
Time Resolved Operando X-ray Techniques in Catalysis, a Case Study: CO Oxidation by O2 over Pt Surfaces and Alumina Supported Pt Catalysts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
“As yet, apparently, very few chemists have awakened to the wonderful opportunities that lie open to them on all sides when they attack the problems of chemistry by the new methods which the physicists have developed. The physicist on the other hand, in gradually beginning to extend his investigations into the field of the chemist and we may hope, if the chemist will meet him but halfway, that there will be a new physical chemistry which will have an even more far-reaching effect on our ordinary chemical conceptions than has the physical chemistry of the last decades.”
2. Fast X-ray Methods as Applied to the Study of CO Oxidation over Pt Surfaces and Nanoparticulate Catalysts
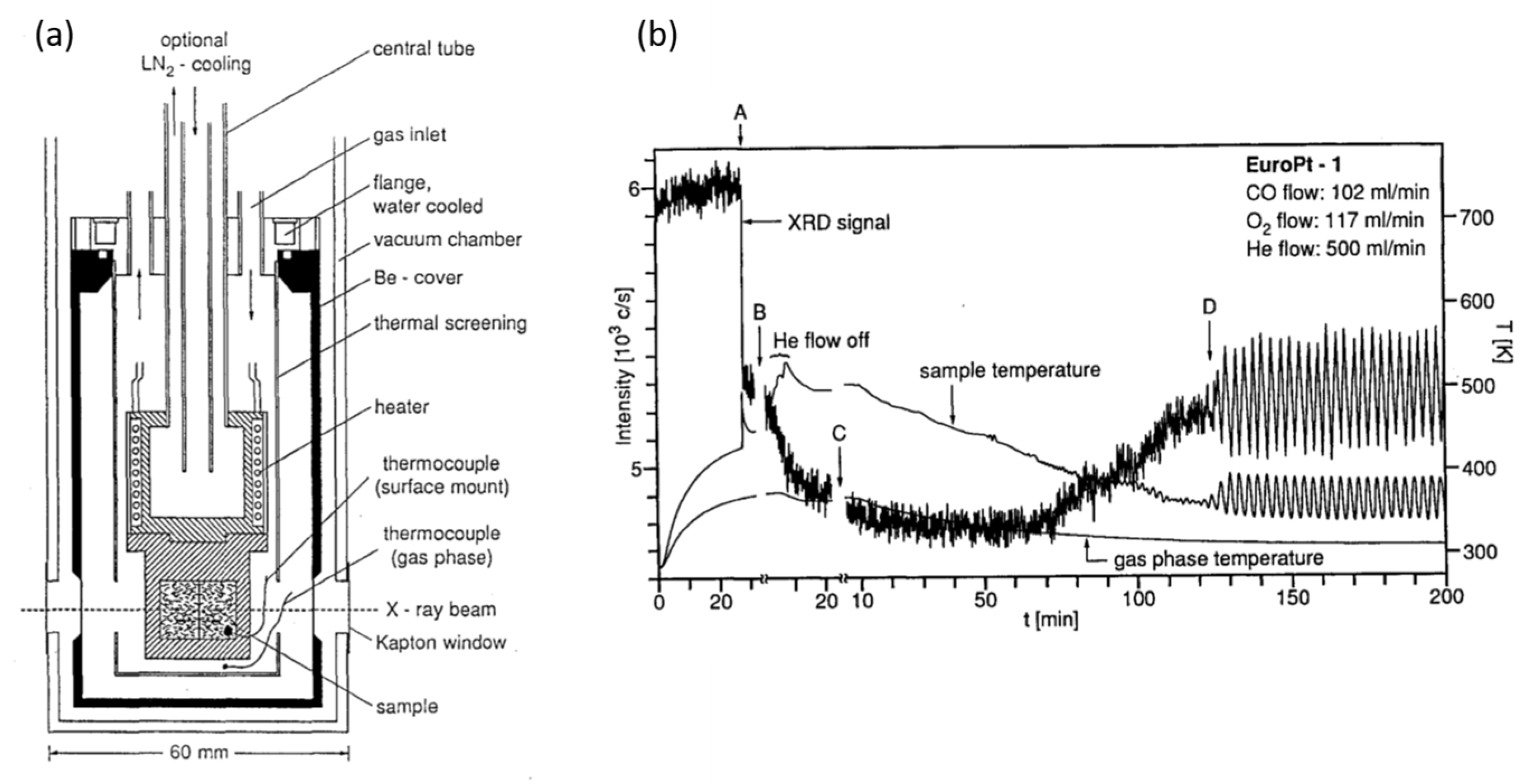
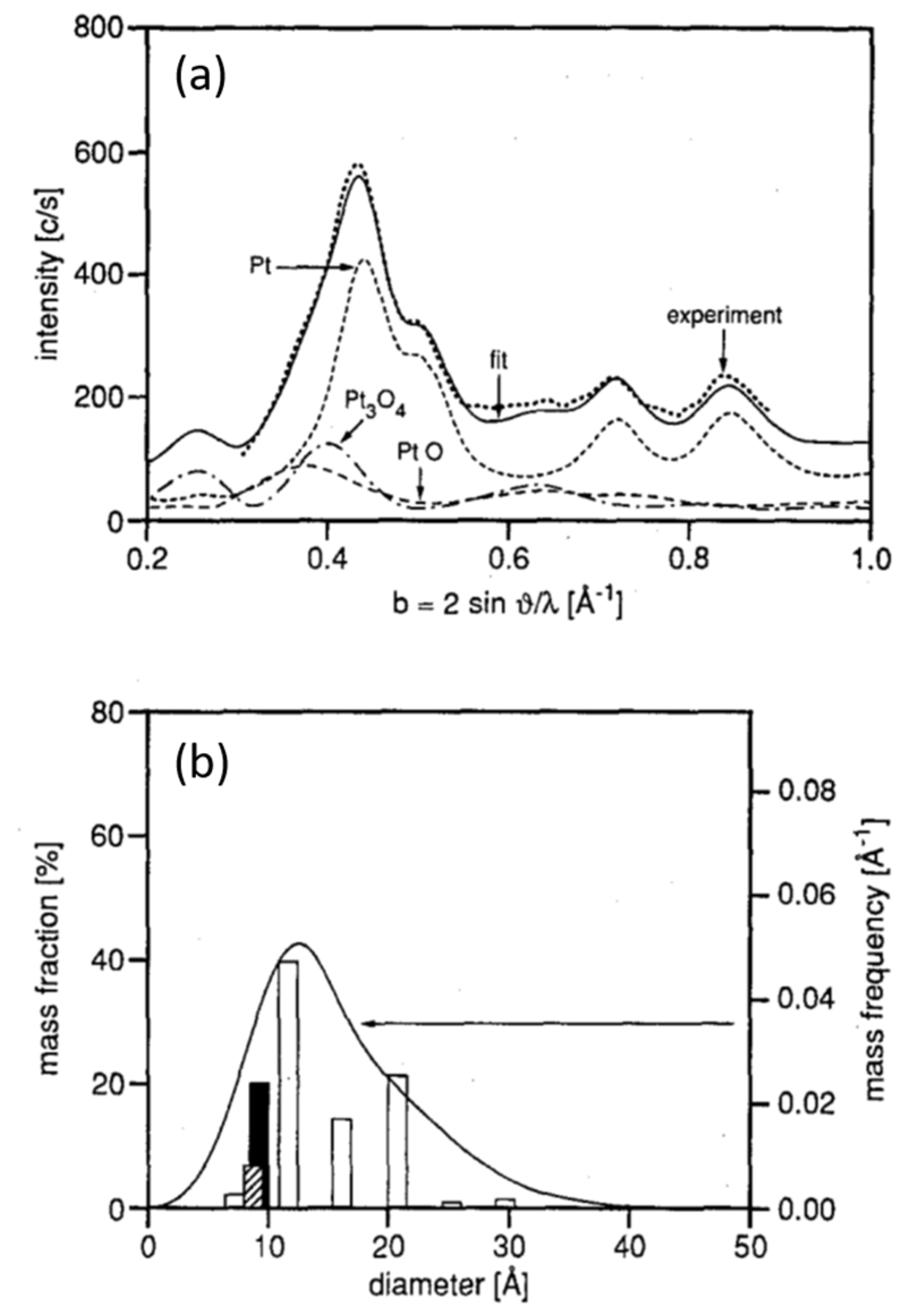
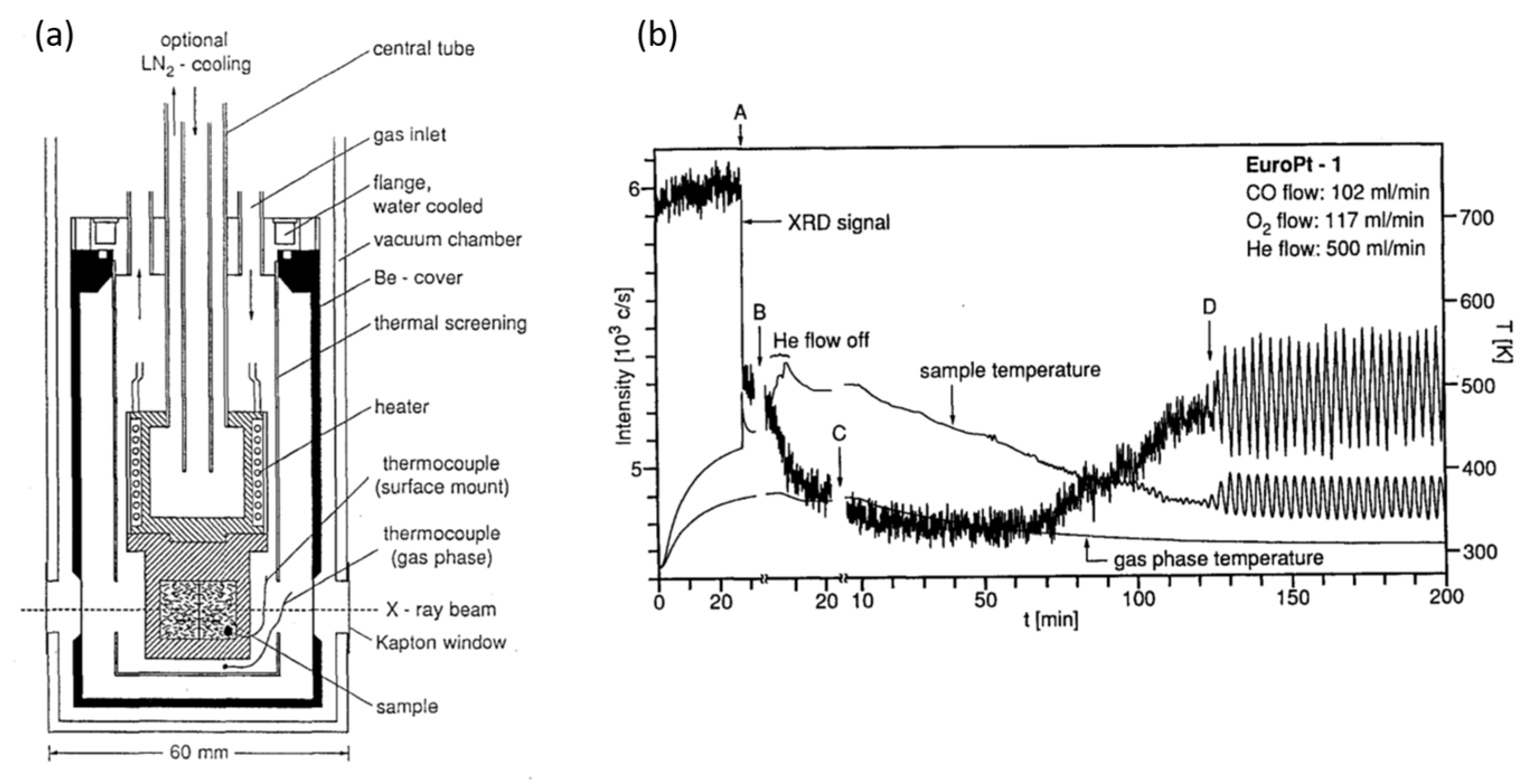
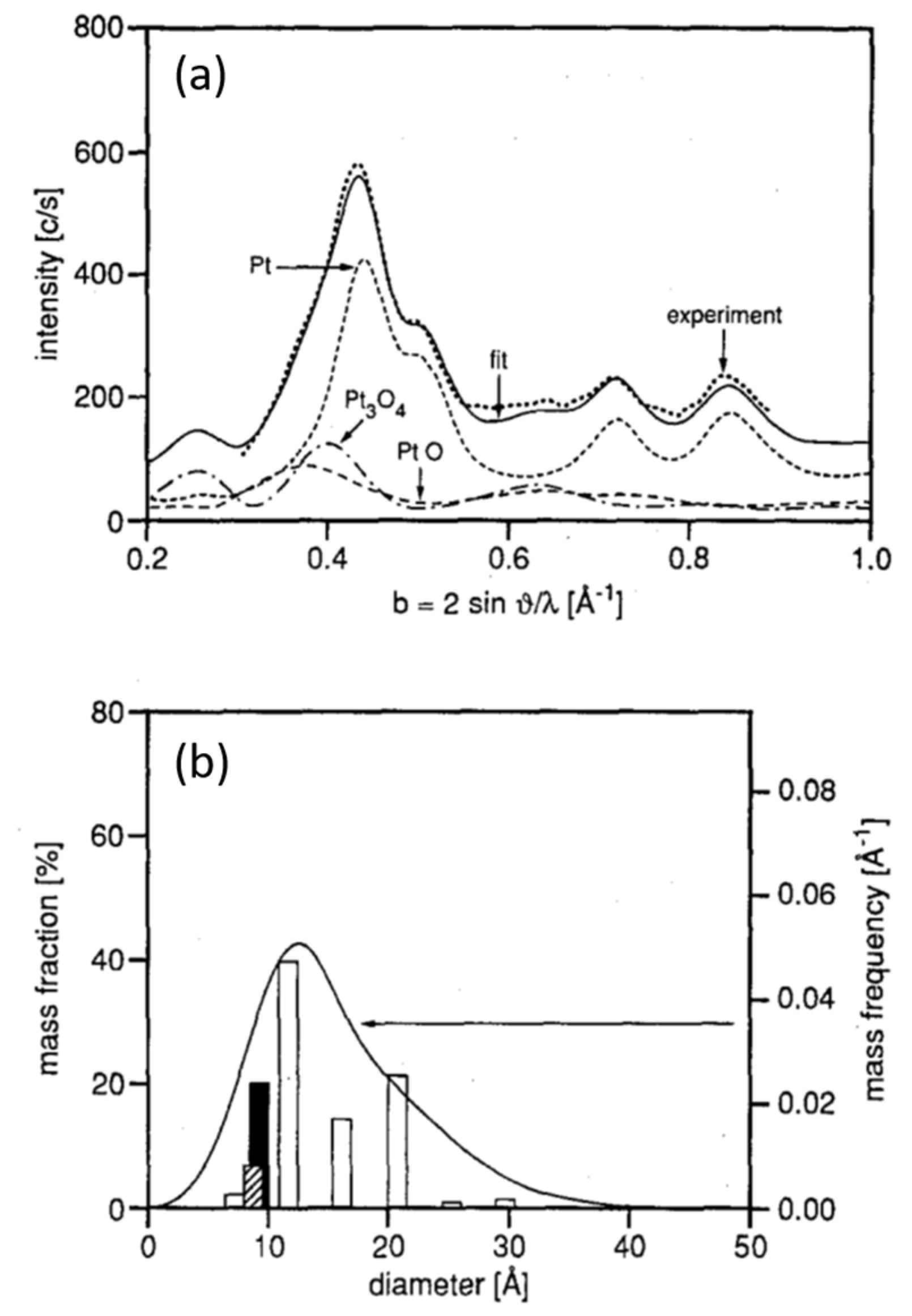
2.1. Fast X-ray Diffraction (XRD) and Debye-Analysis of Oscillations CO Oxidation over EuroPt-1
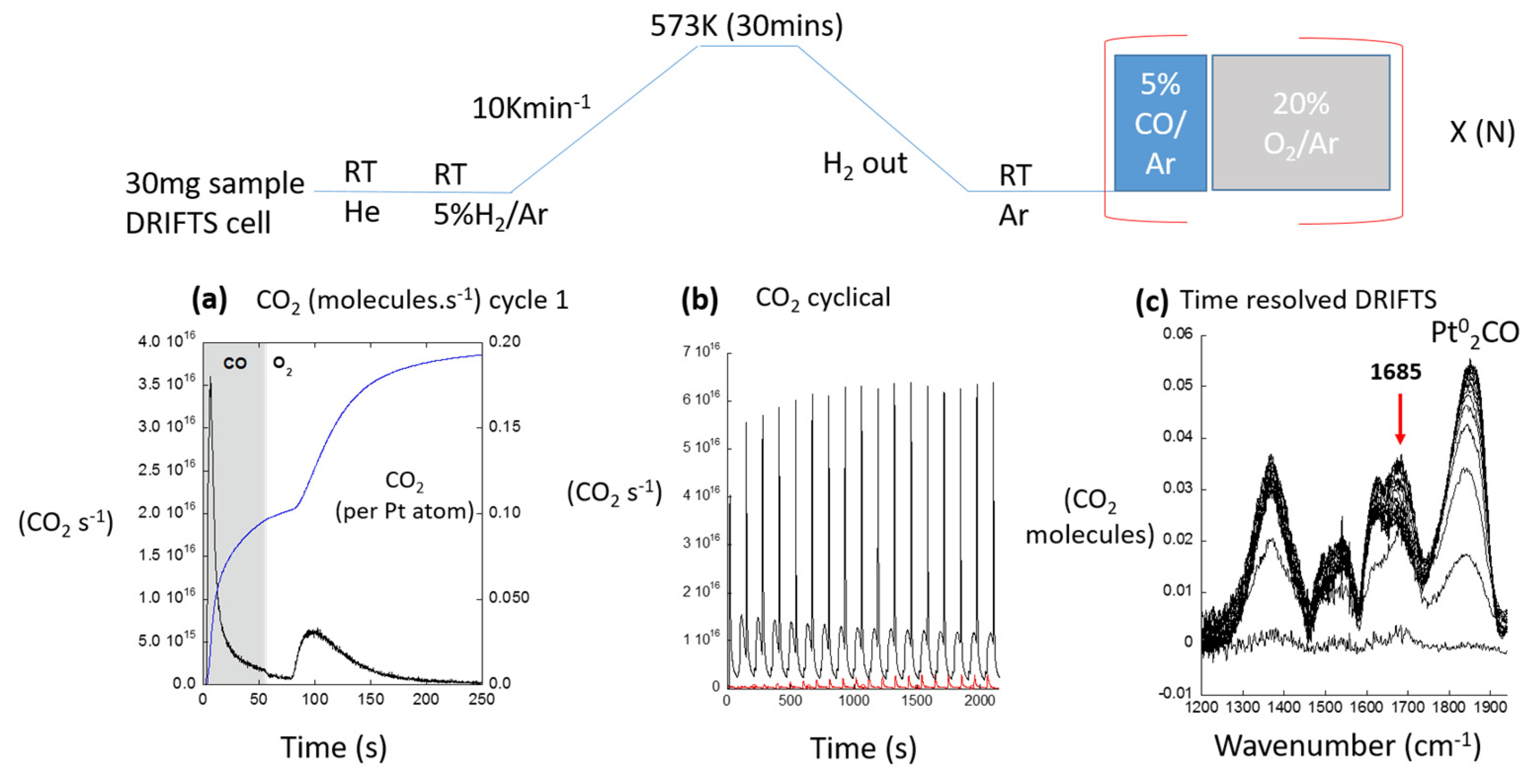
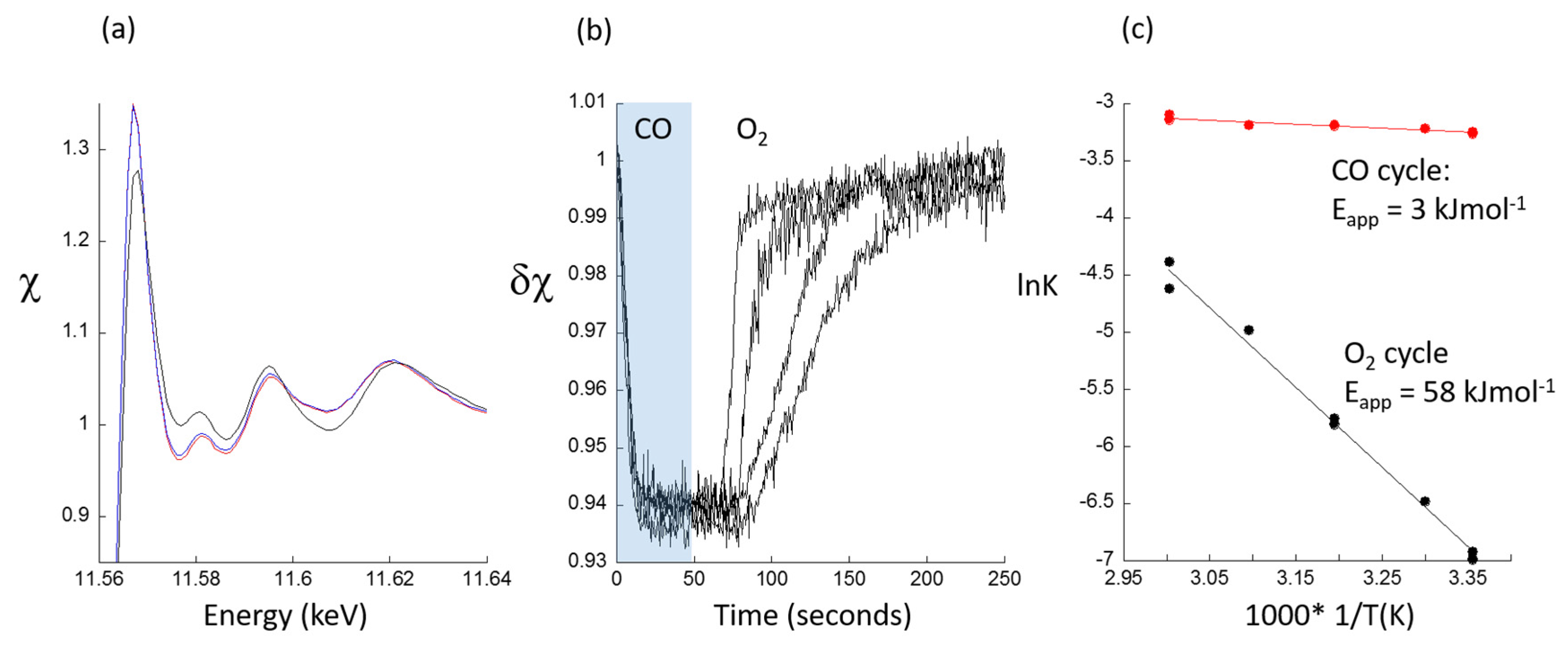
2.2. A Transient Approach to CO Oxidation over Pt/Al2O3 Catalysts Using Fast (Energy Dispersive) X-ray Absorption Spectroscopy (DXAFS)
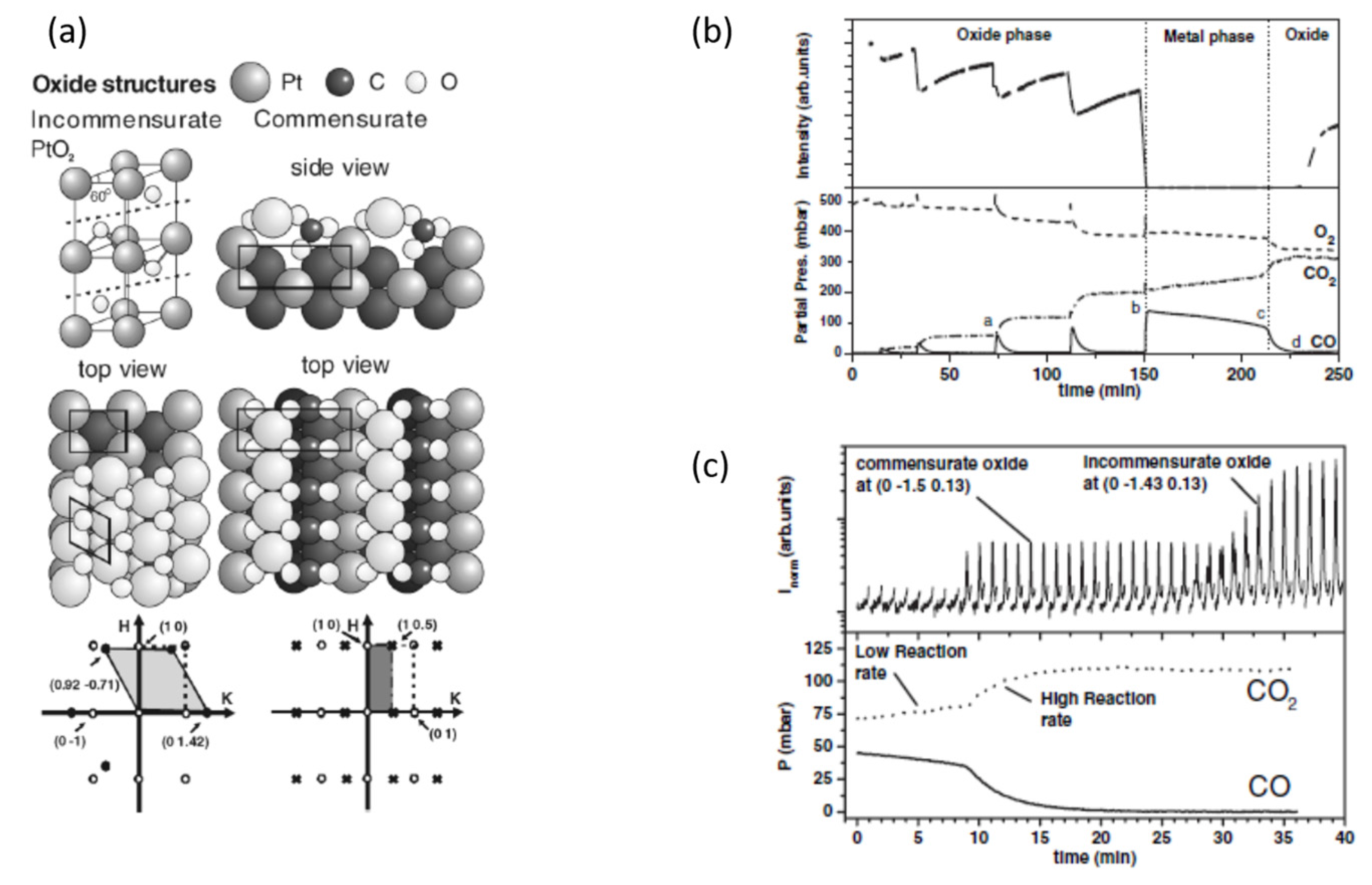
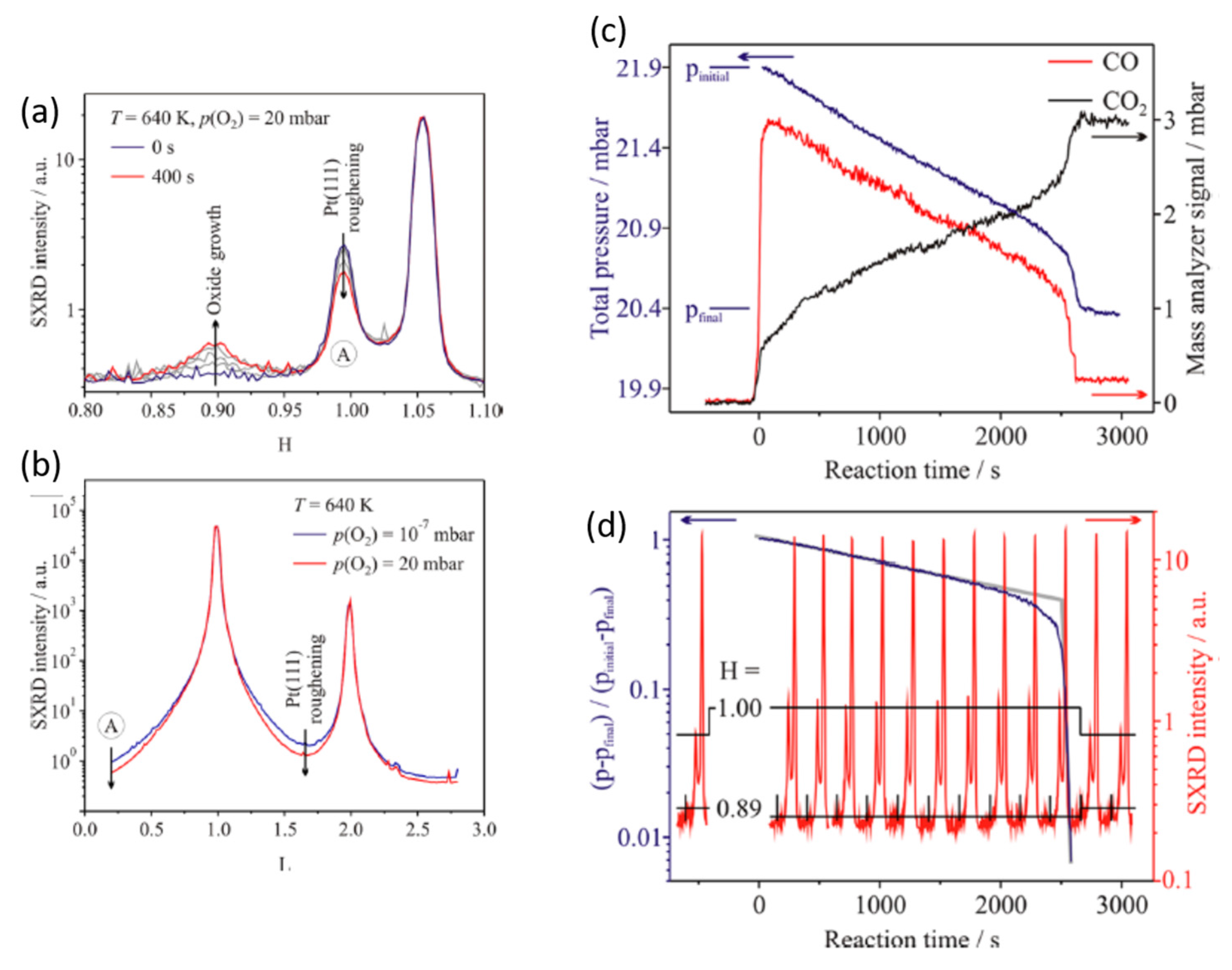
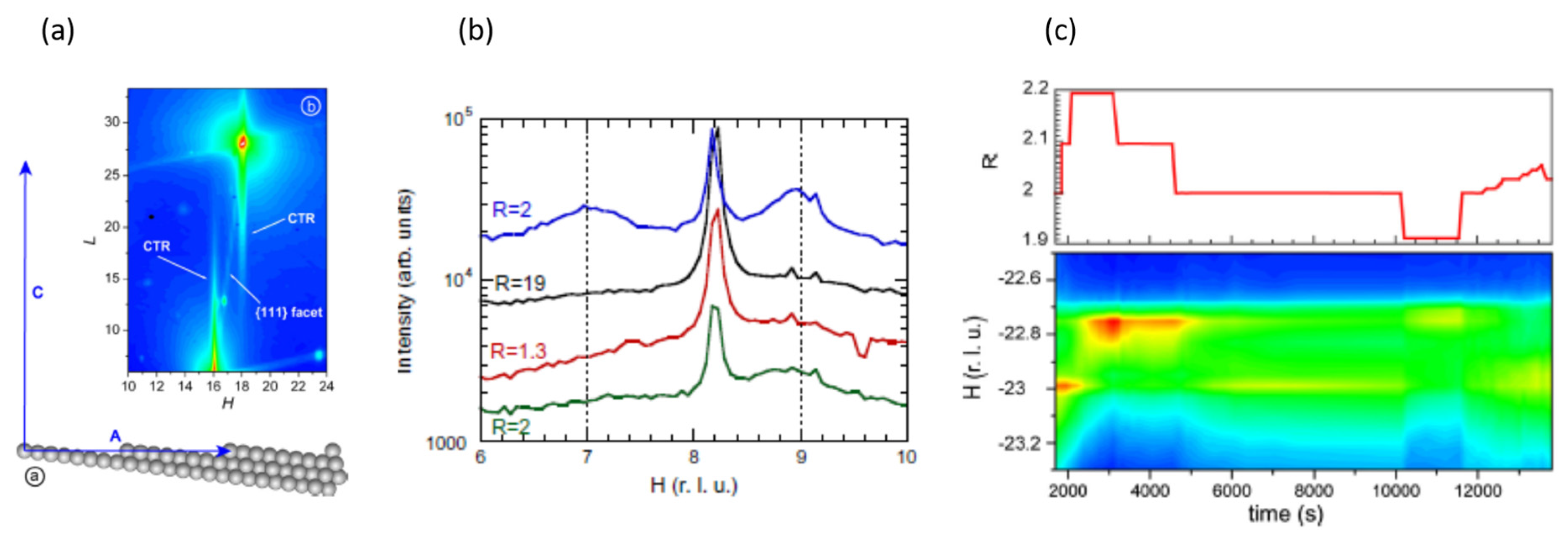
2.3. A Return to Pt Single Crystals: Operando Surface X-ray Diffraction (SXRD) at Elevated (Ambient) Pressures and the Role of Pt Surface Oxides
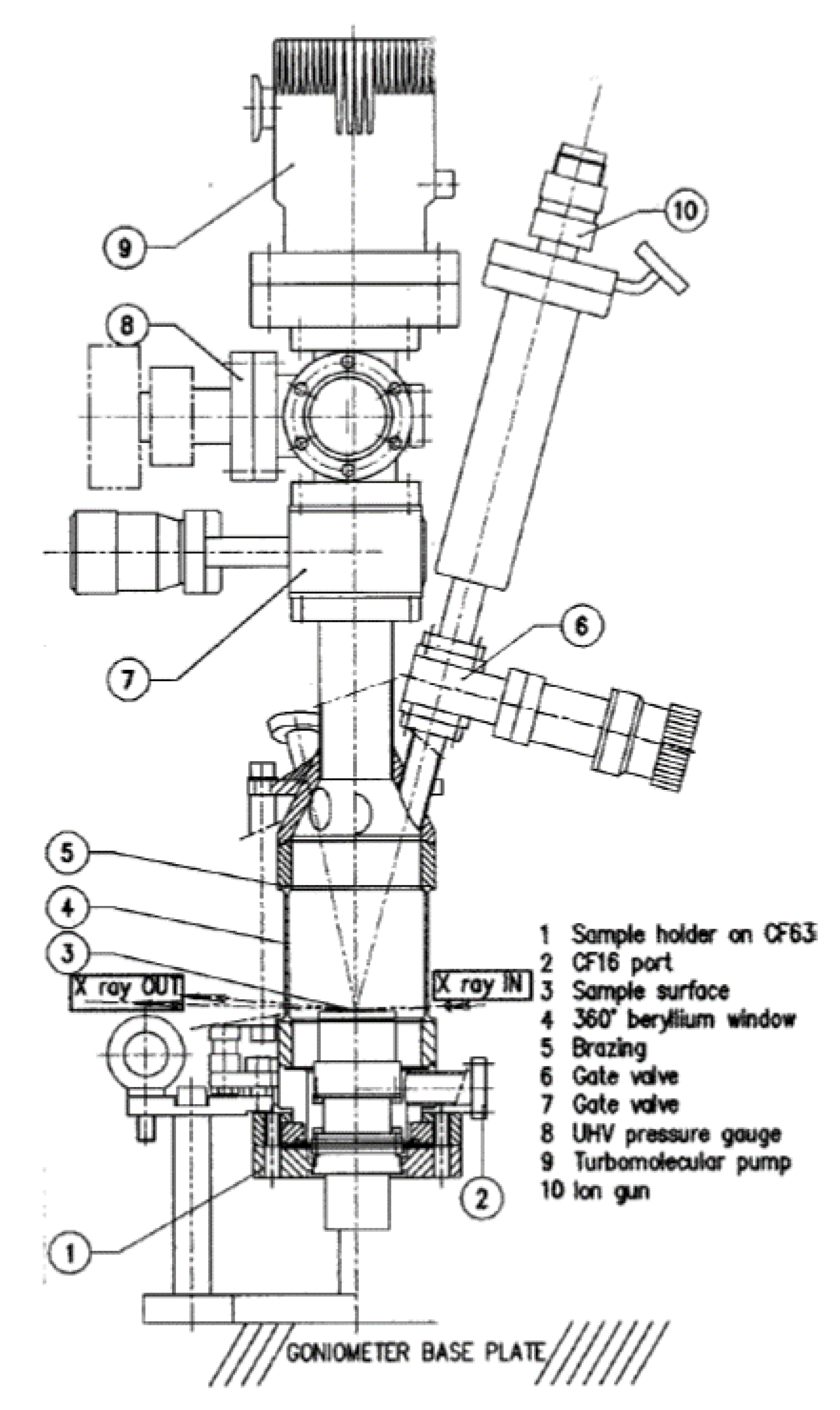
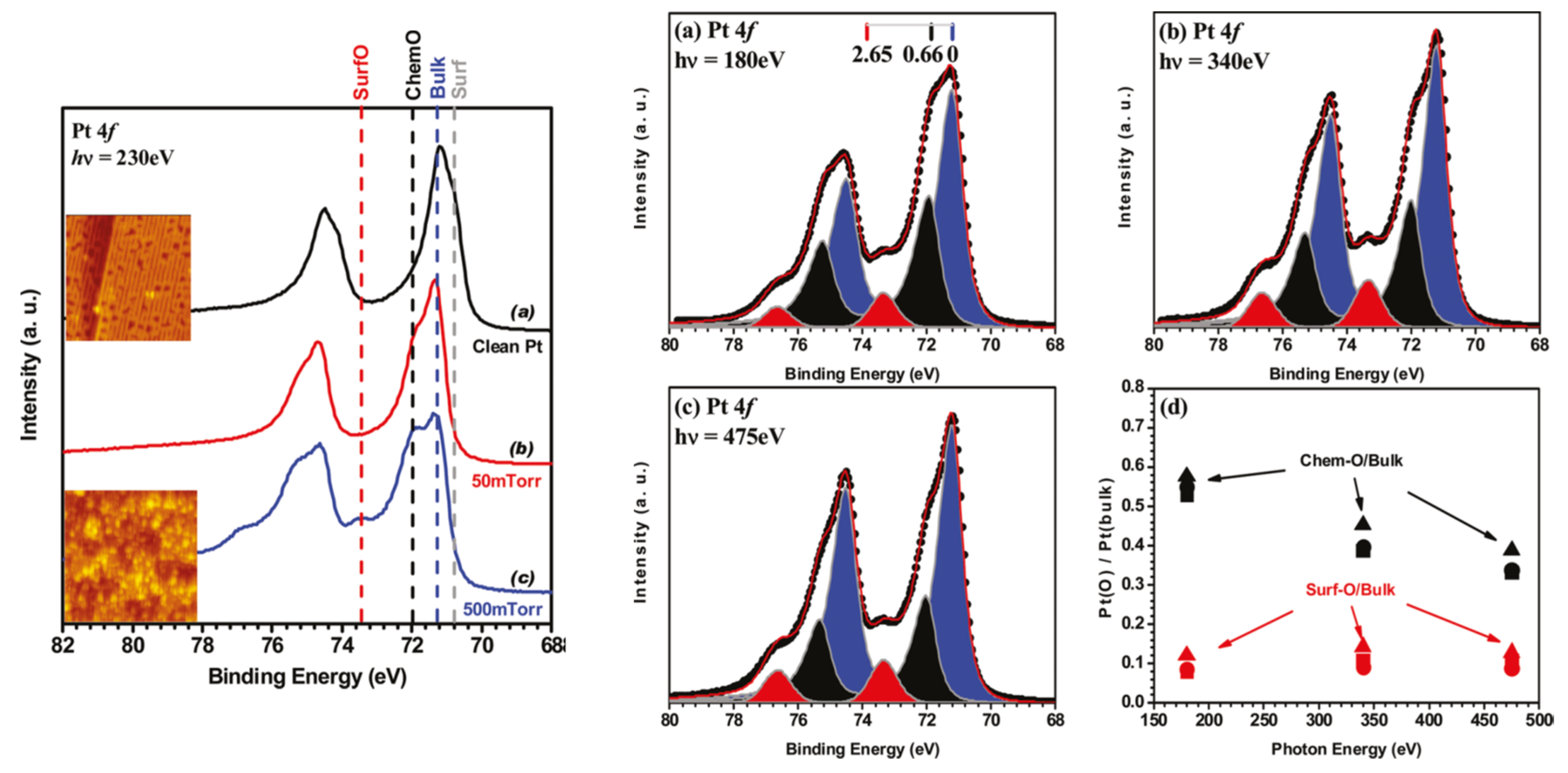
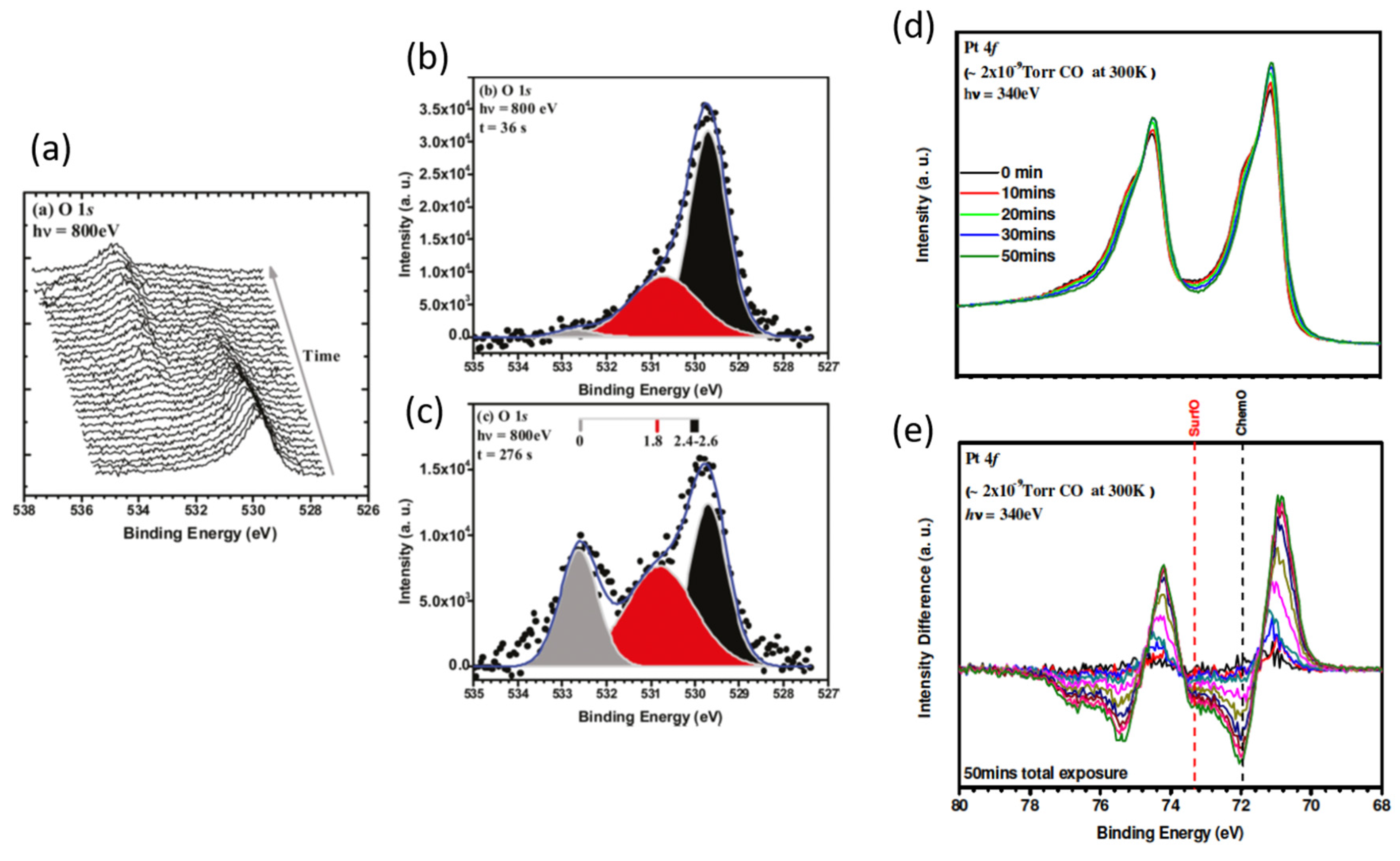
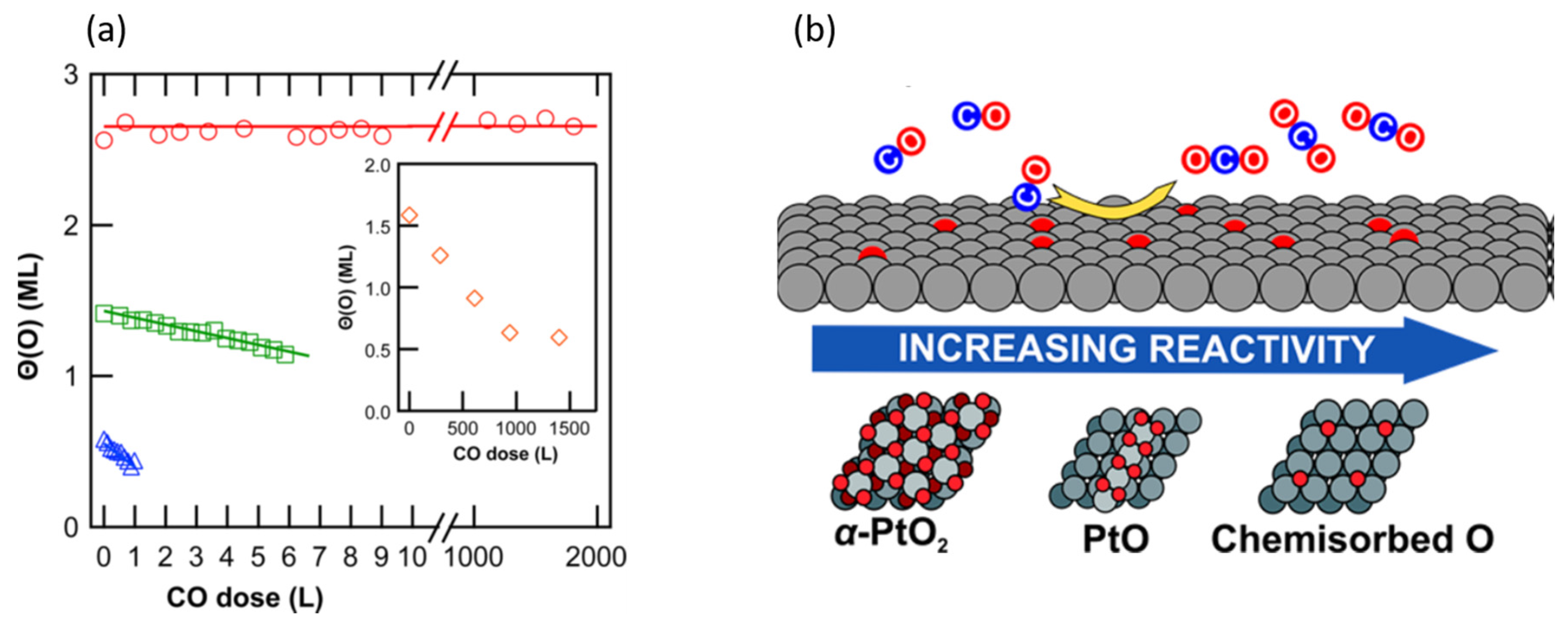
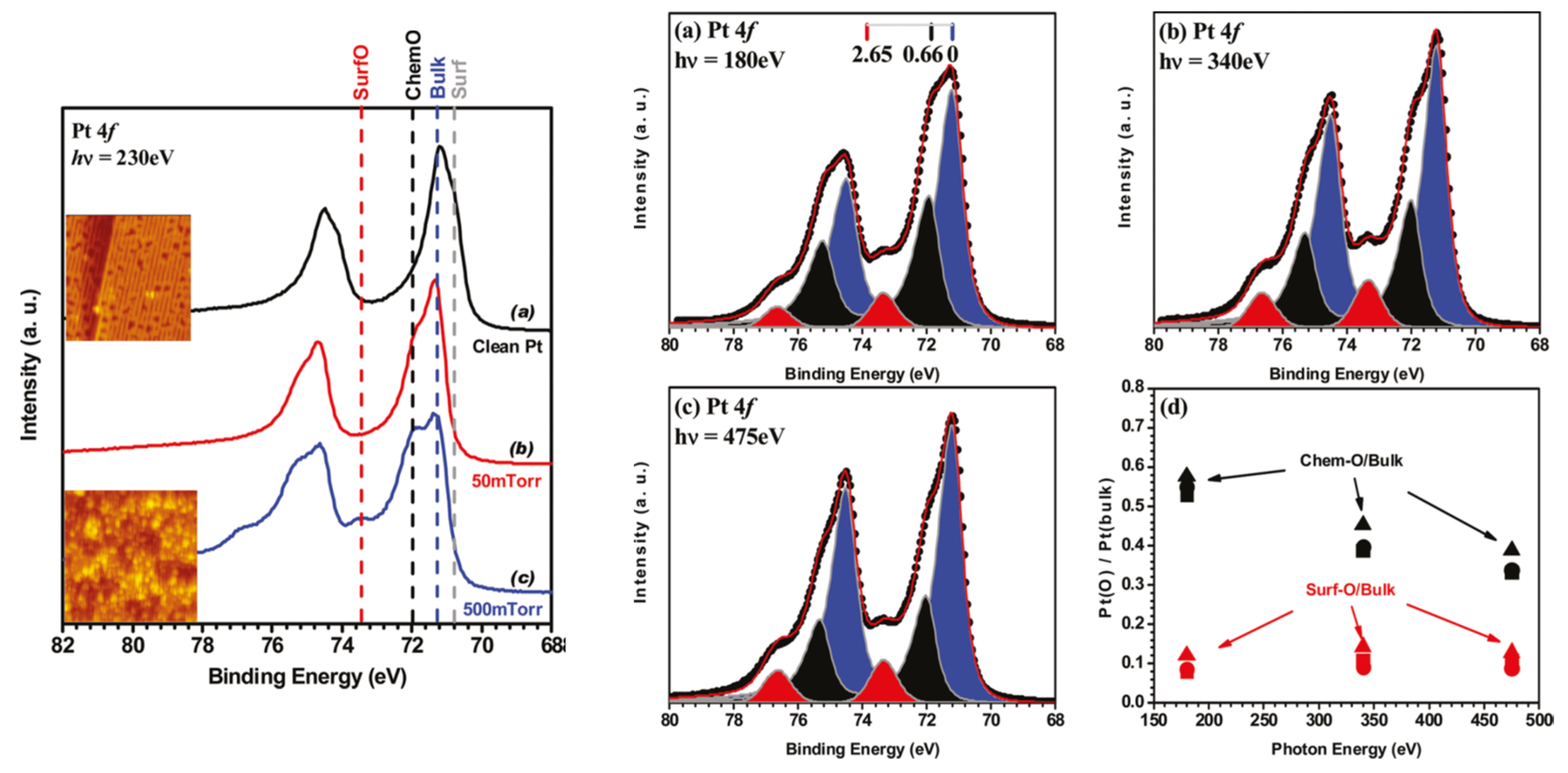
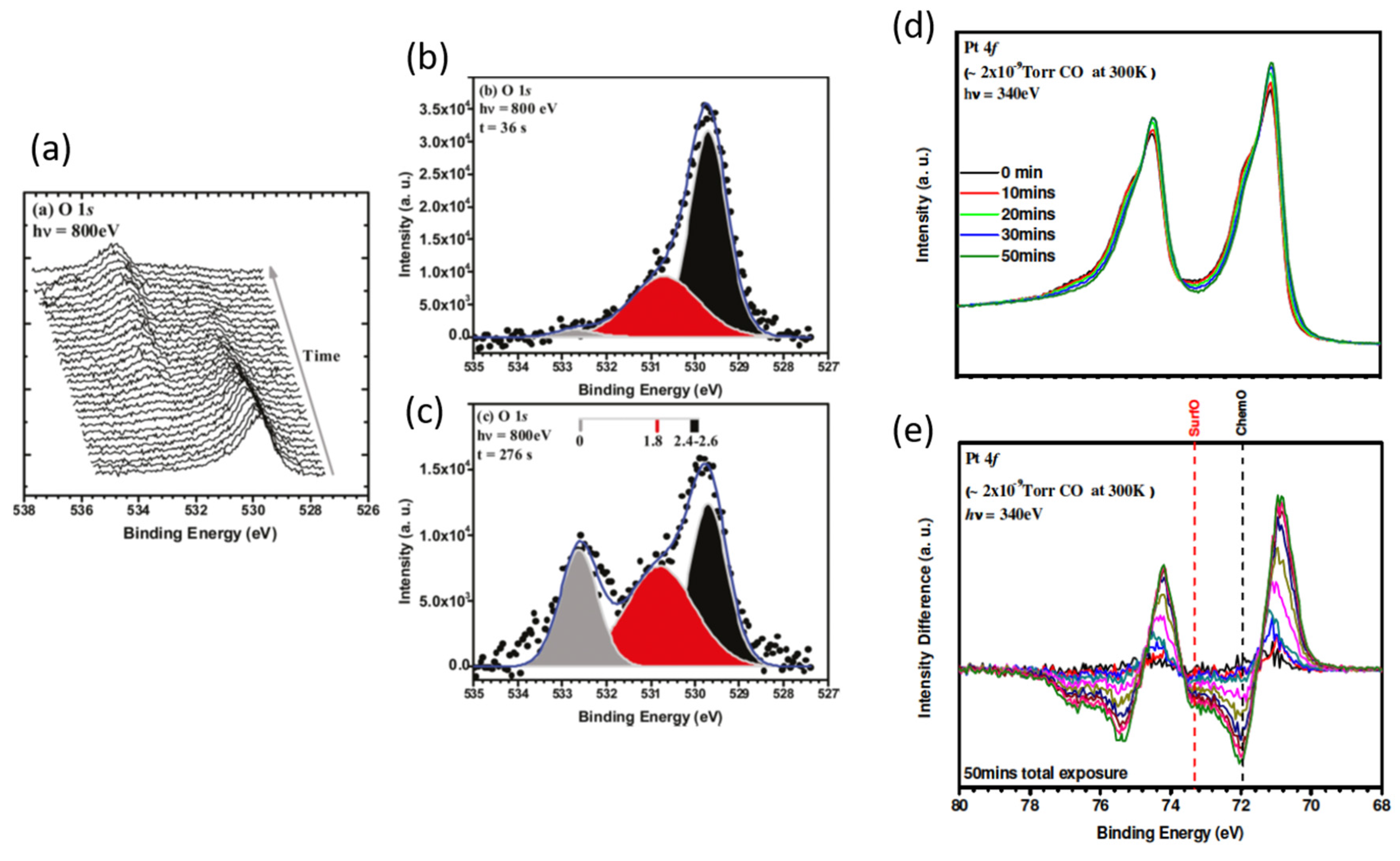
2.4. Ambient Pressure XPS: The Relative Reactivity of Differing Surface Oxides in CO Oxidation over Pt Single Crystal Surfaces
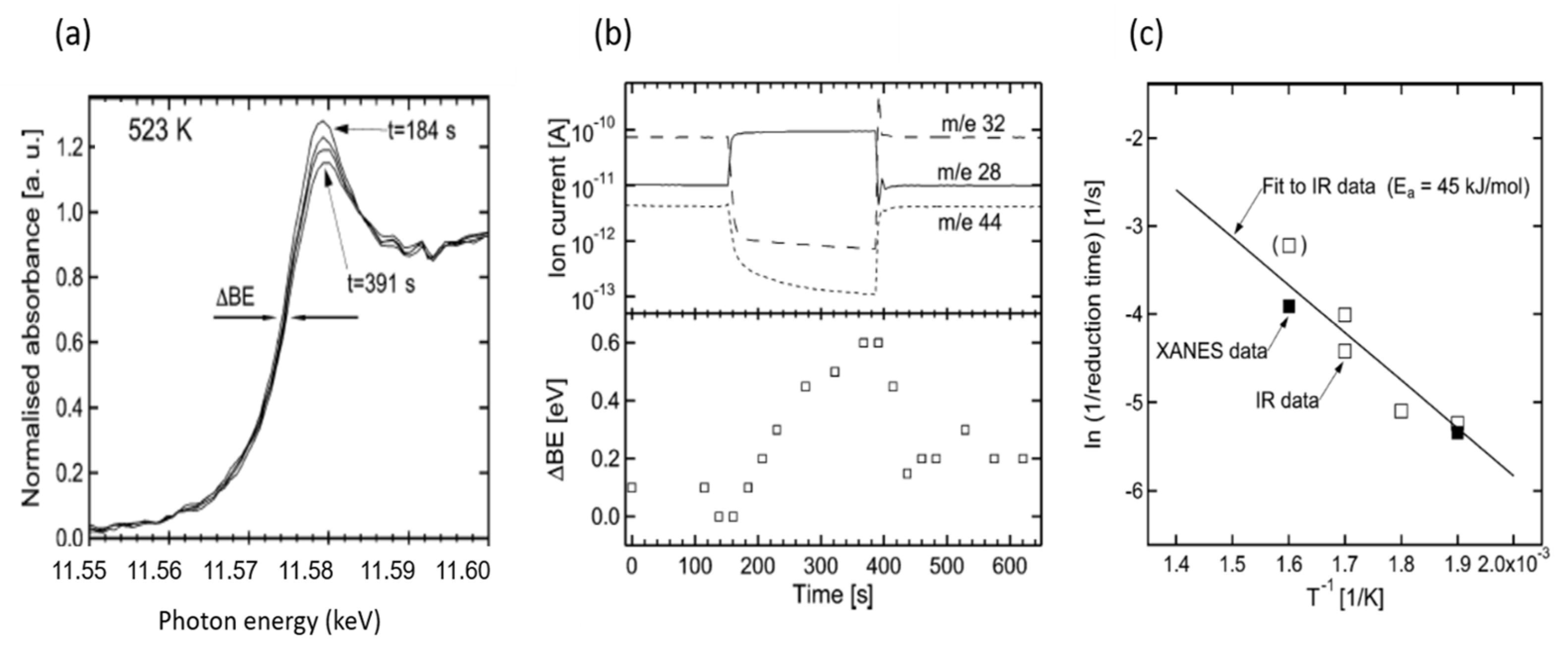
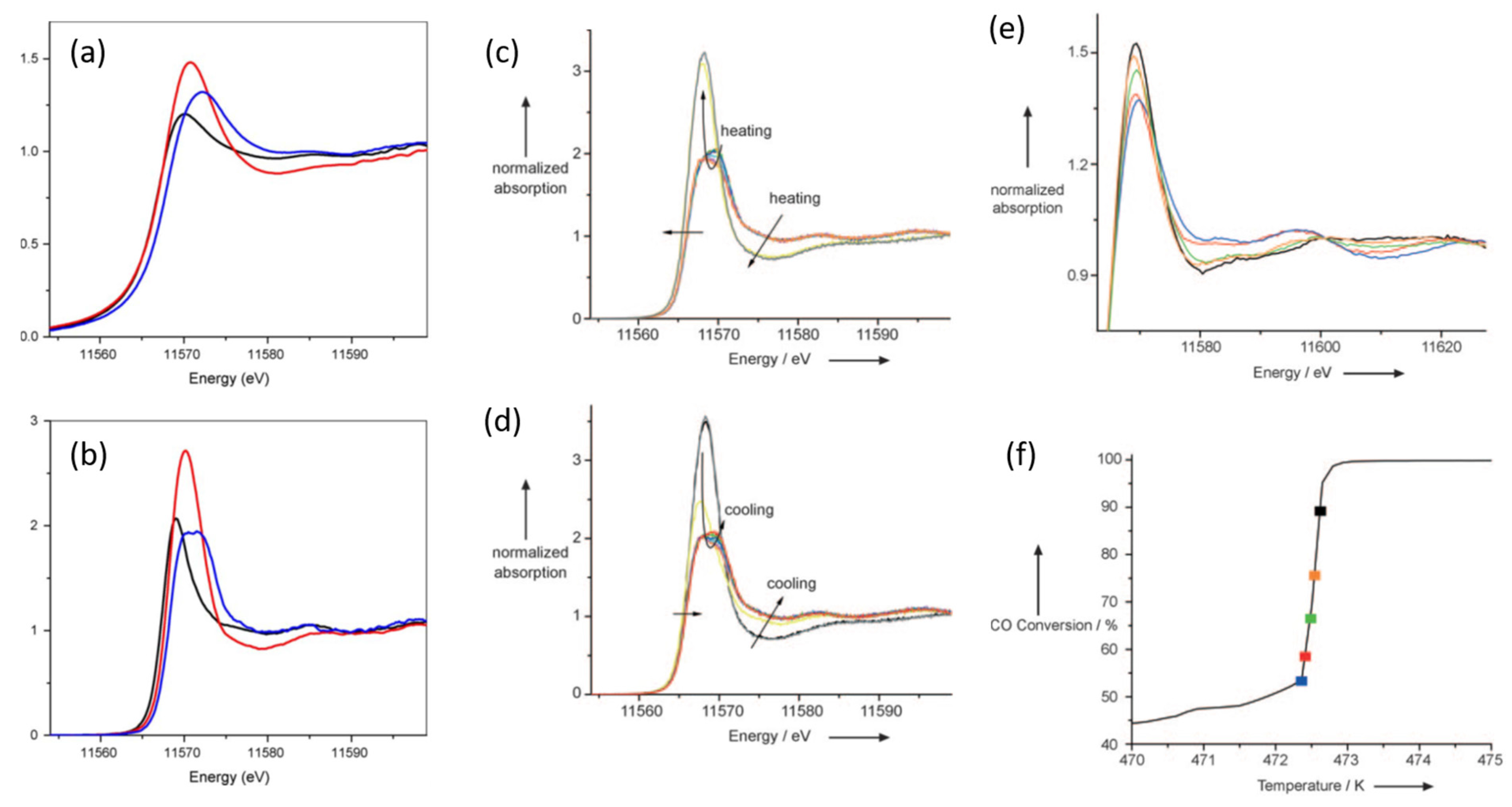
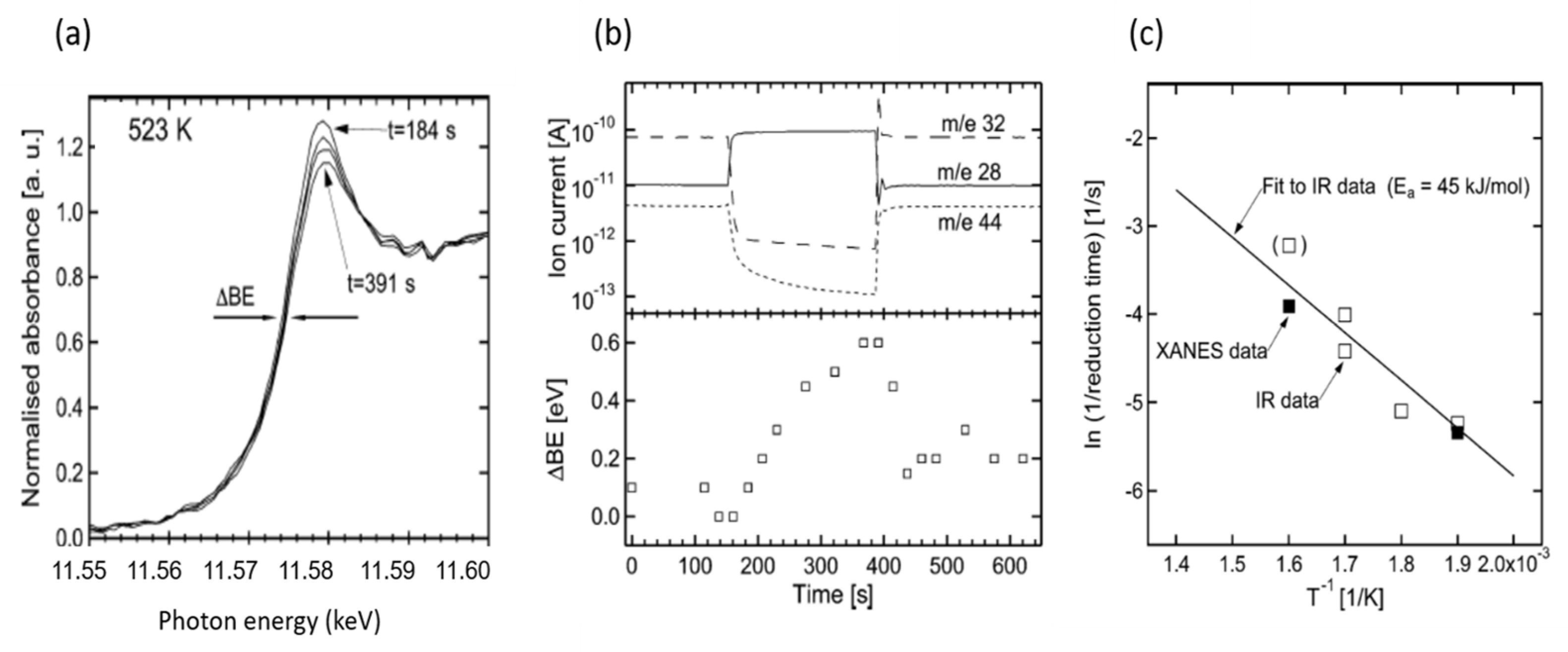
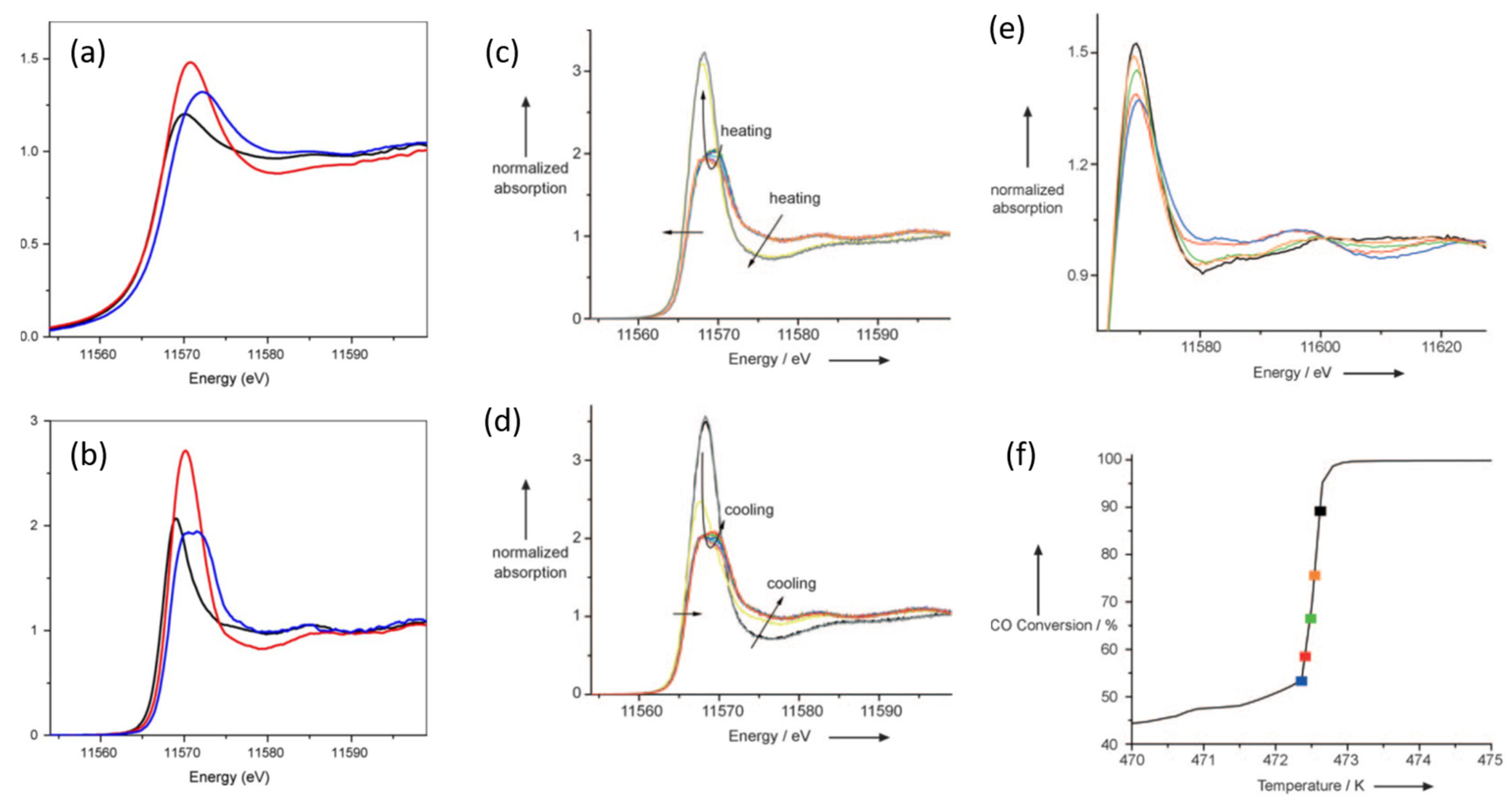
2.5. High Energy Resolution XANES (HERFD) Studies of the Nature of Pt Active in CO Oxidation over Pt/Al2O3 Catalysts
2.6. Oscillations Again: Spatio-Temporal Operando Quick EXAFS Investigations in Plug Flow Microreactors
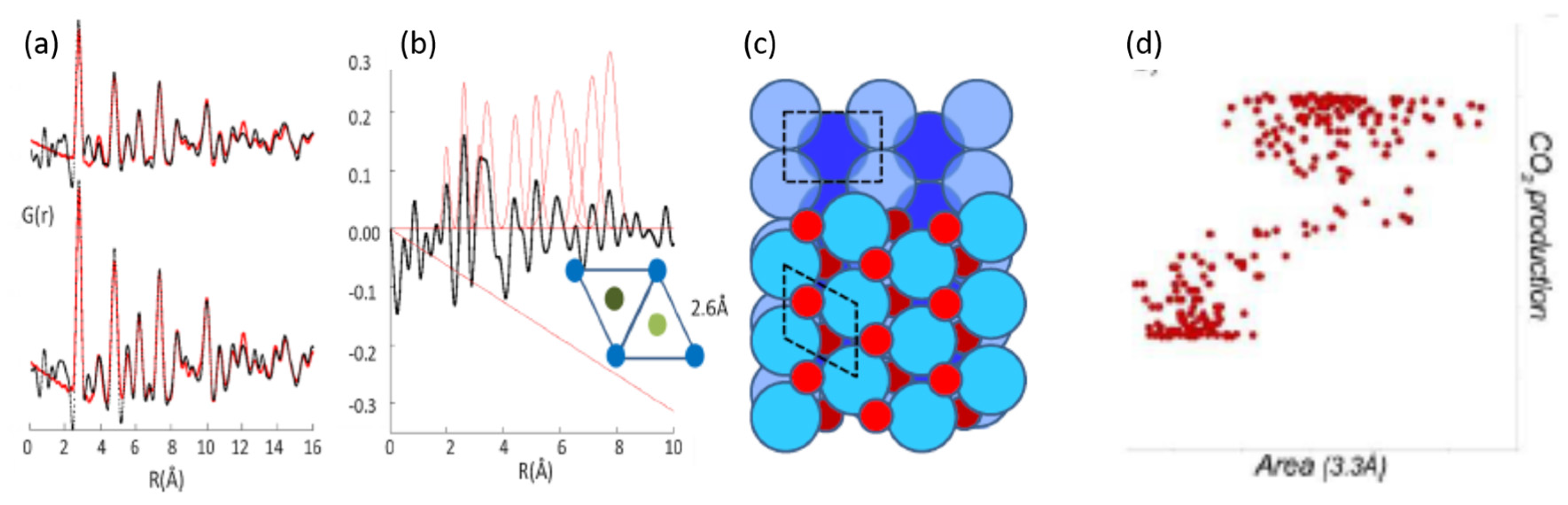
2.7. Time Resolved High Energy X-ray Total Scattering/Pair Distribution Analysis as Applied to CO Oxidation over 1 wt % Pt/Al2O3: Identifying a Reactive Surface Oxide in a Real Catalyst
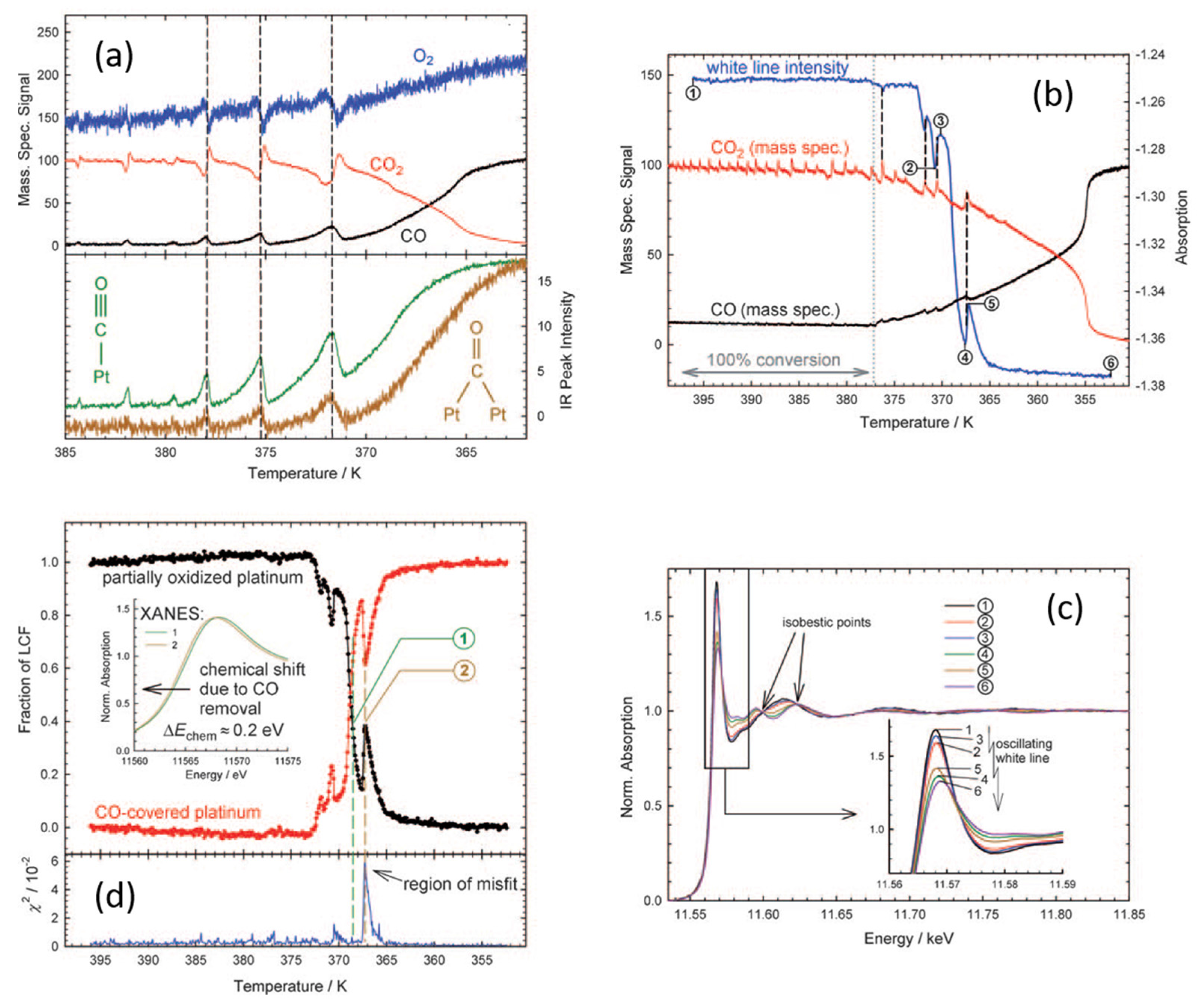
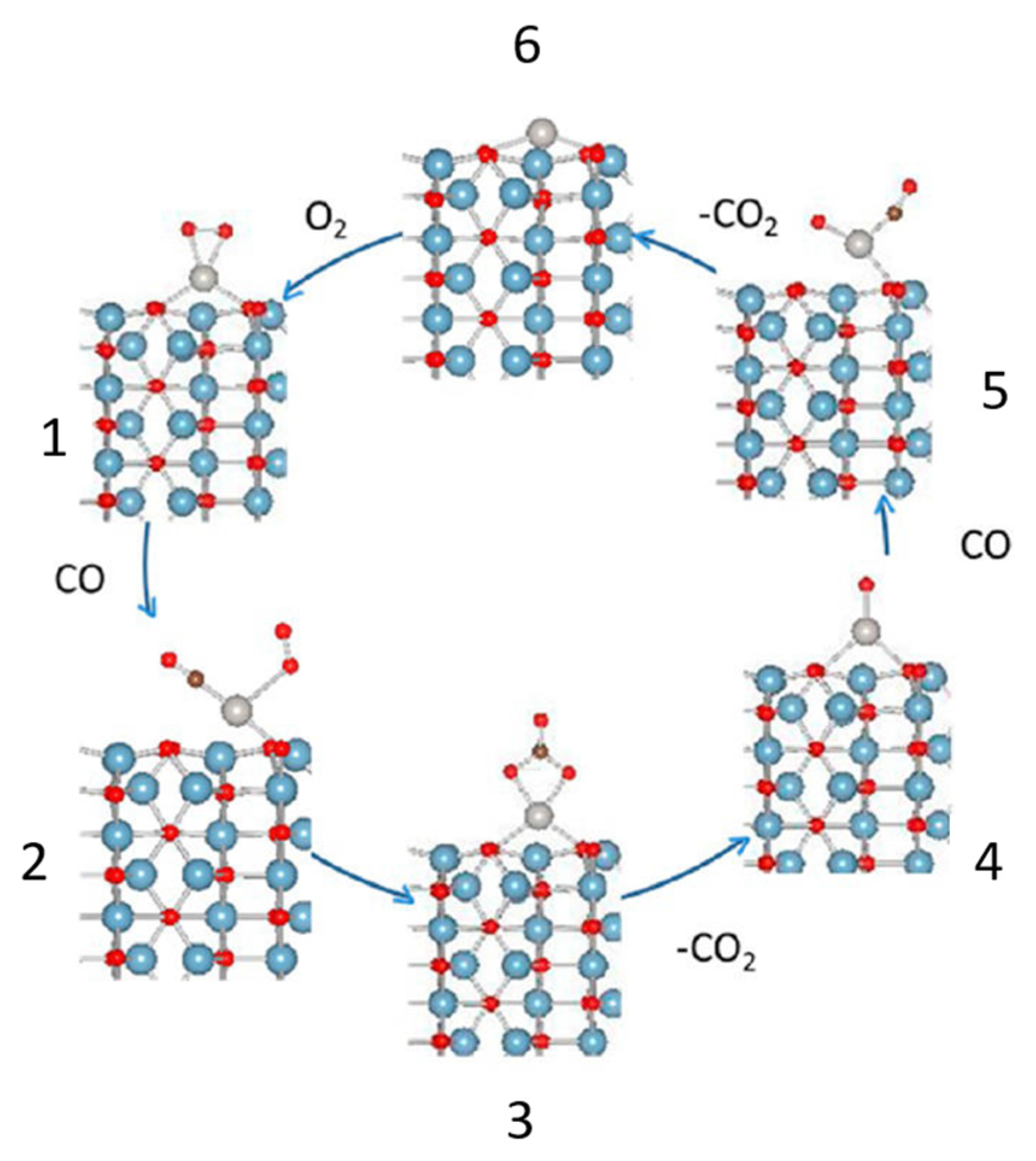
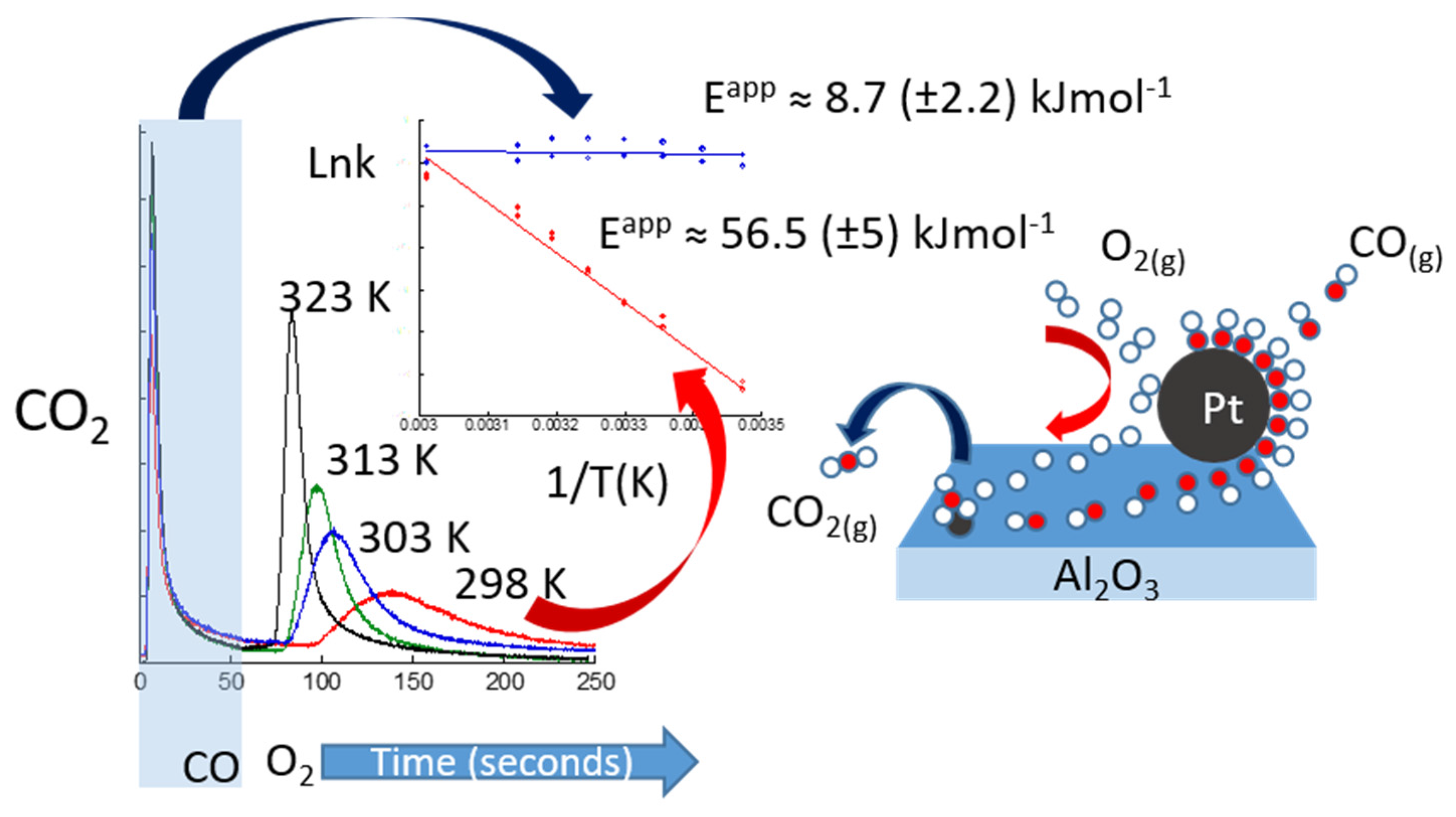
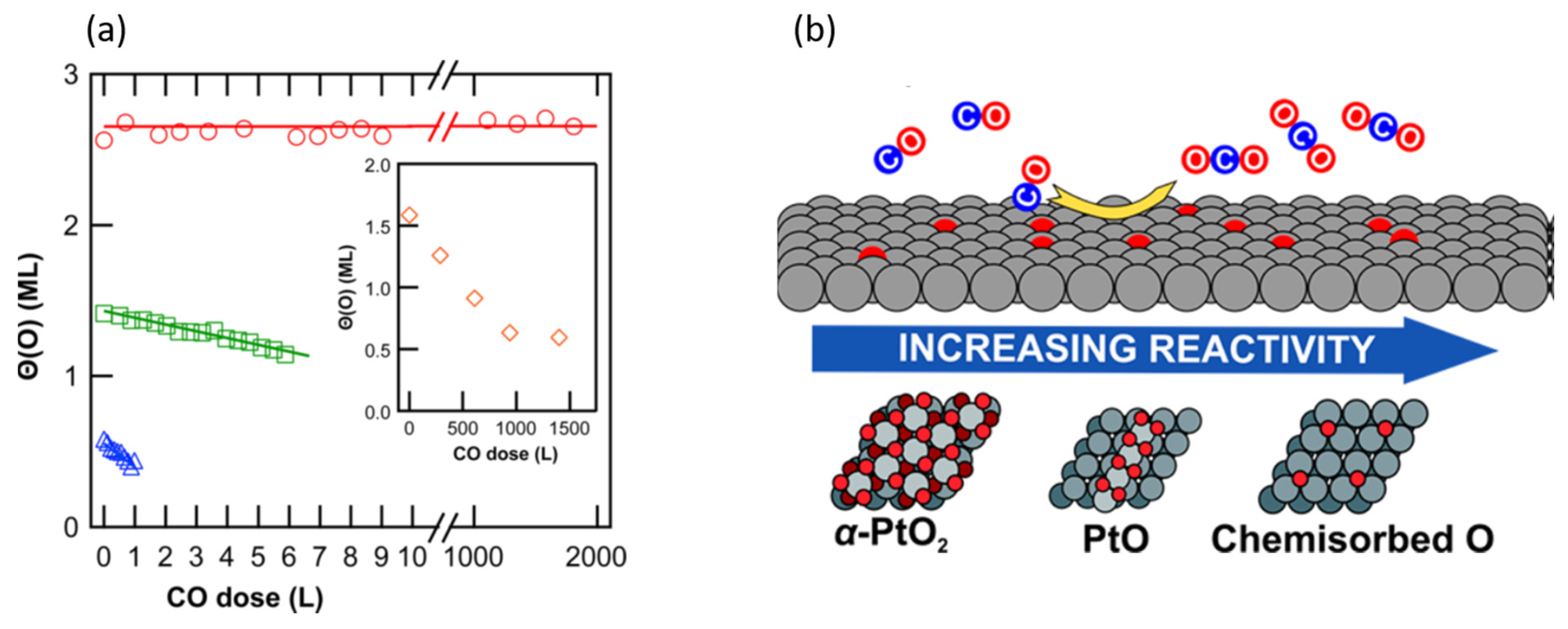
3. A New Mechanistic Possibility for CO Oxidation at Room Temperature Using “Simple” Pt/Al2O3 Catalysts?
4. A Brief Survey of Other Available X-ray Techniques, a Posteriori Methods of Data Treatment, and Caveats Relating X-ray Induced Effects in Operando Catalysis
4.1. XAFS and XES
4.2. Methods Based Upon the Scattering of X-rays: SAXS, GISAXS, and High Energy Scattering Techniques —SXRD and Total X-ray Scattering/PDF
4.2.1. SAXS and GISAXS
4.2.2. Approaches Using High Energy (>50 keV): SXRD and Total X-ray Scattering/PDF
4.3. A Posteriori Approaches to Data Treatment and What They May Have to Offer
4.3.1. Modulation Excitation Spectroscopy (MES).
4.3.2. Modelling of Near Edge Structure (XANES) and Wavelet Analysis in X-ray Spectroscopy
More In-Depth Use of XANES: Analysis and Modelling
Wavelet (WT) Analysis as Applied to EXAFS
4.4. Potential Pitfalls and Caveats: X-ray Induced Effects in Catalytic Study
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Rogers, T.H.; Piggot, C.S.; Balke, W.H.; Jennings, J.M. The catalytic oxidation of carbon monoxide. J. Am. Chem. Soc. 1921, 43, 1743. [Google Scholar] [CrossRef]
- Merrill, D.R.; Scalione, C.C. The Catalytic oxidation of carbon monoxide at ordinary temperatures. J. Am. Chem. Soc. 1921, 43, 1982–2002. [Google Scholar] [CrossRef]
- Lamb, A.B.; Bray, W.C.; Frazer, J.C.W. The removal of carbon dioxide from air. Ind. Eng. Chem. 1920, 12, 213–221. [Google Scholar] [CrossRef]
- Platinum. Available online: http://www.rsc.org/periodic-table/element/78/platinum (accessed on 12 February 2017).
- Platinum Quaterly Q4. Available online: https://www.platinuminvestment.com/files/WPIC_Platinum_Quarterly_Q4_2014.pdf (accessed on 12 February 2017).
- Dallabetta, R.A.; McCune, R.C.; Sprys, J.W. Relative importance of thermal and chemical deactivation of noble-metal automotive oxidation catalysts. Ind. Eng. Chem. Prod. Res. Dev. 1976, 15, 169–172. [Google Scholar] [CrossRef]
- Lamy, C.; Leger, J.M. Electrocatalytic oxidation of small organic materials at platinum single crystal surfaces. J. Chim. Phys. Phys. Chim. Biol. 1991, 88, 1649–1671. [Google Scholar]
- Carrette, L.; Friedrich, K.A.; Stimming, U. Fuel cells: Principles, types, fuels, and applications. Chem. Phys. Chem. 2000, 1, 162–193. [Google Scholar] [PubMed]
- Markovic, N.M.; Ross, P.N. Surface science studies of model fuel cell electrocatalysts. Surf. Sci. Rep. 2002, 45, 121–229. [Google Scholar] [CrossRef]
- Cheng, X.; Shi, Z.; Glass, M.; Zhang, L.; Zhang, J.J.; Song, D.T.; Liu, Z.S.; Wang, H.J.; Shen, J. A review of PEM hydrogen fuel cell contamination: Impacts, mechanisms, and mitigation. J. Power Sources 2007, 165, 739–756. [Google Scholar] [CrossRef]
- Langmuir, I. Chemical reactions at low pressures. J. Am. Chem. Soc. 1915, 37, 1139–1167. [Google Scholar] [CrossRef]
- Langmuir, I. The mechanism of the catalytic action of platinum in the reactions 2CO + O2 = CO2, and 2H2 + O2. Trans. Faraday Soc. 1922, 17, 621–654. [Google Scholar] [CrossRef]
- Eley, D.D.; Rideal, E.K. Parahydrogen conversion on Tungsten. Nature 1940, 146, 401–402. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. Spec. Suppl. 1954, 3, 41–50. [Google Scholar] [CrossRef]
- Laidler, K.J. Chemical Kinetics; Harper & Row: New York, NY, USA, 1987. [Google Scholar]
- Royer, S.; Duprez, D. Catalytic oxidation of carbon monoxide over transition metal oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Haruta, M. Size- and support-dependency in the catalysis of gold. Catal. Today 1997, 36, 153–166. [Google Scholar] [CrossRef]
- Thormählen, P.; Skoglundh, M.; Fridell, E.; Andersson, B. Low-temperature CO oxidation over platinum and cobalt oxide catalysts. J. Catal. 1999, 188, 300–310. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Catalysis by gold. Catal. Rev. Sci. Eng. 1999, 41, 319–388. [Google Scholar] [CrossRef]
- Carretin, S.; Concepcion, P.; Corma, A.; Nieto, J.M.L.; Puntes, V.F. Nanocrystalline CeO2 increases the activity of Au for CO oxidation by two orders of magnitude. Angew. Chem. Int. Ed. 2004, 43, 2538–2540. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Hutchings, G.J. Gold catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.Y. The oxidation of hydrocarbons and CO over metal oxides: III. Co3O4. J. Catal. 1974, 33, 108–122. [Google Scholar] [CrossRef]
- Perti, D.; Kabel, R.L. Kinetics of CO oxidation over Co3O4/γ-Al2O3. Part I: Steady state. AIChE J. 1985, 31, 1420–1426. [Google Scholar] [CrossRef]
- Xie, X.; Liu, Z.-Q.; Haruta, M.; Shen, W. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Vayssilov, G.N.; Lykhach, Y.; Migani, A.; Staudt, T.; Petrova, G.P.; Tsud, N.; Skala, T.; Bruix, A.; Illas, F.; Prince, K.C.; et al. Support nanostructure boosts oxygen transfer to catalytically active platinum nanoparticles. Nat. Mater. 2011, 10, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Wang, A.; Yang, X.F.; Allard, L.F.; Jiang, Z.; Cui, Y.T.; Liu, J.Y.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.X.; Zhao, Y.; Fu, G.; Duschene, P.N.; Gu, L.; Zheng, Y.P.; Weng, X.F.; Chen, M.S.; Zhang, P.; Pao, C.W.; et al. Interfacial effects in iron-nickel hydroxide—Platinum nanoparticles enhance catalytic oxidation. Science 2014, 344, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.J.; De La Riva, A.T.; Lin, S.; Johnson, R.S.; Guo, H.; Miller, H.T.; Kwak, J.H.; Peden, C.H.F.; Kiefer, B.; Allard, L.F.; et al. Low-temperature carbon monoxide oxidation catalysed by regenerable atomically dispersed palladium on alumina. Nat. Commun. 2014, 5, 4885. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Lin, J.; Qiao, B.; Yang, X.; Li, L.; Miao, S.; Liu, J.; Wang, A.; Wang, X.; Zhang, T. Catalytically active Rh sub-nanoclusters on TiO2 for CO oxidation at cryogenic temperatures. Angew. Chem. Int. Ed. 2016, 55, 2820–2824. [Google Scholar] [CrossRef] [PubMed]
- Jakubith, M. Isothermal oscillation in CO-oxidation on platinum mesh. Chem. Ing. Tech. 1970, 42, 943. [Google Scholar]
- Hugo, P.; Jakubith, M. Dynamic behaviour and kinetics of carbon-monoxide on platinum catalysts. Chem. Ing. Tech. 1972, 44, 383. [Google Scholar] [CrossRef]
- Beusch, H.; Wicke, E.; Fieguth, P. Thermally and kinetically produced instabilities in reaction behaviour of individual catalyst grains. Chem. Ing. Tech. 1972, 44, 445. [Google Scholar] [CrossRef]
- Dauchot, J.P.; Vancaken, J. Oscillations during catalytic oxidation of carbon monoxide. Nat. Phys. Sci. 1973, 246, 61–63. [Google Scholar] [CrossRef]
- McCarthy, E.; Zaharadnik, J.; Kuczynski, G.C.; Carberry, J.J. Some unique aspects of CO oxidation on supported Pt. J. Catal. 1975, 39, 29–35. [Google Scholar] [CrossRef]
- Sheintuch, M.; Schmitz, R.A. Oscillations in catalytic reactions. Catal. Rev. Sci. Eng. 1977, 15, 107–172. [Google Scholar] [CrossRef]
- Varghese, P.; Carberry, J.J.; Wolf, E.E. Spurious limit-cycles and related phenomena during CO oxidation on supported platinum. J. Catal. 1978, 55, 76–87. [Google Scholar] [CrossRef]
- Slin’ko, M.G.; Slin’ko, M.M. Self-oscillations of heterogeneous catalytic reaction rates. Catal. Rev. Sci. Eng. 1978, 17, 119–153. [Google Scholar] [CrossRef]
- Plichta, R.T.; Schmitz, R.A. Oscillations in the oxidation of carbon-monoxide on a platinum foil. Chem. Eng. Commun. 1979, 3, 387–398. [Google Scholar] [CrossRef]
- Turner, J.E.; Sales, B.C.; Maple, M.B. Oscillatory oxidation of a Pt catalyst. Surf. Sci. 1981, 103, 54–74. [Google Scholar] [CrossRef]
- Sales, B.C.; Turner, J.E.; Maple, M.B. The oxidation and CO reduction kinetics of a platinum surface. Surf. Sci. 1981, 112, 272–280. [Google Scholar] [CrossRef]
- Sales, B.C.; Turner, J.E.; Maple, M.B. Oscillatory oxidation of CO over Pt, Pd, and Ir catalysts—Theory. Surf. Sci. 1982, 114, 381–394. [Google Scholar] [CrossRef]
- Ertl, G.; Norton, P.R.; Rustig, J. Kinetic oscillations in the platinum-catalyzed oxidation of CO. Phys. Rev. Lett. 1982, 42, 177–180. [Google Scholar] [CrossRef]
- Turner, J.E.; Maple, M.B. Oxide formation and reduction over Pt, Pr, and Ir—A driving mechanism oscillations in the CO oxidation reaction. Surf. Sci. 1984, 147, 647–662. [Google Scholar] [CrossRef]
- Tsai, P.K.; Wu, M.G.; Maple, M.B. Oscillatory oxidation of CO over Pt at pressures from 10 to 760 Torr. J. Catal. 1991, 127, 512–523. [Google Scholar] [CrossRef]
- Lynch, D.T.; Wanke, S.E. Oscillations during CO oxidation over supported metal catalysts: I. Influence of catalyst history on activity. J. Catal. 1984, 88, 333–344. [Google Scholar] [CrossRef]
- Lynch, D.T.; Wanke, S.E. Oscillations during CO oxidation over supported metal catalysts: II. Effects of reactor operating conditions on oscillatory behaviour for a Pt-Pd/Al2O3 catalyst. J. Catal. 1984, 88, 345–354. [Google Scholar] [CrossRef]
- Imbihl, R.; Cox, M.P.; Ertl, G. Kinetic oscillations in the catalytic CO oxidation on Pt(100)—Theory. J. Chem. Phys. 1985, 83, 1578–1587. [Google Scholar] [CrossRef]
- Cox, M.P.; Ertl, G.; Imbihl, R.P. Spatial self-organization of surface-structure during and oscillating catalytic reaction. Phys. Rev. Lett. 1985, 54, 1725–1728. [Google Scholar] [CrossRef] [PubMed]
- Galwey, A.K.; Gray, P.; Griffiths, J.F.; Hasko, S.M. Surface retexturing of Pt wires during the catalytic oxidation of CO. Nature 1985, 313, 668–671. [Google Scholar] [CrossRef]
- Yeates, R.C.; Turner, J.E.; Gellman, A.J.; Somorjai, G.A. The oscillatory behaviour of the CO oxidation reaction at atmospheric pressure over platinum single crystals—Surface analysis and pressure dependent mechanisms. Surf. Sci. 1985, 149, 175–190. [Google Scholar] [CrossRef]
- Lynch, D.T.; Emig, G.; Wanke, S.E. Oscillations during CO oxidation over supported metal catalysts: III. Mathematical modelling of the observed phenomena. J. Catal. 1986, 97, 456–468. [Google Scholar] [CrossRef]
- Imbihl, R.; Cox, M.P.; Ertl, G. Kinetic oscillations in the catalytic CO oxidation on Pt(100)—Experiments. J. Chem. Phys. 1986, 84, 3519–3534. [Google Scholar] [CrossRef]
- Razon, L.F.; Chang, S.-M.; Schmitz, R.A. Chaos during the oxidation of carbon monoxide on platinum—Experiments and analysis. Chem. Eng. Sci. 1986, 41, 1561–1576. [Google Scholar] [CrossRef]
- Razon, L.F.; Schmitz, R.A. Intrinsically instable behaviour during the oxidation of carbon monoxide on platinum. Catal. Rev. Sci. Eng. 1986, 28, 89–164. [Google Scholar] [CrossRef]
- Razon, L.F.; Schmitz, R.A. Multiplicities and instabilities in chemically reacting systems. Catal. Rev. Sci. Eng. 1987, 42, 1005–1047. [Google Scholar] [CrossRef]
- Burrows, V.A.; Sundaresan, S.; Chabal, Y.J.; Christman, S.B. Studies of self-sustained reaction rate oscillations: I. Real-time infrared measurements during oscillatory oxidation of carbon-monoxide on platinum. Surf. Sci. 1985, 160, 122–138. [Google Scholar] [CrossRef]
- Burrows, V.A.; Sundaresan, S.; Chabal, Y.J.; Christmann, S.B. Studies of self-sustained reaction rate oscillations: II. The role of carbon and oxides in the oscillatory oxidation of carbon-monoxide on platinum. Surf. Sci. 1987, 180, 110–135. [Google Scholar] [CrossRef]
- Collins, N.A.; Sundaresan, S.; Chabal, Y.J. Studies of self-sustained reaction rate oscillations: III. The carbon model. Surf. Sci. 1987, 180, 136–152. [Google Scholar] [CrossRef]
- Schuth, F.; Wicke, E. IR spectroscopic investigations during oscillations of the CO/NO and CO/O2 reaction on Pt and Pt catalysts: I. Platinum. Ber. Bunsenges. Phys. Chem. 1989, 93, 191–201. [Google Scholar] [CrossRef]
- Eiswirth, M.; Moller, P.; Wetzl, K.; Imbihl, R.; Ertl, G. Mechanisms of spatial self-organisation in isothermal kinetic oscillations during the catalytic CO oxidation on Pt single crystal surfaces. J. Chem. Phys. 1989, 90, 510–521. [Google Scholar] [CrossRef]
- Ertl, G. Oscillatory kinetics and spatio-temporal self-organisation in reactions at solid surfaces. Science 1991, 254, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Engel, W.; Kordesch, M.E.; Rotermund, H.H.; Kubala, S.; von Oertzen, A. A UHV compatible photoelectron emission microscope for applications in surface science. Ultramicroscopy 1991, 36, 148–153. [Google Scholar] [CrossRef]
- Rotermund, H.H.; Engel, W.; Kordesch, M.; Ertl, G. Imaging of spatiotemporal pattern evolution during carbon-monoxide oxidation on platinum. Nature 1990, 343, 355–357. [Google Scholar] [CrossRef]
- Krischer, K.; Eiswirth, M.; Ertl, G. Oscillatory CO oxidation on Pt(110)—Modelling of temporal self-organisation. J. Chem. Phys. 1992, 96, 9161–9172. [Google Scholar] [CrossRef]
- Lauterbach, J.; Haas, G.; Rotermund, H.H.; Ertl, G. Spatio temporal pattern formation on polycrystalline platinum surfaces during catalytic CO oxidation. Surf. Sci. 1993, 294, 116–130. [Google Scholar] [CrossRef]
- Gorodetskll, V.; Lauterbach, J.; Rotermund, H.H.; Block, J.H.; Ertl, G. Coupling between adjacent crystal places in heterogeneous catalysis by propagating reaction-diffusion waves. Nature 1994, 370, 276–279. [Google Scholar] [CrossRef]
- Eiswirth, M.; Burger, J.; Strasser, P.; Ertl, G. Oscillating Langmuir-Hinshelwood mechanisms. J. Phys. Chem. 1996, 100, 19118–19123. [Google Scholar] [CrossRef]
- Winterlin, J.; Volkening, S.; Janssens, T.V.W.; Zambelli, T.; Ertl, G. Atomic and macroscopic reaction rates of a surface catalysed reaction. Science 1997, 278, 1931–1934. [Google Scholar] [CrossRef]
- Che, M.; Bennett, C.O. The influence of particle size on the catalytic properties of supported metals. Adv. Catal. 1989, 36, 55–172. [Google Scholar]
- Cuenya, B.R. Synthesis and catalytic properties of metal nanoparticles: Size, shape, support composition, and oxidation effects. Thin Solid Films 2010, 518, 3127–3150. [Google Scholar] [CrossRef]
- Guerrero-Perez, M.O.; Banares, M.A. Operando Raman study of alumina supported Sb-V-O catalyst during propene ammoxidation to acrylonitrile with on-line activity measurement. Chem. Commun. 2002, 1292–1293. [Google Scholar] [CrossRef]
- Banares, M.A.; Guerrero-Perez, M.O.; Fierro, J.L.G.; Cortez, G.G. Raman spectroscopy during catalytic operations with on-line activity measurement (operando spectroscopy): A method for understanding the active centres of cations supported on porous materials. J. Mater. Chem. 2002, 12, 3337–3342. [Google Scholar] [CrossRef]
- Newton, M.A.; Jyoti, B.; Dent, A.J.; Fiddy, S.G.; Evans, J. Synchronous, time resolved, diffuse reflectance FT-IR, energy dispersive EXAFS (EDE) and mass spectrometric investigation of Rh catalysts during NO reduction by CO. Chem. Commun. 2004, 2382–2384. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, N.S.; Wang, Q.; Frenkel, A.I. In situ diffuse reflectance IR spectroscopy and X-ray absorption spectroscopy for fast catalytic processes. J. Synchrotron Radiat. 2011, 18, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.A.; Di MIchiel, M.; Kubacka, A.; Fernández-García, M. Combining time resolved hard X-ray diffraction (XRD) and diffuse reflectance infrared spectroscopy (DRIFTS) to illuminate CO dissociation and transient carbon storage by supported Pd nanoparticles during CO/NO cycling. J. Am. Chem. Soc. 2010, 132, 4540–4541. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.A.; Di Michiel, M.; Kubacka, A.; Iglesias-Juez, A.; Fernández-García, M. Seeing oxygen storage and release in action and elucidating synergies between noble metal nanoparticles and promoter oxides. Angew. Chem. Int. Ed. 2012, 51, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Chiarello, G.L.; Nachtegaal, M.; Marchionni, V.; Quaroni, L.; Ferri, D. Adding diffuse reflectance infrared Fourier transform spectroscopy capability to extended X-ray absorption fine structure in a new cell to study solid catalysts in combination with a modulation approach. Rev. Sci. Instrum. 2014, 85, 074102. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.A.; Zhao, H.; Borkiewicz, O.J.; Newton, M.A.; Chupas, P.J.; Chapman, K.W. Simultaneous diffuse reflection infrared spectroscopy and X-ray pair distribution function measurements. J. Appl. Cryst. 2014, 47, 95–101. [Google Scholar] [CrossRef]
- Brieger, C.; Melke, J.; van der Bosch, N.; Reinholz, U.; Riesemeier, H.; Buzanich, A.G.; Kayarkatte, M.K.; Derr, I.; Schökel, A.; Roth, C. A combined in-situ XAS-DRIFTS study unraveling adsorbate induced changes on the Pt nanoparticle structure. J. Catal. 2016, 339, 57–67. [Google Scholar] [CrossRef]
- Bando, K.K.; Wada, T.; Miyamoto, T.; Miyazaki, K.; Takakusagi, S.; Gott, T.; Yamaguchi, A.; Nomura, M.; Oyama, S.T.; Asakura, K. In situ FTIR and XANES studies of thiophene hydrodesulpurisation on Ni2P/MCM-41. J. Phys. C 2009, 190, 012158. [Google Scholar]
- Bando, K.K.; Wada, T.; Miymoto, T.; Miyazaki, K.; Takakusgi, S.; Koike, Y.; Inada, Y.; Nomura, M.; Yamaguchi, A.; Oyama, S.T.; et al. Combined in situ QXAFS and FTIR analysis of a Ni phosphide catalyst under hydrodesulphurisation conditions. J. Catal. 2012, 286, 165–171. [Google Scholar] [CrossRef]
- Beale, A.M.; van der Eerden, A.M.J.; Kervinen, K.; Newton, M.A.; Weckhuysen, B.M. Adding a third dimension to operando spectroscopy: A combined UV-Vis, Raman, and XAFS set up to study heterogeneous catalysts under working conditions. Chem. Commun. 2005, 3015–3017. [Google Scholar] [CrossRef] [PubMed]
- Briois, V.; Belin, S.; Villain, F.; Bouamrane, F.; Lucas, H.; Lescouezec, R.; Julve, M.; Verdaguer, M.; Tokumoto, M.S.; Santilli, C.V.; et al. New insights for materials science characterisation using different complentary techniques combined with X-ray absorption spectroscopy. Phys. Scr. 2005, T115, 38–44. [Google Scholar] [CrossRef]
- Briois, V.; Lutzenkirchen-Hecht, D.; Villain, F.; Fonda, E.; Belin, S.; Griesebock, B.; Frahm, R. Time-resolved study of the oxidation of ethanol by cerium(IV) using combined Quick-EXAFS, UV-Vis, and Raman spectroscopies. J. Phys. Chem. A 2005, 109, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, D.; Beale, A.M.; Petukhov, A.V.; Weckhuysen, B.M. Unraveling the crystallisation mechanism of CoAPO-5 molecular seives under hydrothermal conditions. J. Am. Chem. Soc. 2005, 127, 14454–14465. [Google Scholar] [CrossRef] [PubMed]
- De Smit, E.; Cinquini, F.; Beale, A.M.; Safonova, O.V.; van Beek, W.; Sautet, P.; Weckhuysen, B.M. Stability and reactibity of ε-χ-θ iron carbide catalyst phases in Fischer-Tropsch synthesis: Controlling μc. J. Am. Chem. Soc. 2010, 132, 14928–14941. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Juez, A.; Beale, A.M.; Maajien, L.; Weng, T.C.; Glatzel, P.; Weckhuysen, B.M. A combined in situ time resolved UV-Vis, Raman, and high energy resolution X-ray absorption spectroscopy study on the deactivation behaviour of Pt and Pt-Sn propane dehydrogenation catalyts under industrial reaction conditions. J. Catal. 2010, 276, 268–279. [Google Scholar] [CrossRef]
- Newton, M.A.; van Beek, W. Combining vibrational spectroscopies with synchrotron based X-ray techniques for the in situ study of heterogeneous catalysts: A view from a bridge. Chem. Soc. Rev. 2010, 39, 4845–4863. [Google Scholar] [CrossRef] [PubMed]
- Bentrup, U. Combining in situ characterisation methods in on set-up: Looking with ore eyes into the intricate chemistry of the synthesis and working of heterogeneous catalysts. Chem. Soc. Rev. 2010, 39, 4718–4730. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.A.; Fernandez-Garcia, M. Combining infrared spectroscopy with X-ray techniques for the investigation of working heterogeneous catalysts. In In-Situ Characterization of Heterogeneous Catalysts; Chupas, P.J., Hanson, J., Rodriguez, J.A., Eds.; Wiley: Hoboken, NJ, USA, 2013; Chapter 14; pp. 369–409. [Google Scholar]
- Van Beek, W.; Ruiz-Martinez, J.; Milanesio, M. XRD-Raman and modulation excitation spectroscopy. In In-Situ Characterization of Heterogeneous Catalysts; Chupas, P.J., Hanson, J., Rodriguez, J.A., Eds.; Wiley: Hoboken, NJ, USA, 2013; Chapter 15; pp. 411–440. [Google Scholar]
- Hartmann, N.; Imbihl, R.; Vogel, W. Experimental evidence for an oxidation/reduction mechanism in rate oscillations of catalytic CO oxidation on Platinum. Catal. Lett. 1994, 28, 373–381. [Google Scholar] [CrossRef]
- Gnutzmann, V.; Vogel, W. Structural sensitivity of the standard Pt/SiO2 catalyst EuroPt-1 to hydrogen and oxygen exposure by in situ X-ray diffraction. J. Phys. Chem. 1990, 94, 4991–4997. [Google Scholar] [CrossRef]
- Carlsson, P.-A.; Österlund, L.; Thormählen, P.; Palmqvist, A.; Fridell, E.; Jansson, J.; Skoglundh, M. A transient FTIR and XANES study of CO oxidation over Pt/Al2O3 catalysts. J. Catal. 2004, 226, 422–434. [Google Scholar] [CrossRef]
- Phizackerley, R.P.; Rek, Z.U.; Stephenson, G.B.; Conradson, S.D.; Hodgson, K.O.; Matsushita, T.; Oyanagi, H. An energy dispersive spectrometer for the rapid measurement of X-ray absorption spectra using synchrotron radiation. J. Appl. Cryst. 1983, 16, 220–232. [Google Scholar] [CrossRef]
- Frahm, R. QEXAFS—X-ray absorption studies in seconds. Physica B 1989, 158, 342–343. [Google Scholar] [CrossRef]
- Couves, J.W.; Thomas, J.M.; Waller, D.; Jones, R.H.; Dent, A.J.; Derbyshire, G.E.; Greaves, G.N. Tracing the conversion of aurichalcite to a copper catalyst by combined X-ray absorption and diffraction. Nature 1991, 354, 465–468. [Google Scholar] [CrossRef]
- Coulston, G.W.; Bare, S.R.; Kung, H.; Birkeland, K.; Bethke, G.K.; Harlow, R.; Herron, N.; Lee, P. The kinetic significance of V5+ in n-butane oxidation catalysed by vanadium phosphates. Science 1997, 275, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Ressler, T.; Hagelstein, M.; Hatje, U.; Metz, W. In situ X-ray absorption spectroscopy studies on chemical oscillations in the CO/O2 system on supported Pd catalysts. J. Phys. Chem. B 1997, 101, 6680–6687. [Google Scholar] [CrossRef]
- Fiddy, S.G.; Newton, M.A.; Dent, A.J.; Salvini, G.; Corker, J.M.; Turin, S.; Campbell, T.; Evans, J. In situ energy dispersive EXAFS (EDE) of low loaded Pt(acac)2/HI SiO2 catalyst precursors on a timescale of seconds and below. Chem. Commun. 1999, 851–852. [Google Scholar] [CrossRef]
- Newton, M.A.; Dent, A.J.; Evans, J. Bringing time resolution to EXAFS: Recent developments and application to chemical systems. Chem. Soc. Rev. 2002, 31, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Clausen, B.S.; Grabaek, L.; Steffensen, G.; Hansen, P.L.; Topsoe, H. A combined QEXAFS XRD method for online in situ studies of catalysts—Examples of dynamics measurements of Cu based methanol catalysts. Catal. Lett. 1993, 20, 23–26. [Google Scholar] [CrossRef]
- Als-Nielsen, J.; Grubel, G.; Clausen, B.S. QEXAFS in seconds at an undulator source. Nucl. Instrum. Methods B 1995, 97, 522–525. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Molenbroek, A.M.; Topsoe, N.Y.; Topsoe, H.; Clausen, B.S. In situ investigations of structural changes in Cu/ZnO catalysts. J. Catal. 2000, 194, 452–460. [Google Scholar] [CrossRef]
- Graham, W.R.C.; Lynch, D.T. CO oxidation on Pt—Variable phasing of inputs during forced composition cycling. AICHe 1990, 36, 1796–1806. [Google Scholar] [CrossRef]
- Carlsson, P.-A.; Skoglundh, M.; Fridell, E.; Jobson, E.; Andersson, B. Induced low temperature catalytic ignition by transient changes in the gas composition. Catal. Today 2001, 73, 307–313. [Google Scholar] [CrossRef]
- Carlsson, P.-A.; Thormahlen, P.; Skoglundh, M.; Persson, H.; Fridell, E.; Jobson, E.; Andersson, B. Periodic control for improved low temperature catalytic activity. Top. Catal. 2001, 16, 343–347. [Google Scholar] [CrossRef]
- Bazin, D.; Sayers, S.; Rehr, J.J.; Mottet, C. Numerical simulation of the platinum LIII edge white line relative to nanometer scale clusters. J. Phys. Chem. B 1997, 101, 5332–5336. [Google Scholar] [CrossRef]
- Nagai, Y.; Hirabayashi, T.; Dohmae, K.; Takagi, N.; Minami, H.; Shinjoh, S.; Matsumoto, S. Sintering inhibition mechanism of platinum supported on ceria-based and Pt-oxide-support interaction. J. Catal. 2006, 242, 103–109. [Google Scholar] [CrossRef]
- Nagai, Y.; Dohmae, K.; Ikeda, Y.; Takagi, N.; Tanabe, T.; Hara, N.; Guilera, G.; Pascarelli, S.; Newton, M.A.; Kuno, O.; et al. In situ redsiperson Pt autoexhaust catalsts; an online approach to increasing catalyst lifetimes. Angew. Chem. Int. Ed. 2008, 48, 9303–9306. [Google Scholar] [CrossRef] [PubMed]
- Ankudinov, A.L.; Rehr, J.J.; Low, J.J.; Bare, S.R. Sensitivity of Pt X-ray absorption near edge structure to the morphology of small Pt clusters. J. Chem. Phys. 2002, 116, 1911–1919. [Google Scholar] [CrossRef]
- Newton, M.A. Beamsize related phenomena and effective normalisation in energy dispersive EXAFS for the study of heterogeneous catalysts, powder materials, and the processes they mediate: Observations, and (some) solutions. J. Synchrotron Radiat. 2007, 14, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.A.; Dent, A.J. Energy dispersive EXAFS: Principles, application, and possibilities for the understanding the dynamic behaviour of heterogeneous catalysts. In In-Situ Characterization of Heterogeneous Catalysts; Chupas, P.J., Hanson, J., Rodriguez, J.A., Eds.; Wiley: Hoboken, NJ, USA, 2013; Chapter 3; pp. 75–118. [Google Scholar]
- Mueller, O.; Nachtegaal, M.; Just, J.; Luetzenkirchen-Hecht, D.; Frahm, R. Quick-EXAFS setup at the SuperXAS beamline for in situ X-ray absorption spectroscopy with 10 ms time resolution. J. Synchrotron Radiat. 2016, 23, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Mueller, O.; Luetzenkirchen-Hecht, D.; Frahm, R. Quick scanning monochromator for millisecond in situ and in operando X-ray absorption spectroscopy. Rev. Sci. Instrum. 2015, 86, 093905. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.D.; Pedersen, T.M.; Hendriksen, B.L.M.; Robach, O.; Bobaru, S.C.; Popa, I.; Quiros, C.; Kim, H.; Hammer, B.; Ferrer, S.; et al. Structure and reactivity of surface oxides on Pt(110) during catalytic CO oxidation. Phys. Rev. Lett. 2005, 95, 255505. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.; Peters, K.; Alvarez, J.; Ferrer, S. Ultrahigh vacuum high pressure chamber for surface X-ray diffraction experiments. Rev. Sci. Instrum. 1999, 70, 1478–1480. [Google Scholar] [CrossRef]
- Balmes, O.; van Rijn, R.; Wermeille, D.; Resta, A.; Petit, L.; Isern, H.; Dufrane, T.; Felici, R. The ID 03 surface diffraction beamline for in situ investigations of catalytic reactions at surfaces. Catal. Today 2009, 145, 220–226. [Google Scholar] [CrossRef]
- High Pressure SXRD Reactor. Available online: http://lpmwww.physics.leidenuniv.nl/uploads/datasheets/sxrd_flyer_web.pdf (accessed on 12 February 2017).
- Blomberg, S.; Zhou, J.; Gustafson, J.; Zetterberg, J.; Lundgren, E. 2D and 3D imaging of the gas phase close to an operating model catalyst by planar laser induced fluorescence. J. Phys. Condens. Matter 2016, 28, 453002. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Österlund, L.; Vestergaard, E.K.; Vang, R.T.; Matthiesen, J.; Pedersen, T.M.; Laegsgaard, E.; Hammer, B.; Besenbacher, F. Oxidation of Pt(110). Phys. Rev. Lett. 2004, 93, 146104. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, B.L.M.; Frenken, J.M.W. CO oxidation on Pt(110) scanning tunnelling microscopy inside a high pressure flow reactor. Phys. Rev. Lett. 2002, 89, 046101. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hammer, B. Reactivity of a gas/metal/metal-oxide three phase boundary: CO oxidation at the Pt(111)-c(4 × 2)-2CO/α-PtO2 phase boundary. Chem. Phys. Lett. 2005, 492, 183–186. [Google Scholar] [CrossRef]
- Farkas, A.; Zalewska-Wierzbicka, K.; Bachmann, C.; Goritzka, J.; Langsdorf, D.; Balmes, O.; Janek, J.; Over, H. High pressure carbon monoxide oxidation over platinum (111). J. Phys. Chem. C 2013, 117, 9932–9942. [Google Scholar] [CrossRef]
- Ellinger, C.; Stierle, A.; Robinson, I.K.; Nefedov, A. Atmospheric pressure oxidation of Pt(111). J. Phys. C 2008, 20, 184013. [Google Scholar] [CrossRef]
- Miller, D.J.; Öberg, H.; Kaya, S.; Casalongue, H.S.; Fribel, D.; Anniyev, T.; Ogasawara, H.; Bluhm, H.; Pettersson, L.G.M.; Nilsson, A. Oxidation of Pt(111) under near-ambient conditions. Phys. Rev. Lett. 2011, 107, 195502. [Google Scholar] [CrossRef] [PubMed]
- Balmes, O.; Prevot, G.; Torrelles, X.; Lundgren, E.; Ferrer, S. Generation of surface steps on Pt(977) induced by the catalytic oxidation of CO. J. Catal. 2014, 309, 33–37. [Google Scholar] [CrossRef]
- Salmeron, M.; Schlogl, R. Ambient pressure photoelectron spectroscopy: A new tool for surface science and nanotechnology. Surf. Sci. Rep. 2008, 63, 169–199. [Google Scholar] [CrossRef]
- Knop-Gericke, A.; Kleimenov, E.; Haevacker, M.; Blume, R.; Teschner, D.; Zafeiratos, S.; Schlogl, R.; Bukhtivarov, V.I.; Kaichev, V.V.; Prosvirin, I.P.; et al. X-ray photoelectron spectroscopy for investigation of heterogeneous catalytic processes. Adv. Catal. 2009, 52, 213–272. [Google Scholar]
- Papp, C.; Steinruck, H.-P. In situ high resolution X-ray photoelectron spectroscopy—Fundamental insights into surface reactions. Surf. Sci. Rep. 2013, 68, 446–487. [Google Scholar] [CrossRef]
- Toyoshima, R.; Kondoh, H. In-situ observations of catalytic surface reactions with soft X-rays under working conditions. J. Phys. C 2015, 27, 083003. [Google Scholar] [CrossRef] [PubMed]
- Tao, F.; Dag, S.; Wang, L.-W.; Liu, Z.; Butcher, D.R.; Bluhm, H.; Salmeron, M.; Somorjai, G.A. Breakup of stepped platinum surfaces by high CO coverage. Science 2010, 327, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Hollins, P. Influence of surface defects on the infrared spectra of adsorbed species. Surf. Sci. Rep. 1992, 16, 51–94. [Google Scholar] [CrossRef]
- Butcher, D.R.; Grass, M.E.; Zeng, Z.; Aksoy, F.; Bluhm, H.; Li, W.X.; Mun, B.S.; Somorjai, G.A.; Liu, Z. In situ oxidation study of Pt(110) and its interaction with CO. J. Am. Chem. Soc. 2011, 133, 20319–20325. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.; Casalongue, H.S.; Bluhm, H.; Ogasawara, H.; Nilsson, A.; Kaya, S. Different reactivity of the various platinum oxides and chemisorbed oxygen in CO oxidation on Pt(111). J. Am. Chem. Soc. 2014, 136, 6340–6347. [Google Scholar] [CrossRef] [PubMed]
- Hamalainen, K.; Siddons, D.P.; Hastings, J.B.; Berman, L.E. Elimination of the inner-shell lifetime broadening in X-ray absorption spectroscopy. Phys. Rev. Lett. 1991, 67, 2850–2853. [Google Scholar] [CrossRef] [PubMed]
- DeGroot, F.M.F.; Krisch, K.H.; Vogel, J. Spectral sharpening of the Pt L edges by high resolution X-ray emission. Phys. Rev. B 2002, 66, 195112. [Google Scholar] [CrossRef]
- Van Bokhoven, J.A.; Louis, C.; Miller, J.T.; Tromp, M.; Safonova, O.V.; Glatzel, P. Activation of oxygen on gold/alumina catalysts: In situ high energy resolution fluorescence and time resolved X-ray spectroscopy. Angew. Chem. Int. Ed. 2006, 45, 4651–4654. [Google Scholar] [CrossRef] [PubMed]
- Safonova, O.V.; Tromp, M.; van Bokhoven, J.A.; DeGroot, F.M.F.; Evans, J.; Glatzel, P. Identification of CO adsorption sites in supported Pt catalysts using high energy resolution fluorescence detection X-ray spectroscopy. J. Phys. Chem. B 2006, 110, 16162–16164. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Alayon, E.M.C.; Tromp, M.M.; Safonova, O.V.; Glatzel, P.; Nachtegaal, M.; Frahm, R.; van Bokhoven, J.A. Generating highly active partially oxidized platinum during oxidation of carbon monoxide over Pt/Al2O3. Angew. Chem. Int. Ed. 2008, 47, 9260–9264. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Tromp, M.; Safonova, O.V.; Glatzel, P.; van Bokhoven, J.A. In situ XAS with high-energy resolution: The changing structure of platinum during the oxidation of carbon monoxide. Catal. Today 2009, 145, 300–306. [Google Scholar] [CrossRef]
- Singh, J.; van Bokhoven, J.A. Structure of alumina supported platinum catalysts of different particle size during CO oxidation using in situ IR and HERFD XAS. Catal. Today 2010, 155, 199–205. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Caravati, M.; Hannemann, S.; Baiker, A. X-ray absorption spectroscopy under reaction conditions: Suitability of different reaction cells for combined characterisation and time resolved studies. Phys. Chem. Chem. Phys. 2004, 6, 3037–3057. [Google Scholar] [CrossRef]
- Urakawa, A.; Maeda, N.; Baiker, A. Space and time resolved combined DRIFT Raman spectroscopy: Monitoring dynamic surface and bulk processes during NOx storage. Angew. Chem. Int. Ed. 2008, 47, 9256–9259. [Google Scholar] [CrossRef] [PubMed]
- Urakawa, A.; Baiker, A. Space resolved profiling relevant in heterogeneous catalysis. Top. Catal. 2009, 52, 1312–1322. [Google Scholar] [CrossRef]
- Korup, O.; Mavlyankariev, S.; Geske, M.; Goldsmith, C.F.; Horn, R. Measurement and analysis of spatial reactor profiles in high temperature catalysis research. Chem. Eng. Proc. 2011, 50, 998–1009. [Google Scholar] [CrossRef]
- Touitou, J.J.; Morgan, K.; Burch, R.; Hardacre, C.; Goguet, A. An in situ spatially resolved method to probe gas phase reactions through a fixed bed catalyst. Catal. Sci. Technol. 2012, 2, 1811–1813. [Google Scholar] [CrossRef]
- Morgan, K.; Touitou, J.J.; Choi, J.S.; Coney, C.; Hardacre, C.; Pihl, J.A.; Stere, C.E.; Kim, M.Y.; Stewart, C.; Goguet, A.; et al. Evolution and enabling capacities of spatially resolved techniques for the characterisation of heterogeneously catalysed reactions. ACS Catal. 2016, 6, 1356–1381. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Baiker, A. Axial variation of the oxidation state of Pt-Rh/Al2O3 during partial methane oxidation in affixed bed reactor: An in situ X-ray absorption spectroscopy study. Catal. Lett. 2005, 99, 5–12. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Wagner, J.B.; Dunin-Borkowski, R.E. Imaging catalysts at work: A hierarchical approach from the macro- to the meso- and nano-scale. ChemCatChem 2013, 5, 62–80. [Google Scholar] [CrossRef]
- Newton, M.A.; Jyoti, B.; Dent, A.J.; Diaz-Moreno, S.; Fiddy, S.G.; Evans, J. Rapid monitoring of the nature and interconversion of supported catalyst phases and their influence upon performance: CO oxidation to CO2 by γ-Al2O3 supported Rh catalysts. Chem. Eur. J. 2006, 12, 1975–1985. [Google Scholar] [CrossRef]
- Singh, J.; Nachtegaal, M.; Alayon, E.M.C.; Stötzel, J.; van Bokhoven, J.A. Dynamic structure changes of a heterogeneous catalyst within a reactor: Oscillations in CO oxidation over a supported platinum catalyst. ChemCatChem 2010, 2, 653–657. [Google Scholar] [CrossRef]
- Gänzler, A.M.; Casapu, M.; Boubnov, A.; Müller, O.; Conrad, S.; Lichtenberg, H.; Frahm, R.; Grunwaldt, J.D. Operando spatially and time-resolved X-ray absorption spectroscopy and infrared thermography during oscillatory CO oxidation. J. Catal. 2015, 328, 216–224. [Google Scholar] [CrossRef]
- Campbell, C.T.; Ertl, G.; Kuipers, H.; Segner, J. A molecular beam study of the catalytic oxidation of CO on a Pt(111) surface. J. Phys. Chem. 1980, 73, 5862–5873. [Google Scholar] [CrossRef]
- Warren, B.E. X-ray determination of the stricture of a glass. J. Am. Ceram. Soc. 1934, 17, 249–254. [Google Scholar] [CrossRef]
- Warren, B.E.; Krutter, H.; Morningstar, O. Fourier analysis of X-ray patterns of vitreous SiO2 and B2O2. J. Am. Ceram. Soc. 1936, 19, 202–206. [Google Scholar] [CrossRef]
- Franklin, R.E. The interpretation of diffuse X-ray diagrams of carbon. Acta Cryst. 1950, 3, 107–121. [Google Scholar] [CrossRef]
- Franklin, R.E. The structure of graphitic carbons. Acta Cryst. 1951, 4, 253. [Google Scholar] [CrossRef]
- Gallezot, P. X-ray techniques in catalysis. In Catalysis; Anderson, J.R., Boudart, M., Eds.; Springer: Beriln/Heidelberg, Germany, 1979; Chapter 4; pp. 221–273. [Google Scholar]
- Chupas, P.J.; Chapman, K.W.; Lee, P.L. Applications of an amorphous silicon detector for high resolution, high sensitivity and pair distribution function measurements. J. Appl. Cryst. 2007, 40, 463–470. [Google Scholar] [CrossRef]
- Liang, K.S.; Laderman, S.S.; Sinfelt, J.H. Structural study of small catalytic particles using differential anomalous scattering. J. Chem. Phys. 1987, 86, 2352–2355. [Google Scholar] [CrossRef]
- Chupas, P.J.; Chapman, K.W.; Jennings, G.; Lee, P.L.; Grey, C.P. Watching nanoparticles grow: The mechanism and kinetics for the formation of TiO2 supported platinum nanoparticles. J. Am. Chem. Soc. 2007, 129, 13822. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.A.; Chapman, K.W.; Thompsett, D.; Chupas, P.J. Chasing changing nanoparticles with time-resolved pair distribution function measurements. J. Am. Chem. Soc. 2012, 134, 5036–5039. [Google Scholar] [CrossRef]
- Chupas, P.J.; Chapman, K.W.; Kurtz, C.; Hanson, J.C.; Lee, P.L.; Grey, C.P. A versatile sample environment for non-ambient X-ray scattering experiments. J. Appl. Cryst. 2008, 41, 822–824. [Google Scholar] [CrossRef]
- Narula, C.K.; Stocks, G.M. Ab initio density functional calculations of adsorption of transition metals on the θ-Al2O3 (010) surface. J. Phys. Chem. C 2012, 116, 5628–5636. [Google Scholar] [CrossRef]
- Narula, C.K.; Allard, L.F.; Stocks, G.M.; Moses-Debusk, M. Remarkable NO oxidation on single supported platinum atoms. Sci. Rep. 2014, 4, 7238. [Google Scholar] [CrossRef] [PubMed]
- Moses-DeBusk, M.; Yoon, M.; Allard, L.F.; Mullins, D.R.; Wu, Z.L.; Yang, X.F.; Veith, G.; Stocks, G.M.; Narula, C.K. CO oxidation on supported single Pt atoms: Experimental and ab initio density functional studies of CO interaction with Pt atom on θ-Al2O3 (010) surface. J. Am. Chem. Soc. 2013, 135, 12634–12645. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.A.; Ferri, D.; Smolentsev, G.; Marchionni, V.; Nachtegaal, M. Room temperature carbon monoxide oxidation by oxygen over Pt/Al2O3 mediated by reactive platinum carbonates. Nat. Commun. 2015, 6, 8675. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.A.; Ferri, D.; Smolentsev, G.; Marchionni, V.; Nachtegaal, M. Kinetic studies of Pt carbonate mediated, room temperature, oxidation of carbon monoxide by oxygen over Pt/Al2O3 using combined, time resolved, XAFS, DRIFTS and mass spectrometry. J. Am. Chem. Soc. 2016, 138, 13930–13940. [Google Scholar] [CrossRef] [PubMed]
- Allian, D.; Takanabe, K.; Fujdala, L.; Hao, X.; Truex, T.; Cai, J.; Buda, C.; Neurock, M.; Iglesia, E. Chemisorption of CO and mechnism of CO oxidation on supported metal nanoclusters. J. Am. Chem. Soc. 2011, 133, 4498–4517. [Google Scholar] [CrossRef]
- Wartnaby, C.E.; Stuck, A.; Yeo, Y.Y.; King, D.A. Microcalorimetric heats of adsorption of CO, NO, and oxygen on Pt(110). J. Phys. Chem. 1996, 100, 12483–12488. [Google Scholar] [CrossRef]
- Yeo, Y.Y.; Vattuone, L.; King, D.A. Calorimetric heats for CO and oxygen adsorption and for the catalytic CO oxidation reaction on Pt(111). J. Chem. Phys. 1997, 106, 392–401. [Google Scholar] [CrossRef]
- Szlacheto, J.; Ferri, D.; Marchionni, V.; Kambolis, A.; Safonova, O.V.; Milne, C.J.; Kröcher, O.; Nachtegaal, M.; Sá, J. Subsecond and in situ chemical speciation of Pt/Al2O3 during oxidation-reduction cycles monitored by high-energy resolution off-resonant X-ray spectroscopy. J. Am. Chem. Soc. 2013, 135, 19071–19074. [Google Scholar] [CrossRef] [PubMed]
- Blachuki, W.; Szlachetko, J.; Hoszowska, J.; Dousse, J.C.; Kayser, Y.; Nachtegaal, M.; Sa, J. High energy resolution off-resonant spectroscopy for X-ray absorption spectra free of self-absorption effects. Phys. Rev. Lett. 2014, 112, 173003. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, S.; Behafarid, D.; Croy, J.R.; Ono, L.K.; Li, L.; Yang, J.C.; Frenkel, A.I.; Roldan-Cuenya, B. Shape-dependent catalytic properties of Pt nanoparticles. J. Am. Chem. Soc. 2010, 132, 15714–15719. [Google Scholar] [CrossRef] [PubMed]
- Chaâbane, N.; Lazzari, R.; Jupille, J.; Renaud, G.; Soares, E.A. CO-induced scavenging of supported Pt nanoclusters: A GISAXS study. J. Phys. Chem. C 2012, 116, 23362–23370. [Google Scholar] [CrossRef]
- Gustafson, J.; Shipilin, M.; Zhang, C.; Stierle, A.; Hejral, U.; Ruett, U.; Gutowski, O.; Carlsson, P.A.; Skoglundh, M.; Lundgren, E. High-energy surface X-ray diffraction for fast surface structure determination. Science 2014, 343, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Shipilin, M.; Gustafson, J.; Zhang, C.; Merte, L.R.; Stierle, A.; Hejral, U.; Ruett, U.; Gutowski, O.; Skoglundh, M.; Carlsson, P.A.; et al. Transient structures of PdO during CO oxidation over Pd(100). J. Phys. Chem. C 2015, 119, 15469–15476. [Google Scholar] [CrossRef]
- Nolte, P.; Stierle, A.; Jin-Phillipp, N.Y.; Kasper, N.; Schulli, T.U.; Dosch, H. Shape changes of supported Rh nanoparticles during oxidation and reduction cycles. Science 2008, 321, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Nolte, P.; Stierle, A.; Kasper, N.; Jin-Phillipp, N.Y.; Reichert, H.; Ruhm, A.; Okasinski, J.; Dosch, H.; Schoder, S. Combinatorial high-energy X-ray microbeam study of the size-dependent oxidation of Pd nanoparticles on MgO(100). Phys. Rev. B 2008, 77, 115444. [Google Scholar] [CrossRef]
- Nolte, P.; Stierle, A.; Kasper, N.; Jin-Phillipp, N.Y.; Jeutter, N.; Dosch, H. Reversible shape changes of Pd nanoparticles on MgO(100). Nano Lett. 2011, 11, 4697–4700. [Google Scholar] [CrossRef] [PubMed]
- Heiral, U.; Muller, P.; Balmes, O.; Pontoni, D.; Stierle, A. Tracking the shape-dependent sintering of platinum-rhodium model catalysts under operando conditions. Nat. Commun. 2016, 7, 10964. [Google Scholar]
- Di Michiel, M.; (ESRF). Private Communication, 2016.
- Baurecht, D.; Fringeli, U.P. Quantitative modulated excitation Fourier transform infrared spectroscopy. Rev. Sci. Instrum. 2001, 72, 3782–3792. [Google Scholar] [CrossRef]
- Urakawa, A.; Wirz, R.; Burgi, T.; Baiker, A. ATR-IR flow-through cell for concentration modulation excitation spectroscopy: Diffusion experiments and simulations. J. Phys. Chem. B 2003, 107, 13061–13068. [Google Scholar] [CrossRef]
- Urakawa, A.; Burgi, T.; Schlapfer, H.P.; Baiker, A. Simultaneous in situ monitoring of surface and gas species and surface properties by modulation excitation polarization-modulation infrared reflection-absorption spectroscopy: CO oxidation over Pt film. J. Chem. Phys. 2006, 124, 054717. [Google Scholar] [CrossRef] [PubMed]
- Urakawa, A.; Burgi, T.; Baiker, A. Sensitivity enhancement and dynamic behavior analysis by modulation excitation spectroscopy: Principle and application in heterogeneous catalysis. Chem. Eng. Sci. 2008, 63, 4902–4909. [Google Scholar] [CrossRef]
- Burgi, T.; Baiker, A. In situ infrared spectroscopy of catalytic solid-liquid interfaces using phase-sensitive detection: Enantioselective hydrogenation of a pyrone over Pd/TiO2. J. Phys. Chem. B 2002, 106, 10649–10658. [Google Scholar] [CrossRef]
- Ferri, D.; Kumar, M.S.; Eyssler, A.; Korsak, O.; Hug, P.; Weidenkaff, A.; Newton, M.A. First steps in combining concentration modulation techniques with synchronous dispersive EXAFS/DRIFTS/mass spectrometry for in situ time resolved study of heterogeneous catalysts. Phys. Chem. Chem. Phys. 2010, 12, 5634–5646. [Google Scholar] [CrossRef] [PubMed]
- Ferri, D.; Newton, M.A.; Nachtegaal, M. Modulation excitation X-ray absorption spectroscopy to probe surface species on heterogeneous catalysts. Top. Catal. 2011, 54, 1070–1078. [Google Scholar] [CrossRef]
- Koenig, C.F.J.; van Bokhoven, J.A.; Schildhauer, T.J.; Nachtegaal, M. Quantitative analysis of modulated excitation X-ray absorption spectra: Enhanced precision of EXAFS fitting. J. Phys. Chem. C 2012, 116, 19857–19866. [Google Scholar] [CrossRef]
- Marchionni, V.; Newton, M.A.; Kambolis, A.; Kumar, S.M.; Weidenkaff, A.; Ferri, D. A modulated excitation ED-EXAFS/DRIFTS study of hydrothermal ageing of Rh/Al2O3. Catal. Today 2014, 229, 80–87. [Google Scholar] [CrossRef]
- Nilsson, J.; Carlsson, P.A.; Fouladvand, S.; Martin, N.M.; Gustafson, J.; Newton, M.A.; Lundgren, E.; Grönbeck, H.; Skoglundh, M. Chemistry of supported palladium nanoparticles during methane oxidation. ACS Catal. 2015, 5, 2481–2489. [Google Scholar] [CrossRef]
- Urakawa, A.; Van Beek, W.; Monrabal-Capilla, M.; Galan-Mascaros, J.R.; Palin, L.; Milanesio, M. Combined, modulation enhanced X-ray powder diffraction and Raman spectroscopic study of structural transitions in the spin crossover material [Fe(Htrz)2(trz)](BF4). J. Phys. Chem. C 2011, 115, 1323–1329. [Google Scholar] [CrossRef]
- Chernyshov, D.; Van Beek, W.; Emerich, H.; Milanesio, M.; Urakawa, A.; Viterbo, D.; Palin, L.; Caliandro, R. Kinematic diffraction on a structure with periodically varying scattering function. Acta Crystallogr. A 2011, 67, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Chernyshov, D.; Dyadin, V.; Van Beek, W.; Urakawa, A. Frequency analysis for modulation-enhanced powder diffraction. Acta Crystallogr. A 2016, 72, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Voronov, A.; Urakawa, A.; Van Beek, W.; Tsakoumis, N.E.; Emerich, H.; Ronning, M. Multivariate curve resolution applied to in situ X-ray absorption spectroscopy data: An efficient tool for data processing and analysis. Anal. Chim. Acta 2014, 840, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Ferri, D.; Newton, M.A.; Di Michiel, M.; Chiarello, G.L.; Yoon, S.; Lu, Y.; Andrieux, J.; Kumar, M.S.; Weidenkaff, A. Synchrotron hard X-ray methods coupled to phase sensitive analysis to characterize aging of solid catalysts with enhanced sensitivity. Phys. Chem. Chem. Phys. 2013, 15, 8629–8639. [Google Scholar] [CrossRef] [PubMed]
- Ferri, D.; Newton, M.A.; Di Michiel, M.; Chiarello, G.L.; Yoon, S.; Lu, Y.; Andrieux, J. Seeing further into the dynamic structure of complex solid catalysts using X-ray diffraction combined with modulated excitation spectroscopy. Angew. Chem. Int. Ed. 2014, 53, 8890–8894. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Garcia, M. XANES analysis of catalytic systems under reaction conditions. Catal. Rev. Sci. Eng. 2002, 44, 59–121. [Google Scholar] [CrossRef]
- Iglesias-Juez, A.; Kubacka, A.; Fernández-Garcia, M.; Di Michiel, M.; Newton, M.A. Nanoparticulate Pd supported catalysts: Size-dependent formation of Pd(I)/Pd(0) and their role in CO elimination. J. Am. Chem. Soc. 2011, 133, 4484–4489. [Google Scholar] [CrossRef] [PubMed]
- Ankudinov, A.L.; Ravel, B.; Rehr, J.J.; Conradson, S.D. Real-space multiple-scattering calculation and interpretation of X-ray-absorption near-edge structure. Phys. Rev. B 1998, 58, 7565–7576. [Google Scholar] [CrossRef]
- Ankudinov, A.L.; Bouldin, C.E.; Rehr, J.J.; Sims, J.; Hung, H. Parallel calculation of electron multiple scattering using Lanczos algorithms. Phys. Rev. B 2002, 65, 104107. [Google Scholar] [CrossRef]
- Rehr, J.J.; Ankudinov, A.L. Progress in the theory and interpretation of XANES. Coord. Chem. Rev. 2005, 249, 131–140. [Google Scholar] [CrossRef]
- Smolentsev, G.; Soldatov, A. Quantitative local structure refinement from XANES: Multi-dimensional interpolation approach. J. Synchrotron Radiat. 2006, 13, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Benfatto, M.; Della Longa, S. MXAN: New improvements for potential and structural refinement. JPCS 2009, 190, 012031. [Google Scholar] [CrossRef]
- Lima, F.A.; Bjornsson, R.; Weyhermuller, T.; Chandrasekran, P.; Glatzel, P.; Neese, F.; DeBeer, S. High-resolution molybdenum K-edge X-ray absorption spectroscopy analyzed with time-dependent density functional theory. Phys. Chem. Chem. Phys. 2013, 15, 20911–20920. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Jaime, M.U.; Zhang, K.; Vura-Weiss, J.; de Groot, F.M.F. CTM4DOC: Electronic structure analysis from X-ray spectroscopy. J. Synchrotron Radiat. 2016, 23, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Farges, F.; Argoul, P. Continuous Cauchy wavelet transform of XAFS spectra. Phys. Scr. 2005, T115, 221. [Google Scholar]
- Funke, H.; Scheinost, A.C.; Chukalina, M. Wavelet analysis of extended X-ray absorption fine structure data. Phys. Rev. B 2005, 71, 094110. [Google Scholar] [CrossRef]
- Filez, M.; Redekop, E.A.; Poelman, H.; Galvita, V.V.; Ramachandran, R.K.; Dendooven, J.; Detavernier, C.; Marin, G.B. Unravelling the formation of Pt-Ga alloyed nanoparticles on calcined Ga-modified hydrotalcites by in situ XAS. Chem. Mater. 2014, 26, 5936. [Google Scholar] [CrossRef]
- Filez, M.; Redekop, E.A.; Poelman, H.; Galvita, V.V.; Marin, G.B. Advanced elemental characterization during Pt—In catalyst formation by wavelet transformed X-ray absorption spectroscopy. Anal. Chem. 2015, 87, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Mesu, J.G.; Beale, A.M.; De Groot, F.M.F.; Weckhuysen, B.M. Synchrotron radiation effects on catalytic systems as probed with a combined in situ UV-Vis/XAFS spectroscopic setup. J. Phys. Chem. B 2006, 110, 17671–17677. [Google Scholar] [CrossRef] [PubMed]
- Martis, V.; Nikitenko, S.; Sen, S.; Sankar, G.; van Beek, W.; Filinchuk, Y.; Snigireva, I.; Bras, W. Effects of X-rays on crystal nucleation in lithium disilicate. Cryst. Growth Des. 2011, 11, 2858–2865. [Google Scholar] [CrossRef]
- Peng, J.; Porsgaard, S.; Borondics, F.; Kober, M.; Caballero, A.; Bluhm, H.; Besenbacher, F.; Salmeron, M. Room-temperature reaction of oxygen with gold: An in situ ambient-pressure X-ray photoelectron spectroscopy investigation. J. Am. Chem. Soc. 2010, 132, 2858–2859. [Google Scholar]
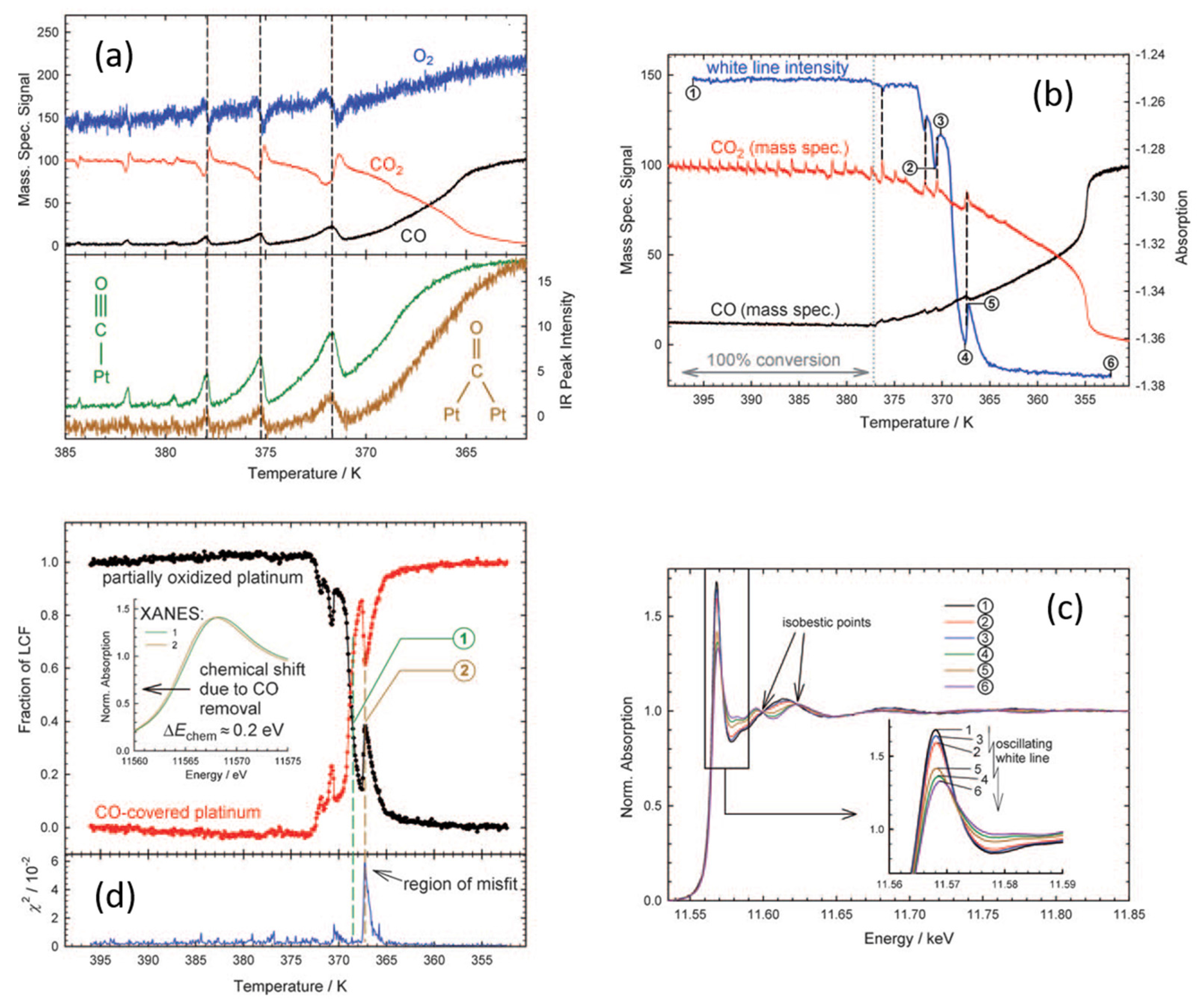
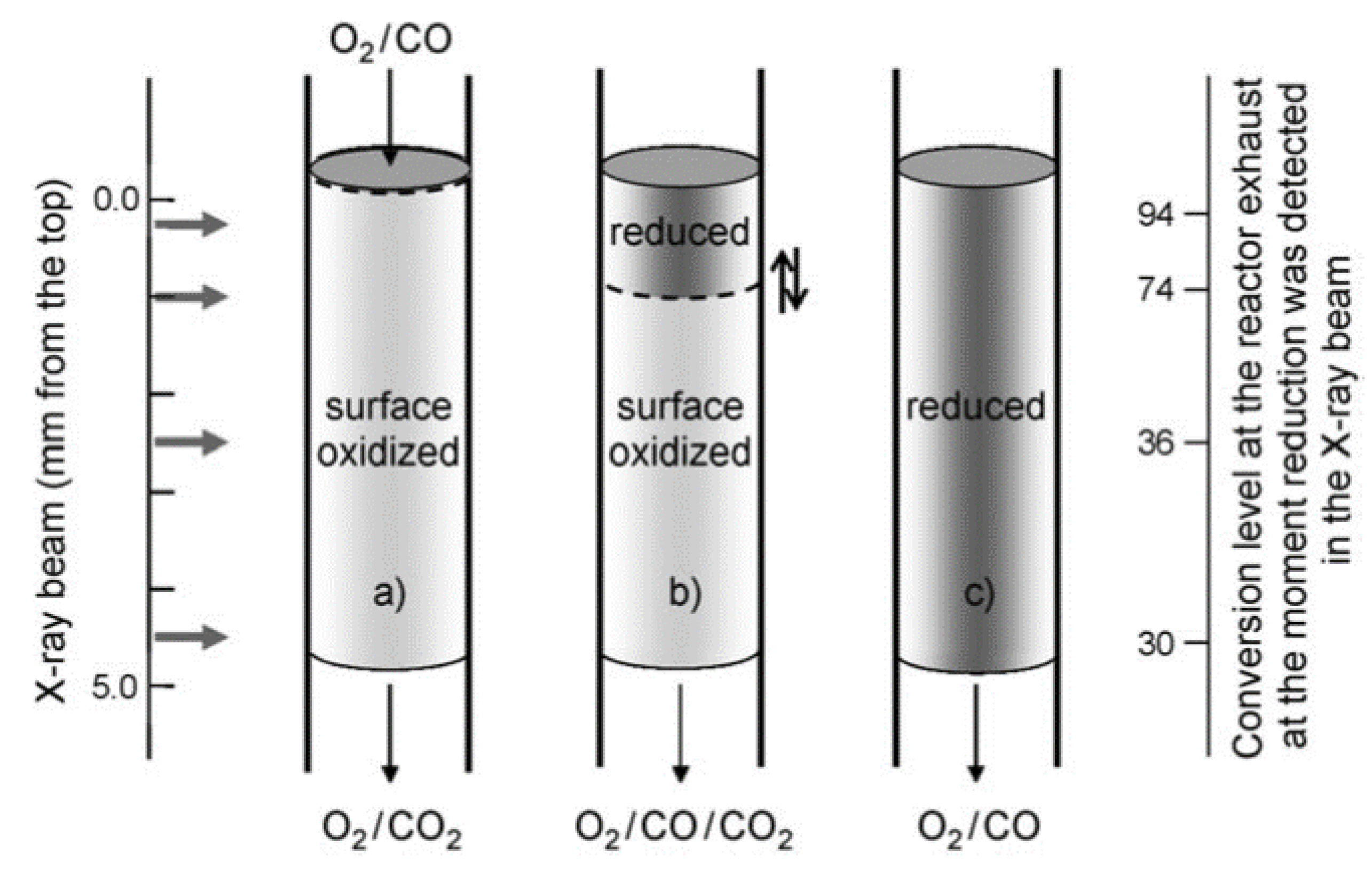
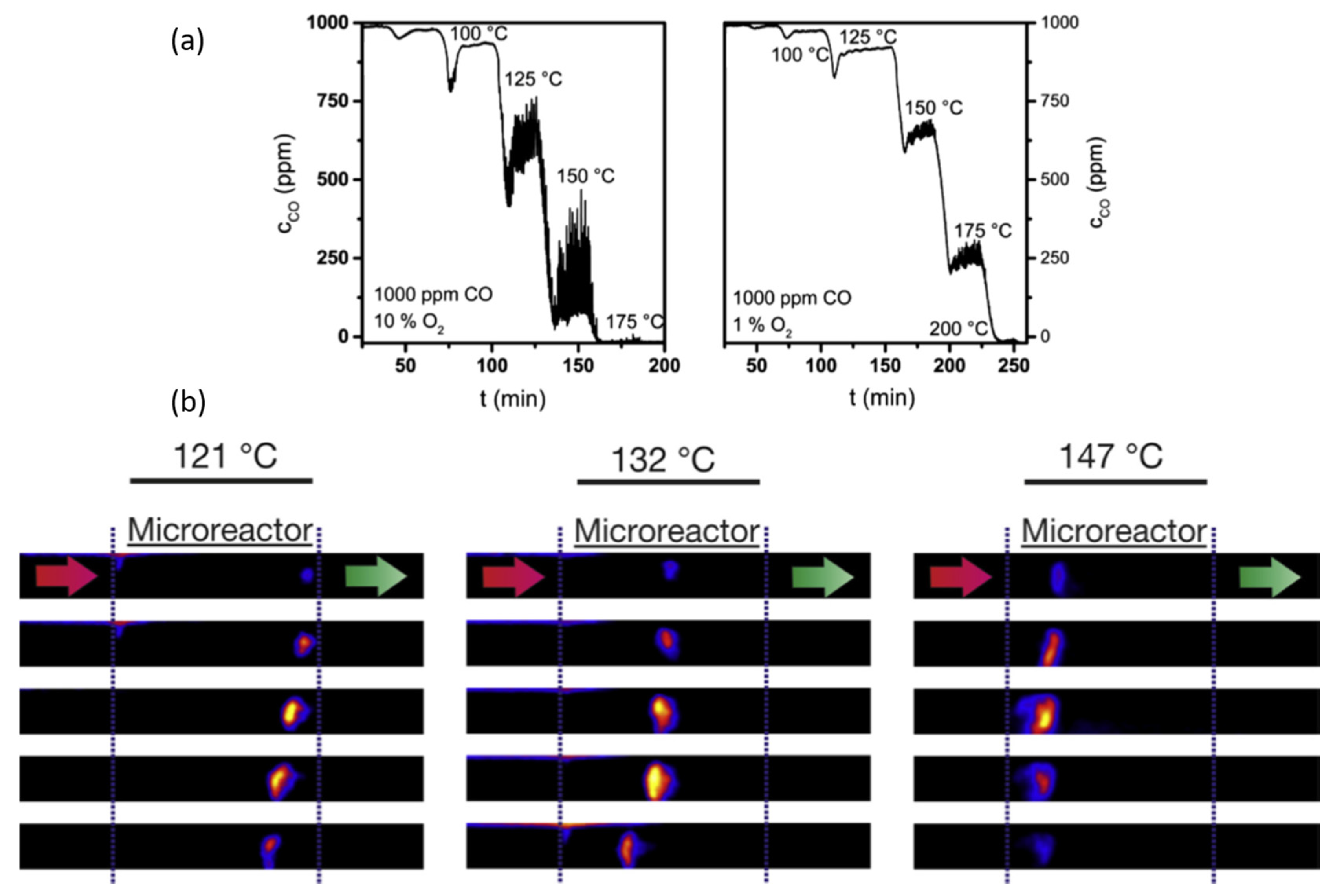
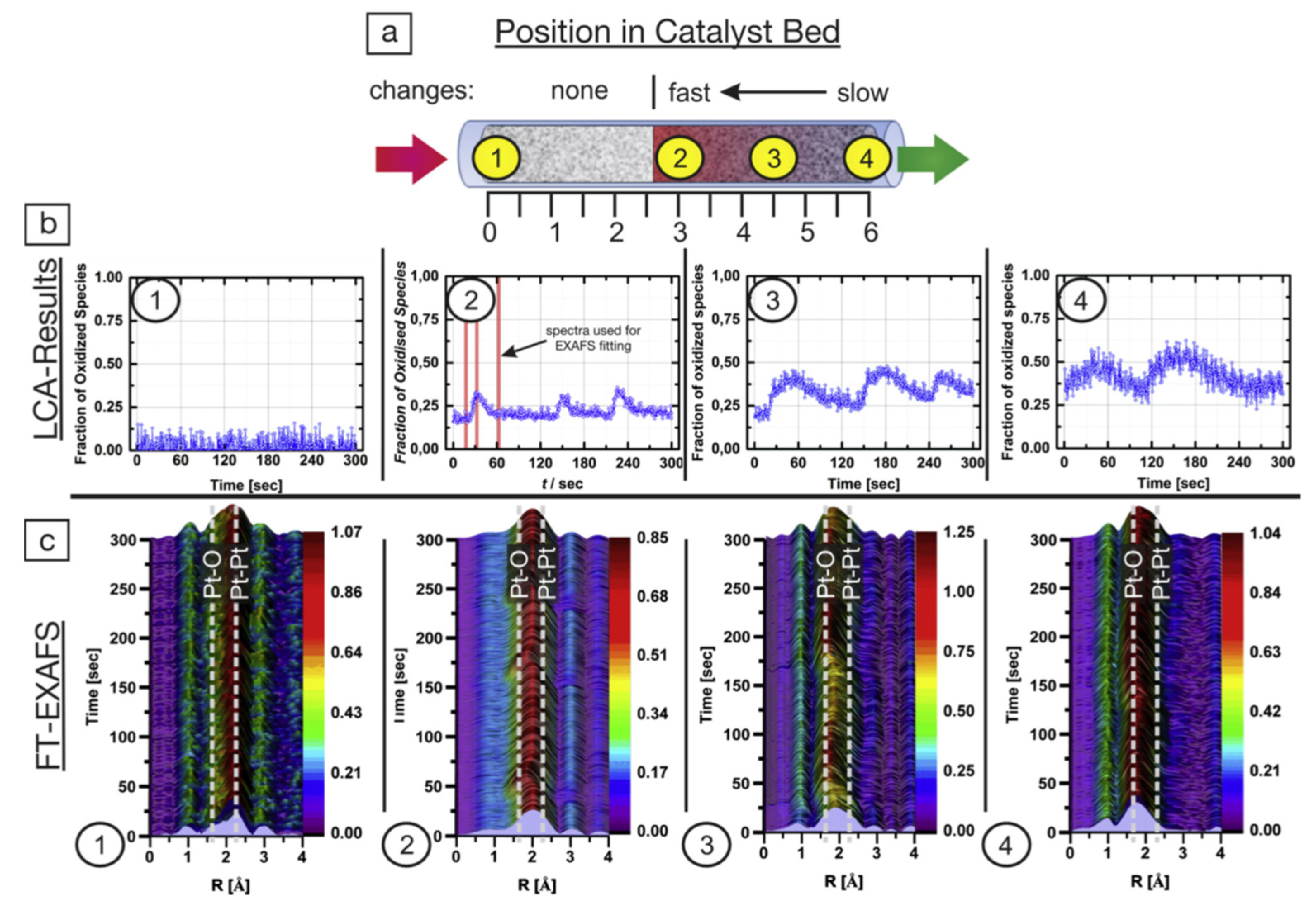
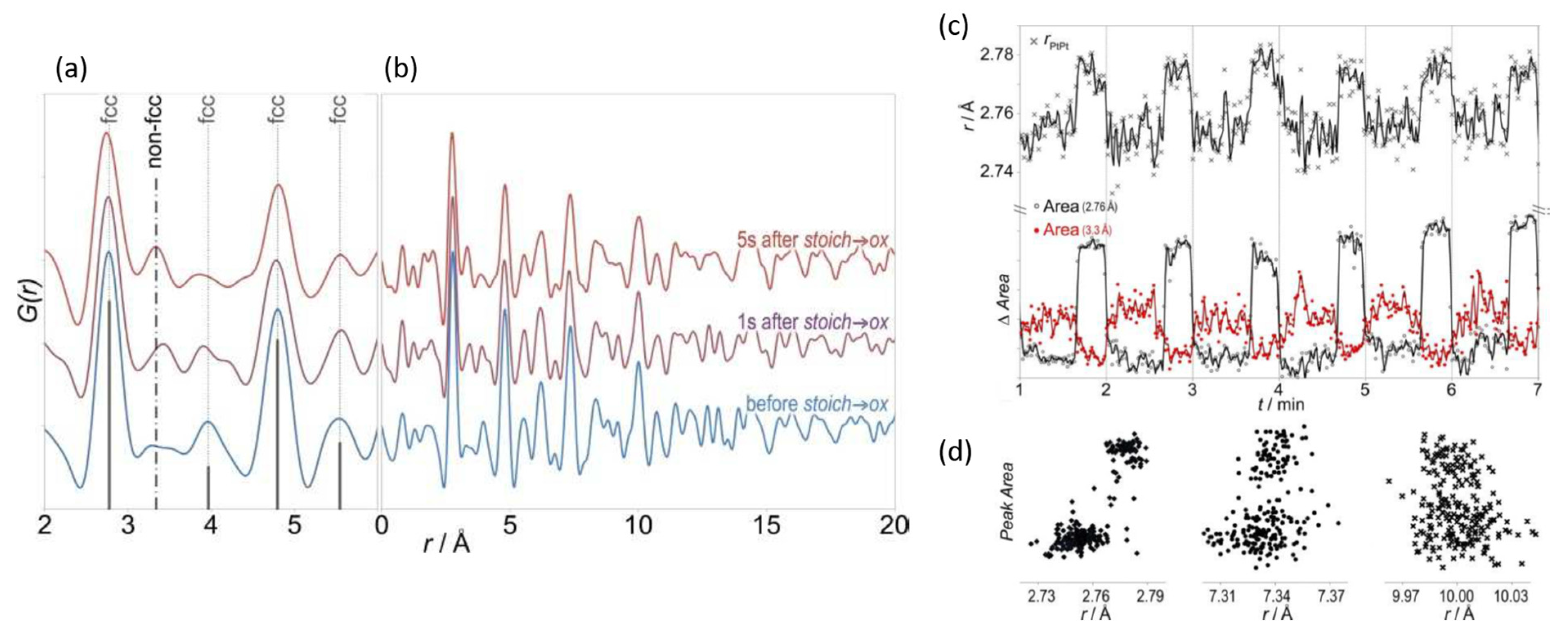
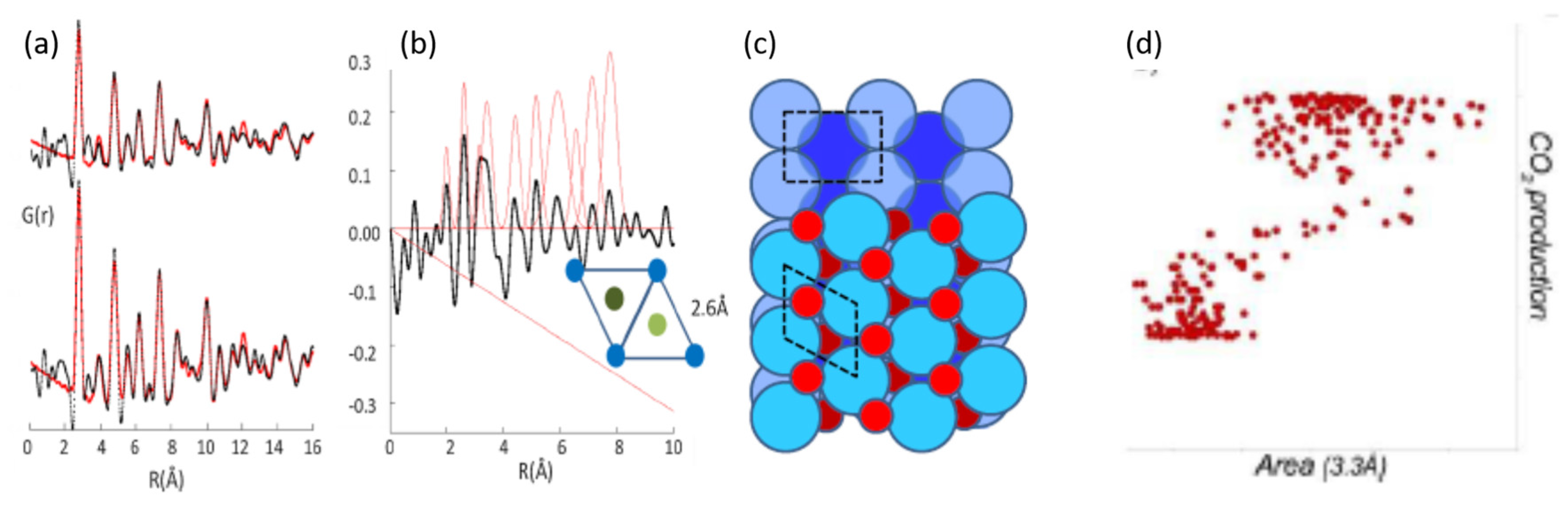
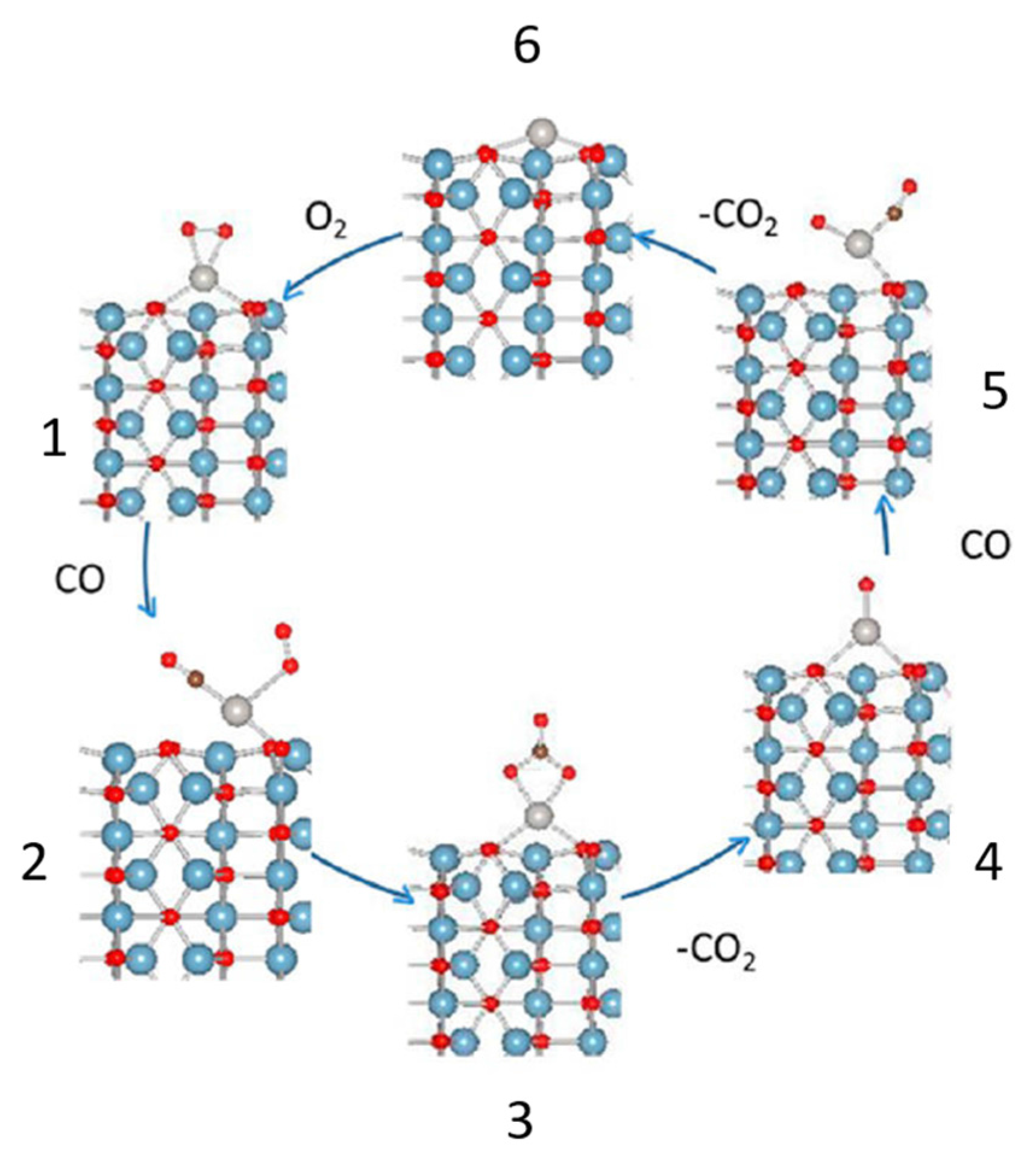
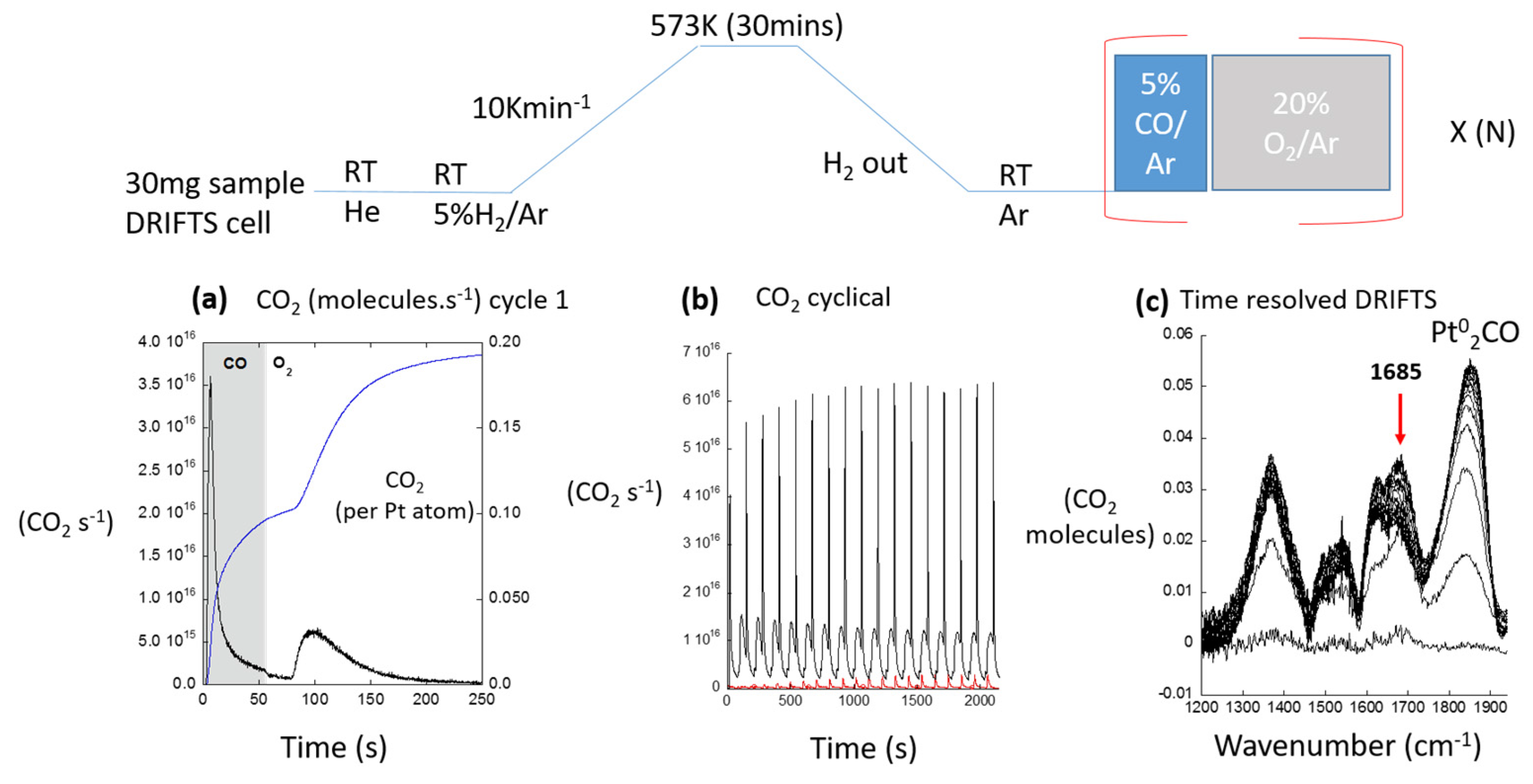
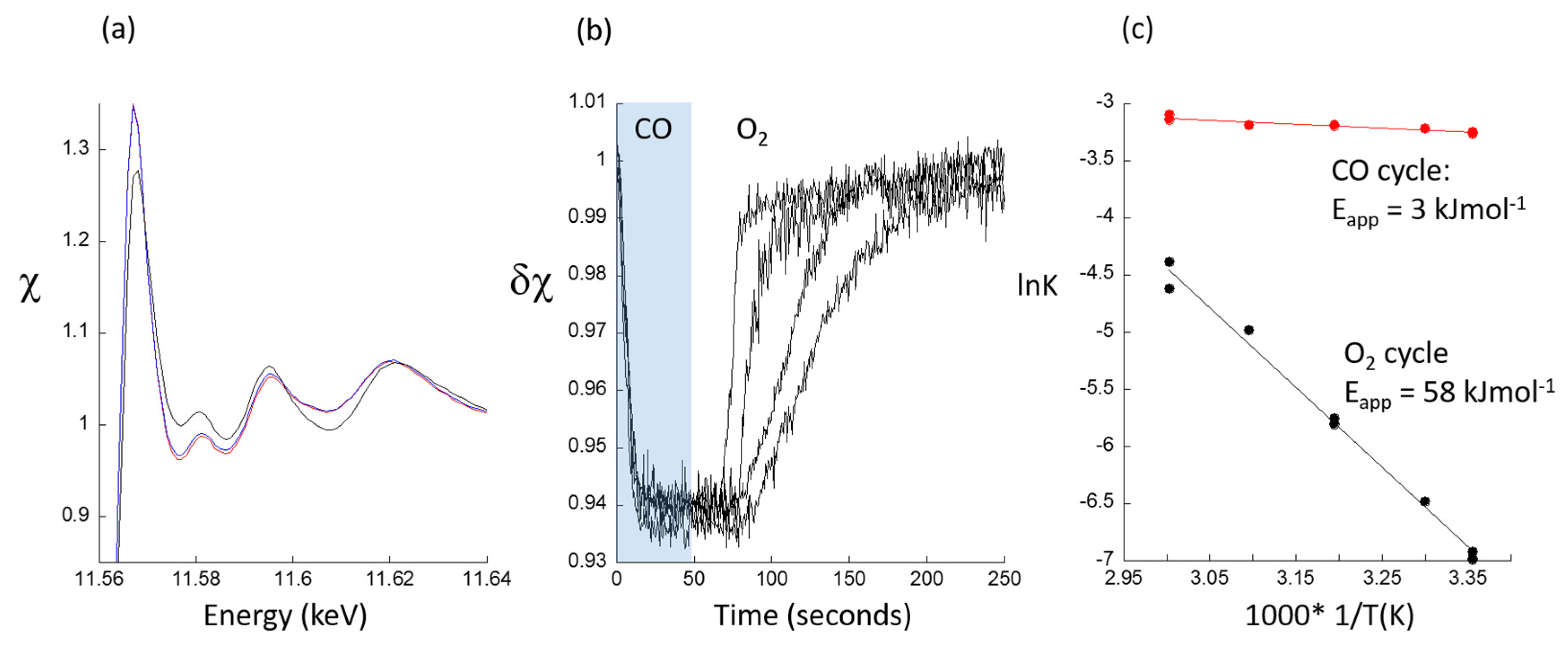
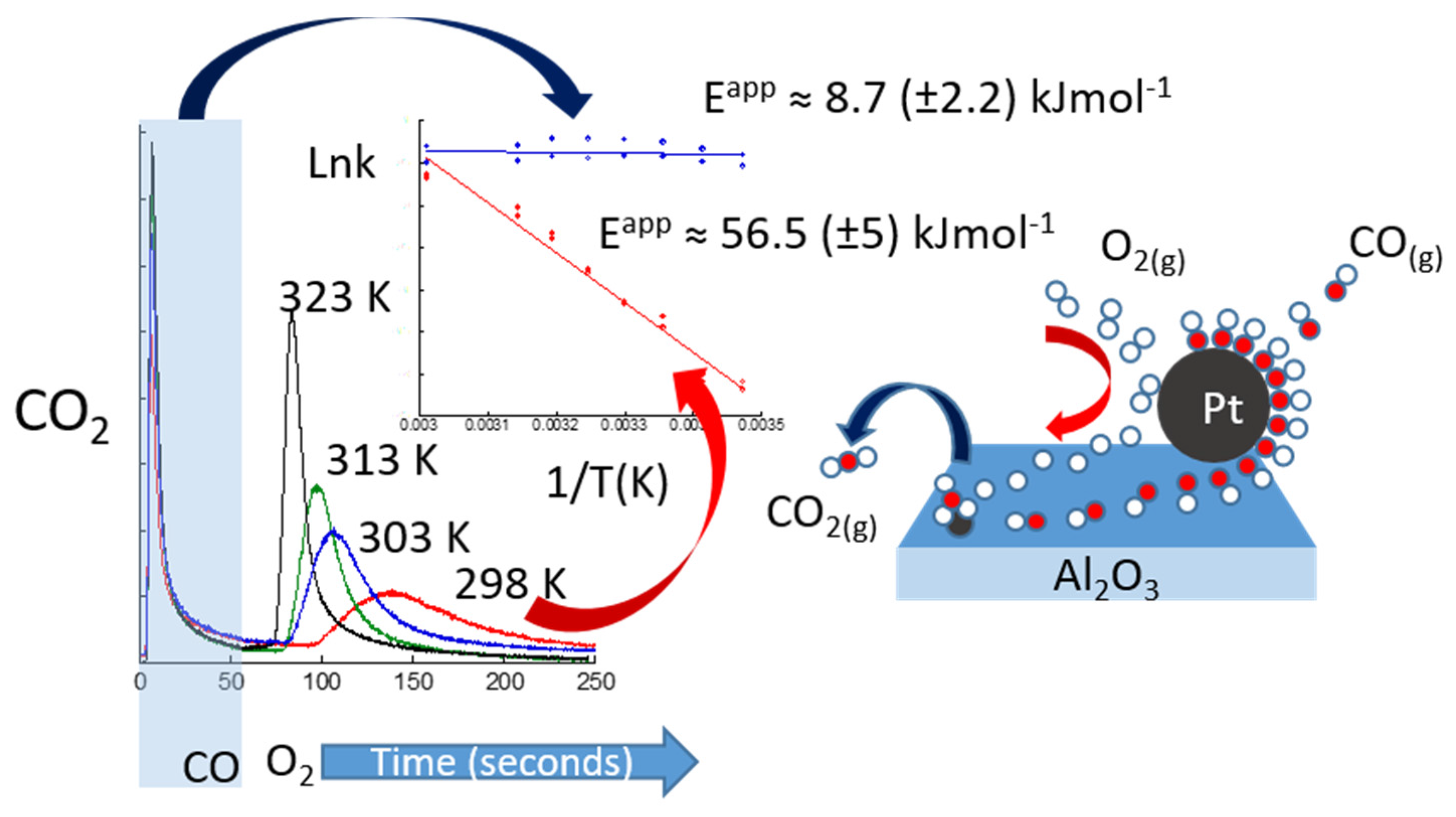
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newton, M.A. Time Resolved Operando X-ray Techniques in Catalysis, a Case Study: CO Oxidation by O2 over Pt Surfaces and Alumina Supported Pt Catalysts. Catalysts 2017, 7, 58. https://doi.org/10.3390/catal7020058
Newton MA. Time Resolved Operando X-ray Techniques in Catalysis, a Case Study: CO Oxidation by O2 over Pt Surfaces and Alumina Supported Pt Catalysts. Catalysts. 2017; 7(2):58. https://doi.org/10.3390/catal7020058
Chicago/Turabian StyleNewton, Mark A. 2017. "Time Resolved Operando X-ray Techniques in Catalysis, a Case Study: CO Oxidation by O2 over Pt Surfaces and Alumina Supported Pt Catalysts" Catalysts 7, no. 2: 58. https://doi.org/10.3390/catal7020058
APA StyleNewton, M. A. (2017). Time Resolved Operando X-ray Techniques in Catalysis, a Case Study: CO Oxidation by O2 over Pt Surfaces and Alumina Supported Pt Catalysts. Catalysts, 7(2), 58. https://doi.org/10.3390/catal7020058