
Controlling Active Site Loop Dynamics in the (β/α)8 Barrel Enzyme Indole-3-Glycerol Phosphate Synthase



Abstract
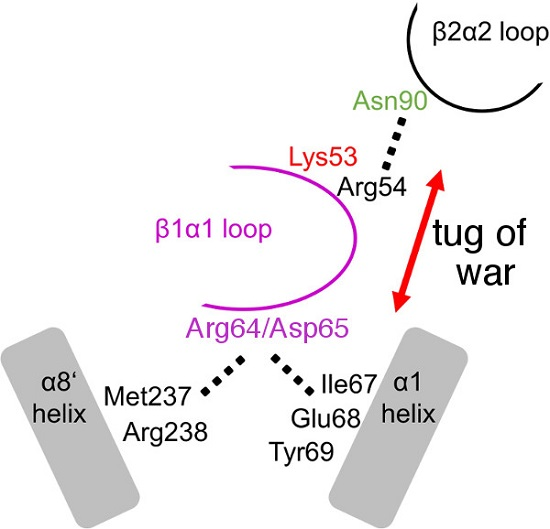
:
1. Introduction
2. Results
2.1. Severing Interactions with the C-Terminal Side of the β1α1 Loop Results in Modest Decreases in Steady-State Kinetics
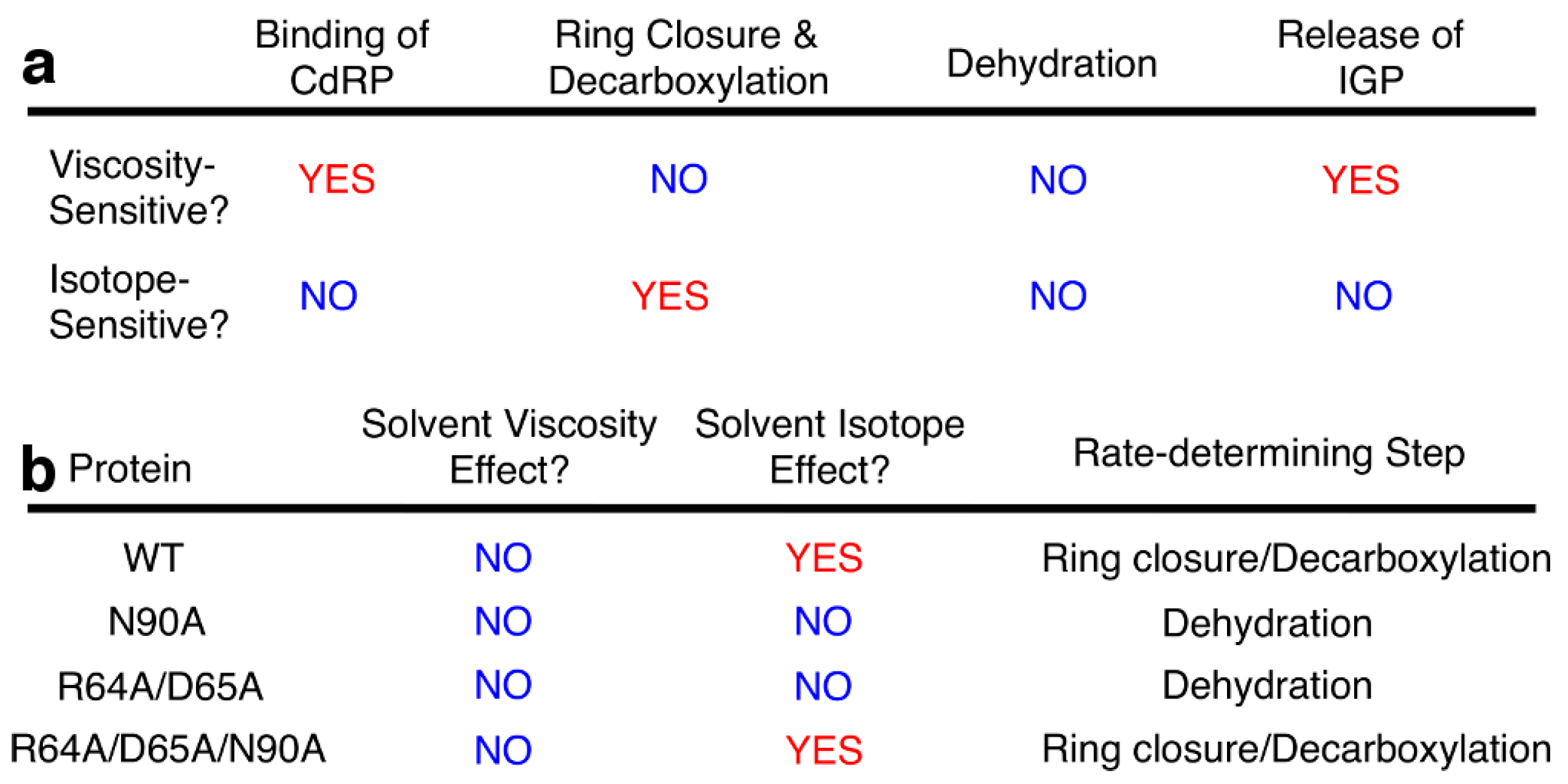
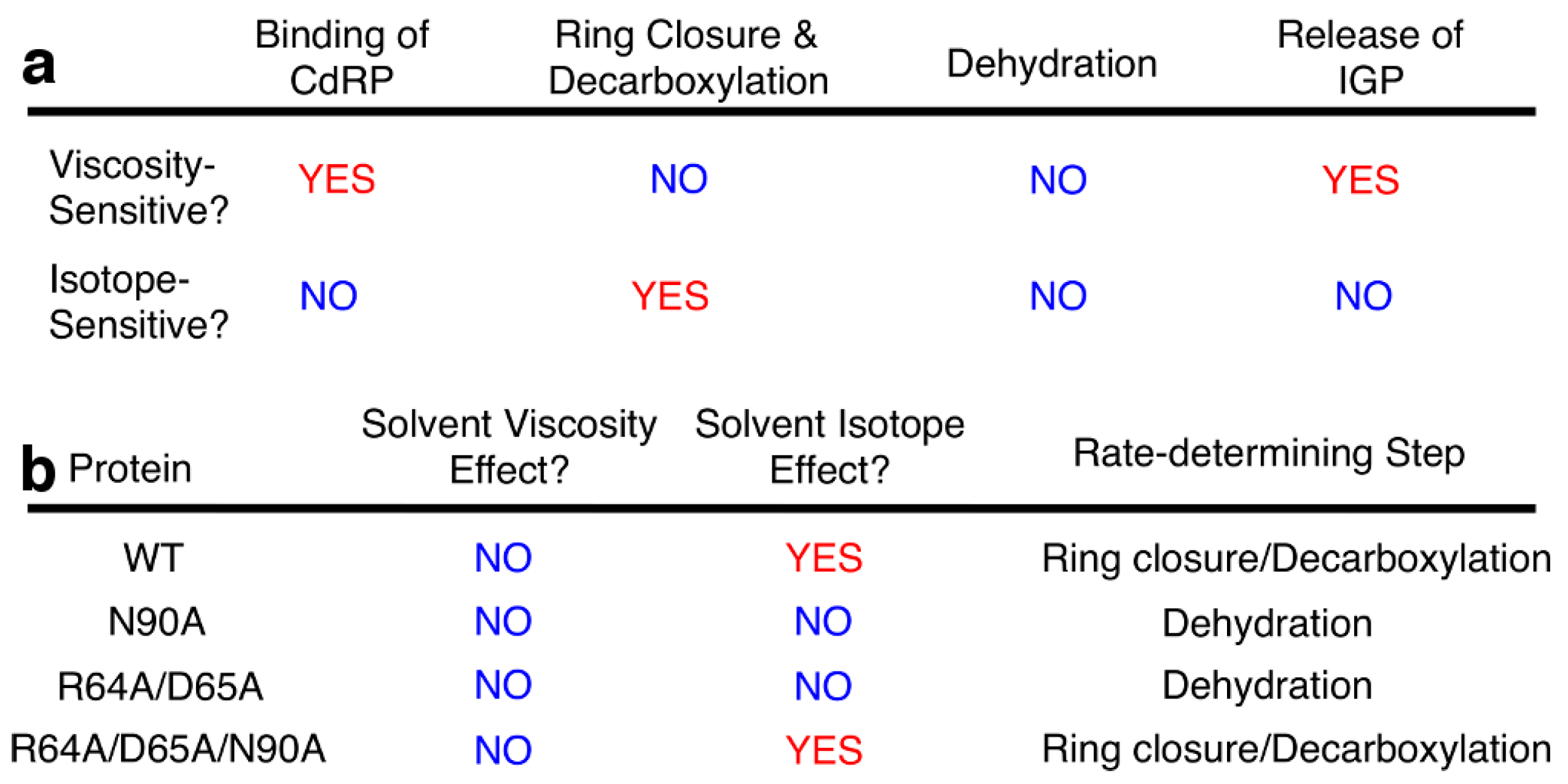
2.2. Severing Interactions with the C-Terminal Side of the β1α1 Loop Changes the Identity of the Rate-Determining Step
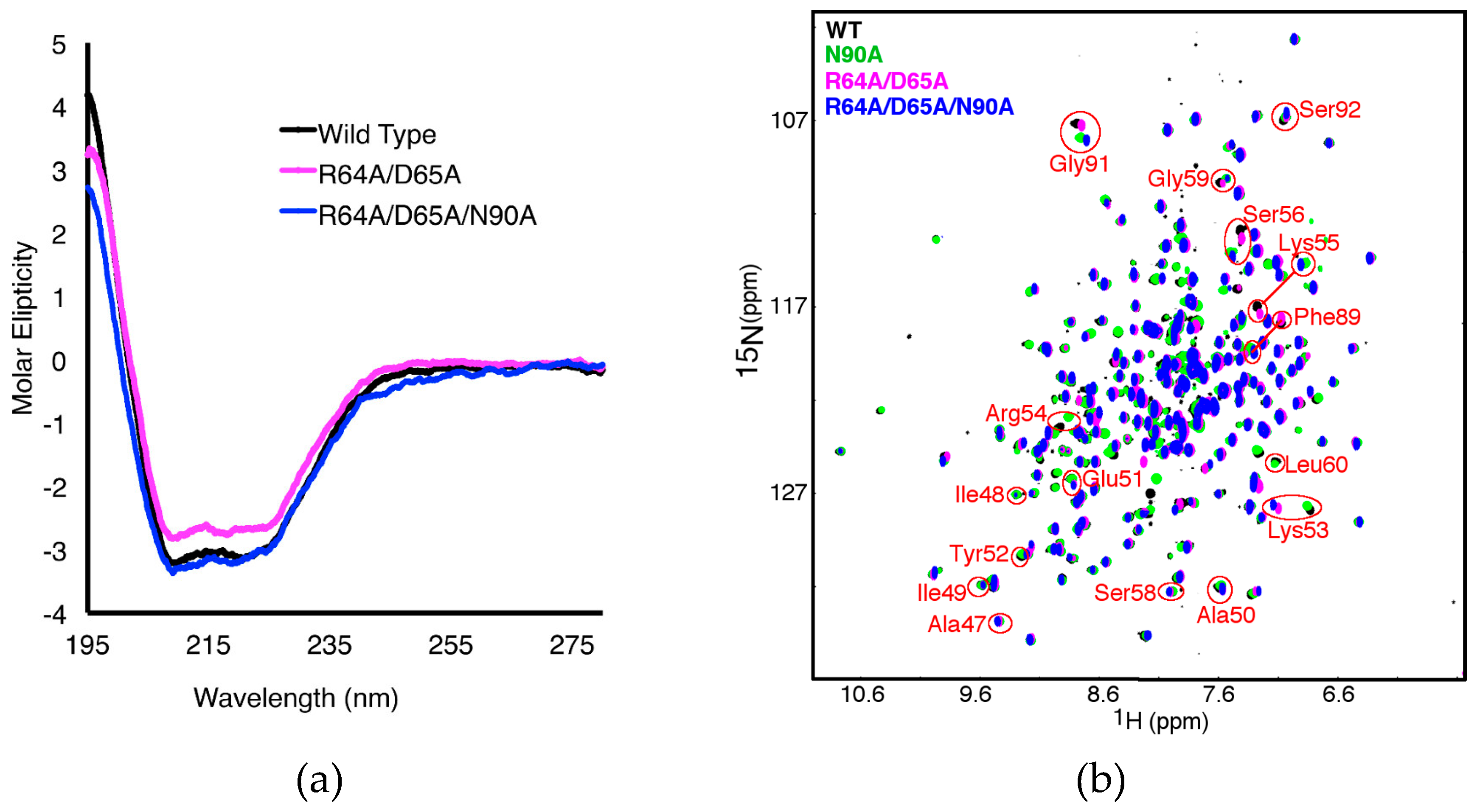
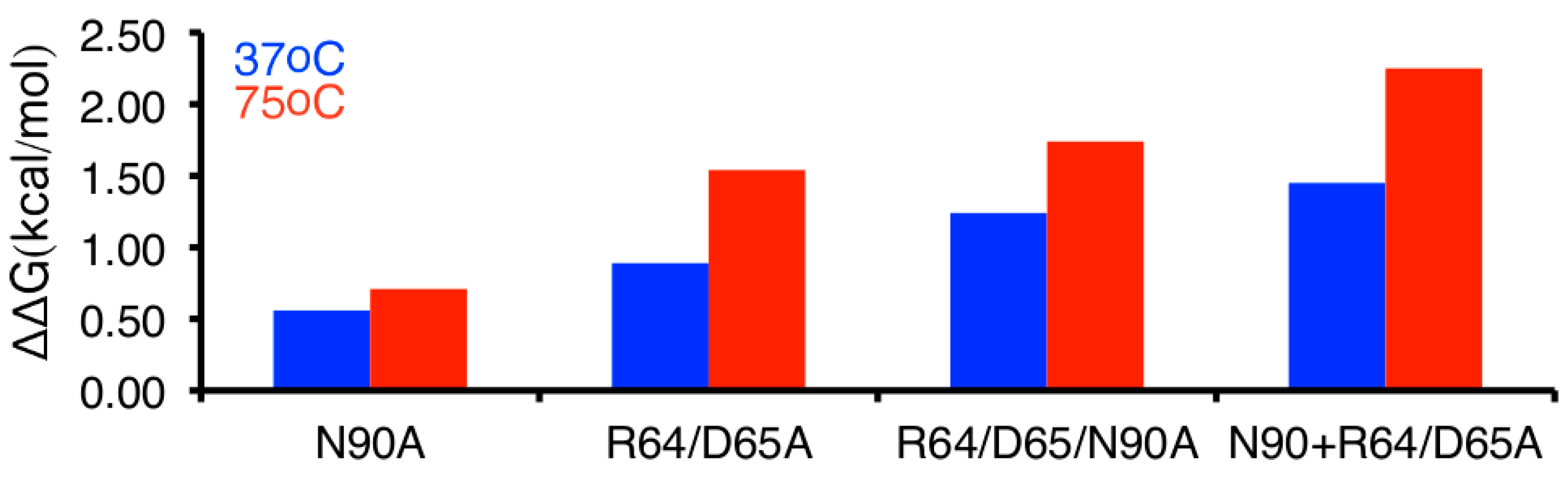
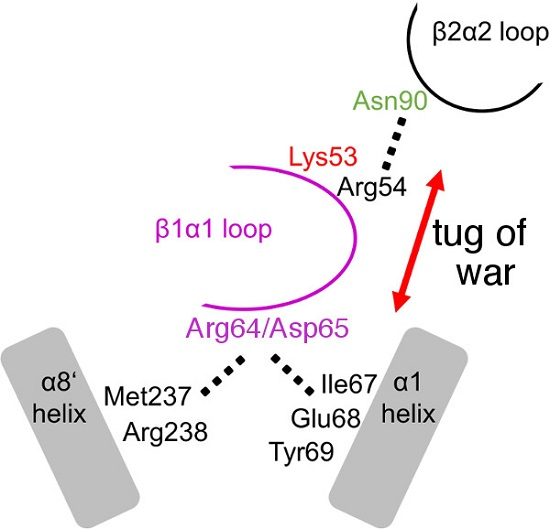
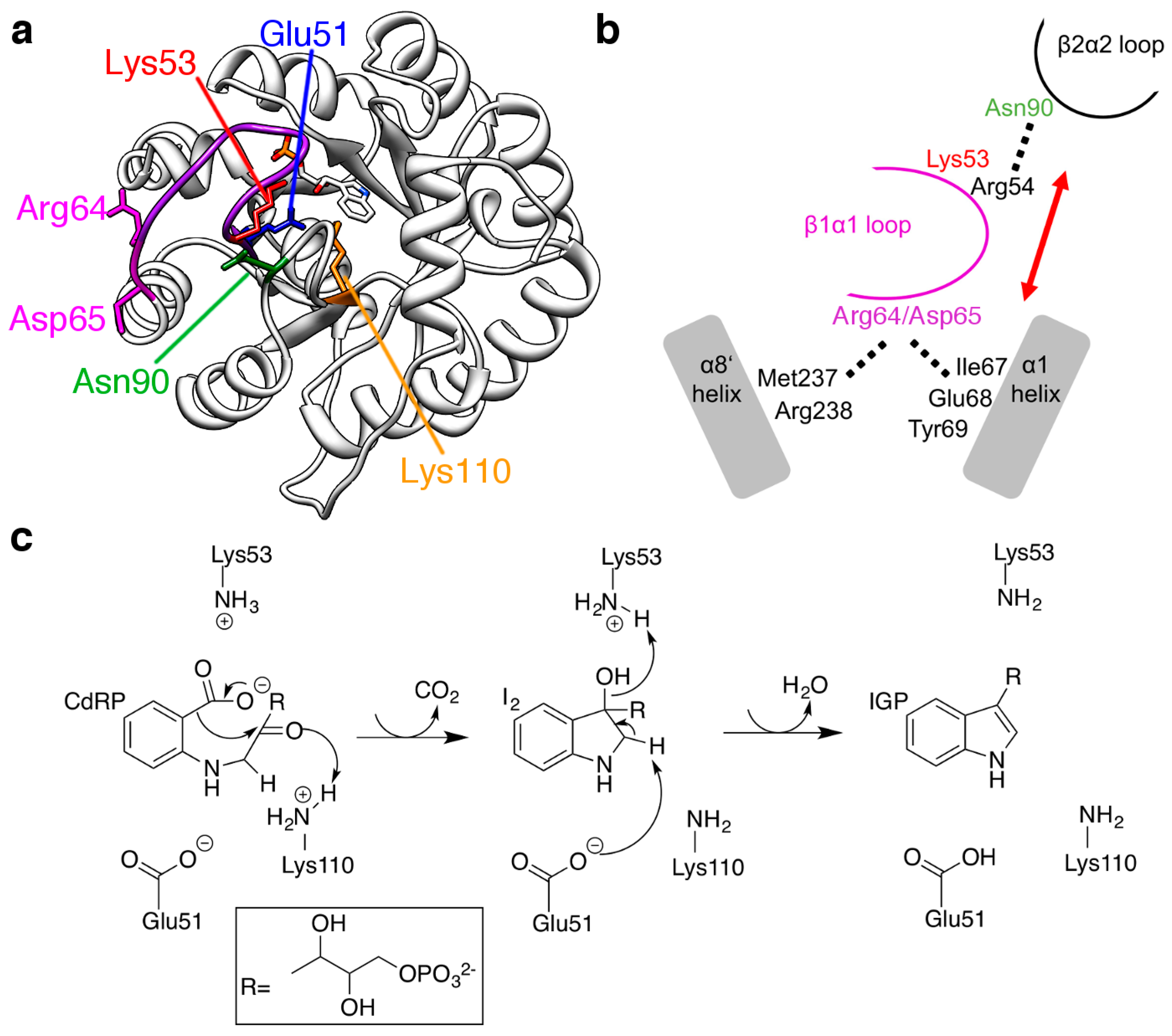
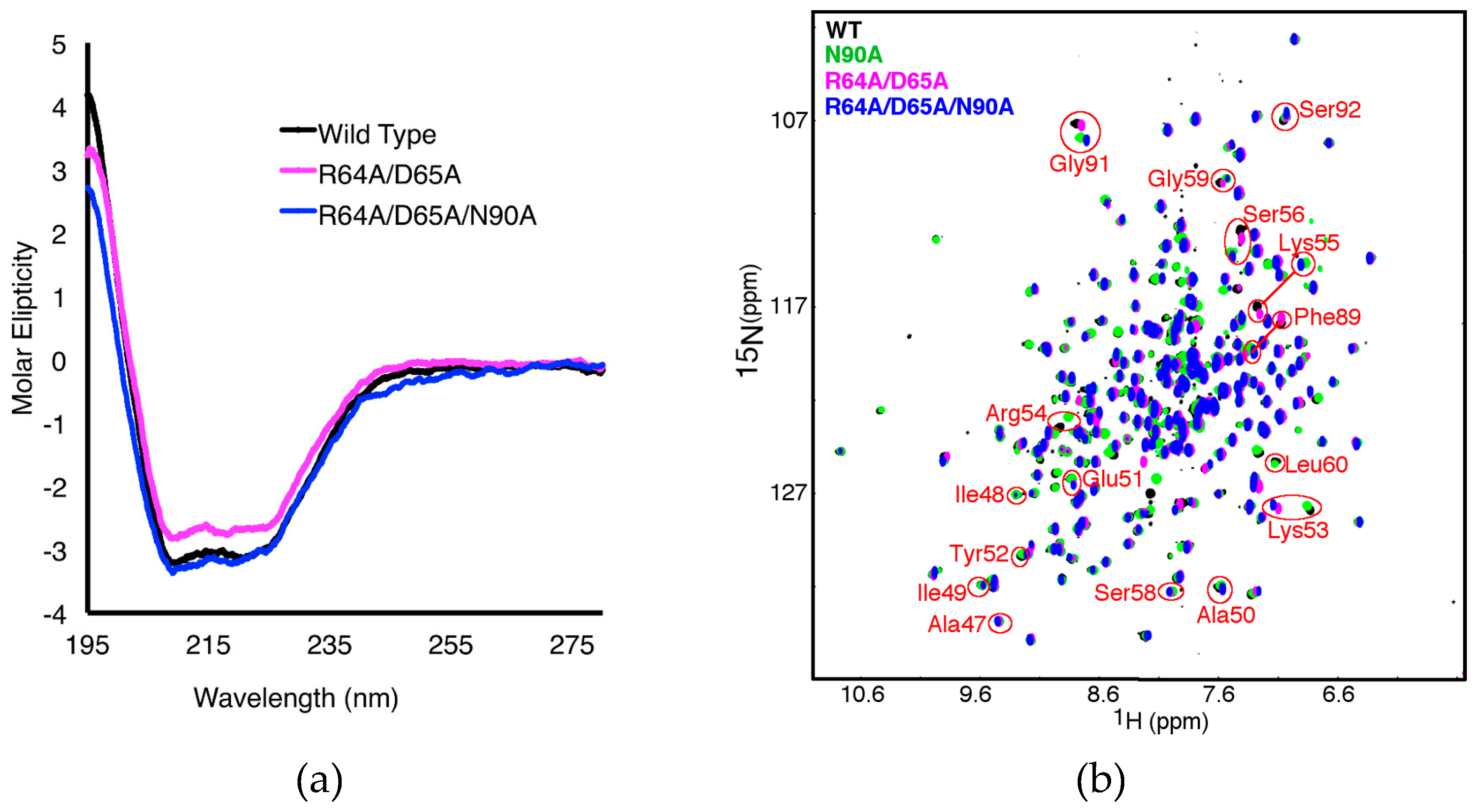
2.3. The R64A/D65A and N90A Substitutions Perturb the Structure of the β1α1 Loop in Different Ways
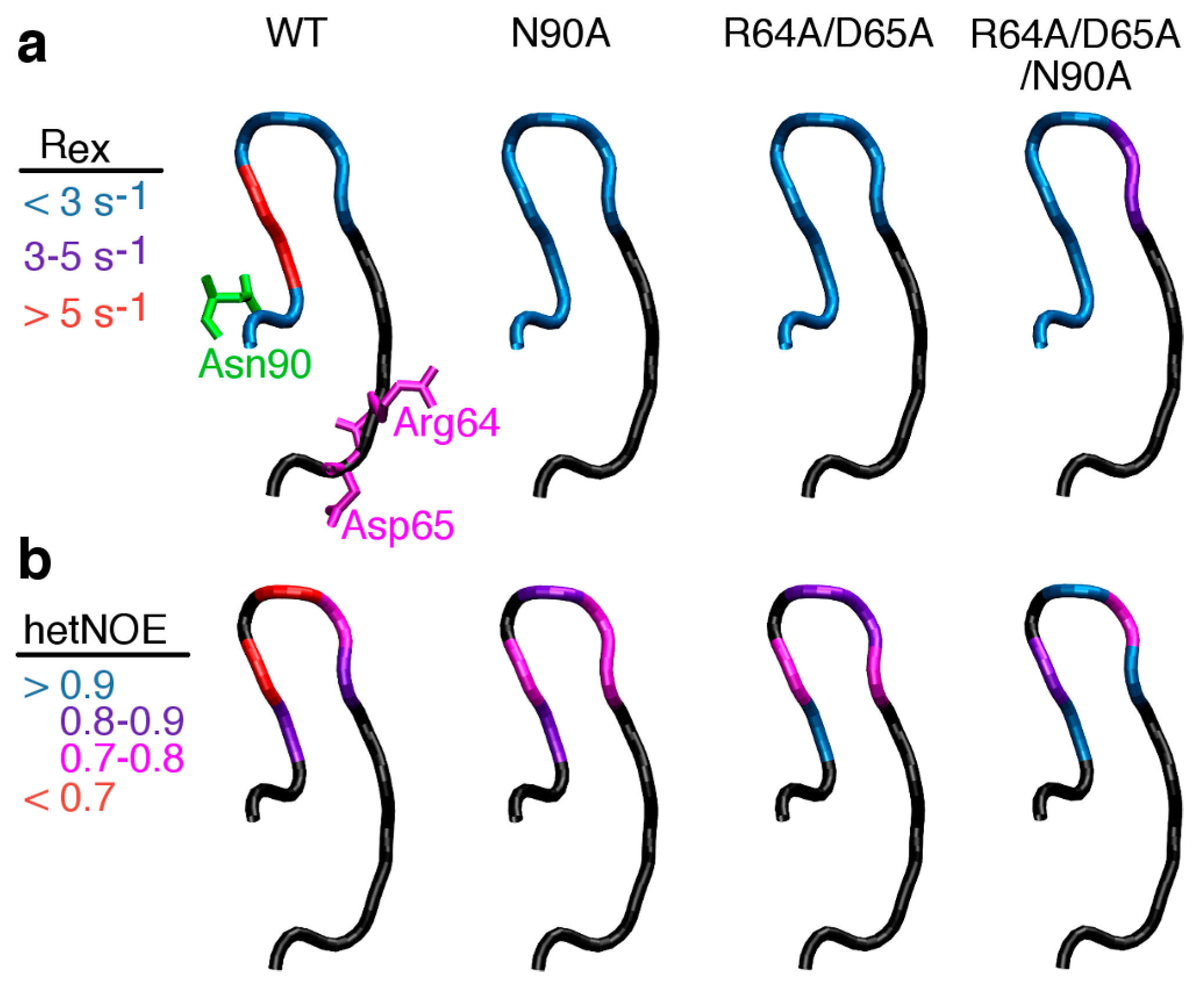
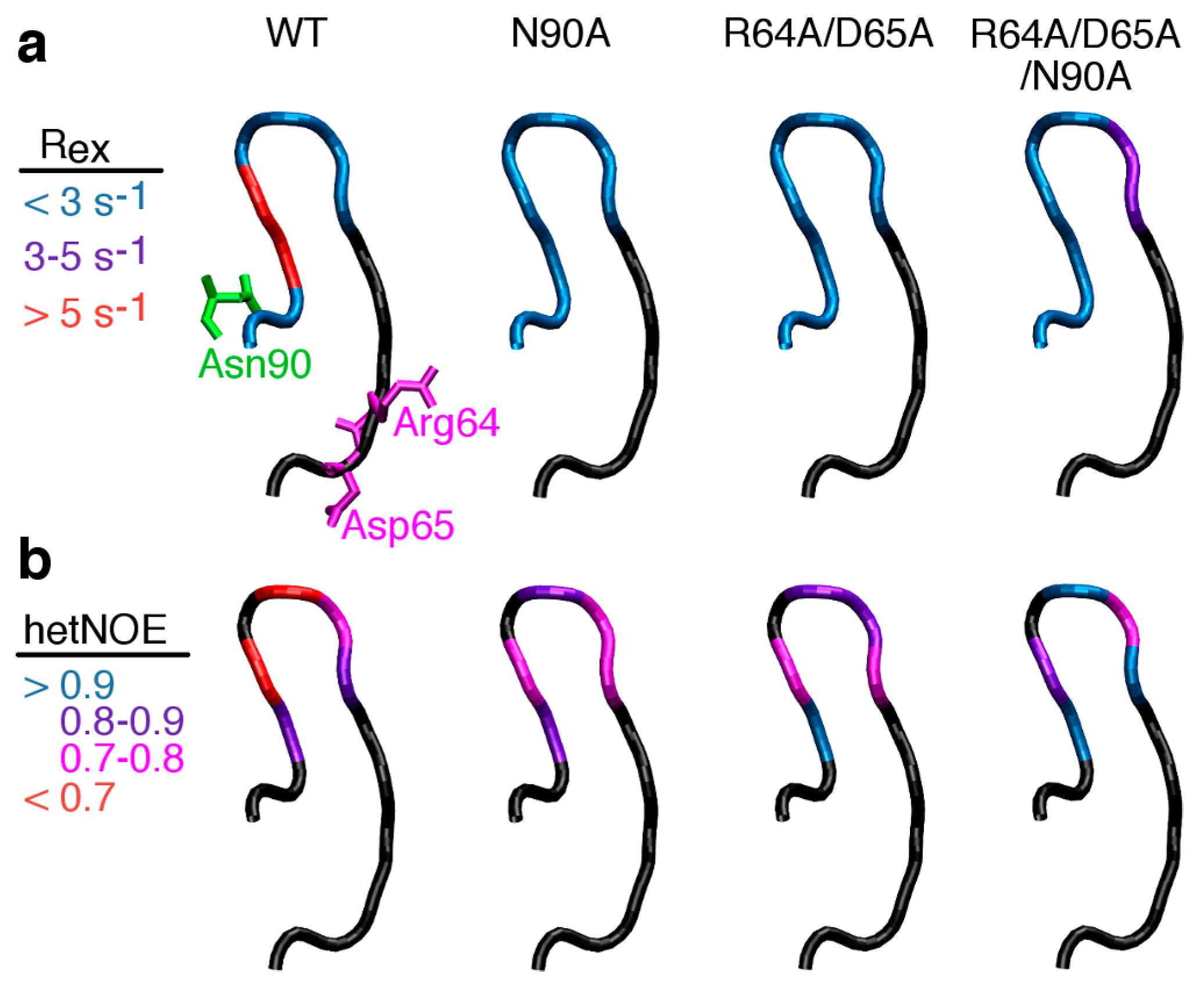
2.4. Severing Interactions with the C-Terminal Side of the β1α1 Loop Quenches μs–ms Timescale Loop Motions
2.5. Severing Interactions with Both the N-Terminal and C-Terminal Sides of the β1α1 Loop Results in a Rate-Determining Step Similar to Wild-Type Enzyme
2.6 Severing Interactions with Both the N-Terminal and C-Terminal Sides of the β1α1 Loop Results in New μs–ms Timescale Loop Dynamics
3. Discussion
4. Materials and Methods
4.1. Site-Directed Mutagenesis, Overexpression, and Purification of ssIGPS Variants
4.2. Steady-State Kinetic Enzyme Assays
4.3. Thermal Inactivation and Circular Dichroism Spectroscopy
4.4. NMR Experiments
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TIM | triose phosphate isomerase |
| IGPS | indole-3-glycerol phosphate synthase |
| ssIGPS | indole-3-glycerol phosphate synthase from Sulfolobus sulfataricus |
| CdRP | 1-(o-carboxyphenylamine)-1-dexoyribulose 5-phosphate |
| IGP | indole-3-glycerol phosphate |
| SCA | statistical coupling analysis |
| MD | molecular dynamics |
| WT | wild-type |
| NMR | nuclear magnetic resonance |
| SVE | solvent viscosity effect |
| SDKIE | solvent deuterium kinetic isotope effect |
| CD | circular dichroism |
| HSQC | heteronuclear single quantum coherence |
| hetNOE | heteronuclear Overhauser effect |
| DHFR | dihydrofolate reductase |
References
- Gutteridge, A.; Thornton, J. Conformational change in substrate binding, catalysis and product release: An open and shut case? FEBS Lett. 2004, 567, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kempner, E.S. Movable lobes and flexible loops in proteins. Structural deformations that control biochemical activity. FEBS Lett. 1993, 326, 4–10. [Google Scholar] [CrossRef]
- Hammes, G.G. Multiple conformational changes in enzyme catalysis. Biochemistry 2002, 41, 8221–8228. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.; Hocker, B. Catalytic versatility, stability, and evolution of the (βα)8-barrel enzyme fold. Chem. Rev. 2005, 105, 4038–4055. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P. A paradigm for enzyme-catalyzed proton transfer at carbon: Triosephosphate isomerase. Biochemistry 2012, 51, 2652–2661. [Google Scholar] [CrossRef] [PubMed]
- Malabanan, M.M.; Nitsch-Velasquez, L.; Amyes, T.L.; Richard, J.P. Magnitude and origin of the enhanced basicity of the catalytic glutamate of triosephosphate isomerase. J. Am. Chem. Soc. 2013, 135, 5978–5981. [Google Scholar] [CrossRef] [PubMed]
- Hennig, M.; Darimont, B.; Sterner, R.; Kirschner, K.; Jansonius, J.N. 2.0 A structure of indole-3-glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus: Possible determinants of protein stability. Structure 1995, 3, 1295–1306. [Google Scholar] [CrossRef]
- Alahuhta, M.; Wierenga, R.K. Atomic resolution crystallography of a complex of triosephosphate isomerase with a reaction-intermediate analog: New insight in the proton transfer reaction mechanism. Proteins 2010, 78, 1878–1888. [Google Scholar] [CrossRef] [PubMed]
- Kursula, I.; Wierenga, R.K. Crystal structure of triosephosphate isomerase complexed with 2-phosphoglycolate at 0.83-Å resolution. J. Biol. Chem. 2003, 278, 9544–9551. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, A.C.; Amyes, T.L.; Richard, J.P. Hydron transfer catalyzed by triosephosphate isomerase. Products of isomerization of (R)-glyceraldehyde 3-phosphate in D2O. Biochemistry 2005, 44, 2610–2621. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, A.C.; Amyes, T.L.; Richard, J.P. Slow proton transfer from the hydrogen-labelled carboxylic acid side chain (Glu-165) of triosephosphate isomerase to imidazole buffer in D2O. Org. Biomol. Chem. 2008, 6, 391–396. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, A.C.; Amyes, T.L.; Richard, J.P. Hydron transfer catalyzed by triosephosphate isomerase. Products of isomerization of dihydroxyacetone phosphate in D2O. Biochemistry 2005, 44, 2622–2631. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Amyes, T.L.; Goryanova, B.; Zhai, X. Enzyme architecture: On the importance of being in a protein cage. Curr. Opin. Chem. Biol. 2014, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Malabanan, M.M.; Amyes, T.L.; Richard, J.P. A role for flexible loops in enzyme catalysis. Curr. Opin. Struct. Biol. 2010, 20, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Lolis, E.; Petsko, G.A. Crystallographic analysis of the complex between triosephosphate isomerase and 2-phosphoglycolate at 2.5-Å resolution: Implications for catalysis. Biochemistry 1990, 29, 6619–6625. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Amyes, T.L.; Malabanan, M.M.; Zhai, X.; Kim, K.J.; Reinhardt, C.J.; Wierenga, R.K.; Drake, E.J.; Gulick, A.M. Structure-function studies of hydrophobic residues that clamp a basic glutamate side chain during catalysis by triosephosphate isomerase. Biochemistry 2016, 55, 3036–3047. [Google Scholar] [CrossRef] [PubMed]
- Loria, J.P.; Berlow, R.B.; Watt, E.D. Characterization of enzyme motions by solution NMR relaxation dispersion. Acc. Chem. Res. 2008, 41, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.G., 3rd. Enzyme dynamics from NMR spectroscopy. Acc. Chem. Res. 2015, 48, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Rozovsky, S.; McDermott, A.E. The time scale of the catalytic loop motion in triosephosphate isomerase. J. Mol. Biol. 2001, 310, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Whittier, S.K.; Hengge, A.C.; Loria, J.P. Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science 2013, 341, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Lisi, G.P.; Loria, J.P. Using NMR spectroscopy to elucidate the role of molecular motions in enzyme function. Prog. Nucl. Mag. Res. Spectrosc. 2016, 92–93, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schlee, S.; Dietrich, S.; Kurcon, T.; Delaney, P.; Goodey, N.M.; Sterner, R. Kinetic mechanism of indole-3-glycerol phosphate synthase. Biochemistry 2012, 52, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Merz, A.; Yee, M.C.; Szadkowski, H.; Pappenberger, G.; Crameri, A.; Stemmer, W.P.C.; Yanofsky, C.; Kirschner, K. Improving the catalytic activity of a thermophilic enzyme at low temperatures. Biochemistry 2000, 39, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Zaccardi, M.J.; Mannweiler, O.; Boehr, D.D. Differences in the catalytic mechanisms of mesophilic and thermophilic indole-3-glycerol phosphate synthase enzymes at their adaptive temperatures. Biochem. Biophys. Res. Commun. 2012, 418, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Zaccardi, M.J.; Yezdimer, E.M.; Boehr, D.D. Functional identification of the general acid and base in the dehydration step of indole-3-glycerol phosphate synthase catalysis. J. Biol. Chem. 2013, 288, 26350–26356. [Google Scholar] [CrossRef] [PubMed]
- Bhabha, G.; Biel, J.T.; Fraser, J.S. Keep on moving: Discovering and perturbing the conformational dynamics of enzymes. Acc. Chem. Res. 2015, 48, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.B.; Xu, F.; Hu, H.R.; Wang, F.F.; Wu, Q.; Huang, Q.; Wang, H.H. Coevolving residues of (β/α)8-barrel proteins play roles in stabilizing active site architecture and coordinating protein dynamics. J. Struct. Biol. 2008, 164, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Hennig, M.; Darimont, B.D.; Jansonius, J.N.; Kirschner, K. The catalytic mechanism of indole-3-glycerol phosphate synthase: Crystal structures of complexes of the enzyme from Sulfolobus solfataricus with substrate analogue, substrate, and product. J. Mol. Biol. 2002, 319, 757–766. [Google Scholar] [CrossRef]
- Zaccardi, M.J.; O’Rourke, K.F.; Yezdimer, E.M.; Loggia, L.J.; Woldt, S.; Boehr, D.D. Loop-loop interactions govern multiple steps in indole-3-glycerol phosphate synthase catalysis. Protein Sci. 2014, 23, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, D.U.; Komives, E.A.; Wolynes, P.G. Frustration in biomolecules. Q. Rev. Biophys. 2014, 47, 285–363. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, L.J.; Mancinelli, R.L. Life in extreme environments. Nature 2001, 409, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Dyson, H.J.; Wright, P.E. An NMR perspective on enzyme dynamics. Chem. Rev. 2006, 106, 3055–3079. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; McElheny, D.; Dyson, H.J.; Wright, P.E. The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Winter, G.; Wilkinson, A.J.; Fersht, A.R. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (bacillus stearothermophilus). Cell 1984, 38, 835–840. [Google Scholar] [CrossRef]
- Ghosh, A.; Sakaguchi, R.; Liu, C.; Vishveshwara, S.; Hou, Y.M. Allosteric communication in cysteinyl tRNA synthetase: A network of direct and indirect readout. J. Biol. Chem. 2011, 286, 37721–37731. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, L.K.; Yamazaki, T.; Torchia, D.A.; Grzesiek, S.; Bax, A.; Stahl, S.J.; Kaufman, J.D.; Wingfield, P.T.; Lam, P.Y.; Jadhav, P.K.; et al. Flexibility and function in HIV-1 protease. Nat. Struct. Biol. 1995, 2, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Zgiby, S.; Plater, A.R.; Bates, M.A.; Thomson, G.J.; Berry, A. A functional role for a flexible loop containing Glu182 in the class II fructose-1,6-bisphosphate aldolase from Escherichia coli. J. Mol. Biol. 2002, 315, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, Z.S. Structure and function of lipases. Adv. Protein Chem. 1994, 45, 1–52. [Google Scholar] [PubMed]
- Lebioda, L.; Stec, B.; Brewer, J.M. The structure of yeast enolase at 2.25-Å resolution. An 8-fold β + α-barrel with a novel β β α α (β α)6 topology. J. Biol. Chem. 1989, 264, 3685–3693. [Google Scholar] [PubMed]
- Lebioda, L.; Stec, B. Mechanism of enolase: The crystal structure of enolase-Mg2(+)-2-phosphoglycerate/phosphoenolpyruvate complex at 2.2-Å resolution. Biochemistry 1991, 30, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Yang, J.; Wu, J.; Haste, N.M.; Radzio-Andzelm, E.; Anand, G. Pka: A portrait of protein kinase dynamics. Biochim. Biophys. Acta 2004, 1697, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Kohen, A. Dihydrofolate reductase as a model for studies of enzyme dynamics and catalysis. F1000Res 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, K.; Ma, B.; Nussinov, R. Triggering loops and enzyme function: Identification of loops that trigger and modulate movements. J. Mol. Biol. 2003, 332, 143–159. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Nussinov, R. Modulating functional loop movements: The role of highly conserved residues in the correlated loop motions. Chembiochem 2004, 5, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Sampson, N.S.; Knowles, J.R. Segmental motion in catalysis: Investigation of a hydrogen bond critical for loop closure in the reaction of triosephosphate isomerase. Biochemistry 1992, 31, 8488–8494. [Google Scholar] [CrossRef] [PubMed]
- Sampson, N.S.; Knowles, J.R. Segmental movement: Definition of the structural requirements for loop closure in catalysis by triosephosphate isomerase. Biochemistry 1992, 31, 8482–8487. [Google Scholar] [CrossRef] [PubMed]
- Berlow, R.B.; Igumenova, T.I.; Loria, J.P. Value of a hydrogen bond in triosephosphate isomerase loop motion. Biochemistry 2007, 46, 6001–6010. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Berlow, R.B.; Loria, J.P. Role of loop-loop interactions in coordinating motions and enzymatic function in triosephosphate isomerase. Biochemistry 2009, 48, 4548–4556. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.R.; Dyson, H.J.; Wright, P.E. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.P.; Wahnon, D.C.; Benkovic, S.J. Interloop contacts modulate ligand cycling during catalysis by Escherichia coli dihydrofolate reductase. Biochemistry 2001, 40, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.P.; Benkovic, S.J. Strength of an interloop hydrogen bond determines the kinetic pathway in catalysis by Escherichia coli dihydrofolate reductase. Biochemistry 1998, 37, 6336–6342. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.E.; Benkovic, S.J. Evidence for a functional role of the dynamics of glycine-121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site-directed mutant. Biochemistry 1997, 36, 15792–15800. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Schnell, J.R.; McElheny, D.; Bae, S.H.; Duggan, B.M.; Benkovic, S.J.; Dyson, H.J.; Wright, P.E. A distal mutation perturbs dynamic amino acid networks in dihydrofolate reductase. Biochemistry 2013, 52, 4605–4619. [Google Scholar] [CrossRef] [PubMed]
- Bhabha, G.; Lee, J.; Ekiert, D.C.; Gam, J.; Wilson, I.A.; Dyson, H.J.; Benkovic, S.J.; Wright, P.E. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science 2011, 332, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Mauldin, R.V.; Sapienza, P.J.; Petit, C.M.; Lee, A.L. Structure and dynamics of the G121V dihydrofolate reductase mutant: Lessons from a transition-state inhibitor complex. PLoS ONE 2012, 7, e33252. [Google Scholar] [CrossRef] [PubMed]
- Vogeli, B.; Bibow, S.; Chi, C.N. Enzyme selectivity fine-tuned through dynamic control of a loop. Angew. Chem. Int. Ed. Engl. 2016, 55, 3096–3100. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Knochel, T.; Darimont, B.; Hennig, M.; Dietrich, S.; Babinger, K.; Kirschner, K.; Sterner, R. Role of the N-terminal extension of the (β/α)8-barrel enzyme indole-3-glycerol phosphate synthase for its fold, stability, and catalytic activity. Biochemistry 2005, 44, 16405–16412. [Google Scholar] [CrossRef] [PubMed]
- Loria, J.P.; Rance, M.; Palmer, A.G., 3rd. A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J. Biomol. NMR 1999, 15, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Axe, J.M.; Boehr, D.D. Long-range interactions in the alpha subunit of tryptophan synthase help to coordinate ligand binding, catalysis, and substrate channeling. J. Mol. Biol. 2013, 425, 1527–1545. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Variant | Temp. (°C) | kcat 1 (s−1) | KM 1 (nM) | kcat/KM (M−1·s−1 × 106) | kcat,WT/kcat,var. | (kcat/KM)WT/(kcat/KM)var. | SVE | SDKIE | 2 kinact. (h−1) |
|---|---|---|---|---|---|---|---|---|---|
| WT | 37 | 1.02 | 173 | 5.80 | - | - | 0.6 3 | 5.8 3 | 4.86 |
| 75 | 1.77 | 89 | 19.9 | - | - | ~0 3 | 3.6 3 | ||
| N90A 4 | 37 | 0.17 | 74 | 2.3 | 6.0 | 2.5 | 0.1 | 5.2 | 2.49 |
| 75 | 0.55 | 79 | 7.0 | 3.2 | 2.8 | ~0 | 1.6 | ||
| R64A/D65A | 37 | 0.535 | 384 | 1.39 | 1.91 | 4.17 | ~0 | 2.0 | 24.9 |
| 75 | 1.65 | 770 | 2.14 | 1.07 | 9.30 | 0.1 | 1.0 | ||
| R64A/D65A/N90A | 37 | 0.375 | 477 | 0.786 | 2.72 | 7.37 | ~0 | 1.0 | 25.5 |
| 75 | 0.965 | 598 | 1.61 | 1.83 | 12.4 | ~0 | 4.3 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Rourke, K.F.; Jelowicki, A.M.; Boehr, D.D. Controlling Active Site Loop Dynamics in the (β/α)8 Barrel Enzyme Indole-3-Glycerol Phosphate Synthase. Catalysts 2016, 6, 129. https://doi.org/10.3390/catal6090129
O’Rourke KF, Jelowicki AM, Boehr DD. Controlling Active Site Loop Dynamics in the (β/α)8 Barrel Enzyme Indole-3-Glycerol Phosphate Synthase. Catalysts. 2016; 6(9):129. https://doi.org/10.3390/catal6090129
Chicago/Turabian StyleO’Rourke, Kathleen F., Aneta M. Jelowicki, and David D. Boehr. 2016. "Controlling Active Site Loop Dynamics in the (β/α)8 Barrel Enzyme Indole-3-Glycerol Phosphate Synthase" Catalysts 6, no. 9: 129. https://doi.org/10.3390/catal6090129
APA StyleO’Rourke, K. F., Jelowicki, A. M., & Boehr, D. D. (2016). Controlling Active Site Loop Dynamics in the (β/α)8 Barrel Enzyme Indole-3-Glycerol Phosphate Synthase. Catalysts, 6(9), 129. https://doi.org/10.3390/catal6090129