Hydroconversion of Waste Cooking Oil into Green Biofuel over Hierarchical USY-Supported NiMo Catalyst: A Comparative Study of Desilication and Dealumination


Abstract
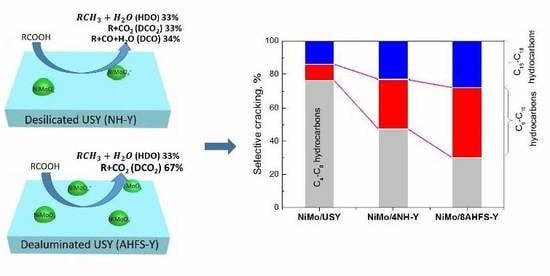
:
1. Introduction
2. Results and Discussion
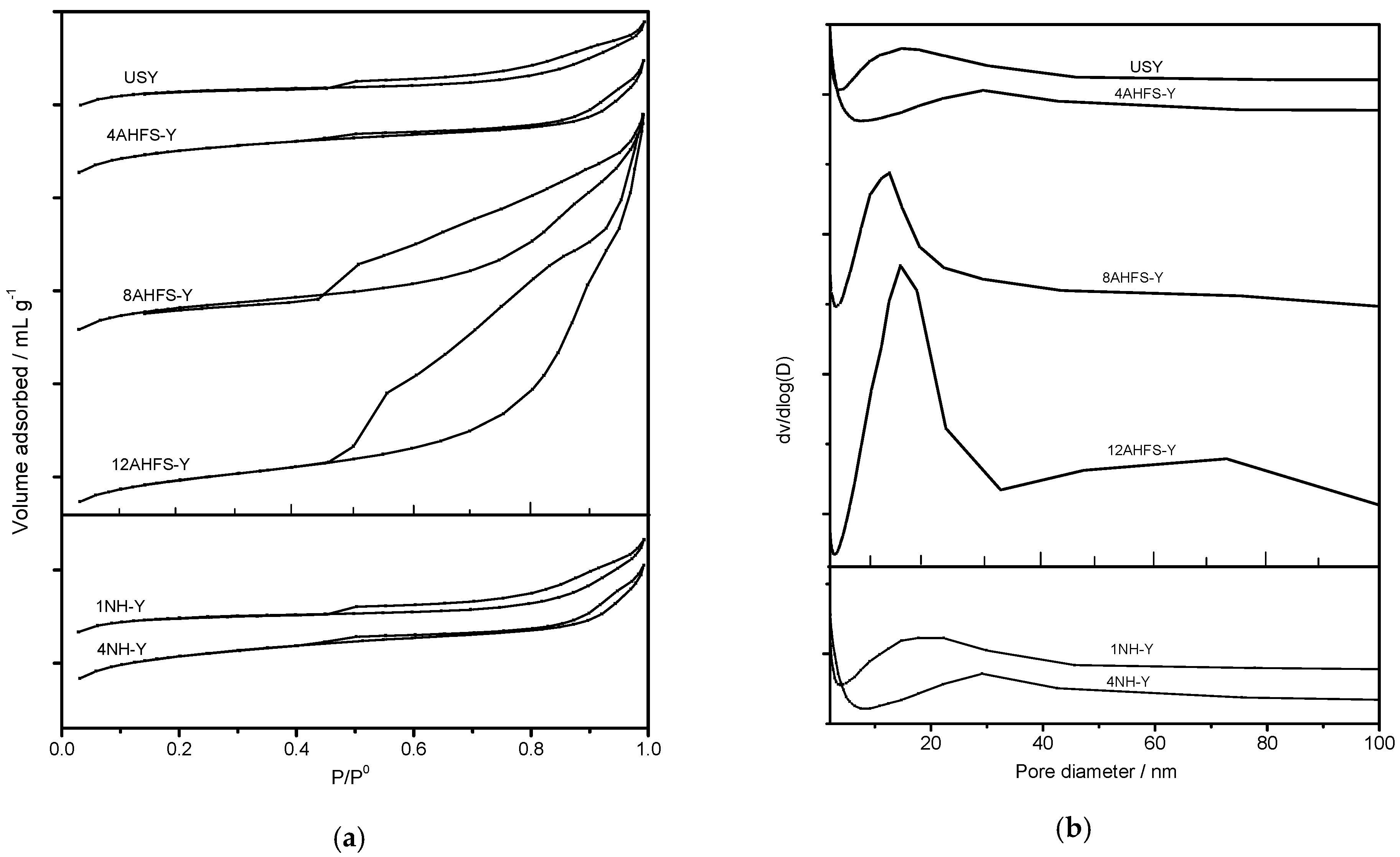
2.1. Textural Structures of Desilicated and Dealuminated USY Zeolites
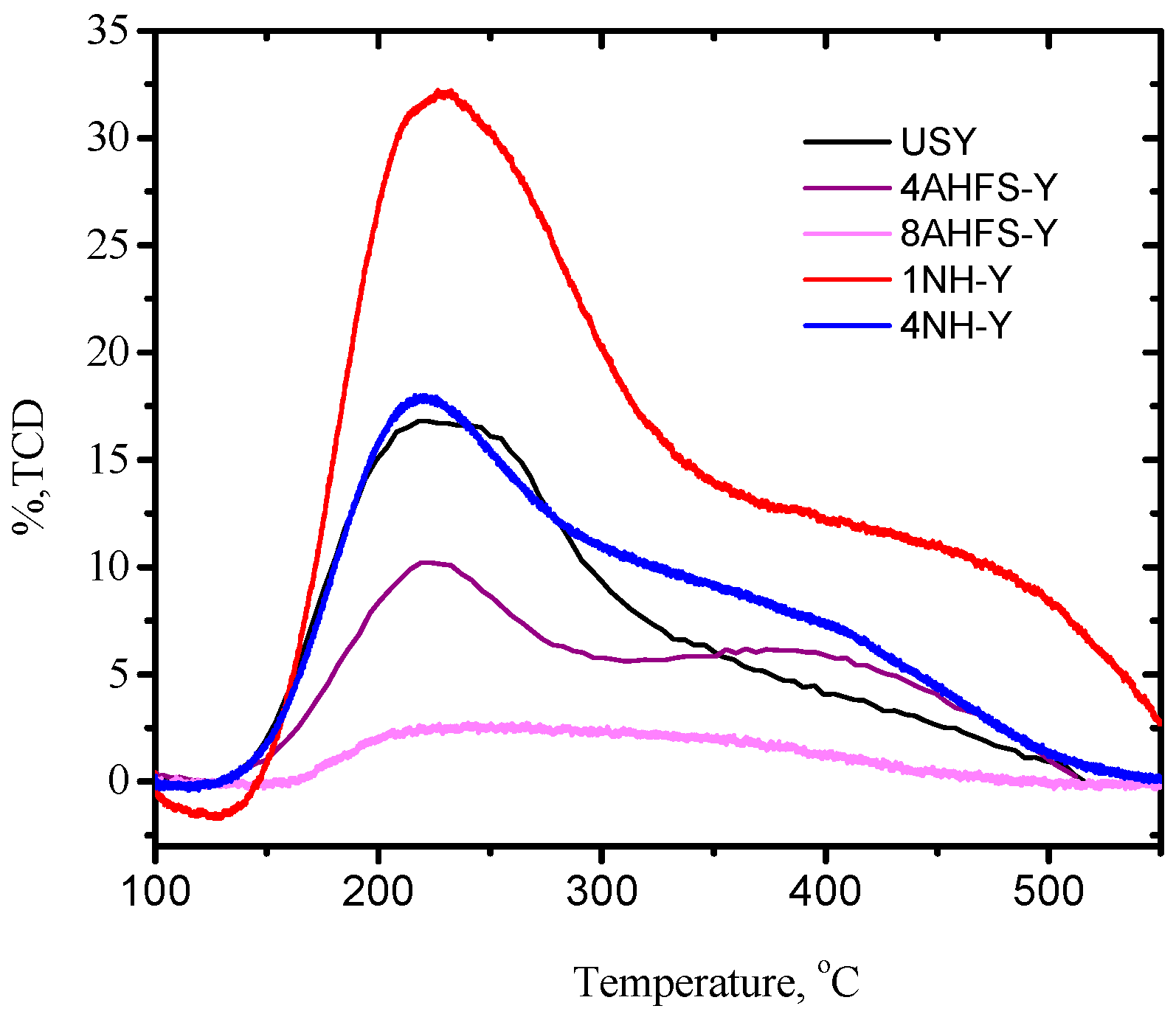
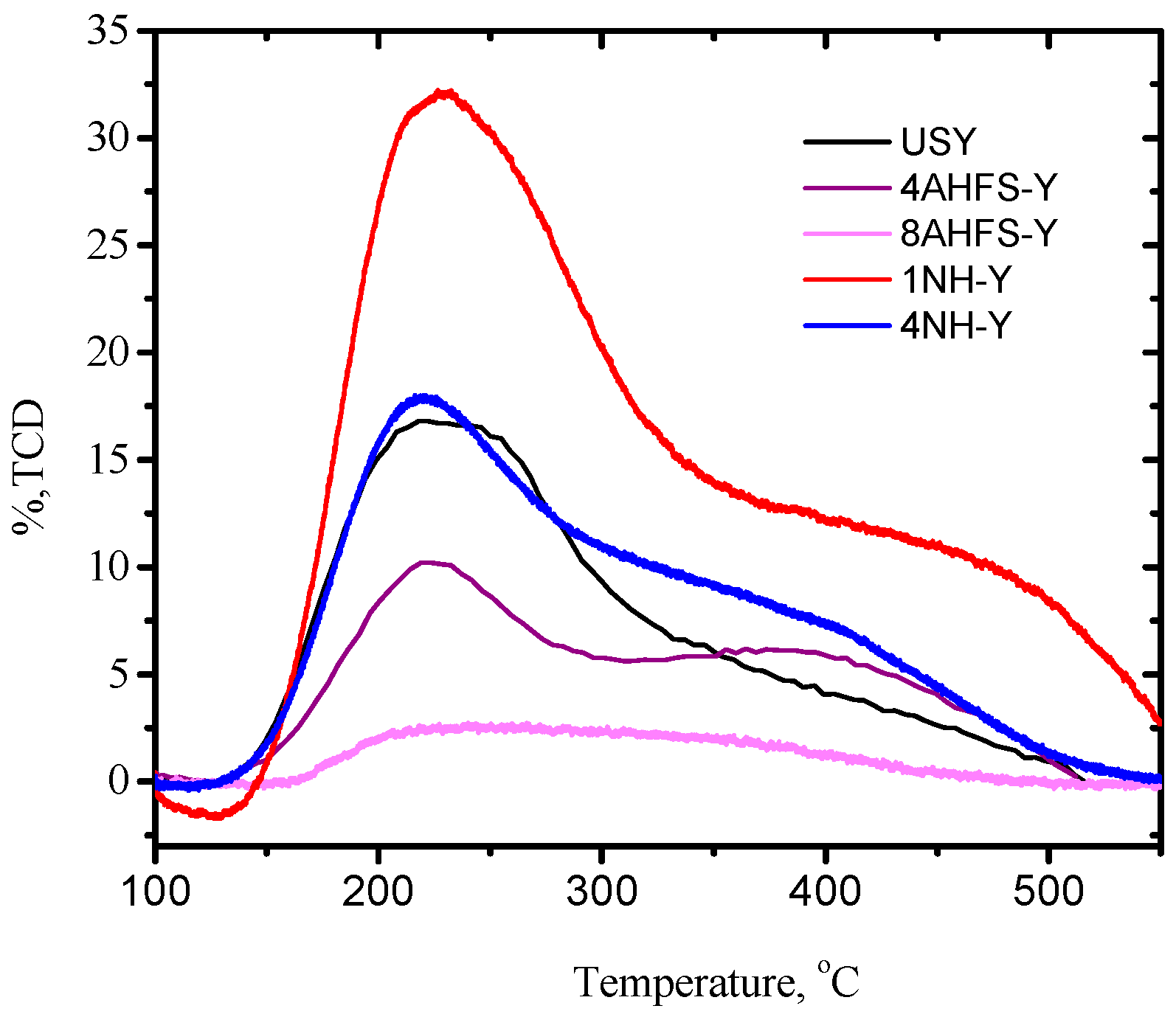
2.2. Acidity Distribution of Desilicated and Dealuminated USY
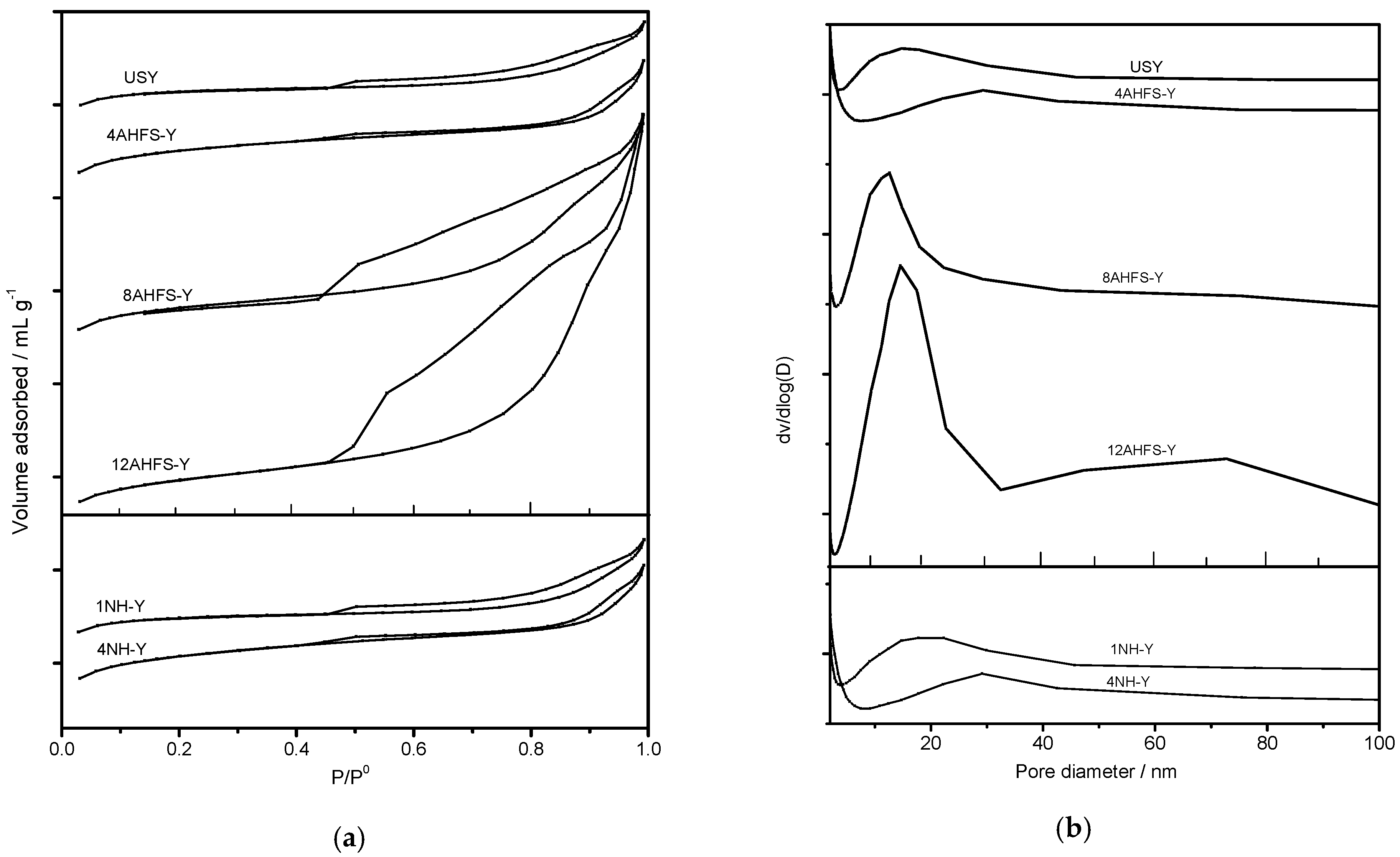
2.3. Pore Size Distribution of Treated USY Zeolites
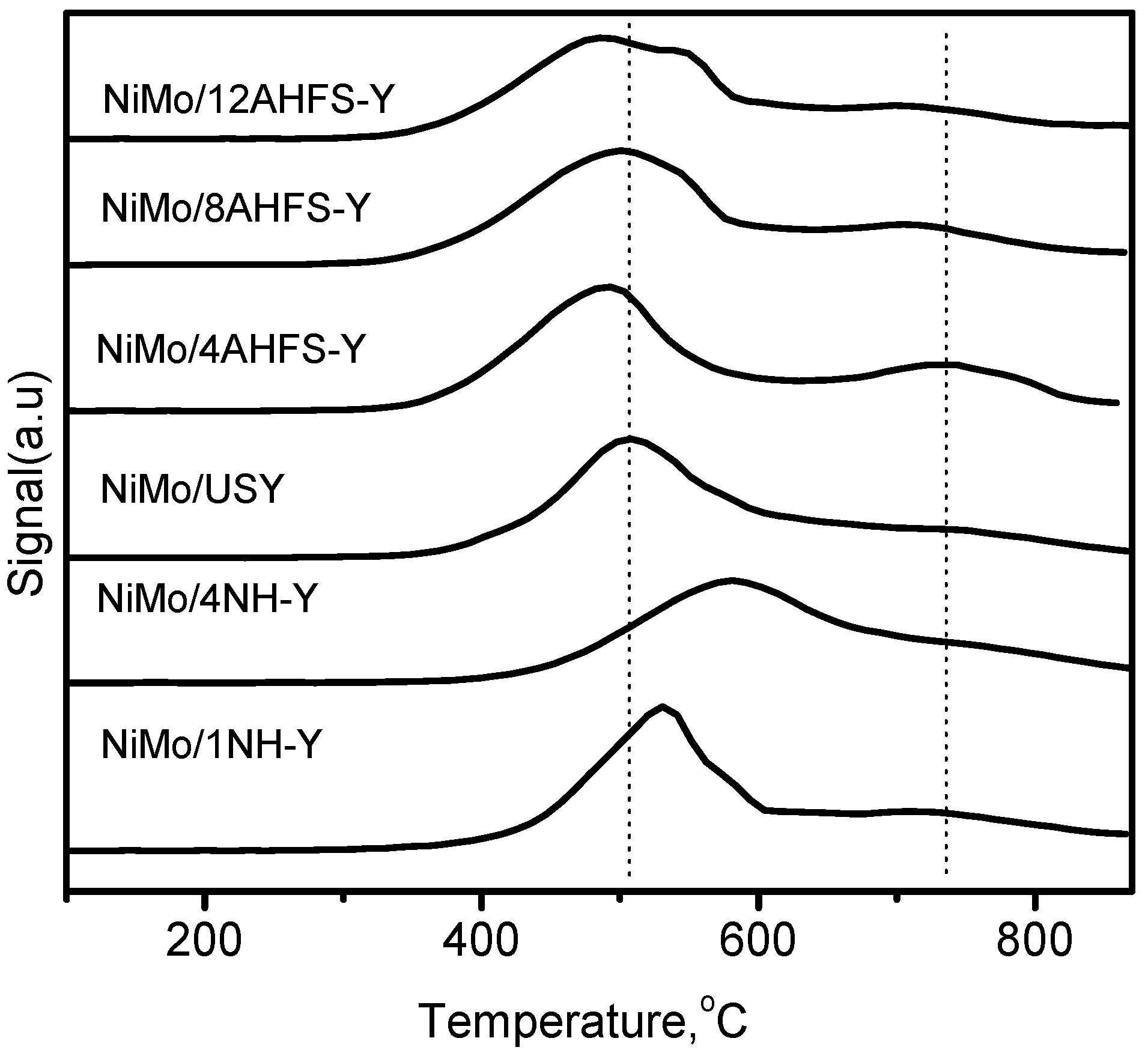
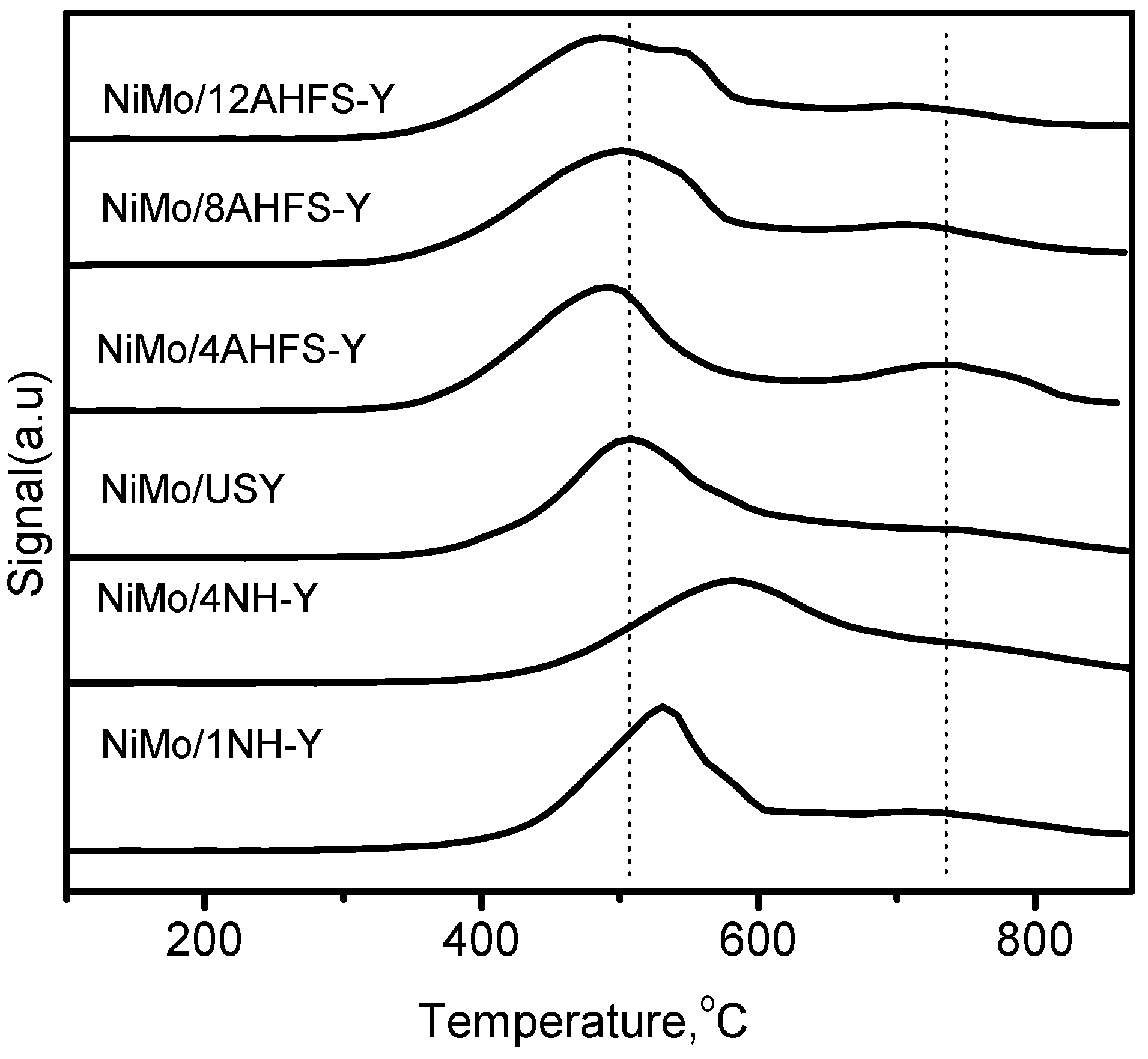
2.4. Influence of Dealumination and Desilication on the Metal State
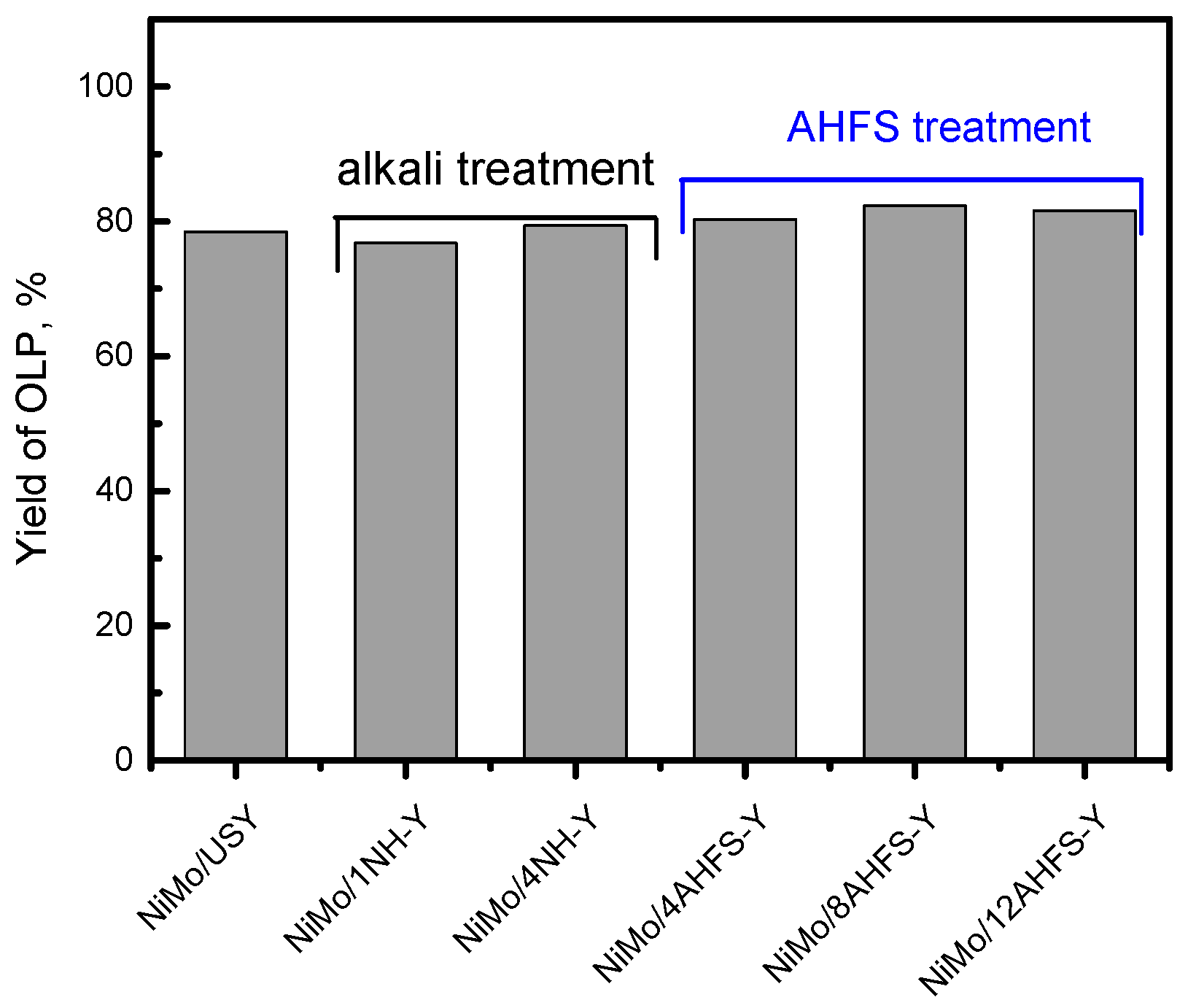
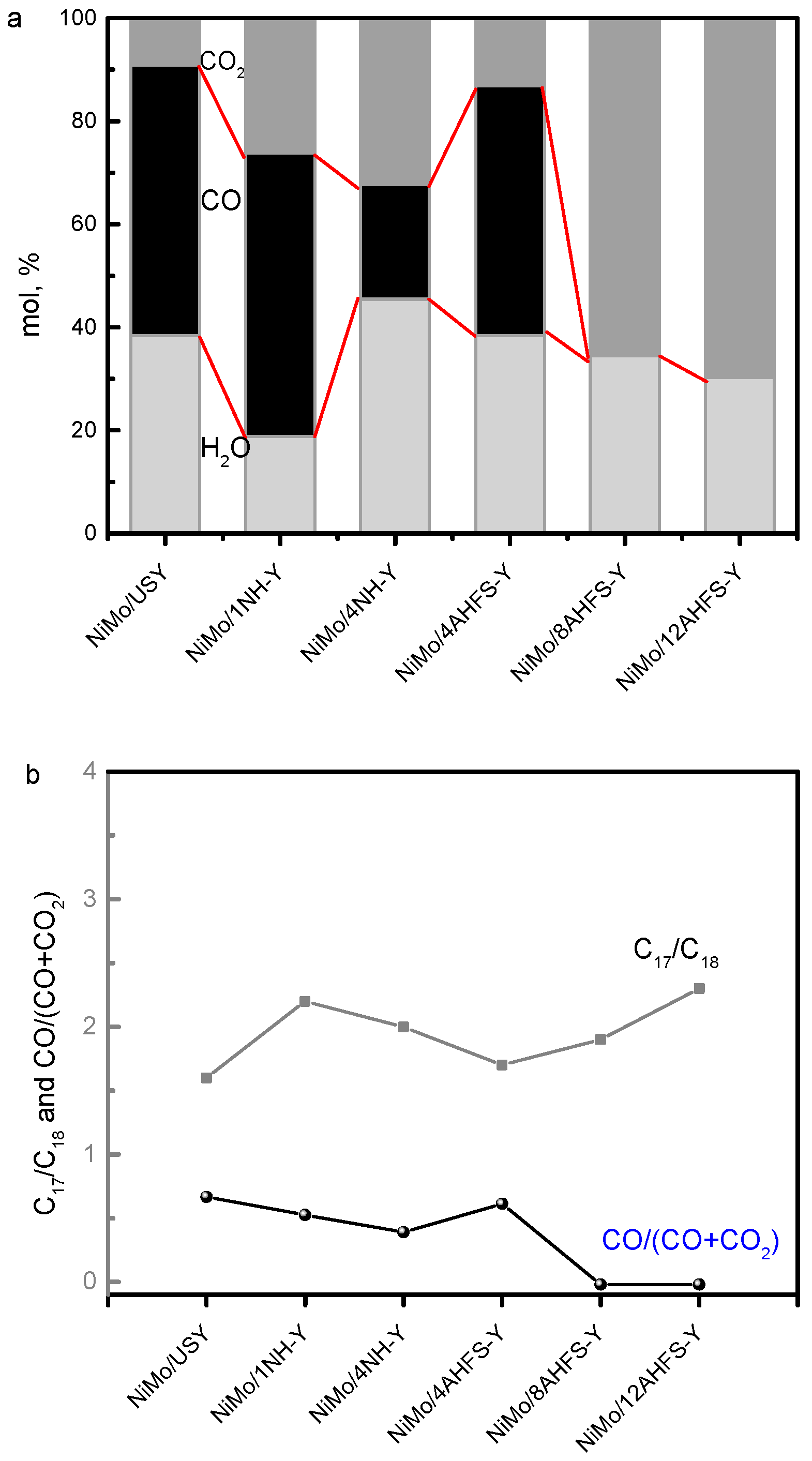
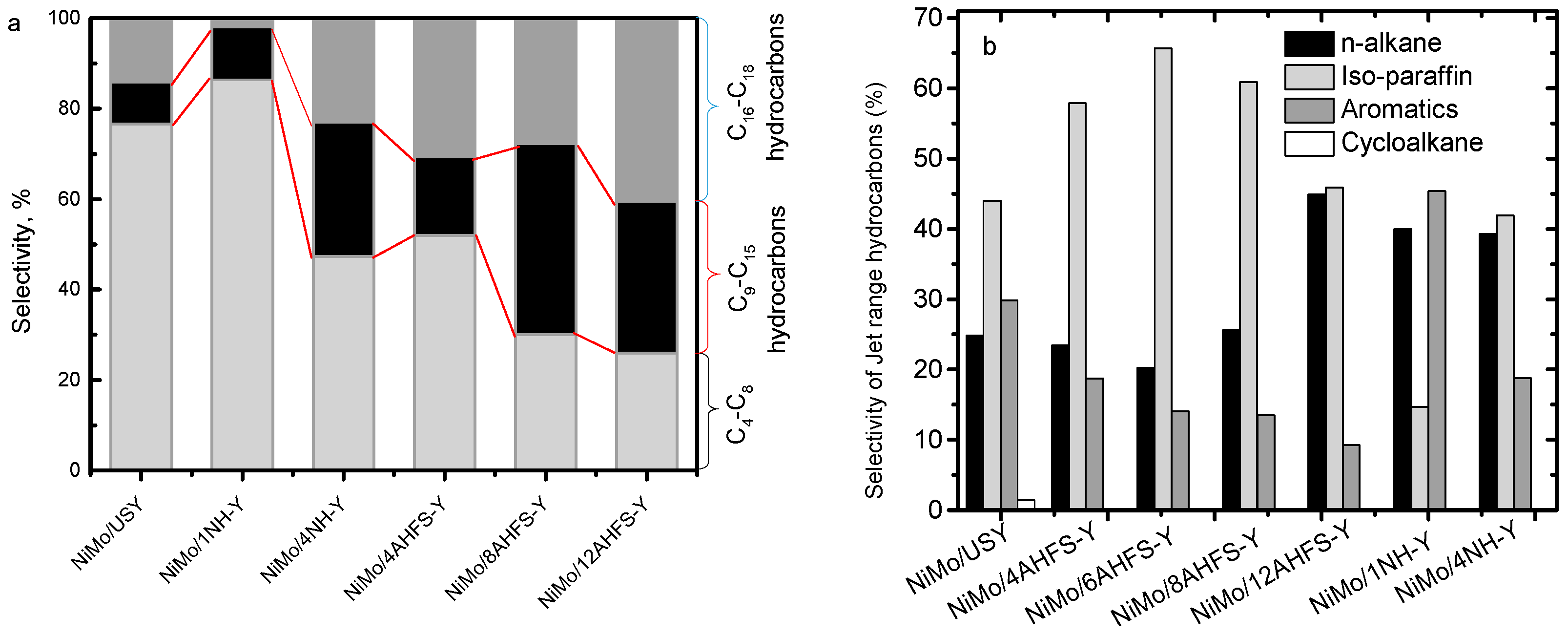
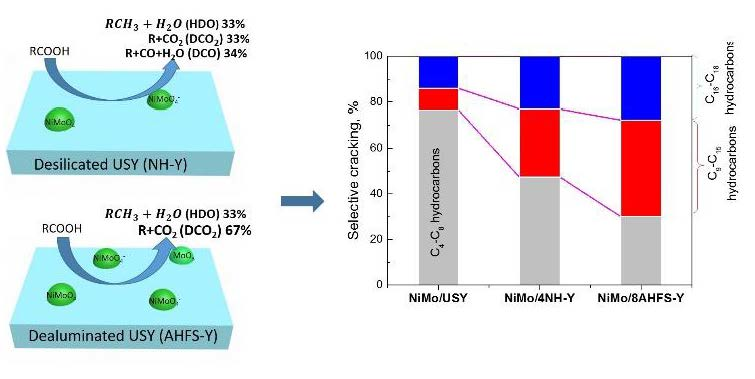
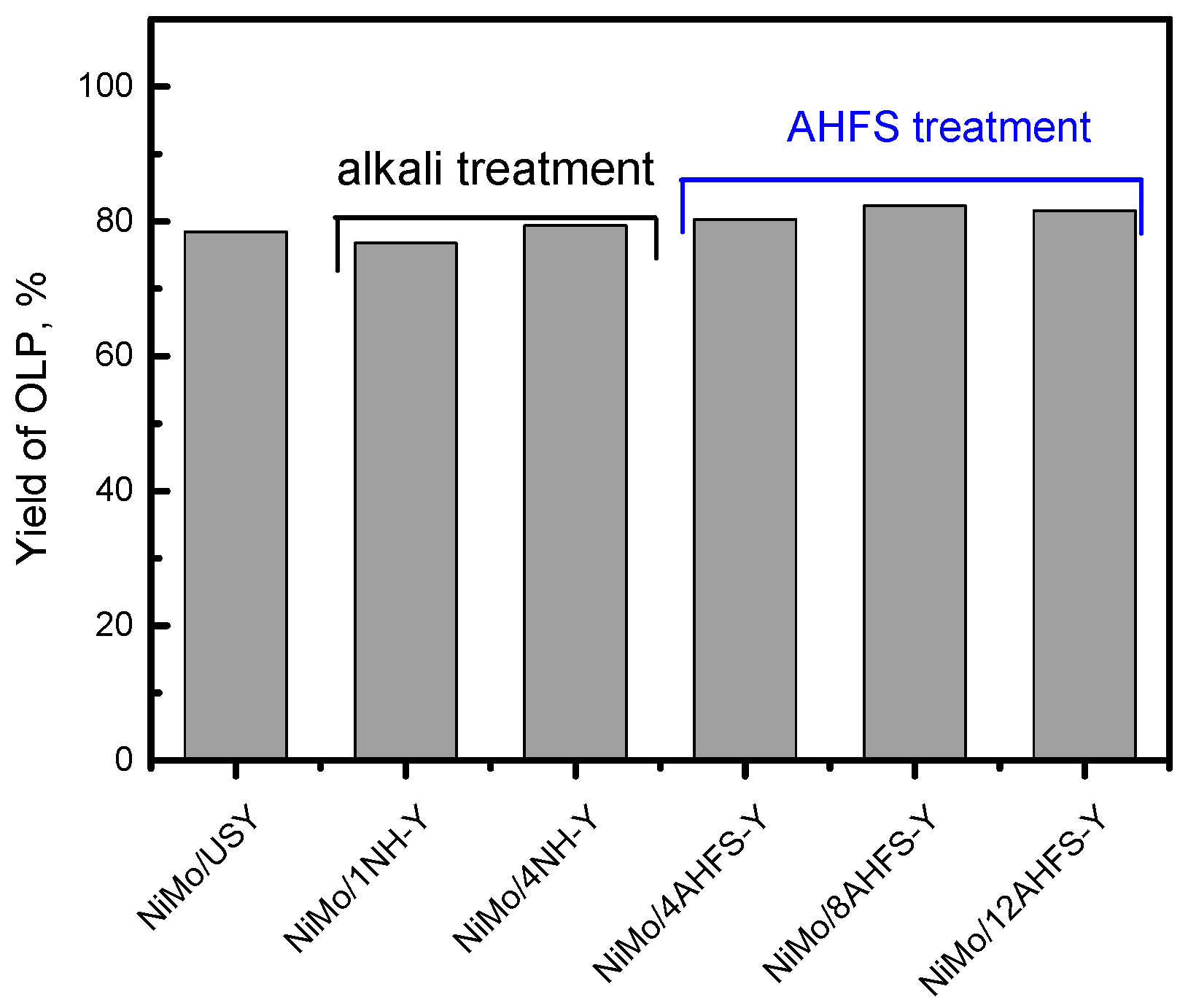
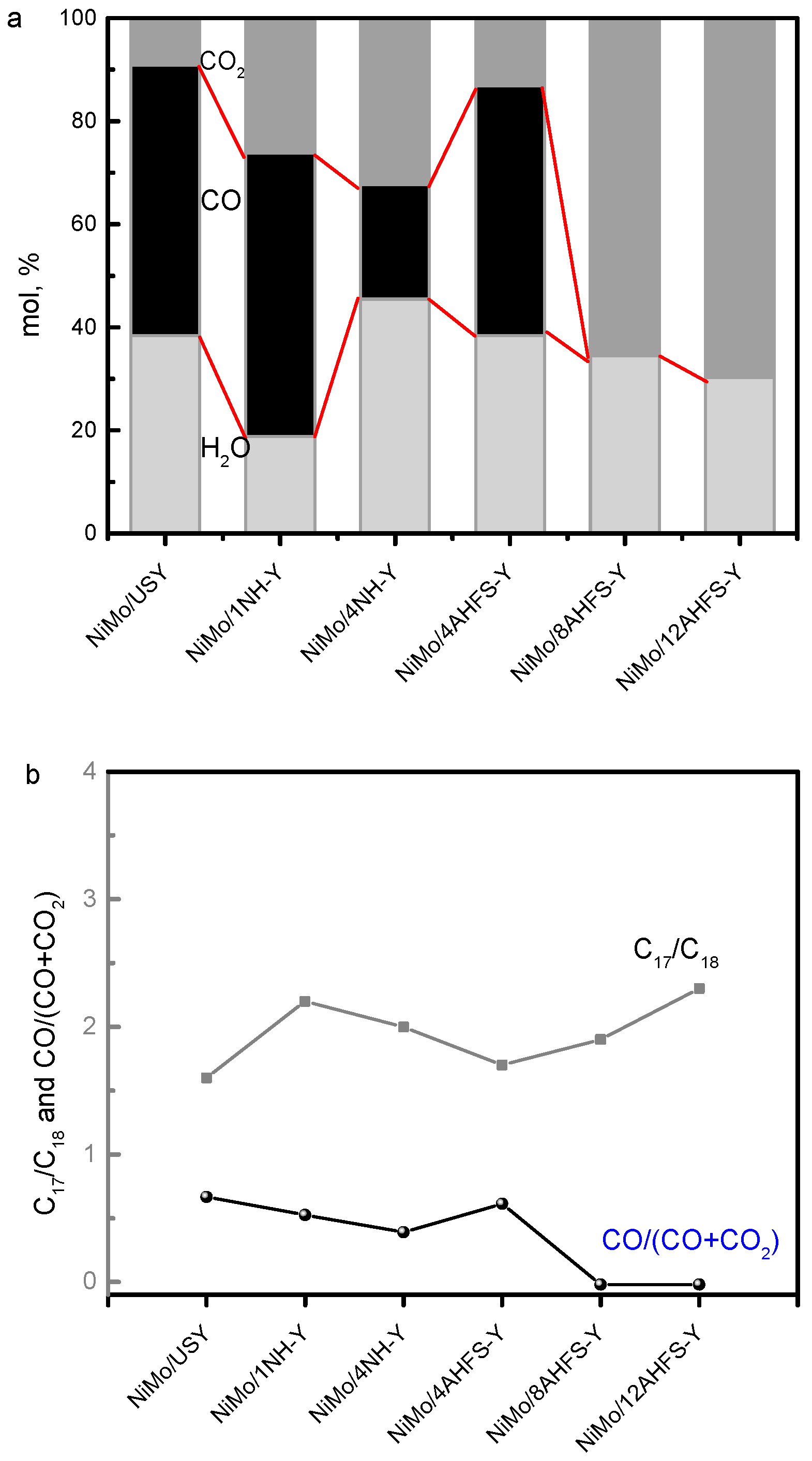
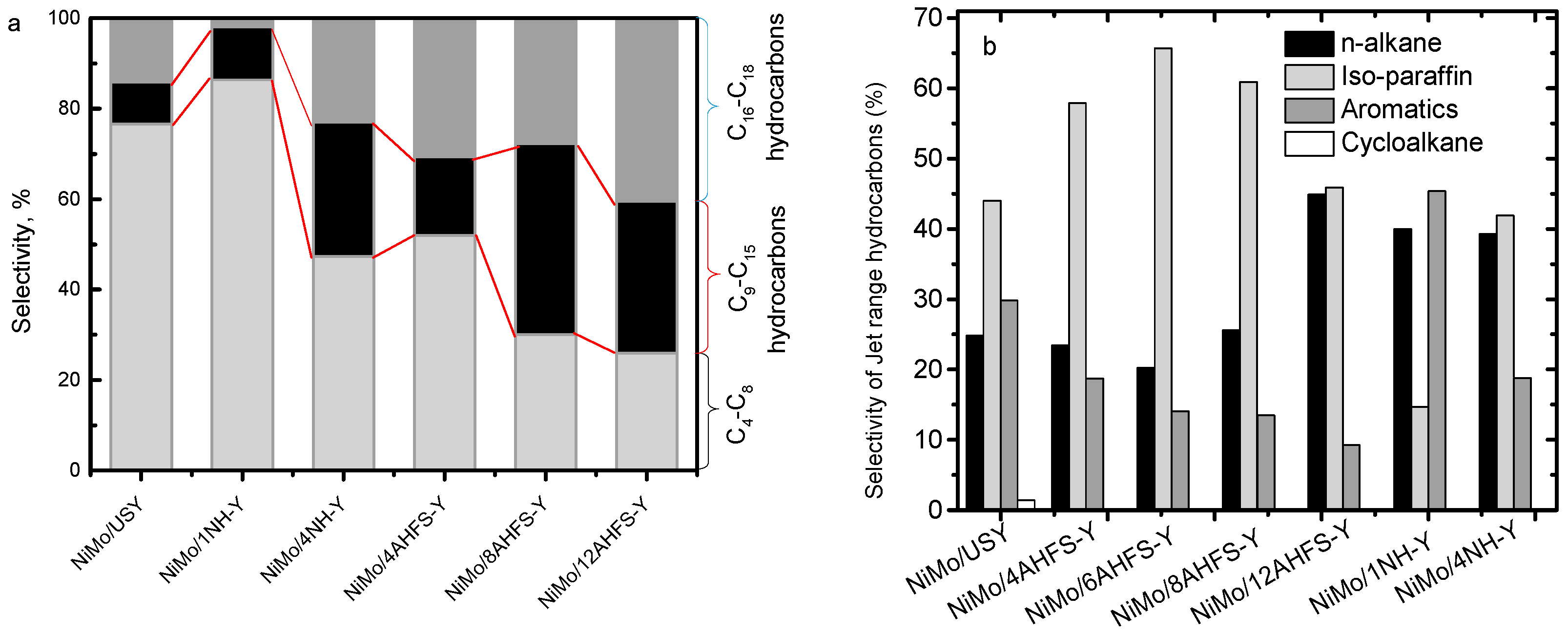
2.5. Hydrotreating of Waste Cooking Oil
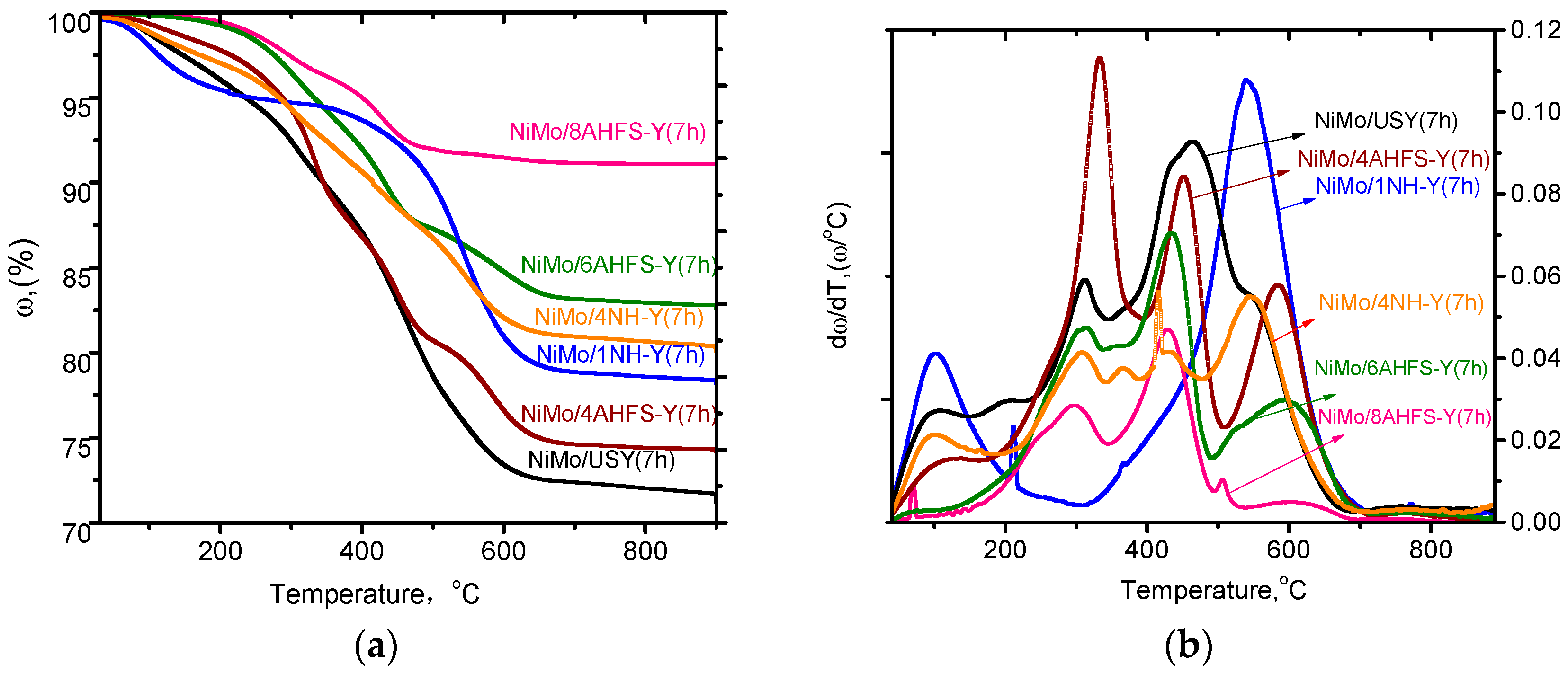
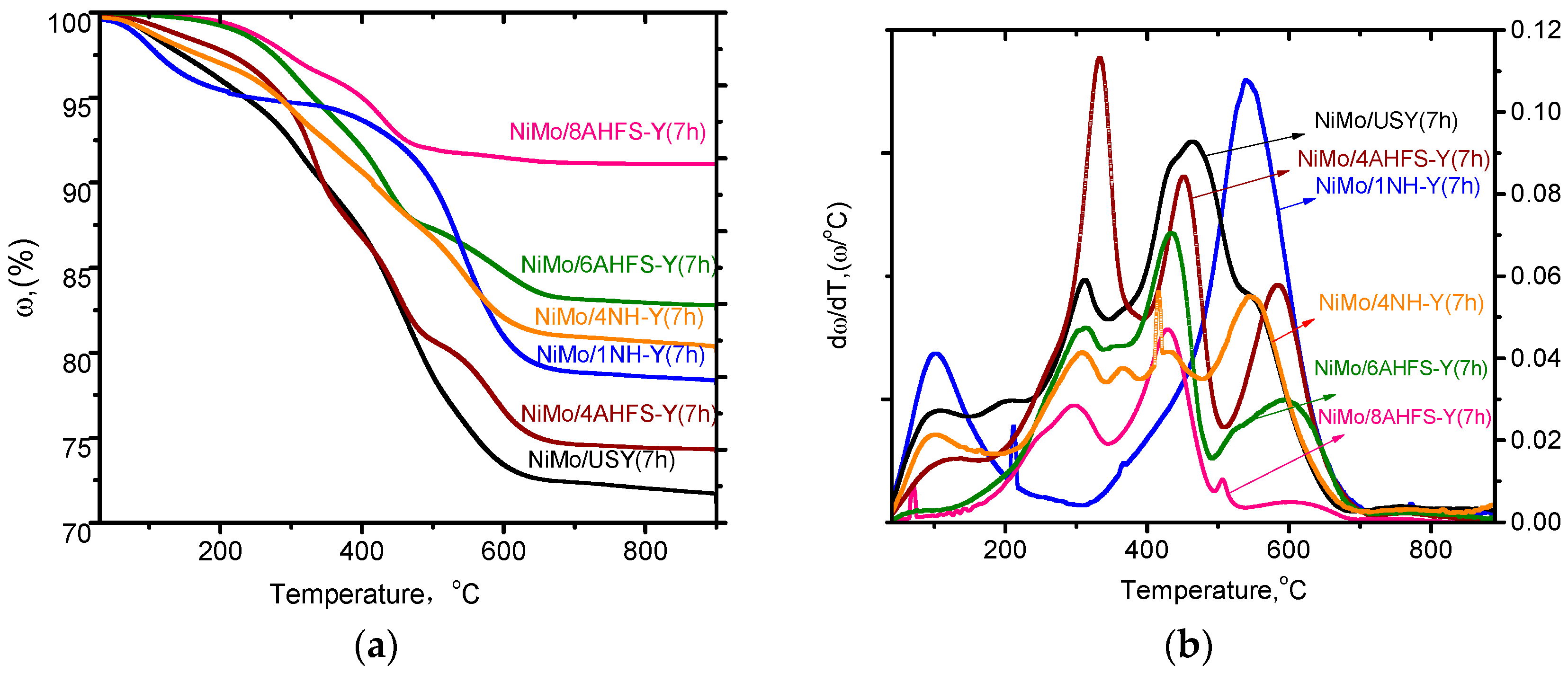
2.6. Stability of Catalysts
3. Experimental
3.1. Materials
3.2. Catalyst Preparation
3.2.1. Alkaline Treatment
3.2.2. AHFS Treatment
3.2.3. Preparation of NiMo/Hierarchical USY
3.3. Characterizations
3.4. Hydrotreating of Waste Cooking Oil
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rye, L.; Blakey, S.; Wilson, C.W. Sustainability of supply or the planet: A review of potential drop-in alternative aviation fuels. Energy Environ. Sci. 2010, 3, 17–27. [Google Scholar] [CrossRef]
- Kon, K.; Onodera, W.; Shimizu, K.I. Selective hydrogenation of levulinic acid to valeric acid and valeric biofuels by a Pt/HMFI catalyst. Catal. Sci. Technol. 2014, 4, 3227–3234. [Google Scholar] [CrossRef]
- Horacek, J.; Homola, F.; Kubickova, I. Lignin to liquids over sulfided catalysts. Catal. Today 2012, 179, 191–198. [Google Scholar] [CrossRef]
- Stumborg, M.; Wong, A.; Hogan, E. Hydroprocessed vegetable oils for diesel fuel improvement. Bioresour. Technol. 1996, 56, 13–18. [Google Scholar] [CrossRef]
- Sandor, K.; Tamas, K.; Artur, T. Fuel production by hydrotreating of triglycerides on NiMo/Al2O3/F catalyst. Chem. Eng. J. 2011, 176, 237–243. [Google Scholar]
- Satyarthi, J.K.; Chiranjeevi, T.; Gokak, D.T.; Viswanathan, P.S. An overview of catalytic conversion of vegetable oils/fats into middle distillates. Catal. Sci. Technol. 2013, 3, 70–80. [Google Scholar] [CrossRef]
- Perego, C.; Ricci, M. Diesel fuel from biomass. Catal. Sci. Technol. 2012, 2, 1776–1786. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Q.; Zhang, X.; Wang, L.; Li, G. Hydrotreating of C18 fatty acids to hydrocarbons on sulphided NiW/SiO2-Al2O3. Fuel Process. Technol. 2013, 116, 165–174. [Google Scholar] [CrossRef]
- Bezergianni, S.; Kalogianni, A. Hydrocracking of used cooking oil for biofuels production. Bioresour. Technol. 2009, 100, 3927–3932. [Google Scholar] [CrossRef] [PubMed]
- Rogelio, S.B.; Fernando, T.Z.; Felipe, J.H.L. Hydroconversion of Triglycerides into Green Liquid Fuels. In Hydrogenation; Karamé, I., Ed.; InTech: Rijeka, Croatia, 2012; pp. 188–216. [Google Scholar]
- Honeywell UOP, Honeywell Green Jet Fuel. Available online: http://www.uop.com/processing-solutions/renewables/green-jet-fuel/#uop-renewable-jet-fuel-process (accessed on 20 October 2016).
- Verma, D.; Kumar, R.; Rana, B.S.; Sinha, A.K. Aviation fuel production from lipids by a single-step route using hierarchical mesoporous zeolites. Energy Environ. Sci. 2011, 4, 1667–1671. [Google Scholar] [CrossRef]
- Verma, D.; Rana, B.S.; Kumar, R.; Sibi, M.G.; Sinha, A.K. Diesel and aviation kerosene with desired aromatics from hydroprocessing of jatropha oil over hydrogenation catalysts supported on hierarchical mesoporous SAPO-11. Appl. Catal. A Gen. 2015, 490, 108–116. [Google Scholar] [CrossRef]
- Rocha Filho, G.N.; Brodzki, D.; Djega-Mariadassou, G. Formation of alkanes, alkylcycloalkanes and alkylbenzenes during the catalytic hydrocracking of vegetable oils. Fuel 1993, 72, 543–549. [Google Scholar] [CrossRef]
- Morais, S.; Mata, T.M.; Martins, A.A.; Pinto, G.A.; Costa, C.A.V. Simulation and life cycle assessment of process design alternatives for biodiesel production from waste vegetable oils. J. Clean. Prod. 2010, 18, 1251–1259. [Google Scholar] [CrossRef]
- Chiaramonti, D.; Prussi, M.; Buffi, M.; Tacconi, D. Sustainable bio kerosene: Process routes and industrial demonstration activities in aviation biofuels. Appl. Energy 2014, 136, 767–774. [Google Scholar] [CrossRef]
- Talebian-Ki akalaieha, A.; Amina, N.A.S.; Mazaheria, H. A review on novel processes of biodiesel production from waste cooking oil. Appl. Energy 2013, 104, 683–710. [Google Scholar] [CrossRef]
- Yaakob, Z.; Mohammad, M.; Alherbawi, M.; Alam, Z.; Sopian, K. Overview of the production of biodiesel from Waste cooking oil. Renew. Sustain. Energy Rev. 2013, 18, 184–193. [Google Scholar] [CrossRef]
- Li, T.; Cheng, J.; Huang, R.; Zhou, J.; Cen, K. Conversion of waste cooking oil to jet biofuel with nickel-based mesoporous zeolite Y catalyst. Bioresour. Technol. 2015, 197, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhu, Q.; Guan, Q.; He, L.; Li, W. Bio-aviation fuel production from hydroprocessing castor oil promoted by the nickel-based bifunctional catalyst. Bioresour. Technol. 2015, 183, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Garralon, G.; Fornes, V.; Corma, A. Faujasites dealuminated with ammonium hexafluorosilicate: Variables affecting the method of preparation. Zeolites 1988, 8, 268–272. [Google Scholar] [CrossRef]
- Silaghi, M.C.; Chizallet, C.; Raybaud, P. Challenges on molecular aspects of dealumination and desilication of zeolites. Microporous Mesoporous Mater. 2014, 191, 82–96. [Google Scholar] [CrossRef]
- Verboekend, D.; Nuttens, N.; Locus, R.; Van Aelst, J.; Verolme, P.; Groen, J.C.; Pérez-Ramírez, J.; Selsa, B.F. Synthesis, characterisation, and catalytic evaluation of hierarchical faujasite zeolites: Milestones, challenges, and future directions. Chem. Soc. Rev. 2016, 45, 3331–3352. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, P.Z.; Yan, B.; Wang, S.; Shen, Y.; Ma, X. Modification of Y Zeolite with Alkaline Treatment: Textural Properties and Catalytic Activity for Diethyl Carbonate Synthesis. Ind. Eng. Chem. Res. 2013, 52, 6349–6356. [Google Scholar] [CrossRef]
- Qin, Z.; Shen, B.; Gao, X.; Lin, F.; Wang, B.; Xu, C. Mesoporous Y zeolite with homogeneous aluminum distribution obtained by sequential desilication–dealumination and its performance in the catalytic cracking of cumene and 1,3,5-triisopropylbenzen. J. Catal. 2011, 278, 266–275. [Google Scholar] [CrossRef]
- Qin, Z.; Shen, B.; Yu, Z.W.; Deng, F.; Zhao, L.; Zhou, S.; Yuan, D.; Gao, X.; Wang, B.; Zhao, H.; et al. A defect-based strategy for the preparation of mesoporous zeolite Y for high-performance catalytic cracking. J. Catal. 2013, 298, 102–111. [Google Scholar] [CrossRef]
- Donk, S.; Janssen, A.H.; Bitter, J.H. Generation, Characterization, and Impact of Mesopores in Zeolite Catalysts. Catal. Rev. 2003, 45, 297–319. [Google Scholar] [CrossRef]
- Corma, A.; Fornes, V.; Rey, F. Extraction of extra-framework aluminium in ultrastable Y zeolites by (NH4)2SiF6 treatments: I. Physicochemical Characterization. Appl. Catal. 1990, 37, 267–274. [Google Scholar] [CrossRef]
- Xu, B.; Bordiga, S.; Prins, R.; Van Bokhoven, J.A. Effect of framework Si/Al ratio and extra-framework aluminum on the catalytic activity of Y zeolite. Appl. Catal. A Gen. 2007, 333, 245–253. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, Y.Z.; Zheng, L.B. Isomorphous subsitution of Faujusite with (NH4)2SiF6: Reaction conditions and properties of products. J. Catal. 1991, 13, 32–37. [Google Scholar]
- Peng, X.; Zhang, Y.; Zheng, L.B. Isomorphous subsitution of Faujusite with (NH4)2SiF6: Reaction mechanism. J. Catal. 1993, 14, 300–306. [Google Scholar]
- Cordero, R.L.; Agudo, A.L. Effect of water extraction on the surface properties of Mo/Al2O3 and NiMo/Al2O3 hydrotreating catalysts. Appl. Catal. A Gen. 2000, 202, 23–35. [Google Scholar] [CrossRef]
- Cordero, R.L.; Llambias, F.J.G.; Agudo, A.L. Temperature-programmed reduction and zeta potential studies of the structure of MoO3/Al2O3 andMoO3/SiO2 catalysts effect of the impregnation pH and molybdenum loading. Appl. Catal. 1991, 74, 125–136. [Google Scholar] [CrossRef]
- Solis, D.; Agudo, A.L.; Ramírez, J.; Klimova, T. Hydrodesulfurization of hindered dibenzothiophenes on bifunctional NiMo catalysts supported on zeolite–alumina composites. Catal. Today 2006, 116, 469–477. [Google Scholar] [CrossRef]
- Gong, S.; Shinozaki, A.; Qian, E.W. Role of Support in Hydrotreatment of Jatropha Oil over Sulfided NiMo Catalysts. Ind. Eng. Chem. Res. 2012, 51, 13953–13960. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Q.; Zhang, X.; Wang, L. Quantitative conversion of triglycerides to hydrocarbons over hierarchical ZSM-5 catalyst. Appl. Catal. B Environ. 2015, 166–167, 327–334. [Google Scholar] [CrossRef]
- Adjaye, J.D.; Katikaneni, S.P.R.; Bakhshi, N.N. Catalytic conversion of a biofuel to hydrocarbons: Effect of mixtures of HZSM-5 and silica-alumina catalysts on product distribution. Fuel Process. Technol. 1996, 48, 115–143. [Google Scholar] [CrossRef]
- Liu, X.J.; Guan, L.L.; Fu, X.N.; Zhao, Y.; Wu, J.D.; Xu, N. Nanocone arrays synthesized by plasma-assisted reaction deposition. Nanoscale Res. Lett. 2014, 9, 550. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, C.S.; Liu, Y.G.; Hou, X.; Zhang, R.; Tang, X. Coke Deposition on Ni/HZSM-5 in Bio-oil Hydrodeoxygenation Processing. Energy Fuels. 2015, 29, 1722–1728. [Google Scholar] [CrossRef]
- Silva, A.O.S.; Souza, M.J.B.; Aquino, J.M.F.B.; Fernandes, V.J., Jr.; Araujo, A.S. Acid properties of the HZSM-12 zeolite with different Si/Al ratio by thermo-programmed desorption. J. Therm. Anal. Calorim. 2004, 76, 783–791. [Google Scholar] [CrossRef]
- Liu, J.X.; Cai, G.; Yang, L.X.; Gao, X.-Y.; Ji, P.; Chen, G.-Q. The coking behaviour of zeolife catalysts during the conversion of methanol to lower olefins. J. Catal. 1985, 6, 238–244. [Google Scholar]
- Breck, D.W.; Flanigen, E.M. Molecular Sieves; Society of Chemical: London, UK, 1968. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ao (A) | Si/AlXRD | Si/AlXRF | Relative Crystallinity (%) |
|---|---|---|---|---|
| USY | 24.515 | 4.1 | 8.8 | 100 |
| 4AHFS-Y | 24.394 | 7.2 | 8.9 | 49 |
| 8AHFS-Y | 24.247 | 28.8 | 10.2 | 37 |
| 12AHFS-Y | 24.246 | 29.3 | 12.3 | 17 |
| 1NH-Y | 24.485 | 4.7 | 6.6 | 50 |
| 4NH-Y | 24.547 | 3.7 | 4.9 | 31 |
| Sample | B | L | Weak Acidity | Strong Acidity | B/L |
|---|---|---|---|---|---|
| USY | 1851 | 634 | 626 | 660 | 2.92 |
| 4AHFS-Y | 1440 | 483 | 402 | 857 | 2.98 |
| 8AHFS-Y | 160 | 55 | 66 | 230 | 2.91 |
| 1NH-Y | 2374 | 602 | 1545 | 1518 | 3.94 |
| 4NH-Y | 1604 | 1254 | 658 | 1130 | 1.28 |
| Samples | Smicro a | Sext b | Vmicro c | Vmeso d | HF e |
|---|---|---|---|---|---|
| m2/g | m2/g | cm3/g | cm3/g | ||
| USY | 609 | 49 | 0.28 | 0.07 | 0.0596 |
| 4AHFS-Y | 598 | 57 | 0.28 | 0.10 | 0.0992 |
| 8AHFS-Y | 381 | 92 | 0.18 | 0.17 | 0.1000 |
| 12AHFS-Y | 196 | 106 | 0.09 | 0.29 | 0.0831 |
| 1NH-Y | 618 | 48 | 0.29 | 0.07 | 0.0581 |
| 4NH-Y | 551 | 101 | 0.26 | 0.10 | 0.1119 |
| Property | Waste Cooking Oil |
|---|---|
| Palimitic acid (C16:0, wt %) | 25.02 |
| Linoleic acid (C18:2, wt %) | 28.85 |
| Oleic acid (C18:1, wt %) | 39.69 |
| Stearic acid (C18:0, wt %) | 6.27 |
| Other acid (wt %) | 0.17 |
| Acid value (mg KOH−1) | 65.47 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Wang, Q.; Chen, H.; Zhang, X. Hydroconversion of Waste Cooking Oil into Green Biofuel over Hierarchical USY-Supported NiMo Catalyst: A Comparative Study of Desilication and Dealumination. Catalysts 2017, 7, 281. https://doi.org/10.3390/catal7100281
Zhang Z, Wang Q, Chen H, Zhang X. Hydroconversion of Waste Cooking Oil into Green Biofuel over Hierarchical USY-Supported NiMo Catalyst: A Comparative Study of Desilication and Dealumination. Catalysts. 2017; 7(10):281. https://doi.org/10.3390/catal7100281
Chicago/Turabian StyleZhang, Zongwei, Qingfa Wang, Hao Chen, and Xiangwen Zhang. 2017. "Hydroconversion of Waste Cooking Oil into Green Biofuel over Hierarchical USY-Supported NiMo Catalyst: A Comparative Study of Desilication and Dealumination" Catalysts 7, no. 10: 281. https://doi.org/10.3390/catal7100281
APA StyleZhang, Z., Wang, Q., Chen, H., & Zhang, X. (2017). Hydroconversion of Waste Cooking Oil into Green Biofuel over Hierarchical USY-Supported NiMo Catalyst: A Comparative Study of Desilication and Dealumination. Catalysts, 7(10), 281. https://doi.org/10.3390/catal7100281