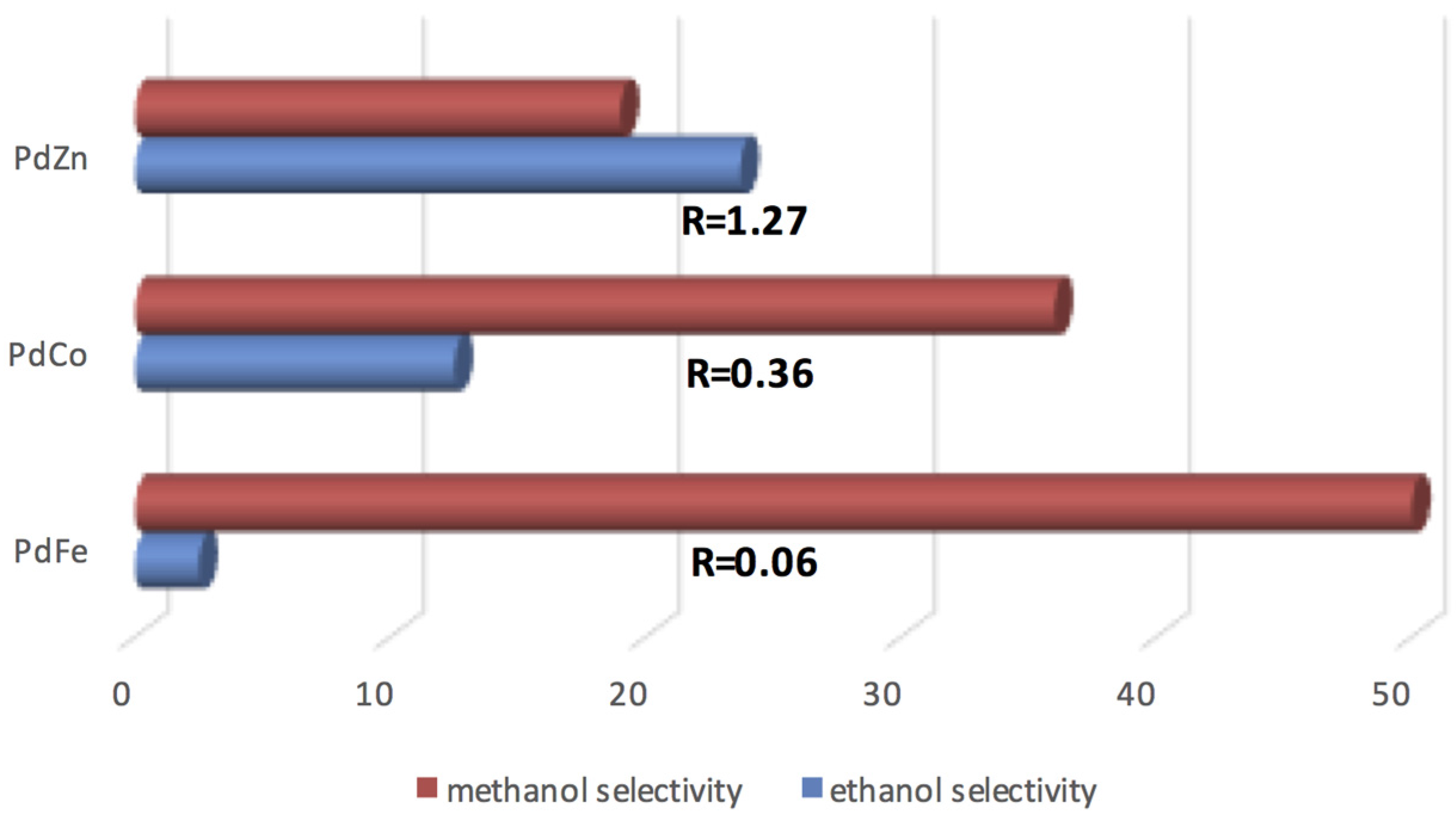
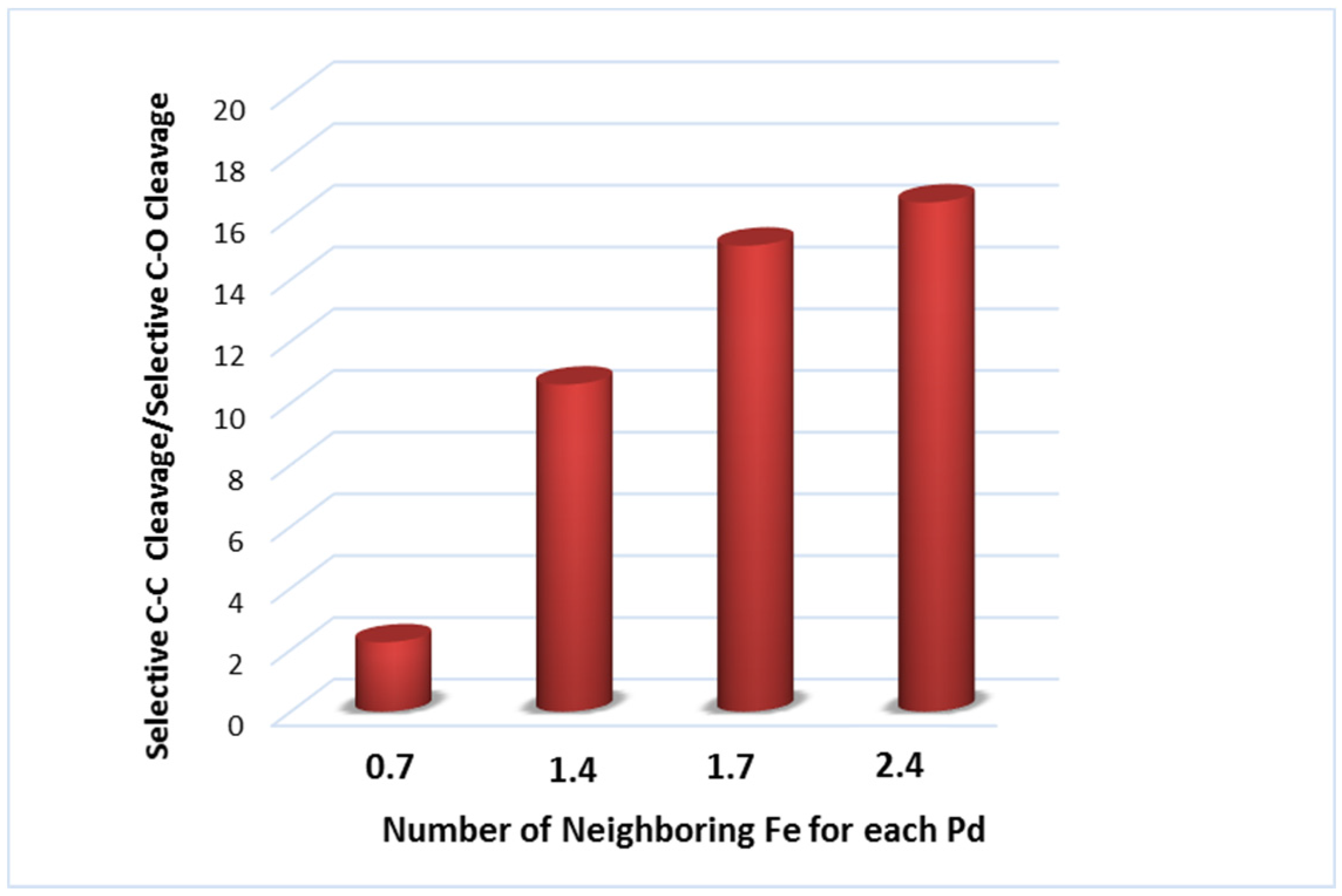
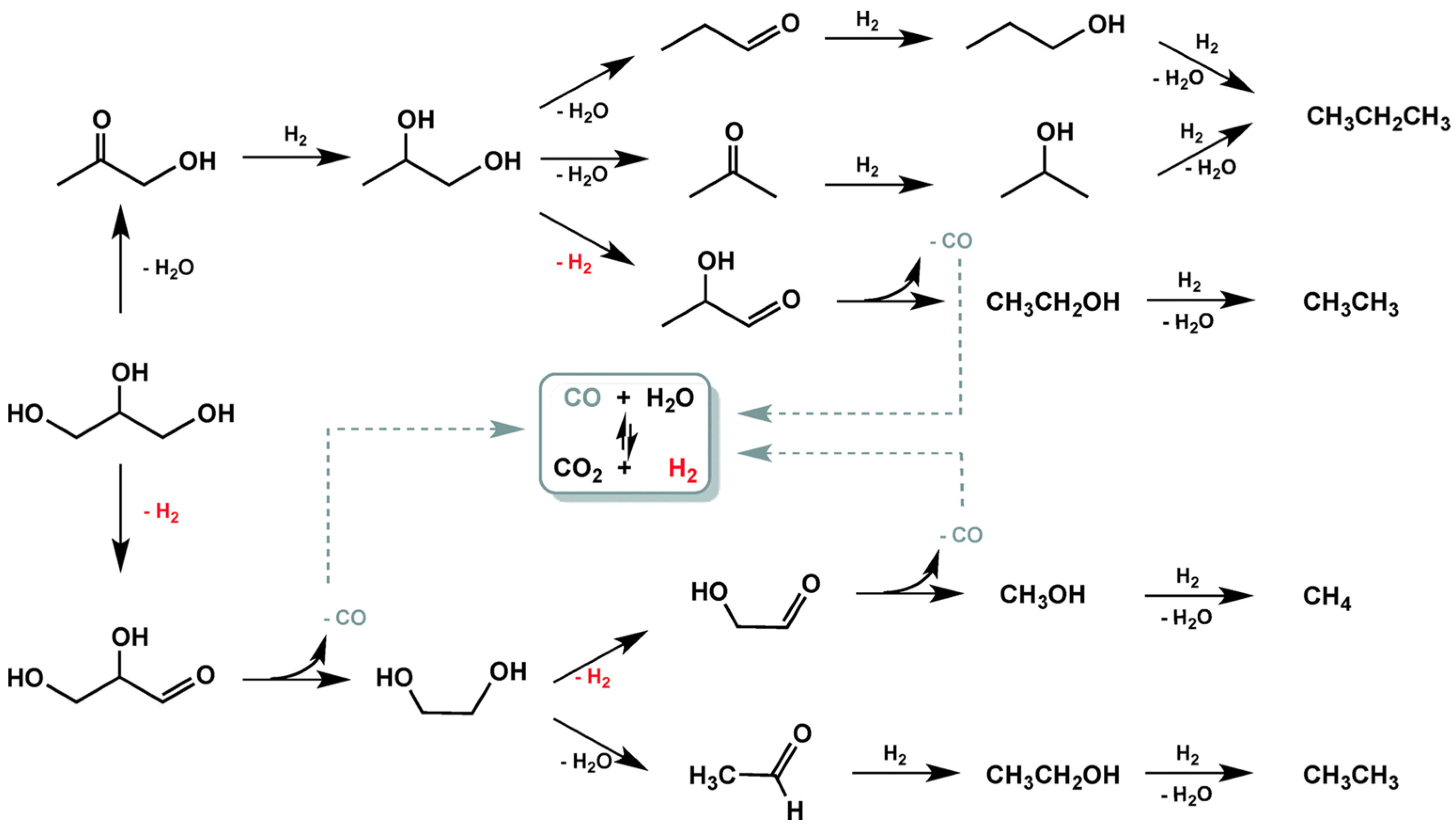
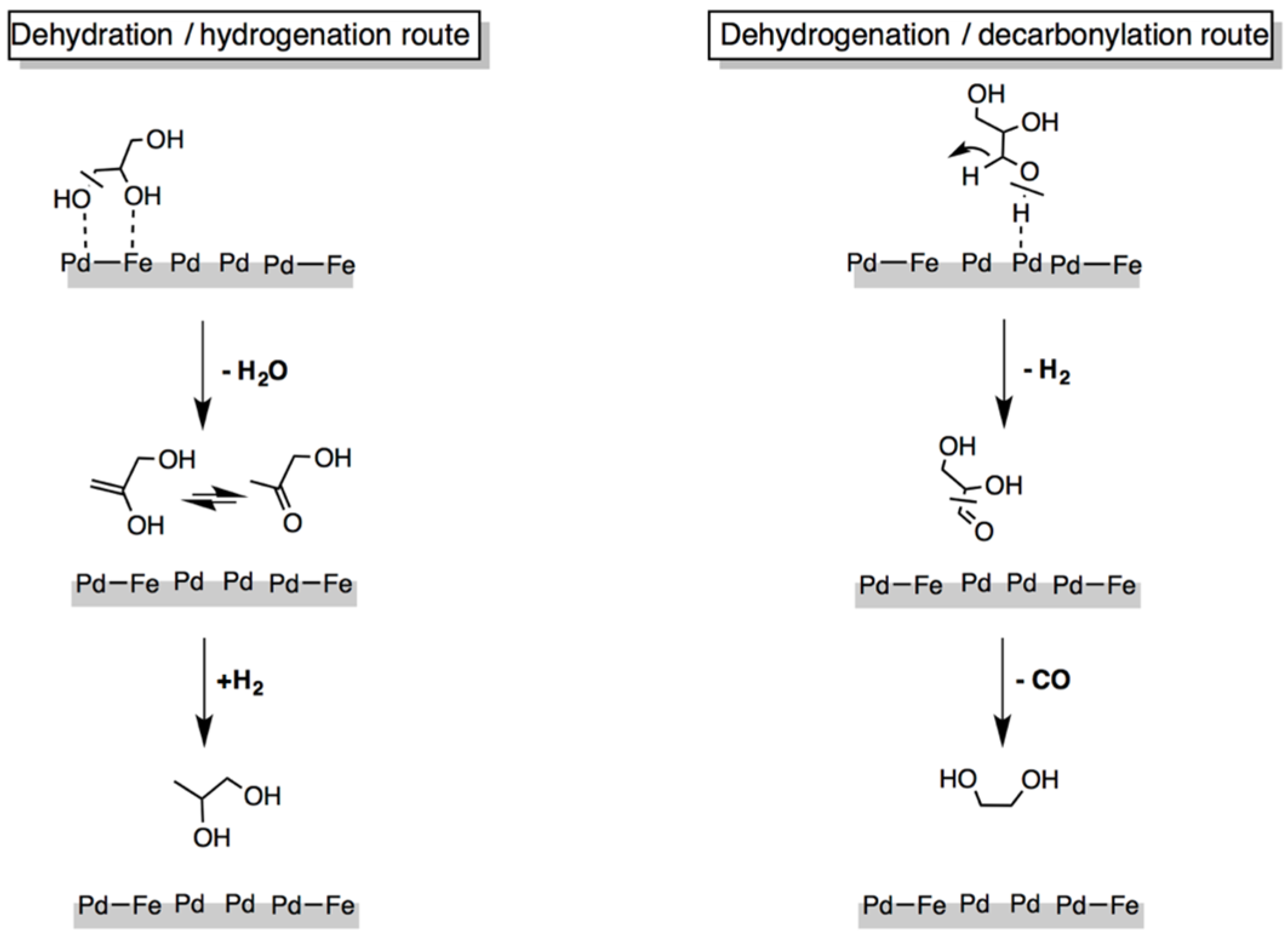
3.1.1. C–C and C–O Bond Breaking in C2–C6 Polyols
3.1.2. Hydrogenation/Hydrogenolysis of Furfural and 5-Hydroxymethylfurfural
3.2. Valorization of Lignin Model Molecules
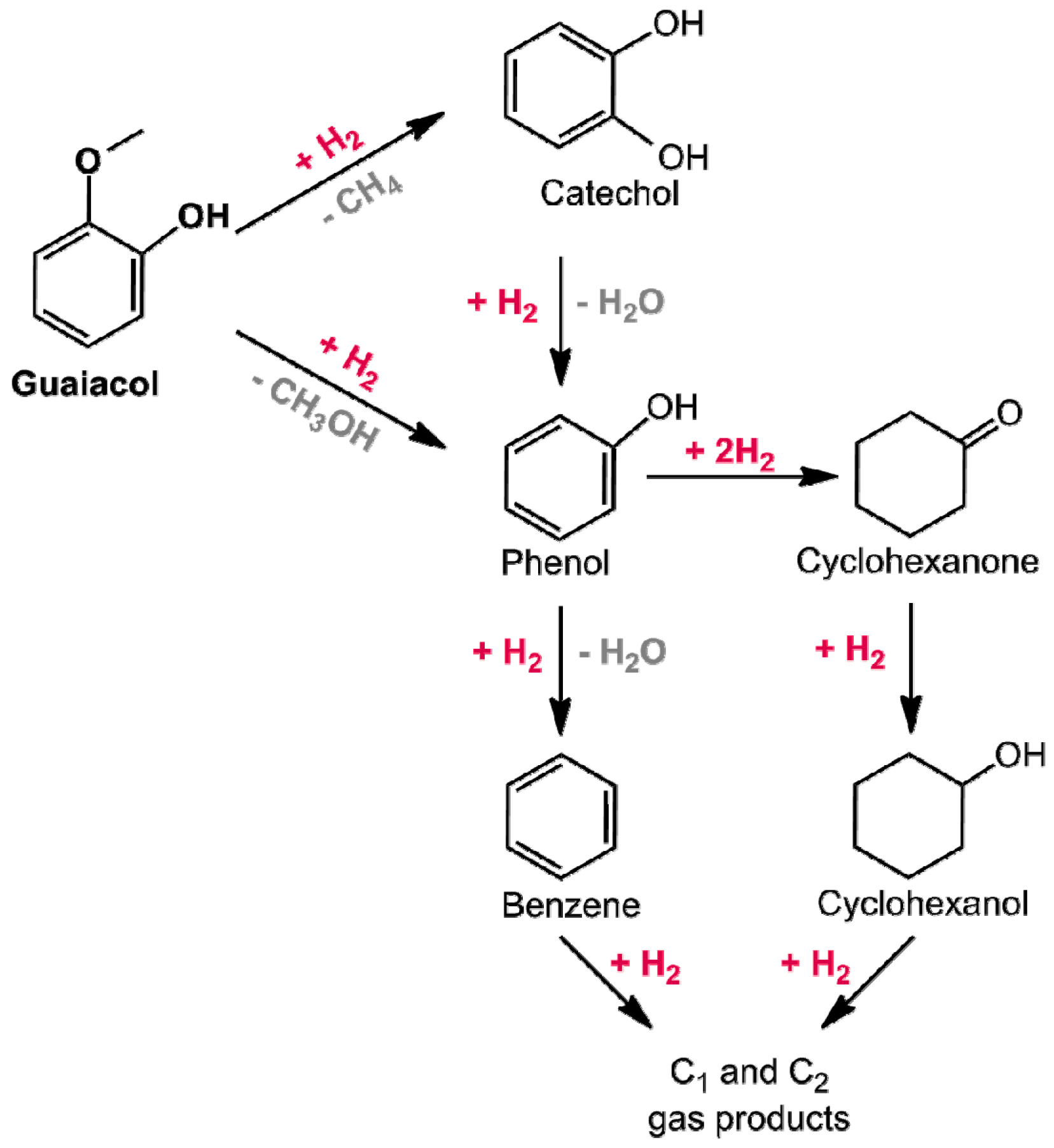
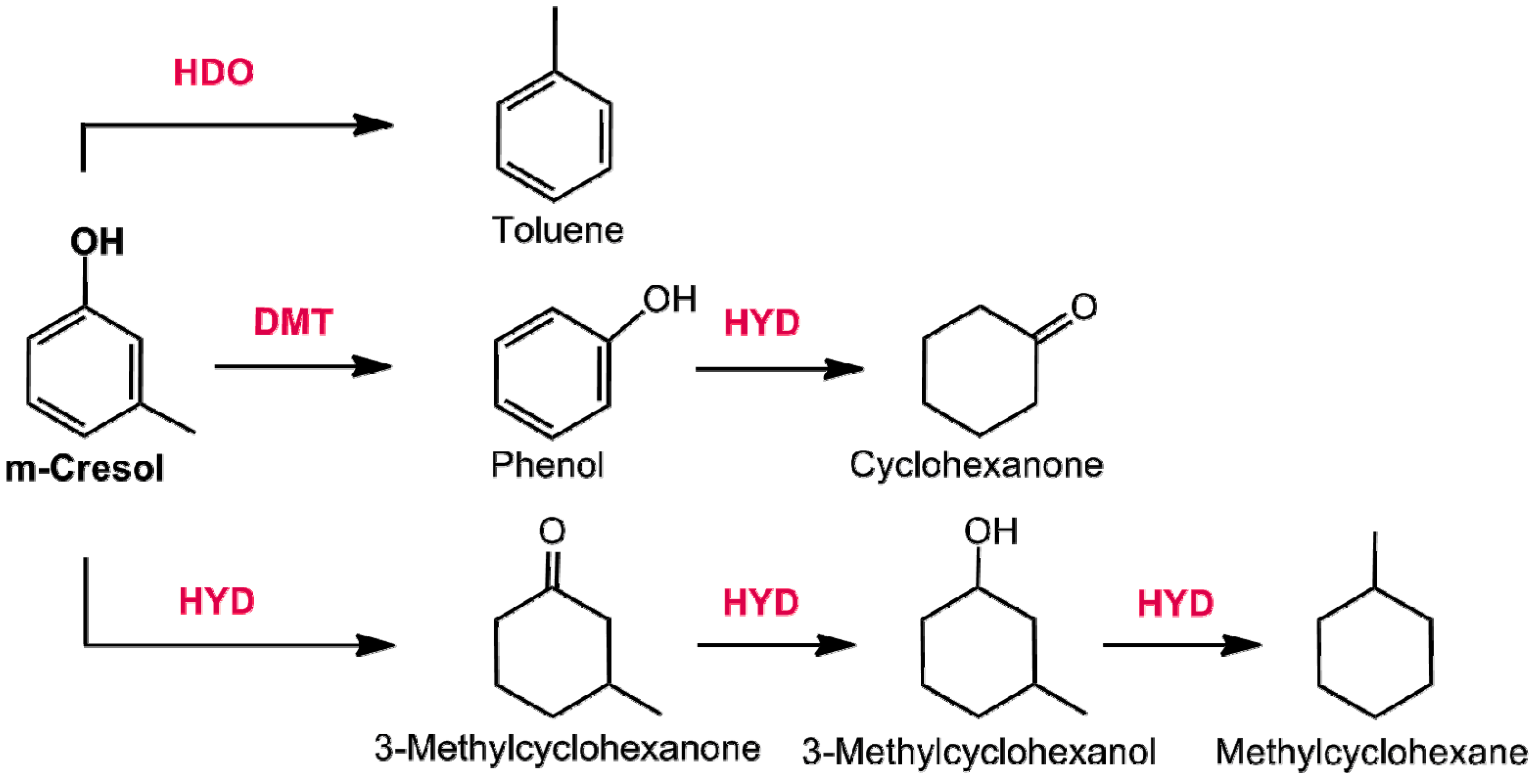
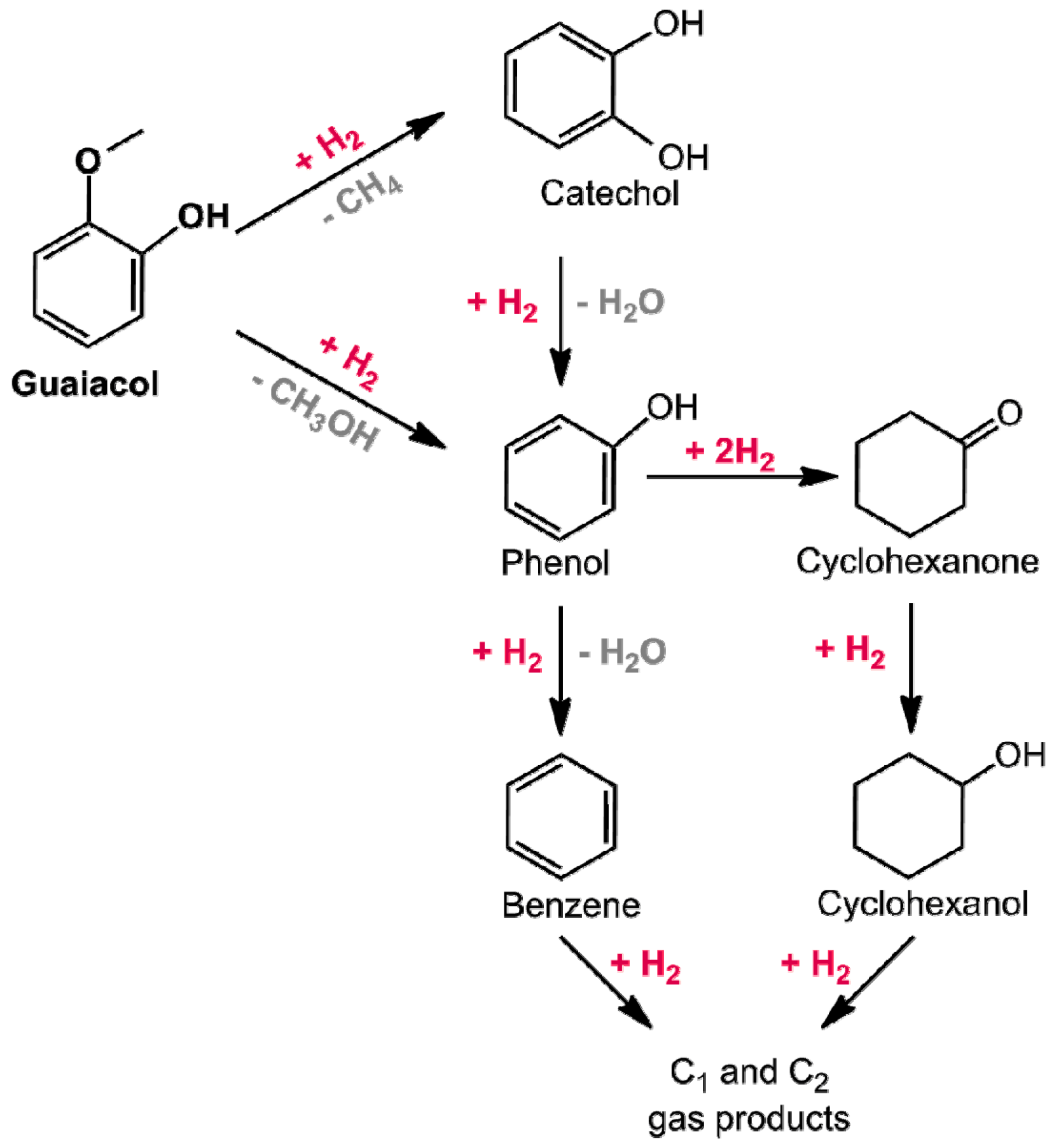
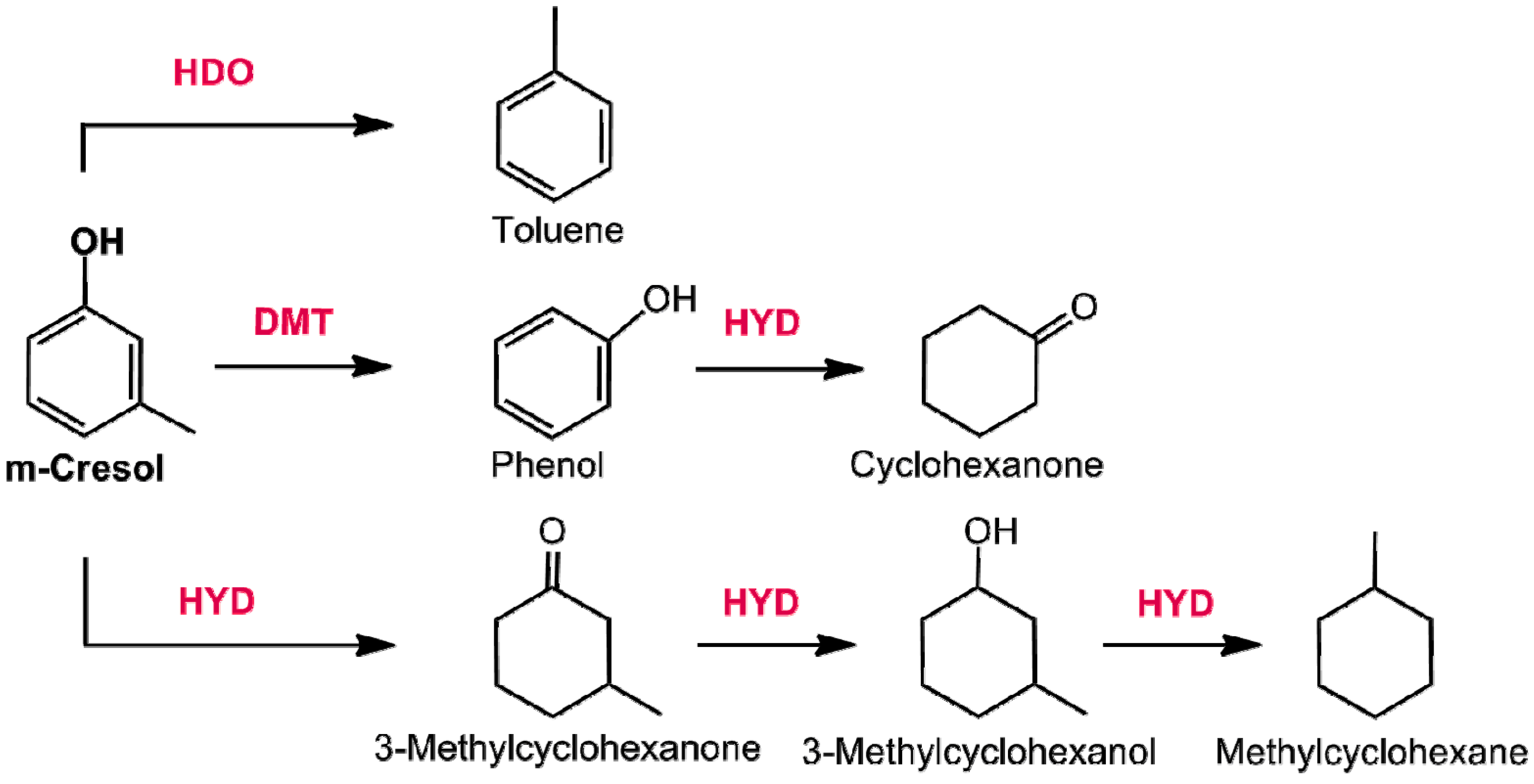
3.2.1. Hydrodeoxygenation of Phenol Derivatives
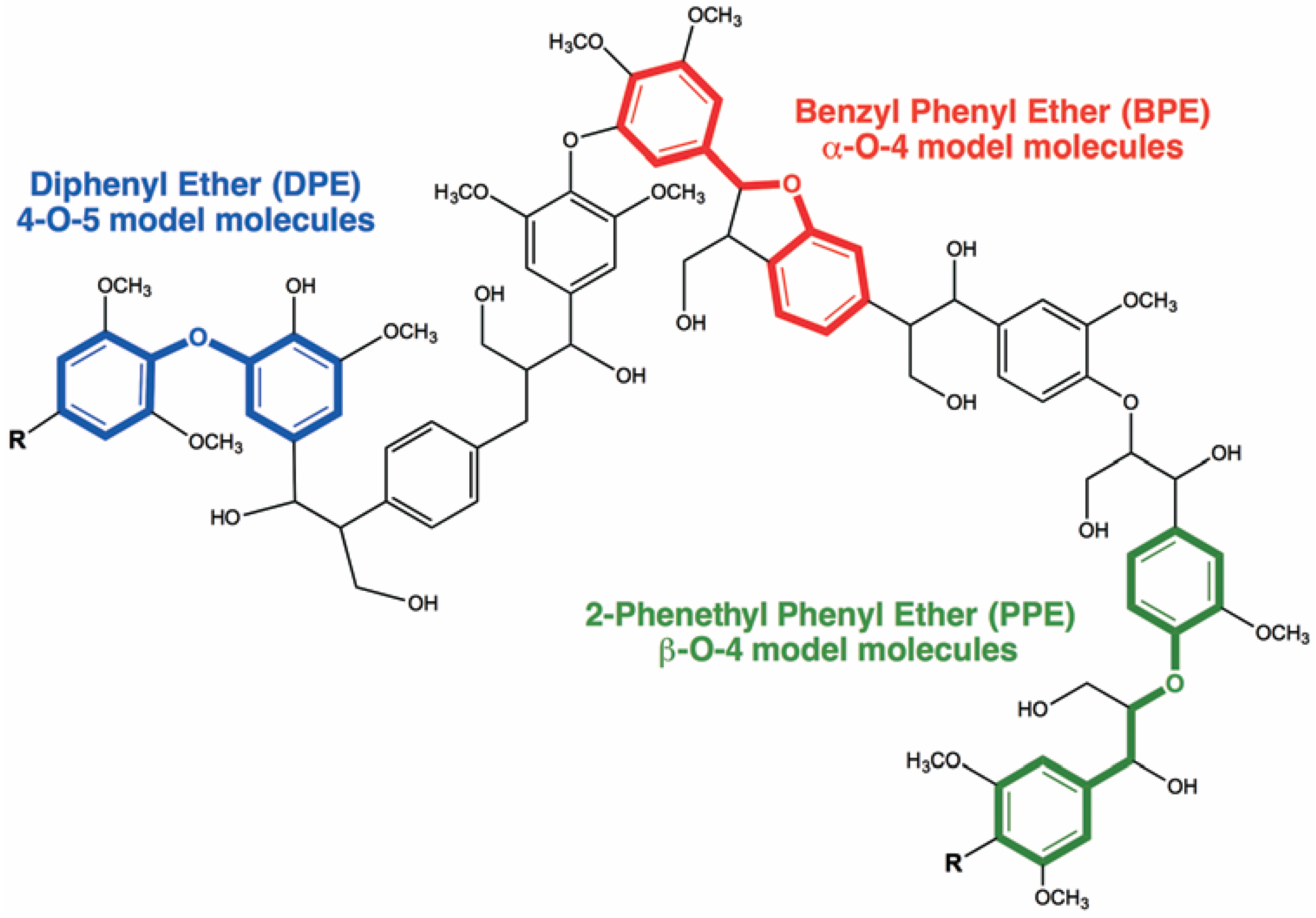
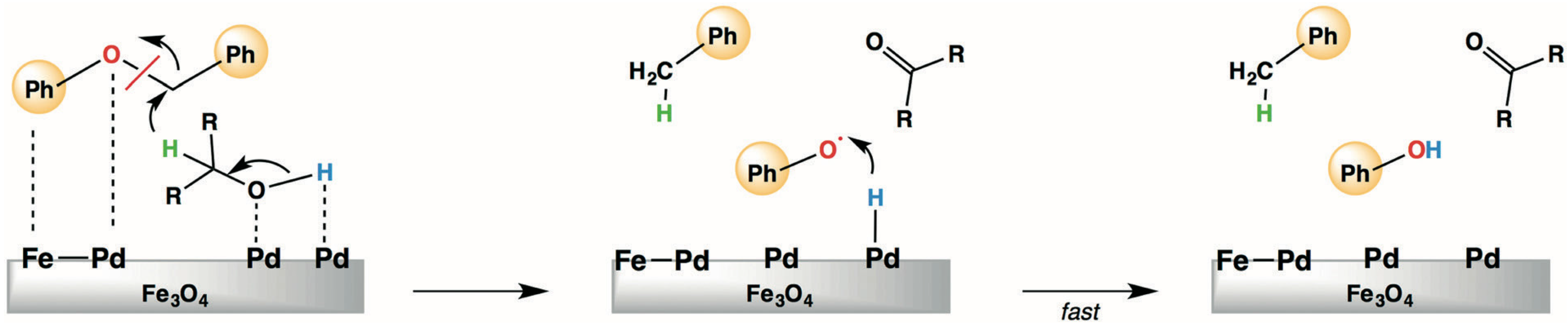
3.2.2. Hydrogenolysis of Benzyl Phenyl Ether, 2-Phenethyl Phenyl Ether and Diphenyl Ether as Model Molecules of α-O-4, β-O-4 and 4-O-5 Ether Bonds in Lignin
4. Conclusions and Outlook
Conflicts of Interest
References
- United Nations. Adoption of the Paris Agreement. FCCC/CP/2015/L.9/Rev.1. 2015, pp. 1–32. Available online: https://unfccc.int/resource/docs/2015/cop21/eng/l09r01.pdf (accessed on 30 December 2016).
- The White House-Washington. National Bio-economy Blueprint. 2012; pp. 1–43. Available online: https://www.whitehouse.gov/sites/default/files/microsites/ostp/national_bioeconomy_blueprint_april_2012 (accessed on 30 December 2016). [Google Scholar]
- European Commission. Innovating for Sustainable Growth: A Bioeconomy for Europe. 2012, pp. 1–9. Available online: http://ec.europa.eu/research/bioeconomy/pdf/official-strategy_en.pdf (accessed on 30 December 2016).
- European Commission. A Roadmap for Moving to a Competitive Low Carbon Economy in 2050. 2011. Available online: http://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX:52011DC0112 (accessed on 30 December 2016).
- European Commission. Bio-Based Economy for Europe: State of Play and Future Potential. 2011. Available online: http://ec.europa.eu/research/bioeconomy/pdf/biobasedeconomyforeuropepart1allbrochureweb.pdf (accessed on 30 December 2016).
- European Commission. Horizon 2020—The Framework Programme for Research and Innovation. 2011, pp. 1–14. Available online: http://eurlex.europa.eu/legalcontent/EN/TXT/PDF/?uri=CELEX:52011DC0808&from=EN (accessed on 30 December 2016).
- Lee, D.-H. Bio-based economies in Asia: Economic analysis of development of bio-based industry in China, India, Japan, Korea, Malaysia and Taiwan. Int. J. Hydrogen Energy 2016, 41, 4333–4346. [Google Scholar] [CrossRef]
- Dey, S. Asian bioeconomy and biobusiness: Current scenario and future prospects. New Biotechnol. 2014, 31, S34. [Google Scholar] [CrossRef]
- Tawfik, M. Asia and bioeconomy: Growing synergies. Asian Biotechnol. Dev. Rev. 2004, 6, 5–8. [Google Scholar]
- Snyder, S.W. Commercializing Biobased Products: Opportunities, Challenges, Benefits, and Risks, 1st ed.; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Thomopoulos, N. Global Markets for Renewable Chemicals Manufacturing. 2016. ENV032A. Available online: http://www.bccresearch.com/market-research/environment/renewable-chemicals-manufacturing-markets-report-env032a.html (accessed on 30 December 2016).
- Global Bio-Based Chemicals Market Insights, Opportunity Analysis, Market Shares and Forecast, 2016–2022; Occams Business Research: Mumbai, India, 2016.
- Somerville, C.; Youngs, H.; Taylor, C.; Davis, S.C.; Long, S.P. Feedstocks for lignocellulosic biofuels. Science 2010, 329, 790–792. [Google Scholar] [CrossRef] [PubMed]
- Tuck, C.O.; Pérez, E.; Horváth, I.T.; Sheldon, R.A.; Poliakoff, M. Valorization of biomass: Deriving more value from waste. Science 2012, 337, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Yang, Y.; Chai, J.; Lu, Y. Catalytic reactions of gamma-valerolactone: A platform to fuels and value-added chemicals. Appl. Catal. B Environ. 2015, 179, 292–304. [Google Scholar] [CrossRef]
- Hu, L.; Lin, L.; Wu, Z.; Zhou, S.; Liu, S. Chemocatalytic hydrolysis of cellulose into glucose over solid acid catalysts. Appl. Catal. B Environ. 2015, 174–175, 225–243. [Google Scholar] [CrossRef]
- Negahdar, L.; Delidovich, I.; Palkovits, R. Aqueous-phase hydrolysis of cellulose and hemicelluloses over molecular acidic catalysts: Insights into the kinetics and reaction mechanism. Appl. Catal. B Environ. 2016, 184, 285–298. [Google Scholar] [CrossRef]
- Sheldon, R.A. Green and sustainable manufacture of chemicals from biomass: State of the art. Green Chem. 2014, 16, 950–963. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, J.; Han, B. Catalytic transformation of lignocellulose into chemicals and fuel products in ionic liquids. Chem. Rev. 2016. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.H.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.P. Lignocellulose conversion: An introduction to chemistry, process and economics. Biofuels Bioprod. Biorefin. 2007, 1, 39–48. [Google Scholar] [CrossRef]
- Stöcker, M. Biofuels and biomass-to-liquid fuels in the biorefinery: Catalytic conversion of lignocellulosic biomass using porous materials. Angew. Chem. Int. Ed. 2008, 47, 9200–9211. [Google Scholar] [CrossRef] [PubMed]
- Petrus, L.; Noordermeer, M.A. Biomass to biofuels, a chemical perspective. Green Chem. 2006, 8, 861–867. [Google Scholar] [CrossRef]
- Chen, H. Chemical composition and structure of natural lignocellulose. In Biotechnology of Lignocellulose: Theory and Practice; Springer Science + Business Media B.V.: Dordrecht, The Netherlands, 2014; pp. 25–71. [Google Scholar]
- Delidovich, I.; Leonhard, K.; Palkovits, R. Cellulose and hemicellulose valorisation: An integrated challenge of catalysis and reaction engineering. Energy Environ. Sci. 2014, 7, 2803–2830. [Google Scholar] [CrossRef]
- Liu, X.; Wang, X.; Yao, S.; Jiang, Y.; Guan, J.; Mu, X. Recent advances in the production of polyols from lignocellulosic biomass and biomass-derived compounds. RSC Adv. 2014, 4, 49501–49520. [Google Scholar] [CrossRef]
- Deng, W.; Zhang, Q.; Wang, Y. Catalytic transformation of cellulose and its derived carbohydrates into chemicals involving C–C bond cleavage. J. Energy Chem. 2015, 24, 595–607. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, G.; Chang, C.; Fang, S.; Liu, Z.; Du, F. Direct Conversion of carbohydrates into ethyl levulinate with potassium phosphotungstate as an efficient catalyst. Catalysts 2015, 5, 1897–1910. [Google Scholar] [CrossRef]
- Antonetti, C.; Licursi, D.; Fulignati, S.; Valentini, G.; Raspolli Galletti, A.M. New frontiers in the catalytic synthesis of levulinic acid: From sugars to raw and waste biomass as starting feedstock. Catalysts 2016, 6, 196. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G.; Aden, A.; Bozell, J.; Holladay, J.; White, J.; Manheim, A.; Eliot, D.; Lasure, L.; Jones, S. Top Value Added Chemicals from Biomass. Results of Screening for Potential Candidates from Sugars and Synthesis Gas; U.S. Department of Energy: Oak Ridge, TN, USA, 2004.
- Pagliaro, M.; Rossi, M. The Future of Glycerol, 2nd ed.; Royal Society of Chemistry: Cambridge, UK, 2010. [Google Scholar]
- Zhou, C.H.; Zhao, H.; Tong, D.S.; Wu, L.M.; Yu, W.H. Recent advances in catalytic conversion of glycerol. Catal. Rev. 2013, 55, 369–453. [Google Scholar] [CrossRef]
- Ciriminna, R.; Della Pina, C.; Rossi, M.; Pagliaro, M. Understanding the glycerol market. Eur. J. Lipid Sci. Technol. 2014, 116, 1432–1439. [Google Scholar] [CrossRef]
- Mauriello, F.; Musolino, M.G.; Pietropaolo, R. Valorization of glycerol in propanediols production by catalytic processes: Production, structure and applications. In Glycerol Production, Structure and Applications; De Santos Silva, M., Ferreira, P.C., Eds.; Nova Science Publishers: New York, NY, USA, 2012; pp. 45–76. [Google Scholar]
- Zakaria, Z.Y.; Amin, N.A.S.; Linnekoski, J. A perspective on catalytic conversion of glycerol to olefins. Biomass Bioenergy 2013, 55, 370–385. [Google Scholar] [CrossRef]
- Tran, N.H.; Kannangara, G.S.K. Conversion of glycerol to hydrogen rich gas. Chem. Soc. Rev. 2013, 42, 9454–9479. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.H.; Beltramini, J.N.; Fan, Y.-X.; Lu, G.Q. Chemo-selective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Tomishige, K. Heterogeneous catalysis of the glycerol hydrogenolysis. Catal. Sci. Technol. 2011, 1, 179–190. [Google Scholar] [CrossRef]
- Scheller, H.V.; Ulvskov, P. Hemicelluloses. Annu. Rev. Plant Biol. 2010, 61, 263–289. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, L.; Chao, Y.; Nawawi, D.S.; Akiyama, T.; Yokoyama, T.; Matsumoto, Y. Relationships between hemicellulose composition and lignin structure in woods. J. Wood Chem. Technol. 2016, 36, 9–15. [Google Scholar] [CrossRef]
- Perez, R.F.; Fraga, M.A. Hemicellulose-derived chemicals: One-step production of furfuryl alcohol from xylose. Green Chem. 2014, 16, 3942–3950. [Google Scholar] [CrossRef]
- Carvalheiro, F.; Duarte, L.C.; Gírio, F.M. Hemicellulose biorefineries: A review on biomass pretreatment. J. Sci. Ind. Res. 2008, 67, 849–864. [Google Scholar]
- Ji, X.-J.; Huang, H.; Nie, Z.-K.; Qu, L.; Xu, Q.; Tsao, G.T. Fuels and chemicals from hemicellulose sugars. Adv. Biochem. Eng./Biotechnol. 2012, 128, 199–224. [Google Scholar]
- Mamman, A.S.; Lee, J.-M.; Kim, Y.-C.; Hwang, I.T.; Park, N.J.; Hwang, Y.K.; Chang, J.-S.; Hwang, J.-S. Furfural: Hemicellulose/xylose-derived biochemical. Biofuels Bioprod. Biorefin. 2008, 2, 438–454. [Google Scholar] [CrossRef]
- Li, X.; Jia, P.; Wang, T. Furfural: A promising platform compound for sustainable production of C4 and C5 chemicals. ACS Catal. 2016, 6, 7621–7640. [Google Scholar] [CrossRef]
- Pizzi, R.; Van Putten, R.-J.; Brust, H.; Perathoner, S.; Centi, G.; Van der Waal, J.C. High-throughput screening of heterogeneous catalysts for the conversion of furfural to bio-based fuel components. Catalysts 2015, 5, 2244–2257. [Google Scholar] [CrossRef]
- Mariscal, R.; Maireles-Torres, P.; Ojeda, M.; Sádaba, I.; Lòpez Granados, M. Furfural: A renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar] [CrossRef]
- Xu, C.; Arancon, R.A.D.; Labidi, J.; Luque, R. Lignin depolymerisation strategies: Towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43, 7485–7500. [Google Scholar] [CrossRef] [PubMed]
- Deuss, P.J.; Barta, K. From models to lignin: Transition metal catalysis for selective bond cleavage reactions. Coord. Chem. Rev. 2016, 306, 510–532. [Google Scholar] [CrossRef]
- Zaheer, M.; Kempe, R. Catalytic Hydrogenolysis of aryl ethers: A key step in lignin valorization to valuable chemicals. ACS Catal. 2015, 5, 1675–1684. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Hensley, J.E.; Medlin, J.W. Bifunctional catalysts for upgrading of biomass-derived oxygenates: A review. ACS Catal. 2016, 6, 5026–5043. [Google Scholar] [CrossRef]
- Sergeev, A.G.; Hartwig, J.F. Selective, nickel-catalyzed hydrogenolysis of aryl ethers. Science 2011, 332, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, A.G.; Webb, J.D.; Hartwig, J.F. A heterogeneous nickel catalyst for the hydrogenolysis of aryl ethers without arene hydrogenation. J. Am. Chem. Soc. 2012, 134, 20226–20229. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhao, C.; Lercher, J.A. Ni-catalyzed cleavage of aryl ethers in the aqueous phase. J. Am. Chem. Soc. 2012, 134, 20768–20775. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lu, L.; Zhao, C.; Mei, D.; Lercher, J.A. Mechanisms of catalytic cleavage of benzyl phenyl ether in aqueous and apolar phases. J. Catal. 2014, 311, 41–51. [Google Scholar] [CrossRef]
- He, J.; Lu, L.; Zhao, C.; Mei, D.; Lercher, J.A. Mechanisms of selective cleavage of C–O bonds in diaryl ethers in aqueous phase. J. Catal. 2014, 309, 280–290. [Google Scholar] [CrossRef]
- Wang, X.; Rinaldi, R. A route for lignin and bio-oil conversion: Dehydroxylation of phenols into arenes by catalytic tandem reactions. Angew. Chem. Int. Ed. 2013, 52, 11499–11503. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rinaldi, R. Solvent effects on the hydrogenolysis of diphenyl ether with raney nickel and their implications for the conversion of lignin. ChemSusChem 2012, 5, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Macala, G.S.; Matson, T.D.; Johnson, C.L.; Lewis, R.S.; Iretskii, A.V.; Ford, P.C. Hydrogen transfer from supercritical methanol over a solid base catalyst: A model for lignin depolymerization. ChemSusChem 2009, 2, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Barta, K.; Matson, T.D.; Fettig, M.L.; Scott, S.L.; Iretskii, A.V.; Ford, P.C. Catalytic disassembly of an organosolv lignin via hydrogen transfer from supercritical methanol. Green Chem. 2010, 12, 1640–1648. [Google Scholar] [CrossRef]
- Galkin, M.V.; Sawadjoon, S.; Rohde, V.; Dawange, M.; Samec, J.S.M. Mild Heterogeneous Palladium-catalyzed cleavage of β-O-4′-ether linkages of lignin model compounds and native lignin in air. ChemCatChem 2013, 6, 179–184. [Google Scholar] [CrossRef]
- Galkin, M.V.; Samec, J.S.M. Selective route to 2-propenyl aryls directly from wood by a tandem organosolv and palladium-catalysed transfer hydrogenolysis. ChemSusChem 2014, 7, 2154–2158. [Google Scholar] [CrossRef] [PubMed]
- Galkin, M.V.; Dahlstrand, C.; Samec, J. Mild and robust redox-neutral Pd/C-catalyzed lignol β-O-4’ bond cleavage through a low-energy-barrier pathway. ChemSusChem 2015, 8, 2187–2192. [Google Scholar] [CrossRef] [PubMed]
- Toledano, A.; Serrano, L.; Labidi, J.; Pineda, A.; Balu, A.M.; Luque, R. Heterogeneously catalysed mild hydrogenolytic depolymerisation of lignin under microwave irradiation with hydrogen-donating solvents. ChemCatChem 2012, 5, 977–985. [Google Scholar] [CrossRef]
- De, S.; Saha, B.; Luque, R. Hydrodeoxygenation processes: Advances on catalytic transformations of biomass-derived platform chemicals into hydrocarbon fuels. Bioresour. Technol. 2015, 178, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Dawes, G.J.S.; Scott, E.L.; Le Nôtre, J.; Sanders, J.P.M.; Bitter, J.H. Deoxygenation of biobased molecules by decarboxylation and decarbonylation—A review on the role of heterogeneous, homogeneous and bio-catalysis. Green Chem. 2015, 17, 3231. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Weinberg, K.; Palkovits, R. Hydrogenolysis goes bio: From carbohydrates and sugar alcohols to platform chemicals. Angew. Chem. Int. Ed. 2012, 51, 2564–2601. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Astruc, D. The golden age of transfer hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.A.W.; Wilby, A.H.; Entwistle, I.D. Heterogeneous catalytic transfer hydrogenation and its relation to other methods for reduction of organic compounds. Chem. Rev. 1985, 85, 129–170. [Google Scholar] [CrossRef]
- Brieger, G.; Nestrick, T.J. Catalytic transfer hydrogenation. Chem. Rev. 1974, 74, 567–580. [Google Scholar] [CrossRef]
- Gilkey, M.J.; Xu, B. Heterogeneous catalytic transfer hydrogenation as an effective pathway in biomass upgrading. ACS Catal. 2016, 6, 1420–1436. [Google Scholar] [CrossRef]
- Knoevenagel, E.; Bergdolt, B. Ueber das Verhalten des Δ2.5-Dihydroterephtalsäuredimethylesters bei höheren Temperaturen und in Gegenwart von Palladiummohr. Eur. J. Inorg. Chem. 1903, 36, 2857–2860. [Google Scholar] [CrossRef]
- Cortright, R.D.; Davda, R.R.; Dumesic, J.A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. Nature 2002, 418, 964–967. [Google Scholar] [CrossRef]
- Davda, R.R.; Dumesic, J.A. Catalytic reforming of oxygenated hydrocarbons for hydrogen with low levels of carbon monoxide. Angew. Chem. Int. Ed. 2003, 42, 4068–4071. [Google Scholar] [CrossRef] [PubMed]
- Metzger, J.O. Production of liquid hydrocarbons from biomass. Angew. Chem. Int. Ed. 2006, 45, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Kunkes, E.L.; Simonetti, D.A.; West, R.M.; Serrano-Ruiz, J.C.; Gärtner, C.A.; Dumesic, J.A. Catalytic conversion of biomass to monofunctional hydrocarbons and targeted liquid-fuel classes. Science 2008, 322, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Davda, R.R.; Shabaker, J.W.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol on silica-supported metal catalysts. Appl. Catal. B Environ. 2003, 43, 13–26. [Google Scholar] [CrossRef]
- Davda, R.R.; Shabaker, J.W.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. A review of catalytic issues and process conditions for renewable hydrogen and alkanes by aqueous-phase reforming of oxygenated hydrocarbons over supported metal catalysts. Appl. Catal. B Environ. 2005, 56, 171–186. [Google Scholar] [CrossRef]
- Huber, G.W.; Shabaker, J.W.; Dumesic, J.A. Raney Ni-Sn catalyst for H2 production from biomass-derived hydrocarbons. Science 2003, 300, 2075–2077. [Google Scholar] [CrossRef] [PubMed]
- Shabaker, J.W.; Davda, R.R.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of methanol and ethylene glycol over alumina-supported platinum catalysts. J. Catal. 2003, 215, 344–352. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Dumesic, J.A. Kinetics of aqueous-phase reforming of oxygenated hydrocarbons: Pt/Al2O3 and Sn-modified Ni catalysts. Ind. Eng. Chem. Res. 2004, 43, 3105–3112. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Huber, G.W.; Davda, R.R.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol over supported platinum catalysts. Catal. Lett. 2003, 88, 1–8. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Huber, G.W.; Dumesic, J.A. Aqueous-phase reforming of oxygenated hydrocarbons over Sn-modified Ni catalysts. J. Catal. 2004, 222, 180–191. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Simonetti, D.A.; Cortright, R.D.; Dumesic, J.A. Sn-modified Ni catalysts for aqueous-phase reforming: Characterization and deactivation studies. J. Catal. 2005, 231, 67–76. [Google Scholar] [CrossRef]
- Frusteri, F.; Italiano, G.; Espro, C.; Arena, F.; Parmaliana, A. Doped Ni thin layer catalysts for catalytic decomposition of natural gas to produce hydrogen. Appl. Catal. A Gen. 2009, 365, 122–129. [Google Scholar]
- Frusteri, F.; Italiano, G.; Espro, C.; Cannilla, C.; Bonura, G. H2 Production by methane decomposition: Catalytic and technological aspects. Int. J. Hydrogen Energy 2012, 37, 16367–16374. [Google Scholar] [CrossRef]
- Kettler, P.B. Platinum group metals in catalysis: Fabrication of catalysts and catalyst precursors. Org. Proc. Res. Dev. 2003, 7, 342–354. [Google Scholar] [CrossRef]
- Wells, P.B.; Wilkinson, A.G. Platinum group metals as heterogeneous enantioselective catalysts. Top. Catal. 1998, 5, 39–50. [Google Scholar] [CrossRef]
- Acres, G.J.K. Platinum group metal catalysis at the end of this century. Platinum Met. Rev. 1984, 28, 150–157. [Google Scholar] [CrossRef]
- Hartley, F. Chemistry of the Platinum Group Metals, 1st ed.; Elsevier Science: Central Milton Keynes, UK, 2008. [Google Scholar]
- Heraeus, Platinum & Palladium Focus 2016. Available online: https://www.heraeus.com/media/media/group/doc_group/precious_metals/Platinum__Palladium_Focus_2016Europe.pdf (accessed on 30 December 2016).
- ETF Securities (US) LLC. Platinum and Palladium: Riding the Wave of an Expanding Auto Market. Available online: https://www.etfsecurities.com/Documents/PP%20Commentary%20submitted%20to%20ALPS.pdf (accessed on 30 December 2016).
- Tungler, A.; Tarnai, T.; Hegediis, L.; Fodor, K. Palladium-mediated heterogeneous catalytic hydrogenations. Platinum Met. Rev. 1998, 42, 108–115. [Google Scholar]
- Astruc, D.; Lu, F.; Aranzaes, J.R. Nanoparticles as recyclable catalysts: The frontier between homogeneous and heterogeneous catalysis. Angew. Chem. Int. Ed. 2005, 48, 7852–7872. [Google Scholar] [CrossRef] [PubMed]
- Brennführer, A.; Neumann, H.; Beller, M. Palladium-catalyzed carbonylation reactions of aryl halides and related compounds. Angew. Chem. Int. Ed. 2009, 48, 4114–4133. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Liebscher, J. Carbon-carbon coupling reactions catalyzed by heterogeneous palladium catalysts. Chem. Rev. 2007, 107, 133–173. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.-U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M. Selective hydrogenation for fine chemicals: Recent trends and new developments. Adv. Synth. Catal. 2003, 346, 103–151. [Google Scholar] [CrossRef]
- Grigg, R.; Mutton, S.P. Pd-catalysed carbonylations: Versatile technology for discovery and process chemists. Tetrahedron 2010, 66, 5515–5548. [Google Scholar] [CrossRef]
- Genet, J.P.; Savignac, M. Recent developments of palladium (0) catalyzed reactions in aqueous medium. J. Organomet. Chem. 1999, 576, 305–317. [Google Scholar] [CrossRef]
- Chen, X.; Engle, K.M.; Wang, D.-H.; Jin-Quan, Y. Palladium(II)-catalyzed C–H activation/C–C cross-coupling reactions: Versatility and practicality. Angew. Chem. Int. Ed. 2009, 48, 5094–5115. [Google Scholar] [CrossRef] [PubMed]
- Beccalli, E.M.; Broggini, G.; Martinelli, M.; Sottocornola, S. C–C, C–O, C–N bond formation on sp2 carbon by Pd(II)-catalyzed reactions involving oxidant agents. Chem. Rev. 2007, 107, 5318–5365. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D. Palladium nanoparticles as efficient green homogeneous and heterogeneous carbon-carbon coupling precatalysts: A unifying view. Inorg. Chem. 2007, 46, 1884–1894. [Google Scholar] [CrossRef] [PubMed]
- Antolini, E. Palladium in fuel cell catalysis. Energy Environ. Sci. 2009, 2, 915–931. [Google Scholar] [CrossRef]
- Kiviaho, J.; Hanaoka, T.; Kubota, Y.; Sugi, Y. Heterogeneous palladium catalysts for the Heck reaction. J. Mol. Catal. A Chem. 1995, 101, 25–31. [Google Scholar] [CrossRef]
- Pagliaro, M.; Pandarus, V.; Ciriminna, R.; Béland, F.; Carà, P.D. Heterogeneous versus homogeneous palladium catalysts for cross-coupling reactions. ChemCatChem 2012, 4, 432–445. [Google Scholar] [CrossRef]
- Hartings, M. Reactions coupled to palladium. Nat. Chem. 2012, 4, 764. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Lo, T.W.B.; Tsang, S.C.E. Recent developments in palladium-based bimetallic catalysts. ChemCatChem 2015, 7, 1998–2014. [Google Scholar] [CrossRef]
- Coq, B.; Figueras, F. Bimetallic palladium catalysts: Influence of the co-metal on the catalyst performance. J. Mol. Catal. A Chem. 2001, 173, 117–134. [Google Scholar] [CrossRef]
- Bond, G.C. Metal-support and metal-additive effects in catalysis. Platinum Met. Rev. 1983, 21, 16–18. [Google Scholar]
- Banadaki, A.D.; Kajbafvala, A. Recent advances in facile synthesis of bimetallic nanostructures: An overview. J. Nanomater. 2014, 2014, 985948. [Google Scholar]
- De, S.; Zhang, J.; Luque, R.; Yan, N. Ni-based bimetallic heterogeneous catalysts for energy and environmental applications. Energy Environ. Sci. 2016, 9, 3314–3347. [Google Scholar] [CrossRef]
- Sankar, M.; Dimitratos, N.; Miedziak, P.J.; Wells, P.P.; Kielye, C.J.; Hutchings, G.J. Designing bimetallic catalysts for a green and sustainable future. Chem. Soc. Rev. 2012, 41, 8099–8139. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Bimetallic catalysts for upgrading of biomass to fuels and chemicals. Chem. Soc. Rev. 2012, 41, 8075–8098. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Goodman, D.W. Pd–Au bimetallic catalysts: Understanding alloy effects from planar models and (supported) nanoparticles. Chem. Soc. Rev. 2012, 41, 8009–8020. [Google Scholar] [CrossRef] [PubMed]
- Conant, T.; Karim, A.M.; Lebarbier, V.; Wang, Y.; Girgsdies, F.; Schlögl, R.; Datye, A. Stability of bimetallic Pd–Zn catalysts for the steam reforming of methanol. J. Catal. 2008, 257, 64–70. [Google Scholar] [CrossRef]
- Sarkany, A.; Zsoldos, Z.; Stefler, G.; Hightower, J.W.; Guczi, L. Promoter effect of Pd in hydrogenation of 1,3-butadiene over Co-Pd catalysts. J. Catal. 1995, 157, 179–189. [Google Scholar] [CrossRef]
- Menezes, W.G.; Altmann, L.; Zielasek, V.; Thiel, K.; Bäumer, M. Bimetallic Co–Pd catalysts: Study of preparation methods and their influence on the selective hydrogenation of acetylene. J. Catal. 2013, 300, 125–135. [Google Scholar] [CrossRef]
- Zhang, R.; Shuai, D.; Guy, K.A.; Shapley, J.R.; Strathmann, T.J.; Werth, C.J. Elucidation of Nitrate Reduction Mechanisms on a Pd-In Bimetallic Catalyst using Isotope Labeled Nitrogen Species. ChemCatChem 2013, 5, 313–321. [Google Scholar] [CrossRef]
- Yin, Z.; Zhou, W.; Gao, Y.; Ma, D.; Kiely, C.J.; Bao, X. Supported Pd-Cu bimetallic nanoparticles that have high activity for the electrochemical oxidation of methanol. Chem.-Eur. J. 2012, 18, 4887–4893. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, V.A.; Kalevaru, N.; Martin, A. Bimetallic catalysts containing gold and palladium for environmentally important reactions. Catalysts 2016, 6, 97. [Google Scholar] [CrossRef]
- Iwasa, N.; Masuda, S.; Ogawa, N.; Takezawa, N. Steam reforming of methanol over Pd/ZnO: Effect of the formation of PdZn alloys upon the reaction. Appl. Catal. A Gen. 1995, 125, 145–157. [Google Scholar] [CrossRef]
- Liao, F.; Lo, B.T.; Sexton, D.; Qu, J.; Ma, C.; Chan, R.C.-T.; Lu, Q.; Che, R.; Kwok, W.-M.; He, H.; et al. A new class of tunable hetero-junction by using two support materials for the synthesis of supported bimetallic catalysts. ChemCatChem 2015, 7, 230–235. [Google Scholar] [CrossRef]
- Tao, F.; Grass, M.E.; Zhang, Y.; Butcher, D.R.; Renzas, J.R.; Liu, Z.; Chung, J.Y.; Mun, B.S.; Salmeron, M.; Somorjai, G.A. Reaction-driven restructuring of Rh-Pd and Pt-Pd core-shell nanoparticles. Science 2008, 322, 932–934. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.W.J.; Wilson, O.M.; Oh, S.-K.; Kenik, E.A.; Crooks, R.M. Bimetallic Palladium–Gold dendrimer-encapsulated catalysts. J. Am. Chem. Soc. 2004, 126, 15583–15591. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Yang, H. Synthesis and Oxygen reduction electrocatalytic property of Pt-on-Pd bimetallic hetero-nanostructures. J. Am. Chem. Soc. 2009, 131, 7542–7543. [Google Scholar] [CrossRef] [PubMed]
- Raja, R.; Hermans, S.; Shephard, D.S.; Johnson, B.F.G.; Raja, R.; Sankar, G.; Bromley, S.; Thomas, J.M. Preparation and characterization of a highly active bimetallic (Pd–Ru) nanoparticle heterogeneous catalyst. Chem. Commun. 1999, 16, 1571–1572. [Google Scholar] [CrossRef]
- Liu, J.; Sun, B.; Hu, J.; Pei, Y.; Li, H.; Qiao, M. Aqueous-phase reforming of ethylene glycol to hydrogen on Pd/Fe3O4 catalyst prepared by co-precipitation: Metal–support interaction and excellent intrinsic activity. J. Catal. 2010, 274, 287–295. [Google Scholar] [CrossRef]
- Mauriello, F.; Vinci, A.; Espro, C.; Gumina, B.; Musolino, M.G.; Pietropaolo, R. Hydrogenolysis vs. aqueous phase reforming (APR) of glycerol promoted by a heterogeneous Pd/Fe catalyst. Catal. Sci. Technol. 2015, 5, 4466–4473. [Google Scholar] [CrossRef]
- Huber, G.W.; Shabaker, J.W.; Evans, S.T.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol over supported Pt and Pd bimetallic catalysts. Appl. Catal. B Environ. 2006, 62, 226–236. [Google Scholar] [CrossRef]
- Wu, C.T.; Yu, K.M.K.; Liao, F.; Young, N.; Nellist, P.; Dent, A.; Kroner, A.; Tsang, S.C.E. A non-syn-gas catalytic route to methanol production. Nat. Commun. 2012, 3, 1050. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Lo, T.W.B.; Sexton, D.; Qu, J.; Wu, C.-T.; Tsang, S.C.E. PdFe nanoparticles as selective catalysts for C–C cleavage in hydrogenolysis of vicinal diol units in biomass-derived chemicals. Catal. Sci. Technol. 2015, 5, 887–896. [Google Scholar] [CrossRef]
- Liao, F.; Lo, T.W.B.; Qu, J.; Wu, K.A.; Dent, A.; Tsang, S.C.E. Tunability of catalytic properties of Pd-based catalysts by rational control of strong metal and support interaction (SMSI) for selective hydrogenolyic C–C and C–O bond cleavage of ethylene glycol units in biomass molecules. Catal. Sci. Technol. 2015, 7, 3491–3495. [Google Scholar] [CrossRef]
- Ge, J.; Zeng, Z.; Liao, F.; Zheng, W.; Hong, Z.; Tsang, S.C.E. Palladium on iron oxide nanoparticles: The morphological effect of the support in glycerol hydrogenolysis. Green Chem. 2013, 15, 2064–2069. [Google Scholar] [CrossRef]
- Musolino, M.G.; Scarpino, L.A.; Mauriello, F.; Pietropaolo, R. Glycerol hydrogenolysis promoted by supported Palladium catalysts. ChemSusChem 2011, 4, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huai, Q.; Geng, T.; Ying, W.; Xiao, T.; Cao, F. Catalytic performance of PdNi bimetallic catalyst for glycerol hydrogenolysis biomass and bioenergy. Biomass Bioenergy 2015, 78, 71–79. [Google Scholar] [CrossRef]
- Musolino, M.G.; Scarpino, L.A.; Mauriello, F.; Pietropaolo, R. Selective transfer hydrogenolysis of glycerol promoted by palladium catalysts in absence of hydrogen. Green Chem. 2009, 11, 1511–1513. [Google Scholar] [CrossRef]
- Mauriello, F.; Ariga, H.; Musolino, M.G.; Pietropaolo, R.; Takakusagi, S.; Asakura, K. Exploring the catalytic properties of supported palladium catalysts in the transfer hydrogenolysis of glycerol. Appl. Catal. B Environ. 2015, 166–167, 121–131. [Google Scholar] [CrossRef]
- Scholz, D.; Aellig, C.; Hermans, I. Catalytic transfer hydrogenation/hydrogenolysis for reductive upgrading of furfural and 5-(hydroxymethyl) furfural. ChemSusChem 2014, 7, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.T.; Huber, G.W. Aqueous-phase hydrogenation and hydrodeoxygenation of biomass-derived oxygenates with bimetallic catalysts. Green Chem. 2014, 16, 708–718. [Google Scholar] [CrossRef]
- Sun, J.; Karim, A.M.; Zhang, H.; Kovarik, L.; Li, X.S.; Hensley, A.J.; McEwen, J.-S.; Wang, Y. Carbon-supported bimetallic Pd-Fe catalysts for vapor-phase hydrodeoxygenation of guaiacol. J. Catal. 2013, 306, 47–57. [Google Scholar] [CrossRef]
- Hong, Y.; Zhang, H.; Sun, J.; Ayman, K.M.; Hensley, A.J.R.; Gu, M.; Engelhard, M.H.; McEwen, J.-S.; Wang, Y. Synergistic catalysis between Pd and Fe in gas phase dydrodeoxygenation of m-cresol. ACS Catal. 2014, 4, 3335–3345. [Google Scholar] [CrossRef]
- Kim, J.K.; Lee, J.K.; Kang, K.H.; Song, J.C.; Song, I.K. Selective cleavage of C–O bond in benzyl phenyl ether to aromatics over Pd-Fe bimetallic catalyst supported on ordered mesoporous carbon. Appl. Catal. A Gen. 2015, 498, 142–149. [Google Scholar] [CrossRef]
- Kim, J.K.; Lee, J.K.; Kang, K.H.; Lee, J.W.; Song, I.K. Catalytic decomposition of phenethyl phenyl ether to aromatics over Pd-Fe bimetallic catalysts supported on ordered mesoporous carbon. J. Mol. Catal. A Chem. 2015, 410, 184–192. [Google Scholar] [CrossRef]
- Paone, E.; Espro, C.; Pietropaolo, R.; Mauriello, F. Selective arene production from transfer hydrogenolysis of benzyl phenyl ether promoted by a co-precipitated Pd/Fe3O4 catalyst. Catal. Sci. Technol. 2016, 6, 7937–7941. [Google Scholar] [CrossRef]
- Guczi, L. Bimetallic nano-particles: Featuring structure and reactivity. Catal. Today 2005, 101, 53–64. [Google Scholar] [CrossRef]
- Guczi, L. Structure and promotion of bimetallic catalysts: Activity and selectivity. Catal. Lett. 1990, 7, 205–212. [Google Scholar] [CrossRef]
- Coq, B.; Hub, S.; Figuéras, F.; Tournigant, D. Conversion under hydrogen of dichlorodifluoromethane over bimetallic palladium catalysts. Appl. Catal. A Gen. 1993, 101, 41–50. [Google Scholar] [CrossRef]
- Lebedeva, O.E.; Chiou, W.A.; Sachtler, W.M.H. Chloride induced migration of supported platinum and palladium across phase boundaries. Catal. Lett. 2000, 66, 189–195. [Google Scholar] [CrossRef]
- Li, W.; Haldar, P. Supportless PdFe nanorods as highly active electrocatalyst for proton exchange membrane fuel cell. Electrochem. Commun. 2009, 11, 1195–1198. [Google Scholar] [CrossRef]
- Ghauch, A.; Abou Assi, H.; Bdeir, S. Aqueous removal of diclofenac by plated elemental iron: Bimetallic systems. J. Hazard. Mater. 2010, 182, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; More, K.L.; Sun, K.; Wu, Z.; Li, W. Preparation and characterization of PdFe nanoleaves as electrocatalysts for oxygen reduction reaction. Chem. Mater. 2011, 23, 1570–1577. [Google Scholar] [CrossRef]
- Wang, H.F.; Kaden, W.E.; Dowler, R.; Sterrer, M.; Freund, H.J. Model oxide supported metal catalysts Comparison of ultrahigh vacuum and solution based preparation of Pd nanoparticles on a single crystalline oxide substrate. Phys. Chem. Chem. Phys. 2012, 14, 11525–11533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, C.; Sun, J.; Kou, T.; Bai, Q.; Wang, Y.; Ding, Y. Ultrafine nanoporous PdFe/Fe3O4 catalysts with doubly enhanced activities towards electro oxidation of methanol and ethanol in alkaline media. J. Mater. Chem. A 2013, 1, 3620–3628. [Google Scholar] [CrossRef]
- Han, B.; Xu, C. Nanoporous PdFe alloy as highly active and durable electrocatalyst for oxygen reduction reaction. Int. J. Hydrogen En. 2014, 39, 18247–18255. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; Zhao, D.; Xu, C. Facile fabrication of nanoporous PdFe alloy for nonenzymatic electrochemical sensing of hydrogen peroxide and glucose. Anal. Chim. Acta 2014, 832, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, H.; Fan, D.; Zhao, D.; Xu, C. A glassy carbon electrode modified with nanoporous PdFe alloy for highly sensitive continuous determination of nitrite. Microchim. Acta 2015, 182, 1055–1061. [Google Scholar] [CrossRef]
- Kang, Y.S.; Choi, K.H.; Ahn, D.; Lee, M.J.; Baik, J.; Chung, D.Y.; Kim, M.J.; Lee, S.Y.; Kim, M.; Shin, H.; et al. Effect of post heat-treatment of composition controlled PdFe nanoparticles for oxygen reduction reaction. J. Power Sources 2016, 303, 234–242. [Google Scholar] [CrossRef]
- Zhou, Y.; Du, C.; Han, G.; Gao, Y.; Yin, G. Ultralow Pt decorated PdFe Alloy Nanoparticles for Formic Acid Electro oxidation. Electrochim. Acta 2016, 217, 203–209. [Google Scholar] [CrossRef]
- Leng, L.; Li, J.; Zeng, X.; Song, H.; Shu, T.; Wang, H.; Liao, S. Enhancing the cyclability of Li–O2 batteries using PdM alloy nanoparticles anchored on nitrogen doped reduced graphene as the cathode catalyst. J. Power Sources 2017, 337, 173–179. [Google Scholar] [CrossRef]
- Lieltz, G.; Nimz, M.; Völter, J.; Läzär, K.; Guczi, L. Double promotion of palladium/silica catalysts by iron and magnesium oxide in the synthesis of methanol from carbon monoxide and hydrogen. Appl. Catal. 1988, 45, 71–83. [Google Scholar] [CrossRef]
- Fukuoka, A.; Kimura, T.; Ichikawa, M. Selective Hydrogenation of CO into C1 and C2 Alcohols by SiO2-supported RhFe, PtFe, and PdFe Bimetallic Cluster-derived Catalysts. J. Chem. Soc. Commun. 1988, 428–430. [Google Scholar] [CrossRef]
- Lázár, K.; Nimz, M.; Lietz, G.; Völter, J.; Guczi, L. Formation of PdFe alloys on silica supported catalysts. Hyperfine Interact. 1988, 41, 657–660. [Google Scholar] [CrossRef]
- Felicissimo, M.P.; Martyanov, O.N.; Risse, T.; Freund, H.-J. Characterization of a Pd-Fe bimetallic model catalyst. Surf. Sci. 2007, 601, 2105–2116. [Google Scholar] [CrossRef]
- Fukuoka, A.; Kimura, T.; Rao, L.F.; Ichikawa, M. Heteronuclear CO activation in CO based reactions catalyzed by SiO2 supported RhFe and PdFe bimetallic clusters. Catal. Today 1989, 6, 55–62. [Google Scholar] [CrossRef]
- Fukuoka, A.; Kimura, T.; Kosugi, N.; Kuroda, H.; Minai, Y.; Sakai, Y.; Tominaga, T.; Ichikawa, M. Bimetallic promotion of alcohol production in CO hydrogenation and olefin hydroformylation on RhFe, PtFe, PdFe, and IrFe cluster derived catalysts. J. Catal. 1990, 126, 434–450. [Google Scholar] [CrossRef]
- Pinna, F.; Selva, M.; Signoretto, M.; Strukul, G.; Boccuzzi, F.; Benedetti, A.; Canton, P.; Fagherazzi, G. Pd-Fe/SiO2 Catalysts in the hydrogenation of 2,4-Dinitrotoluene. J. Catal. 1994, 150, 356–367. [Google Scholar] [CrossRef]
- Komatsu, T.; Inaba, K.; Uezono, T.; Onda, A.; Yashima, T. Nanosize particles of palladium intermetallic compounds as catalysts for oxidative acetoxylation. Appl. Catal. A Gen. 2003, 251, 315–326. [Google Scholar] [CrossRef]
- Garten, R.L.; Ollis, D.F. The chemical state of iron in reduced PdFe Al2O3 catalysts. J. Catal. 1974, 35, 232–246. [Google Scholar] [CrossRef]
- Garten, R.L. Direct evidence for bimetallic clusters. J. Catal. 1976, 43, 18–33. [Google Scholar] [CrossRef]
- Kadinov, G.; Bonev, C.; Todorova, S.; Palazov, A.; Lietz, G.; Völter, J. Infrared study of carbon monoxide adsorption and hydrogenation over PdFe catalysts. J. Mol. Catal. 1993, 83, 157–166. [Google Scholar] [CrossRef]
- Pârvulescu, V.I.; Filoti, G.; Pârvulescu, V.; Grecu, N.; Angelescu, E.; Nicolescu, I.V. Styrene hydrogenation on supported Pd, Fe and PdFe/γ-Al2O3 catalysts. J. Mol. Catal. 1994, 89, 267–282. [Google Scholar] [CrossRef]
- Golubina, E.V.; Lokteva, E.S.; Lunin, V.V.; Telegina, N.S.; Stakheev, A.Y.; Tundo, P. The role of Fe addition on the activity of Pd containing catalysts in multiphase hydrodechlorination. Appl. Catal. A Gen. 2006, 302, 32–41. [Google Scholar] [CrossRef]
- Tarasevich, M.R.; Zhutaeva, G.V.; Bogdanovskaya, V.A.; Radina, M.V.; Ehrenburg, M.R.; Chalykh, A.E. Oxygen kinetics and mechanism at electrocatalysts on the base of palladium iron system. Electrochim. Acta 2007, 52, 5108–5118. [Google Scholar] [CrossRef]
- Jin, Y.; Ma, C.; Shi, M.; Chu, Y.; Xu, Y.; Huang, T.; Huang, Q.; Miao, Y. Highly active carbon nanotube supported bimetallic palladium iron electrocatalysts for formic acid electro oxidation. Int. J. Electrochem. Sci. 2012, 7, 3399–3408. [Google Scholar]
- Yeh, Y.C.; Chen, H.M.; Liu, R.S.; Asakura, K.; Lo, M.Y.; Peng, Y.M.; Chan, T.S.; Lee, J.F. PdcFe nanoparticles investigated by X-ray absorption spectroscopy as electrocatalysts for oxygen reduction. Chem. Mater. 2009, 21, 4030–4036. [Google Scholar] [CrossRef]
- Yang, J.; Zhou, W.; Cheng, C.H.; Lee, J.Y.; Liu, Z. Pt decorated PdFe nanoparticles as methanol tolerant oxygen reduction electrocatalyst. ACS Appl. Mater. Interfaces 2010, 2, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, R.; Ji, S.; Feng, H.; Wang, H.; Lei, Z. Pt overgrowth on carbon supported PdFe seeds in the preparation of core shell electrocatalysts for the oxygen reduction reaction. J. Power Sources 2010, 195, 3498–3503. [Google Scholar] [CrossRef]
- Yin, S.; Cai, M.; Wang, C.; Shen, P.K. Tungsten carbide promoted PdFe as alcohol tolerant electrocatalysts for oxygen reduction reactions. Energy Environ. Sci. 2011, 4, 558–563. [Google Scholar] [CrossRef]
- Alexeyeva, N.; Sarapuu, A.; Tammeveski, K.; VidalIglesias, F.J.; SollaGullón, J.; Feliu, J.M. Electro reduction of oxygen on Vulcan carbon supported Pd nanoparticles and PdM nanoalloys in acid and alkaline solutions. Electrochim. Acta 2011, 56, 6702–6708. [Google Scholar] [CrossRef]
- Wang, H.; Ji, S.; Wang, W.; Linkov, V.; Pasupathi, S.; Wang, R. Pt decorated PdFe/C: Extremely high electrocatalytic activity for methanol oxidation International. J. Electrochem. Sci. 2012, 7, 3390–3398. [Google Scholar]
- Pires, F.I.; Villullas, H.M. Pd-based catalysts: Influence of the second metal on their stability and oxygen reduction activity. Int. J. Hydrogen Energy 2012, 37, 17052–17059. [Google Scholar] [CrossRef]
- Rivera Gavidia, L.M.; García, G.; Anaya, D.; Querejeta, A.; Alcaide, F.; Pastor, E. Carbon supported Pt-free catalysts with high specificity and activity toward the oxygen reduction reaction in acidic medium. Appl. Catal. B Environ. 2016, 184, 12–19. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Nadagouda, M.N.; Varma, R.S. Microwave assisted chemistry: A rapid and sustainable route to synthesis of organics and nanomaterials. Aust. J. Chem. 2009, 62, 16–26. [Google Scholar] [CrossRef]
- Holade, Y.; Da Silva, R.G.; Servat, K.; Napporn, T.W.; Canaff, C.; De Andrade, A.R.; Kokoh, K.B. Facile synthesis of highly active and durable PdM/C (M = Fe, Mn) nanocatalysts for the oxygen reduction reaction in an alkaline medium. J. Mater. Chem. A 2016, 4, 8337–8349. [Google Scholar] [CrossRef]
- Homeyer, S.T.; Sheu, L.L.; Zhang, Z.; Sachtler, W.M.H.; Balse, V.R.; Dumesic, J.A. Chemical interaction of palladium particles with iron cations in NaY. Appl. Catal. 1990, 64, 225–241. [Google Scholar] [CrossRef]
- Choudary, B.M.; Lázár, K.; Bogyay, I.; Guczi, L. Promotion effect of Fe on the selectivity of Pd zeoliteX in methanol synthesis. J. Chem. Soc. Faraday Trans. 1990, 86, 419–424. [Google Scholar] [CrossRef]
- Wen, B.; Jia, J.; Sachtler, W.M.H. Chemical anchoring of palladium by Fe Oxo ions in zeolite ZSM-5. J. Phys. Chem. B 2002, 106, 7520–7523. [Google Scholar] [CrossRef]
- Hinokuma, S.; Katsuhara, Y.; Ando, E.; Ikeue, K.; Machida, M. PdFe/CeO2 bimetal catalysts prepared by dual arc-plasma deposition. Catal. Today 2013, 201, 92–97. [Google Scholar] [CrossRef]
- Mizuno, N.; Misono, M. Heterogeneous catalysis. Chem. Rev. 1998, 98, 199–218. [Google Scholar] [CrossRef]
- Campanati, M.; Fornasari, G.; Vaccari, A. Fundamentals in the preparation of heterogeneous catalysts. Catal. Today 2003, 77, 299–314. [Google Scholar] [CrossRef]
- LePage, J.F.; Ertl, G.; Knözinger, H.; Weitkamp, J. Handbook of Heterogeneous Catalysis; Wiley/VCH: New York, NY, USA; Weinheim, Germany, 1997; Volume 1, p. 49. [Google Scholar]
- Musolino, M.G.; Busacca, C.; Mauriello, F.; Pietropaolo, R. Aliphatic carbonyl reduction promoted by palladium catalysts under mild conditions. Appl. Catal. A Gen. 2010, 379, 77–86. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, H.; Zhang, J.; Xia, B. Synthesis of LiNi0.6Co0.2Mn0.2O2 cathode material by a carbonate co-precipitation method and its electrochemical characterization. Solid State Ion. 2006, 177, 3303–3307. [Google Scholar] [CrossRef]
- Cho, T.H.; Shiosaki, Y.; Noguchi, H. Preparation and characterization of layered LiMn1/3Ni1/3Co1/3O2 as a cathode material by an oxalate co-precipitation method. J. Power Sources 2006, 159, 1322–1327. [Google Scholar] [CrossRef]
- Lee, S.H.; Georgii, I.N. Transfer hydrogenation of ketones, nitriles, and esters catalyzed by a half-sandwich complex of Ruthenium. ChemCatChem 2015, 7, 107–113. [Google Scholar] [CrossRef]
- Yang, L.; Chen, X.; Zhou, Z.; Zhang, R.; Li, L.; Cheng, Z.; Fang, X. Magnetic Fe3O4@SiO2/Pd and Fe3O4@SiO2/Pd-M (M = Ag, Cu and Zn) catalysts for selective hydrogenation of phenylacetylene. ChemistrySelect 2016, 1, 5599–5606. [Google Scholar] [CrossRef]
- Deng, H.; Li, X.; Peng, Q.; Wang, X.; Chen, J.; Li, Y. Monodisperse magnetic single-crystal ferrite microspheres. Angew. Chem. Int. Ed. 2005, 44, 2782–2785. [Google Scholar] [CrossRef] [PubMed]
- Bunaciu, A.A.; UdriŞtioiu, E.G.; Aboul-Enein, H.Y. X-ray diffraction: Instrumentation and applications. Crit. Rev. Anal. Chem. 2015, 45, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Thibault, P.; Elser, V. X-ray diffraction microscopy. Annu. Rev. Condens. Matter Phys. 2010, 1, 237–255. [Google Scholar] [CrossRef]
- Datye, A.K.; Smith, D.J. The study of heterogeneous catalysts by high-resolution electron microscopy. Catal. Rev. 1992, 34, 129–178. [Google Scholar] [CrossRef]
- Yang, J.C.; Small, M.W.; Grieshaber, R.V.; Nuzzo, R.G. Recent developments and applications of electron microscopy to heterogeneous catalysis. Chem. Soc. Rev. 2012, 41, 8179–8194. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Midgley, P.A. High-resolution transmission electron microscopy: The ultimate nanoanalytical technique. Chem. Commun. 2004, 1253–1267. [Google Scholar] [CrossRef] [PubMed]
- Datye, A.K. Electron microscopy of catalysts: Recent achievements and future prospects. J. Catal. 2003, 216, 144–154. [Google Scholar] [CrossRef]
- International Center for Diffraction Data. Powder Diffraction Database; International Center for Diffraction Data: Pennsylvania, PA, USA, 1997. [Google Scholar]
- Feio, L.S.F.; Hori, C.E.; Mattos, L.V.; Zanchet, D.; Noronha, F.B.; Bueno, J.M.C. Partial oxidation and autothermal reforming of methane on Pd/CeO2–Al2O3 catalysts. Appl. Catal. A Gen. 2008, 348, 183–192. [Google Scholar] [CrossRef]
- Neri, G.; Rizzo, G.; De Luca, L.; Donato, A.; Musolino, M.G.; Pietropaolo, R. Supported Pd catalysts for the hydrogenation of campholenic aldehyde: Influence of support and preparation method. Appl. Catal. A Gen. 2009, 356, 113–120. [Google Scholar] [CrossRef]
- Braunstein, P.; Devenish, R.; Gallezot, P.; Heaton, B.T.; Hurnphreys, C.J.; Kervennal, J.; Muliey, S.; Ries, M. Silica-supported Fe-Pd bimetallic particles: Formation from mixed-metal clusters and catalytic activity. Angew. Chem. Int. Ed. 1988, 27, 927–929. [Google Scholar] [CrossRef]
- Hensley, A.J.R.; Hong, Y.; Zhang, R.; Zhang, H.; Sun, J.; Wang, Y.; McEwen, J.-S. Enhanced Fe2O3 reducibility via surface modification with Pd: Characterizing the synergy within Pd/Fe catalysts for hydrodeoxygenation reactions. ACS Catal. 2014, 4, 3381–3392. [Google Scholar] [CrossRef]
- Li, Z.Q.; Lu, C.J.; Xia, Z.P.; Zhou, Y.; Luo, Z. X-ray diffraction patterns of graphite and turbostratic carbon. Carbon 2007, 45, 1686–1695. [Google Scholar] [CrossRef]
- Liao, M.; Hu, Q.; Zheng, J.; Li, Y.; Zhou, H.; Zhong, C.-J.; Chen, B.H. Pd decorated Fe/C nanocatalyst for formic acid electro oxidation. Electrochim. Acta 2013, 111, 504–509. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, D.; Du, C.; Zou, Z.; Zhang, X.; Xia, B.; Yang, H.; Akins, D.L. Carbon-supported Pd-Co bimetallic nanoparticles as electrocatalysts for the oxygen reduction reaction. J. Power Sources 2007, 167, 243–249. [Google Scholar] [CrossRef]
- Hurst, N.W.; Gentry, S.J.; Jones, A.; McNicol, B.D. Temperature programmed reduction. Catal. Rev. 1982, 24, 233–309. [Google Scholar] [CrossRef]
- Chou, C.-W.; Chu, S.-J.; Chiang, H.-J.; Huang, C.-Y.; Lee, C.-J.; Sheen, S.-R.; Perng, T.P.; Yeh, C.-T. Temperature-programmed reduction study on calcination of nano-Palladium. J. Phys. Chem. B 2001, 105, 9113–9117. [Google Scholar] [CrossRef]
- Gil, S.; Garcia-Vargas, J.M.; Liotta, L.F.; Pantaleo, G.; Ousmane, M.; Retailleau, L.; Giroir-Fendler, A. Catalytic oxidation of propene over Pd catalysts supported on CeO2, TiO2, Al2O3 and M/Al2O3 Oxides (M = Ce, Ti, Fe, Mn). Catalysts 2015, 5, 671–689. [Google Scholar] [CrossRef]
- Tiernan, M.J.; Barnes, P.A.; Parkes, G.M.B. Reduction of iron oxide catalysts: The investigation of kinetic parameters using rate perturbation and linear heating thermoanalytical techniques. J. Phys. Chem. B 2001, 105, 220–228. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chen, Y.W.; Li, C. The mechanism of reduction of iron oxide by hydrogen. Thermochim. Acta 2003, 400, 61–67. [Google Scholar] [CrossRef]
- Turner, N.H.; Schreifels, J.A. Surface Analysis: X-ray photoelectron spectroscopy and auger electron spectroscopy. Anal. Chem. 1996, 68, 309–332. [Google Scholar] [CrossRef]
- Van der Heide, P. X-ray Photoelectron Spectroscopy: An Introduction to Principles and Practices; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Winograd, N.; Gaarenstroom, S.W. X-ray Photoelectron Spectroscopy, Physical Methods in Modern Chemical Analysis; Academic Press, Inc./Elsevier Inc.: New York, NY, USA, 1980; Volume 2, pp. 115–169. [Google Scholar]
- Sharpe, L.R.; Heineman, W.R.; Elder, R.C. EXAFS spectro-electrochemistry. Chem. Rev. 1990, 90, 705–722. [Google Scholar] [CrossRef]
- Gaur, A.; Shrivastava, B.D.; Nigam, H.L. X-ray Absorption fine structure (XAFS) spectroscopy—A review. Proc. Indian Natl. Sci. Acad. 2013, 79, 921–966. [Google Scholar]
- Asakura, K. Recent developments in the in situ XAFS and related work for the characterization of catalysts in Japan. Catal. Surv. Asia 2003, 7, 177–182. [Google Scholar] [CrossRef]
- Schlaf, M. Selective deoxygenation of sugar polyols to α,ω-diols and other oxygen content reduced materials—A new challenge to homogeneous ionic hydrogenation and hydrogenolysis catalysis. Dalton Trans. 2006, 39, 4645–4653. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Armbruster, U.; Gandarias, I.; Arias, P.L. Glycerol hydrogenolysis into propanediols using in situ generated hydrogen—A critical review. Eur. J. Lipid Sci. Technol. 2013, 115, 9–27. [Google Scholar] [CrossRef]
- Gandarias, I.; Arias, P.L.; Requies, J.; El Doukkali, M.; Güemez, M.B. Liquid-phase glycerol hydrogenolysis to 1,2-propanediol under nitrogen pressure using 2-propanol as hydrogen source. J. Catal. 2011, 282, 237–247. [Google Scholar] [CrossRef]
- Yue, H.; Zhao, Y.; Ma, X.; Gong, J. Ethylene glycol: Properties, synthesis, and applications. Chem. Soc. Rev. 2012, 41, 4218–4244. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Pang, J.; Wang, A.; Zhang, T. One-pot catalytic conversion of cellulose to ethylene glycol and other chemicals: From fundamental discovery to potential commercialization. Chin. J. Catal. 2014, 35, 602–613. [Google Scholar] [CrossRef]
- Wang, A.; Zhang, T. One-pot conversion of cellulose to ethylene glycol with multifunctional Tungsten-based catalysts. Acc. Chem. Res. 2013, 46, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Salciccioli, M.; Yu, W.; Barteau, M.A.; Chen, J.G.; Vlachos, D.G. Differentiation of O–H and C–H bond scission mechanisms of ethylene glycol on Pt and Ni/Pt using theory and isotopic labeling experiments. J. Am. Chem. Soc. 2011, 133, 7996–8004. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.A.; Pang, C.L.; Perkins, N.; Smith, R.D.; Morrall, P.; Kvon, R.I.; Bowker, M. Surface structures in the SMSI state; Pd on (1 × 2) reconstructed TiO2(110). J. Phys. Chem. B 2002, 106, 4688–4696. [Google Scholar] [CrossRef]
- Dulub, O.; Hebenstreit, W.; Diebold, U. Imaging cluster surfaces with atomic resolution: The strong metal-support interaction state of Pt supported on TiO2 (110). Phys. Rev. Lett. 2000, 84, 3646–3649. [Google Scholar] [CrossRef] [PubMed]
- Alhanash, A.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Hydrogenolysis of glycerol to propanediol over Ru: polyoxometalate bifunctional catalyst. Catal. Lett. 2007, 120, 307–311. [Google Scholar] [CrossRef]
- Balaraju, M.; Rekha, V.; Sai Prasad, P.S.; Prasad, R.B.N.; Lingaiah, N. Selective hydrogenolysis of glycerol to 1,2 propanediol over Cu–ZnO catalysts. Catal. Lett. 2008, 126, 119–124. [Google Scholar] [CrossRef]
- Hattori, H. Heterogeneous basic catalysis. Chem. Rev. 1995, 95, 537–558. [Google Scholar] [CrossRef]
- Hattori, H. Solid base catalysts: Generation of basic sites and application to organic synthesis. Appl. Cat. A Gen. 2001, 222, 247–259. [Google Scholar] [CrossRef]
- Bernas, J.H.; Taskinen, A.; Wärnå, J.; Murzin, D.Y. Describing the inverse dependence of hydrogen pressure by multi-site adsorption of the reactant: Hydrogenolysis of hydroxymatairesinol on a Pd/C catalyst. J. Mol. Catal. A Chem. 2009, 306, 33–39. [Google Scholar] [CrossRef]
- Frusteri, F.; Italiano, G.; Espro, C.; Arena, F. CH4 Decomposition on Ni and Co thin layer catalysts to produce H2 for fuel cell. Catal. Today 2011, 171, 60–66. [Google Scholar] [CrossRef]
- Vannice, M.A.; Garten, R.L. Supported Palladium catalysts for methanation. Ind. Eng. Chem. Prod. Res. Dev. 1979, 18, 186–191. [Google Scholar] [CrossRef]
- Deutsch, K.L.; Lahr, D.G.; Shanks, B.H. Probing the ruthenium-catalyzed higher polyol hydrogenolysis reaction through the use of stereoisomers. Green Chem. 2012, 14, 1635–1642. [Google Scholar] [CrossRef]
- Shanks, B.H. Conversion of biorenewable feedstocks: New challenges in heterogeneous catalysis. Ind. Eng. Chem. Res. 2010, 49, 10212–10217. [Google Scholar] [CrossRef]
- Sohounloue, D.K.; Montassier, C.; Barbier, J. Catalytic hydrogenolysis of sorbitol. React. Kinet. Catal. Lett. 1983, 22, 391–397. [Google Scholar] [CrossRef]
- Li, N.; Huber, G.W. Aqueous-phase hydrodeoxygenation of sorbitol with Pt/SiO2–Al2O3: Identification of reaction intermediates. J. Catal. 2010, 270, 48–59. [Google Scholar] [CrossRef]
- Rinaldi, R. (Ed.) Catalytic Hydrogenation for Biomass Valorization; RSC Energy and Environment Series No. 13; The Royal Society of Chemistry: London, UK, 2015; pp. 1–310.
- Moreno, B.M.; Li, N.; Lee, J.; Huber, G.W.; Klein, M.T. Modeling aqueous-phase hydrodeoxygenation of sorbitol over Pt/SiO2–Al2O3. RSC Adv. 2013, 3, 23769–23784. [Google Scholar] [CrossRef]
- Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Renewable alkanes by aqueous-phase reforming of biomass-derived oxygenates. Angew. Chem. Int. Ed. 2004, 43, 1549–1551. [Google Scholar] [CrossRef] [PubMed]
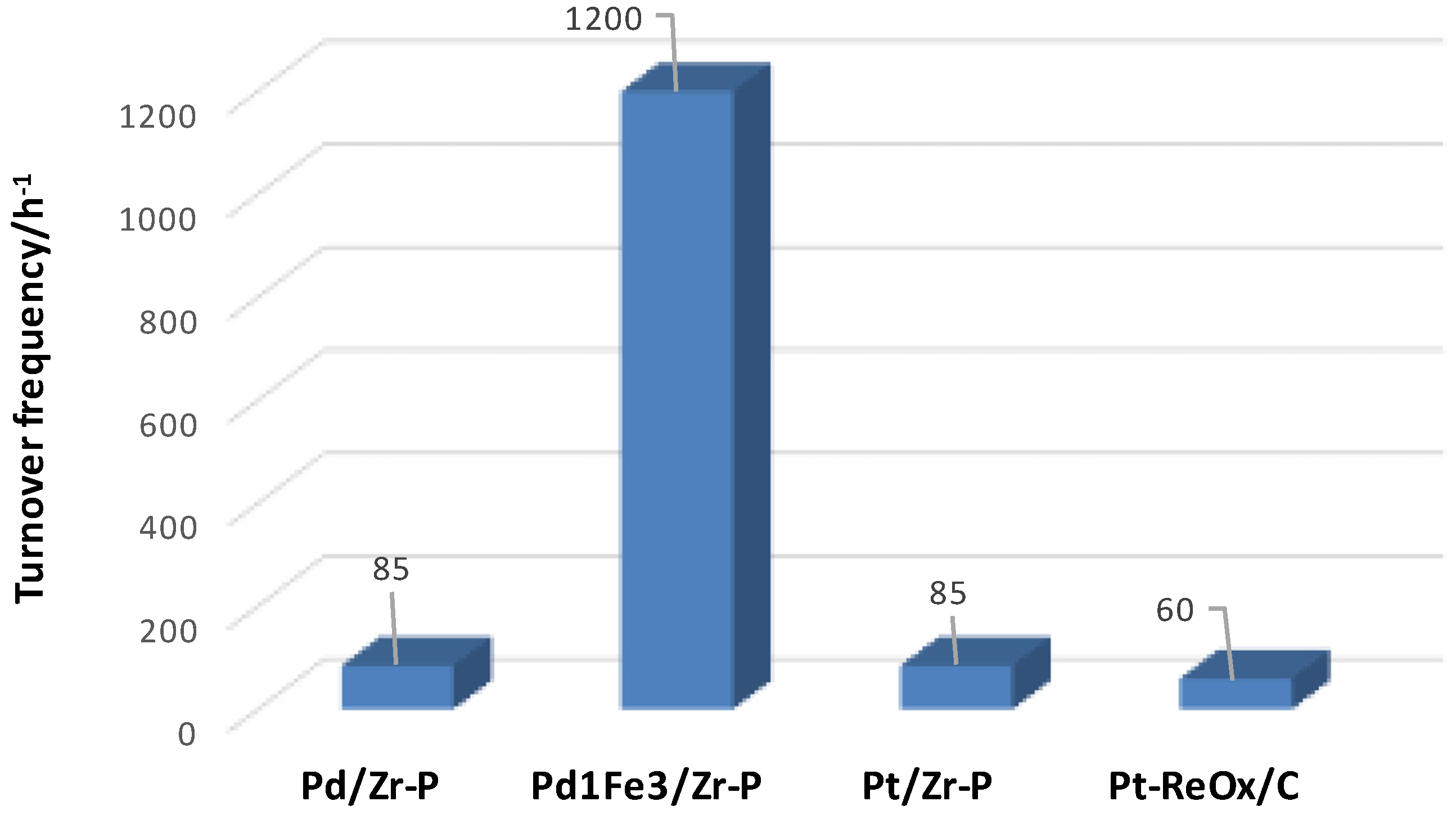
- Kim, Y.T.; Dumesic, J.A.; Huber, G.W. Aqueous-phase hydrodeoxygenation of sorbitol: A comparative study of Pt/Zr phosphate and Pt_ReOx/C. J. Catal. 2013, 304, 72–85. [Google Scholar] [CrossRef]
- Li, B.D.; Wang, J.; Yuan, Y.Z.; Ariga, H.; Takakusagi, S.; Asakura, K. Carbon nanotube-supported RuFe bimetallic nanoparticles as efficient and robust catalysts for aqueous-phase selective hydrogenolysis of glycerol to glycols. ACS Catal. 2011, 1, 1521–1528. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Aloise, A.; Antonucci, P.; Giordano, G.; Nagy, J.B. Effect of support surface on methane dry-reforming catalyst preparation. Catal. Today 2013, 218, 18–29. [Google Scholar] [CrossRef]
- Lo Faro, M.; Frontera, P.; Antonucci, P.; Aricò, A.S. Ni-Cu based catalysts prepared by two different methods and their catalytic activity toward the ATR of methane. Chem. Eng. Res. Des. 2015, 93, 269–277. [Google Scholar] [CrossRef]
- Frontera, P.; Aloise, A.; MacArio, A.; Crea, F.; Antonucci, P.; Giordano, G.; Nagy, J.B. Zeolite-supported Ni catalyst for methane reforming with carbon dioxide. Res. Chem. Intermed. 2015, 37, 267–279. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Hiyoshi, N.; Sato, O.; Shirai, M. Sorbitol dehydration in high temperature liquid water. Green Chem. 2011, 13, 873–881. [Google Scholar] [CrossRef]
- Karinen, R.; Vilonen, K.; Niemelä, M. Biorefining: Heterogeneously catalyzed reactions of carbohydrates for the production of furfural and hydroxymethylfurfural. ChemSusChem 2011, 4, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Binder, J.B.; Blank, J.J.; Cefali, A.V.; Raines, R.T. Synthesis of furfural from xylose and xylan. ChemSusChem 2010, 3, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Aellig, C.; Hermans, I. Continuous D-fructose dehydration to 5-Hydroxymethylfurfural under mild conditions. ChemSusChem 2012, 5, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.Y.; Zhu, Y.X.; Chen, J.G.G. Promoting low-temperature hydrogenation of C=O bonds of acetone and acetaldehyde by using Co–Pt bimetallic catalysts. ChemCatChem 2011, 3, 578–581. [Google Scholar] [CrossRef]
- Zheng, R.Y.; Humbert, M.P.; Zhu, Y.X.; Chen, J.G. Low-temperature hydrogenation of the C=O bond of propanal over Ni–Pt bimetallic catalysts: From model surfaces to supported catalysts. Catal. Sci. Technol. 2011, 1, 638–643. [Google Scholar] [CrossRef]
- Wang, H.; Male, J.; Wang, Y. Recent advances in hydrotreating of pyrolysis bio-oil and its oxygen-containing model compound. ACS Catal. 2013, 3, 1047–1070. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Mohan, D.; Pittman, C.U.; Steele, P.H. Pyrolysis of wood/biomass for bio-oil: A critical review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Shafaghat, H.; Rezaeia, P.S.; Daud, W.M.A.W. Effective parameters on selective catalytic hydrodeoxygenation of phenolic compounds of pyrolysis bio-oil to high-value hydrocarbons. RSC Adv. 2015, 5, 103999–104042. [Google Scholar] [CrossRef]
- Saidi, M.; Samimi, F.; Karimipourfard, D.; Nimmanwudipong, T.; Gates, B.C.; Rahimpour, M.R. Upgrading of lignin-derived bio-oils by catalytic hydrodeoxygenation. Energy Environ. Sci. 2014, 7, 103–129. [Google Scholar] [CrossRef]
- Song, W.; Liu, Y.; Baráth, E.; Zhao, C.; Lercher, J.A. Synergistic effects of Ni and acid sites for hydrogenation and C–O bond cleavage of substituted phenol. Green Chem. 2015, 17, 1204–1218. [Google Scholar] [CrossRef]
- Grilc, M.; Likozar, B.; Levec, J. Hydrodeoxygenation and hydrocracking of solvolysed lignocellulosic biomass by oxide, reduced and sulphide form of NiMo, Ni, Mo and Pd catalysts. Appl. Catal. B Environ. 2014, 150–151, 275–287. [Google Scholar] [CrossRef]
- Hensley, A.J. R.; Wang, Y.; McEwen, J.S. Phenol deoxygenation mechanisms on Fe(110) and Pd(111). ACS Catal. 2015, 5, 523–536. [Google Scholar] [CrossRef]
- Bolognini, M.; Cavani, F.; Scagliarini, D.; Flego, C.; Perego, C.; Saba, M. Heterogeneous basic catalysts as alternatives to homogeneous catalysts: Reactivity of Mg/Al mixed oxides in the alkylation of m-cresol with methanol. Catal. Today 2002, 75, 103–111. [Google Scholar] [CrossRef]
- Gu, G.H.; Mullen, C.A.; Boateng, A.A.; Vlachos, D.G. Mechanism of dehydration of phenols on noble metals via first-principles microkinetic modeling. ACS Catal. 2016, 6, 3047–3055. [Google Scholar] [CrossRef]
- Shafaghat, H.; Rezaei, P.S.; Daud, W.M.A.W. Catalytic hydrogenation of phenol, cresol and guaiacol over physically mixed catalysts of Pd/C and zeolite solid acids. RSC Adv. 2015, 5, 33990–33998. [Google Scholar] [CrossRef]
- Cozzula, D.; Vinci, A.; Mauriello, F.; Pietropaolo, R.; Müller, T.E. Directing the cleavage of ester C–O bonds by controlling the hydrogen availability on the surface of coprecipitated Pd/Fe3O4. ChemCatChem 2016, 8, 1515–1522. [Google Scholar] [CrossRef]
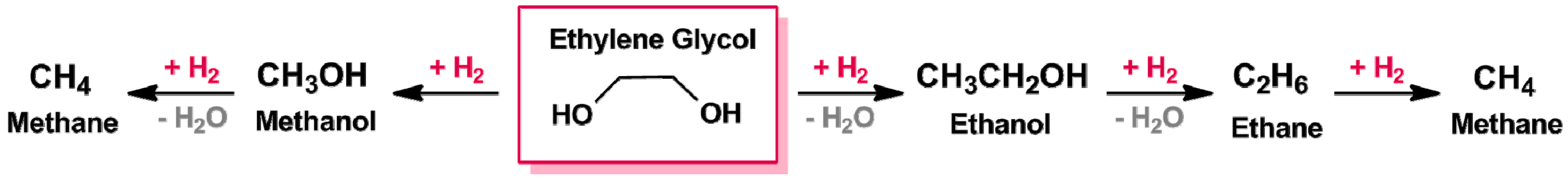
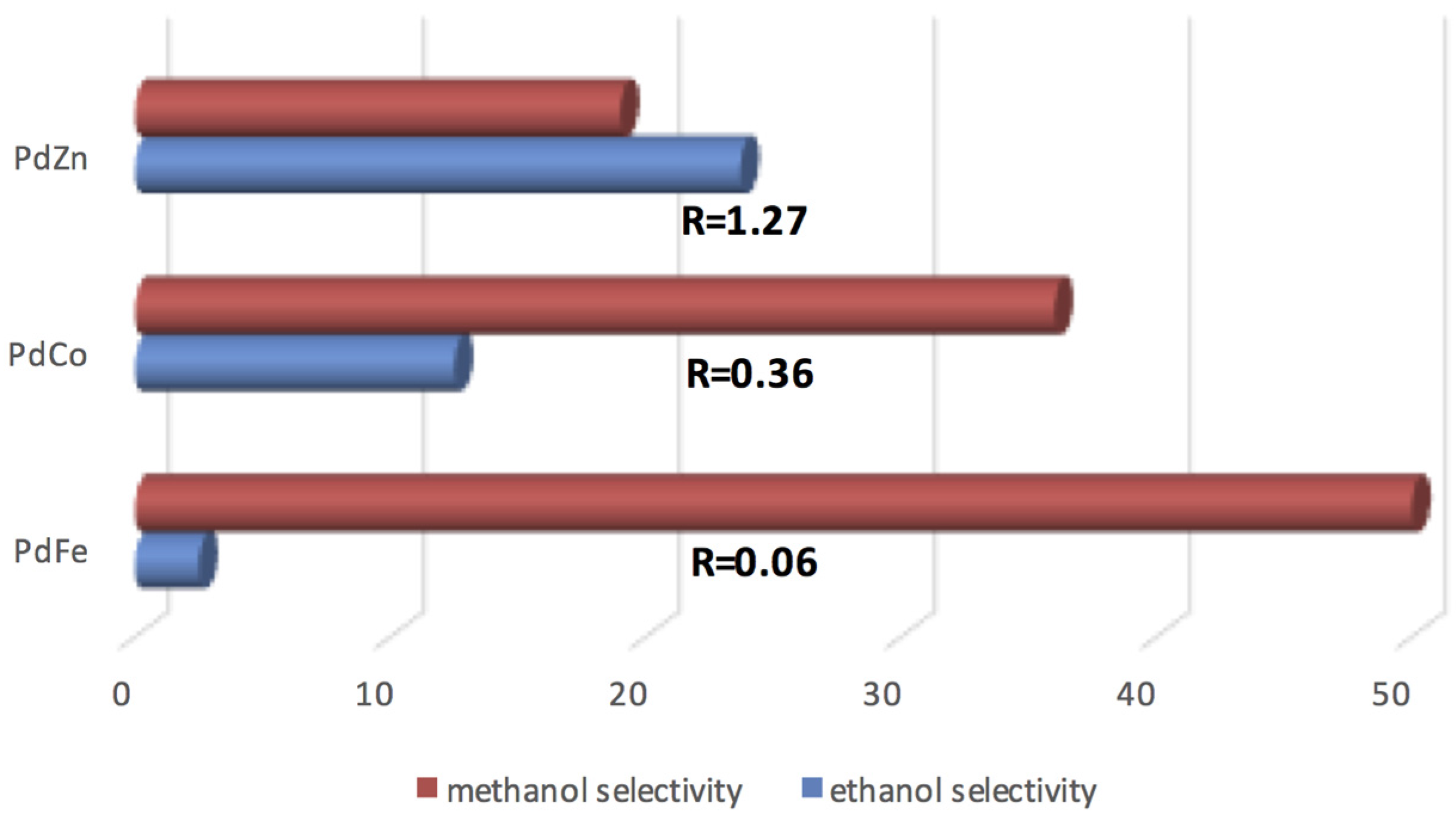
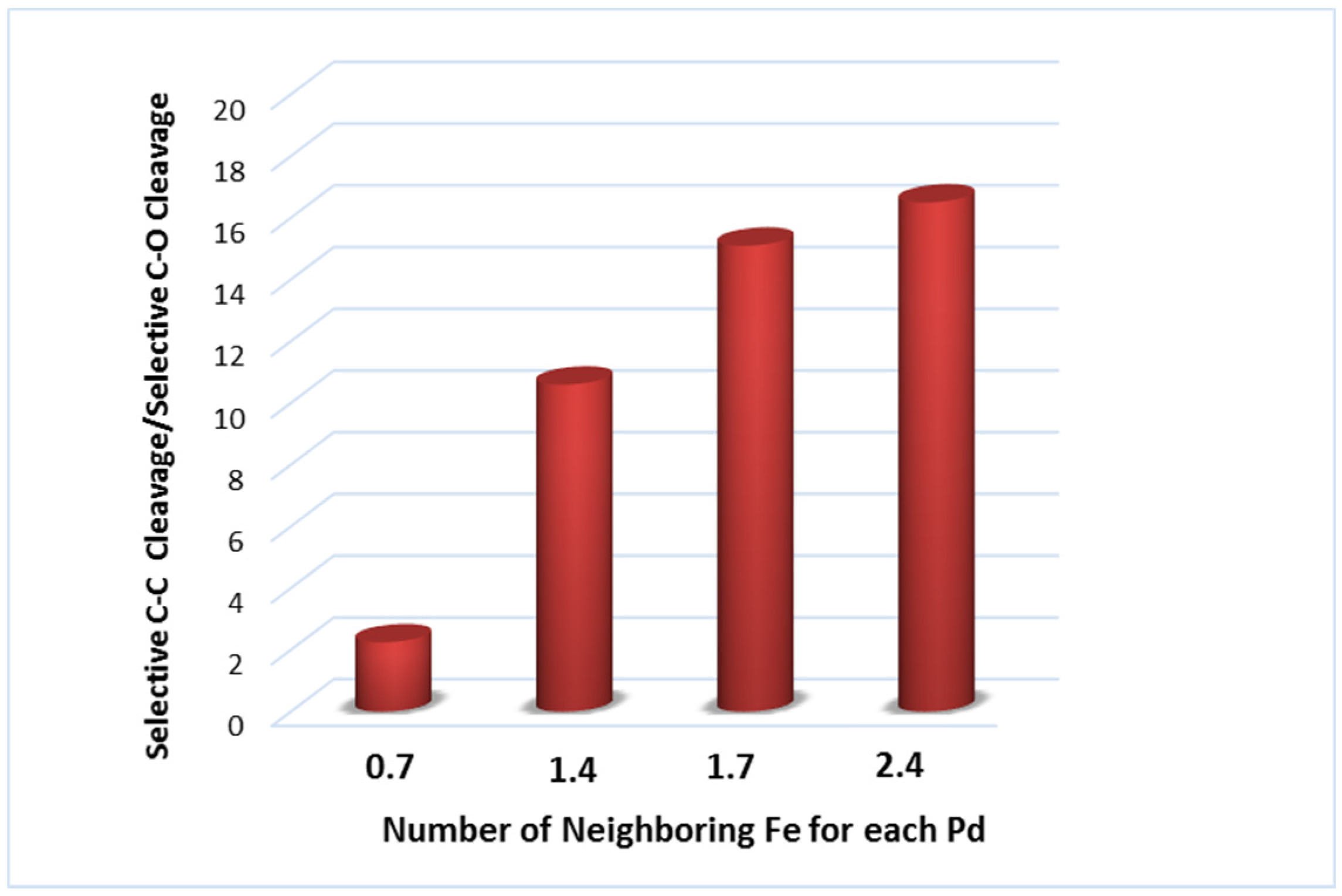
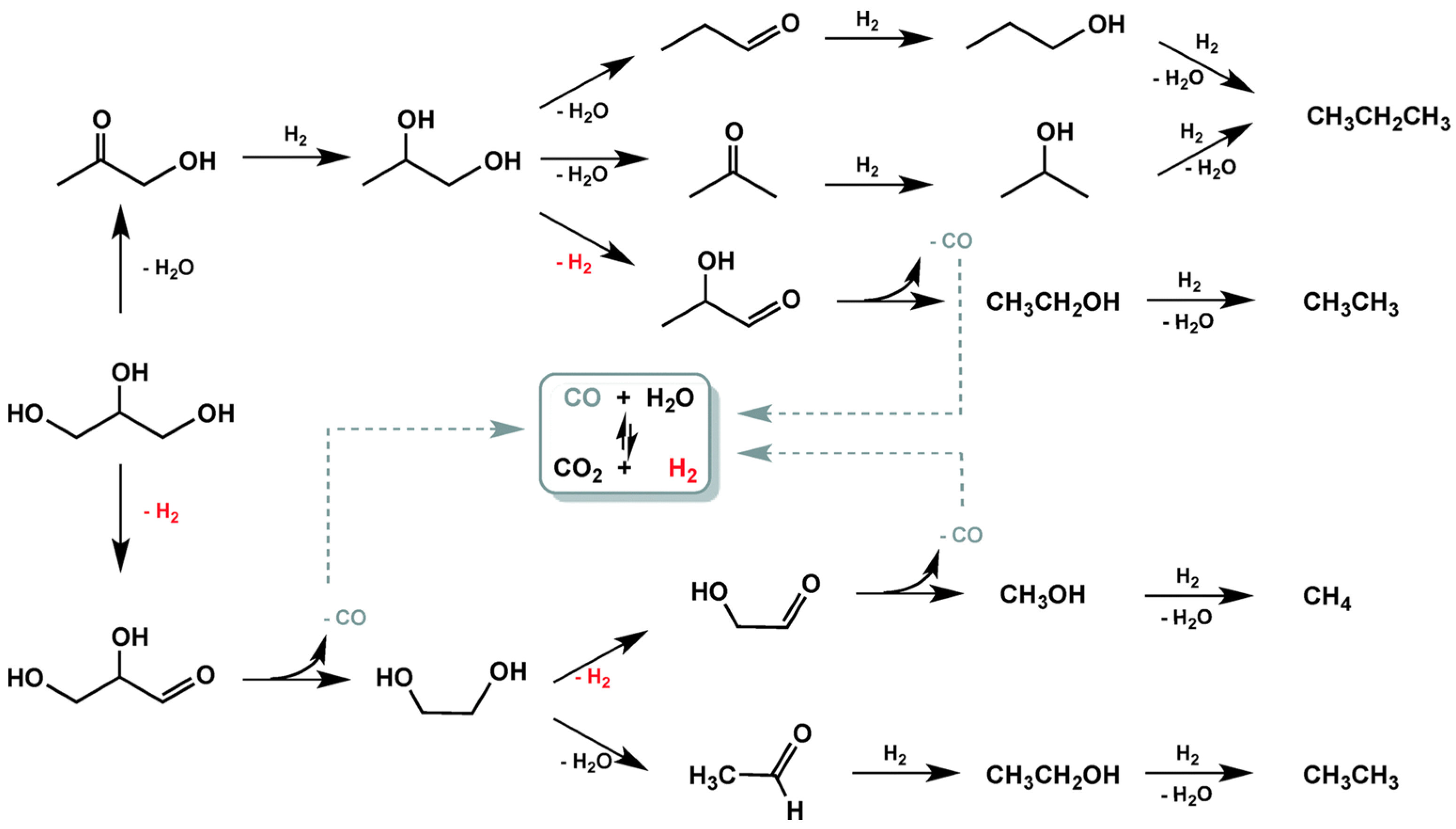
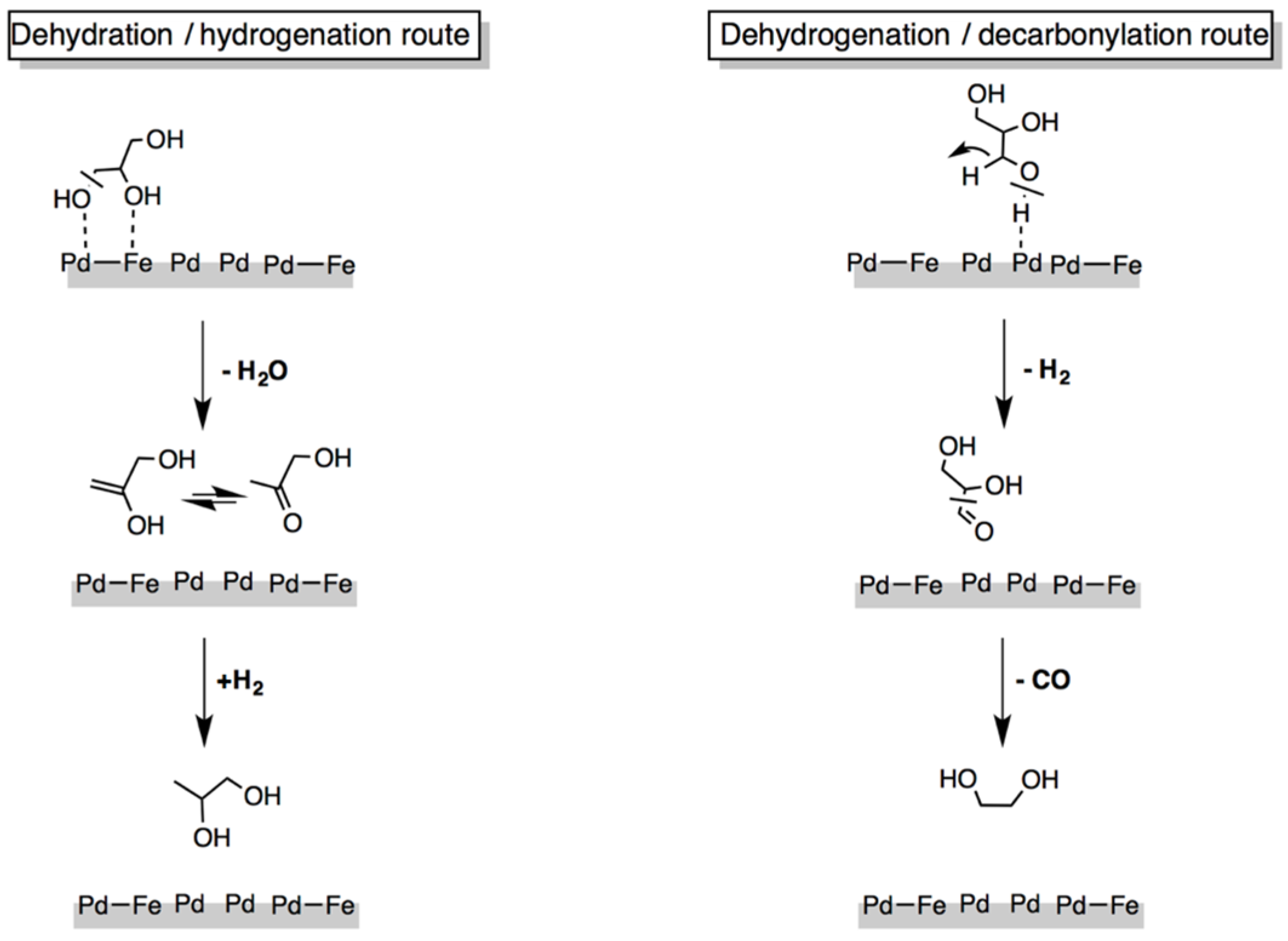


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
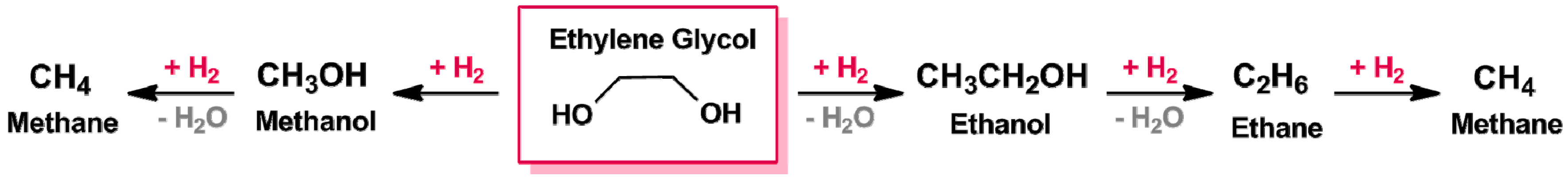{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
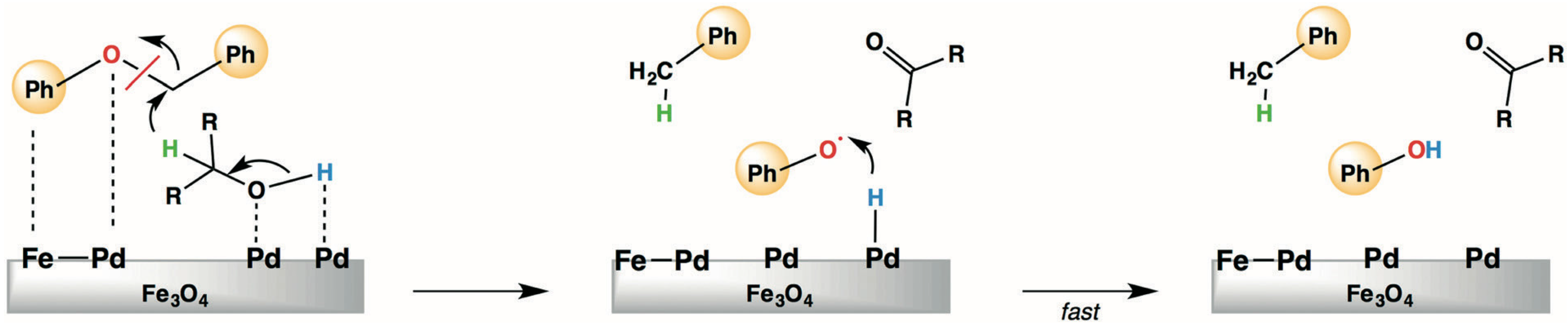{kind=link}
| Catalyst Notation | Preparation Method 1 | Binding Energy (eV) | References | |||
|---|---|---|---|---|---|---|
| Pd 3d5/2 | Fe 2p3/2 | Fe 2p1/2 | Fe 2p3/2sat | |||
| Pd/Fe | CP | 335.2 ± 1 | 710.7 ± 0.8 | 724.4 | - | [131,135,137,138,139] |
| Pd-rod Fe2O3 | DP | 335.9 | 711.2 | - | - | [134] |
| Pd-plate Fe2O3 | DP | 335.7 | 711.4 | - | - | [134] |
| PdCo/Fe2O3 | CP | 334.8 | 710.2 | - | - | [133] |
| Pd-Zn/Fe2O3 | CP | 334.8 | 707.8 | - | - | [132] |
| Pd/Fe2O3 | IWi | 334.8 | 710.4 | 723.8 | 718.4 | [135,138,142] |
| Pd/Fe3O4 | IWi | 334.8 | 710.5 | - | - | [138] |
| Pd-Fe/OMC | IWi | 334.4 | 708.2 | - | - | [143] |
| Pd1–FeX/OMC | IWi | 334.4 | 708.2 | - | - | [144] |
| Catalyst Notation | Scattering Pair | CN * | Bond Length (Å) | R-Factor (%) | Ef (eV) | DW Factor ** (Å2) | k-Range | References |
|---|---|---|---|---|---|---|---|---|
| Pd/C | Pd-Pd | 8.0 (3) | 2.73 (1) | 2.6 | −7.6 | 6.1 (3) | 2–17 | [131] |
| Pd/Fe2O3 (before reduction) | Pd-O | 3.5 (2) | 2.01 (1) | 1.3 | −4.5 | 2.8 (6) | 2–16 | [131] |
| Pd/Fe2O3 (after reduction) | Pd-Fe | 1.0 (2) | 2.59 (1) | 1.6 | −7.4 | 5.1 (4) | 2–12 | [131] |
| Pd-Pd | 1.9 (2) | 2.66 (1) | 7.3 (6) | |||||
| Pd-Pd | 2.4 (2) | 2.75 (1) | 3.6 (3) | |||||
| Pd-Co/Fe2O3 | Pd-Pd | 6.8 (4) | 2.69 (1) | 0.4 | 1.2 | 0.010 (1) | - | [133] |
| Catalyst | Preparation Method 1 | Substrate [wt %] 2 | Solvent | H-Source | Temp. [°C] | Time [h] | Conversion [%] | Main Products [%] 4 | References |
|---|---|---|---|---|---|---|---|---|---|
| Zn-Pd/Fe3O4 | CP | Gly (1%) | H2O | H2 (50 bar) | 250 | 120 | 100 | 1,2-PDO (33%), EtOH (32%) | [132] |
| Pd/Fe3O4 | CP | Gly (1%) | H2O | H2 (50 bar) | 250 | 120 | 100 | 1,2-PDO (36%), EtOH (17%), 1-PO (15%) | [132] |
| Pd/Fe3O4 | CP | Gly (4%) | H2O | H2 (5 bar) | 240 | 24 | 100 | EtOH (70%), POs (10%) | [129] |
| Pd/Fe3O4 | CP | Gly (4%) | H2O | APR (5bar) | 240 | 24 | 100 | EtOH (73%), 1,2-PDO (9.4%)POs (8.7%) | [129] |
| PdO/Fe2O3 | CP | Gly (12%) | 2-PO 3 | 2-PO (5bar) | 180 | 24 | 100 | 1,2-PDO (94%), EG (6%) | [137] |
| Pd/Fe3O4 | CP | Gly (4%) | 2-PO | 2-PO (5bar) | 180 | 24 | 100 | 1,2-PDO (56%), AC (25%) | [138] |
| Pd/Fe3O4 | IWi | Gly (4%) | 2-PO | 2-PO (5bar) | 180 | 24 | 66.5 | 1,2-PDO (48%), AC (44%) | [138] |
| Pd/Fe2O3 | IWi | Gly (4%) | 2-PO | 2-PO (5bar) | 180 | 24 | 40.4 | 1,2-PDO (26.6%), AC (58%) | [138] |
| Catalyst | Preparation Method 1 | Substrate | H-Source | Temp. [°C] | Time [min] | Conversion [%] | Aromatic Yeld [%] | References |
|---|---|---|---|---|---|---|---|---|
| Pd/OMC | STM-IWi | BPE | H2 (10 bar) | 250 | 60 | 93.0 | 36.2 | [143] |
| Fe/OMC | STM-IWi | BPE | H2 (10 bar) | 250 | 60 | 50.1 | 37.2 | [143] |
| Pd-Fe/OMC | STM-IWi | BPE | H2 (10 bar) | 250 | 60 | 88.5 | 74.3 | [143] |
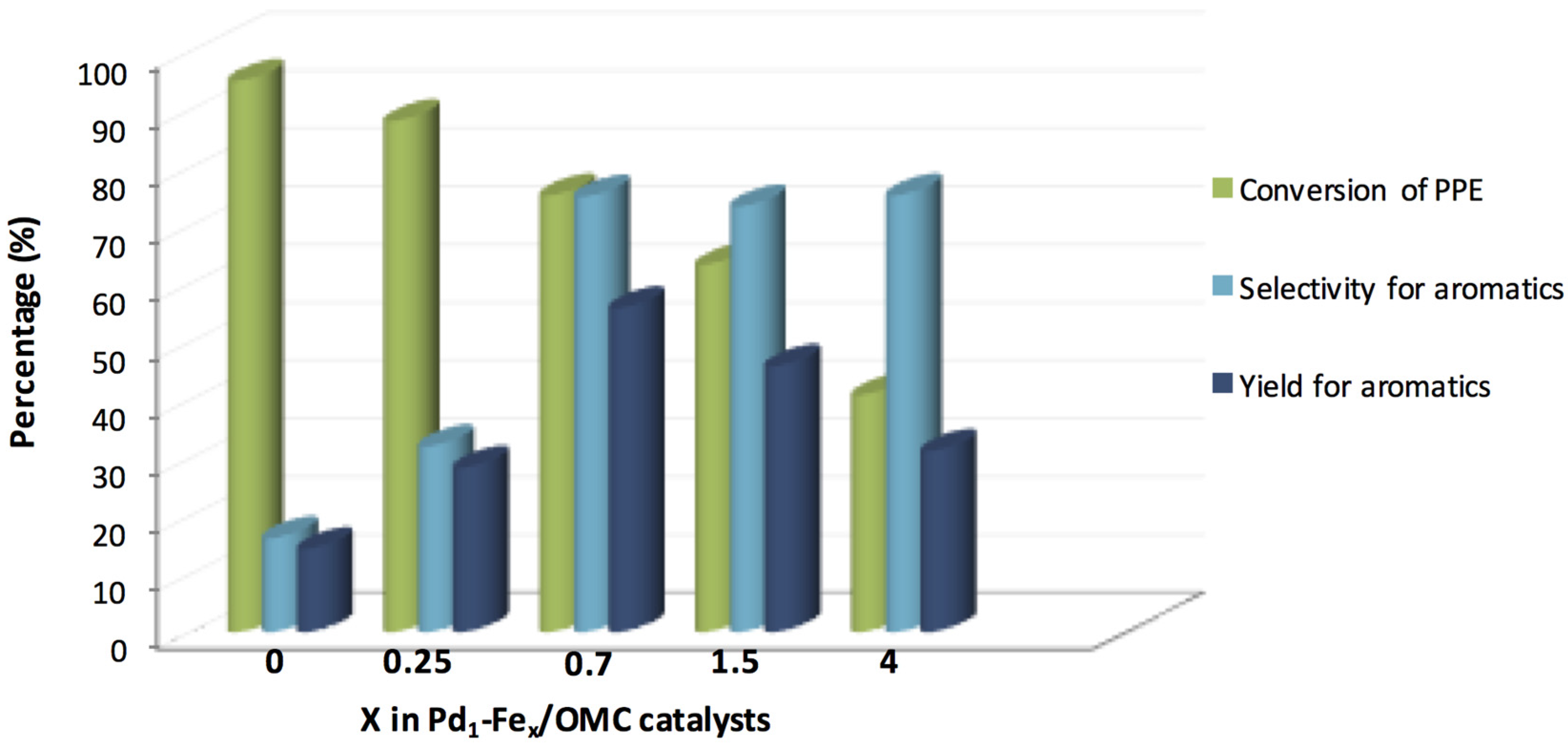
| Pd1-Fe0/OMC | STM-IWi | PPE | H2 (10 bar) | 250 | 180 | 94.1 | 15.1 | [144] |
| Pd1-Fe0.25/OMC | STM-IWi | PPE | H2 (10 bar) | 250 | 180 | 88.3 | 28.3 | [144] |
| Pd1-Fe0.7/OMC | STM-IWi | PPE | H2 (10 bar) | 250 | 180 | 75.6 | 56.7 | [144] |
| Pd1-Fe1.5/OMC | STM-IWi | PPE | H2 (10 bar) | 250 | 180 | 63.2 | 46.6 | [144] |
| Pd1-Fe4/OMC | STM-IWi | PPE | H2 (10 bar) | 250 | 180 | 41.2 | 31.1 | [144] |
| Pd/Fe3O4 | CP | BPE | H2 (10 bar) | 240 | 90 | 75.0 | 75.0 | [145] |
| Pd/Fe3O4 | CP | BPE | H2 (20 bar) | 240 | 90 | 73.0 | 73.0 | [145] |
| Pd/Fe3O4 | CP | BPE | H2 (40 bar) | 240 | 90 | 71.0 | 69.6 | [145] |
| Pd/Fe3O4 | CP | BPE | iPrOH | 180 | 90 | 19.7 | 100 | [145] |
| Pd/Fe3O4 | CP | BPE | iPrOH | 210 | 90 | 49.4 | 100 | [145] |
| Pd/Fe3O4 | CP | BPE | iPrOH | 240 | 90 | 100 | 100 | [145] |
| Pd/Fe3O4 | CP | PPE | iPrOH | 240 | 90 | 22.0 | 100 | [145] |
| Pd/Fe3O4 | CP | DPE | iPrOH | 240 | 90 | <2 | - | [145] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espro, C.; Gumina, B.; Paone, E.; Mauriello, F. Upgrading Lignocellulosic Biomasses: Hydrogenolysis of Platform Derived Molecules Promoted by Heterogeneous Pd-Fe Catalysts. Catalysts 2017, 7, 78. https://doi.org/10.3390/catal7030078
Espro C, Gumina B, Paone E, Mauriello F. Upgrading Lignocellulosic Biomasses: Hydrogenolysis of Platform Derived Molecules Promoted by Heterogeneous Pd-Fe Catalysts. Catalysts. 2017; 7(3):78. https://doi.org/10.3390/catal7030078
Chicago/Turabian StyleEspro, Claudia, Bianca Gumina, Emilia Paone, and Francesco Mauriello. 2017. "Upgrading Lignocellulosic Biomasses: Hydrogenolysis of Platform Derived Molecules Promoted by Heterogeneous Pd-Fe Catalysts" Catalysts 7, no. 3: 78. https://doi.org/10.3390/catal7030078
APA StyleEspro, C., Gumina, B., Paone, E., & Mauriello, F. (2017). Upgrading Lignocellulosic Biomasses: Hydrogenolysis of Platform Derived Molecules Promoted by Heterogeneous Pd-Fe Catalysts. Catalysts, 7(3), 78. https://doi.org/10.3390/catal7030078