
Au/TiO2-CeO2 Catalysts for Photocatalytic Water Splitting and VOCs Oxidation Reactions

 ,
, 



Abstract
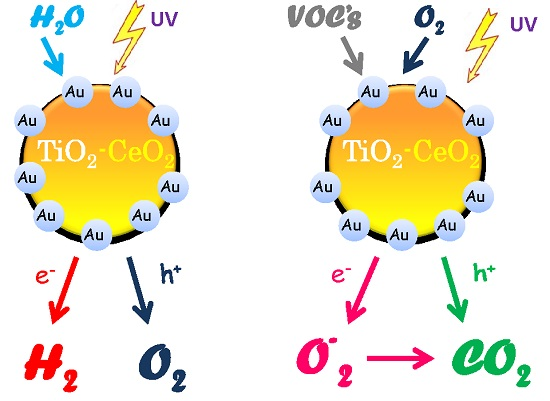
:
1. Introduction
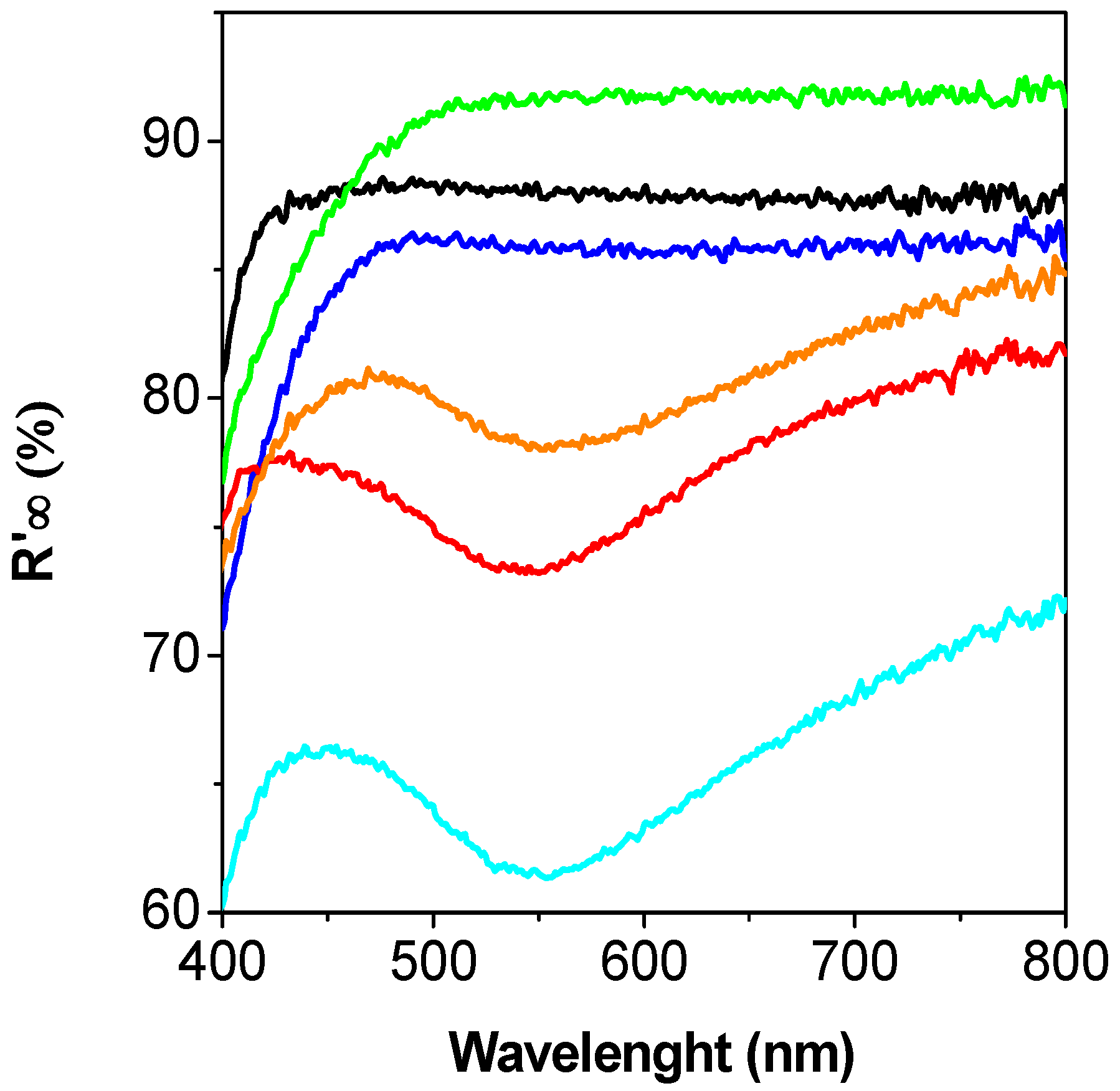
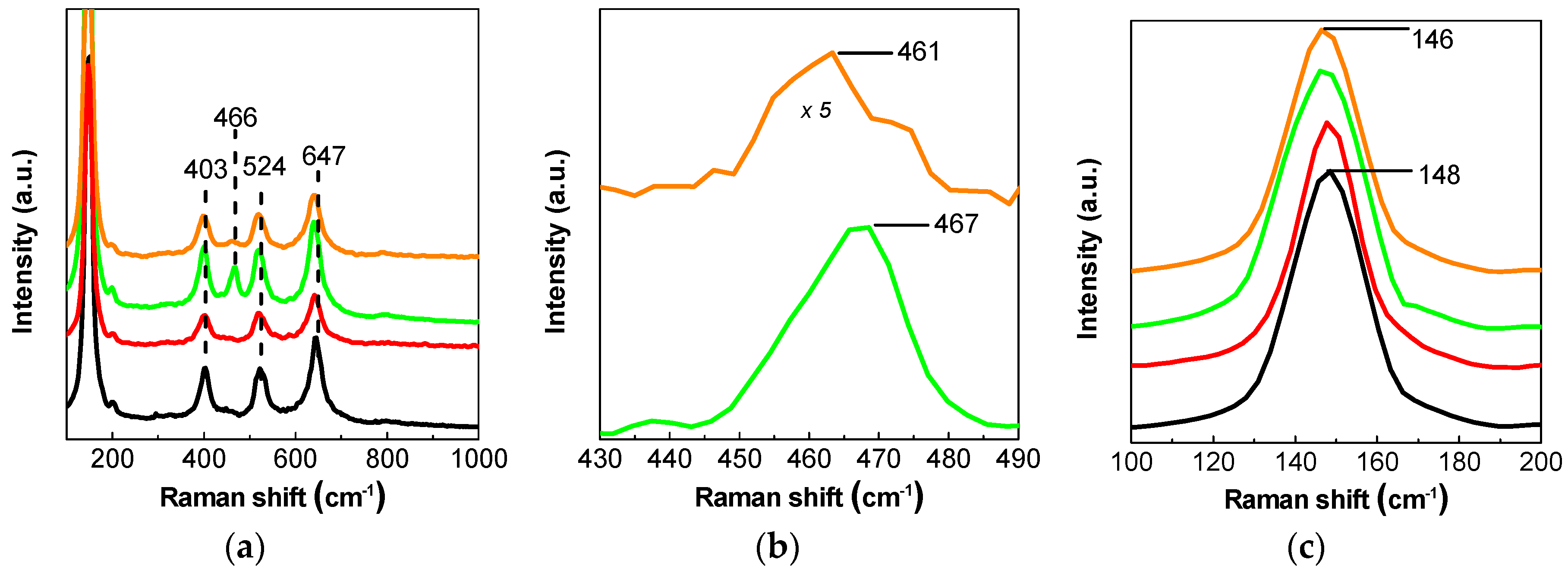
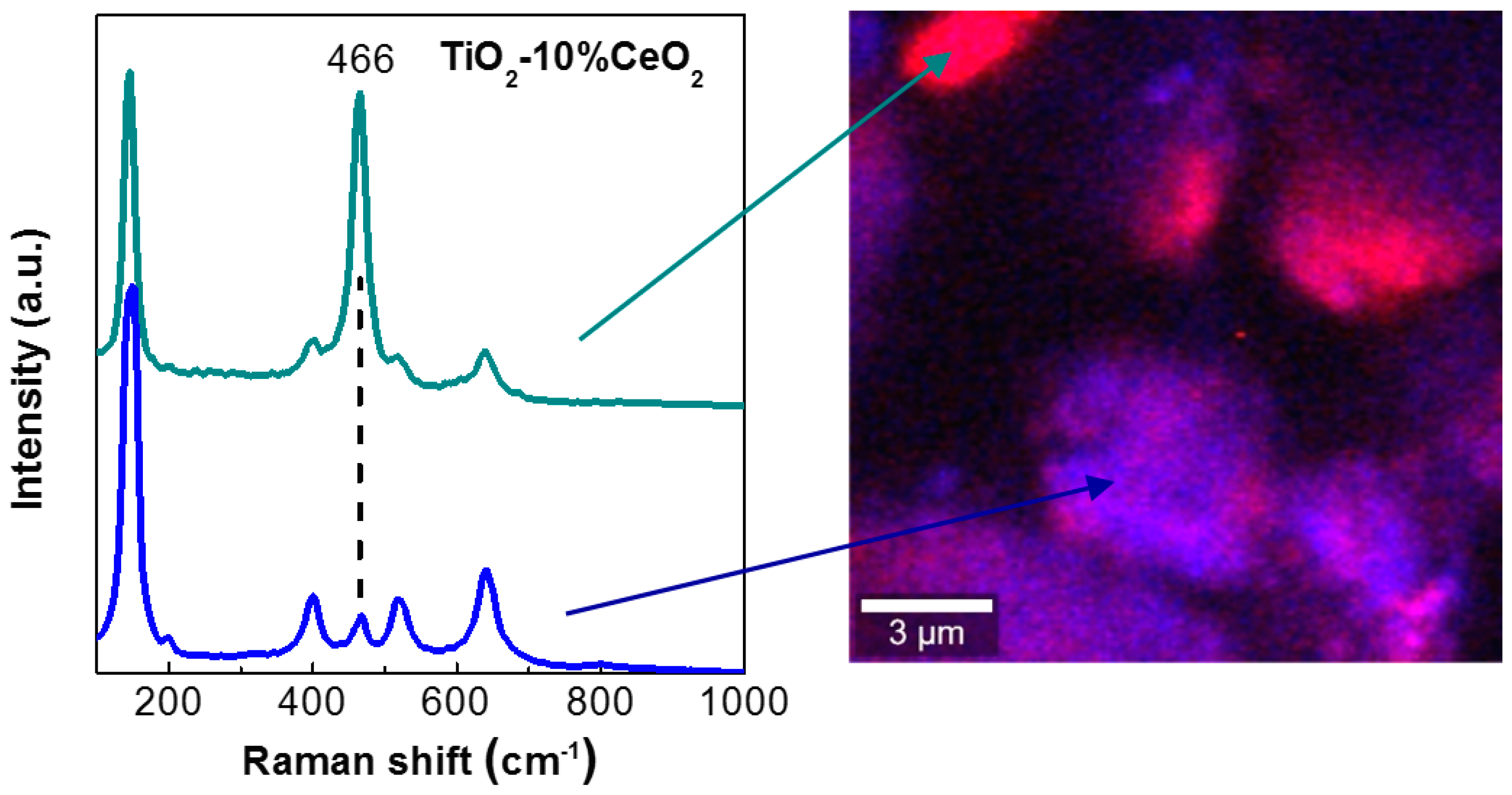
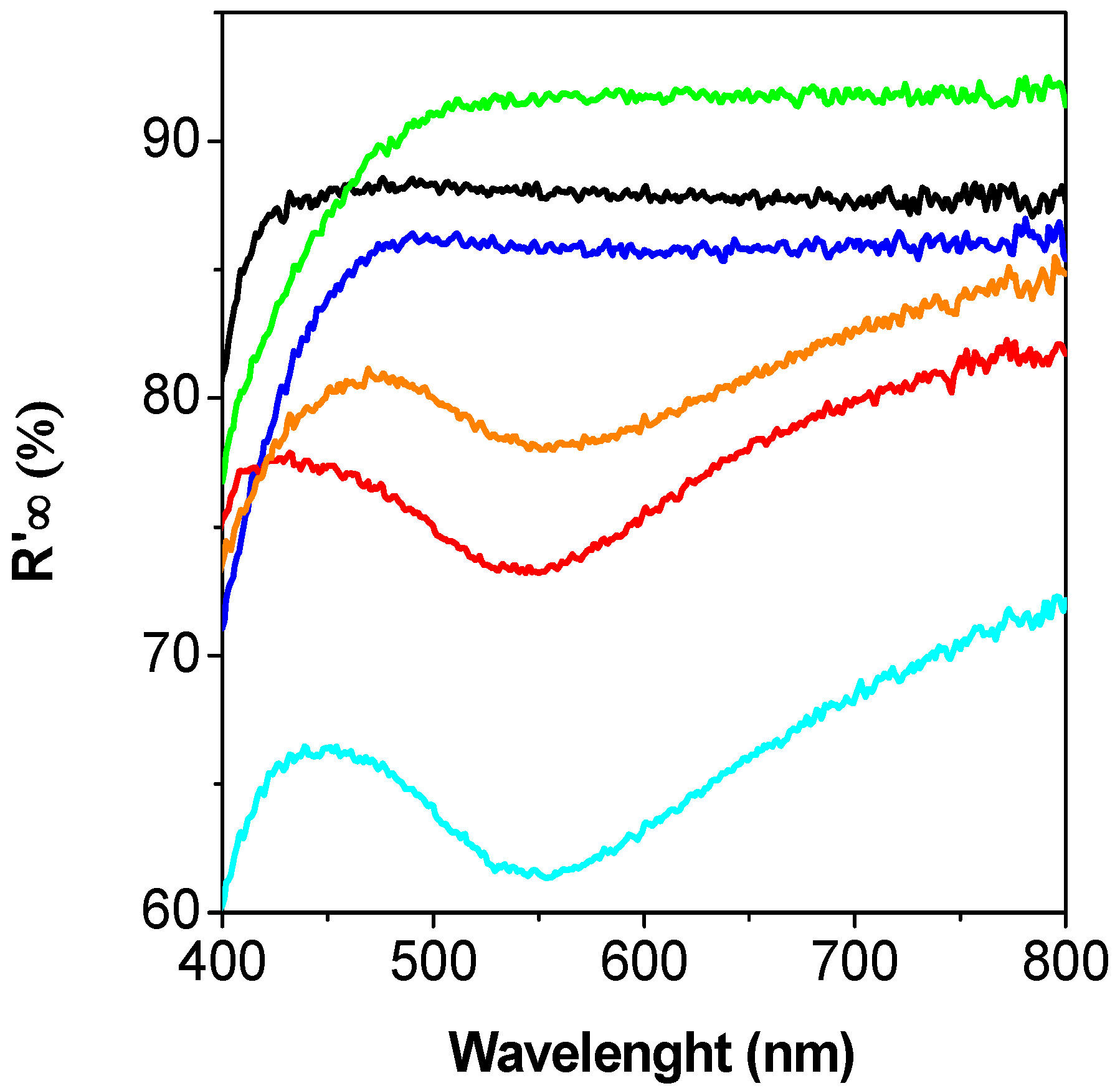
2. Results and Discussion
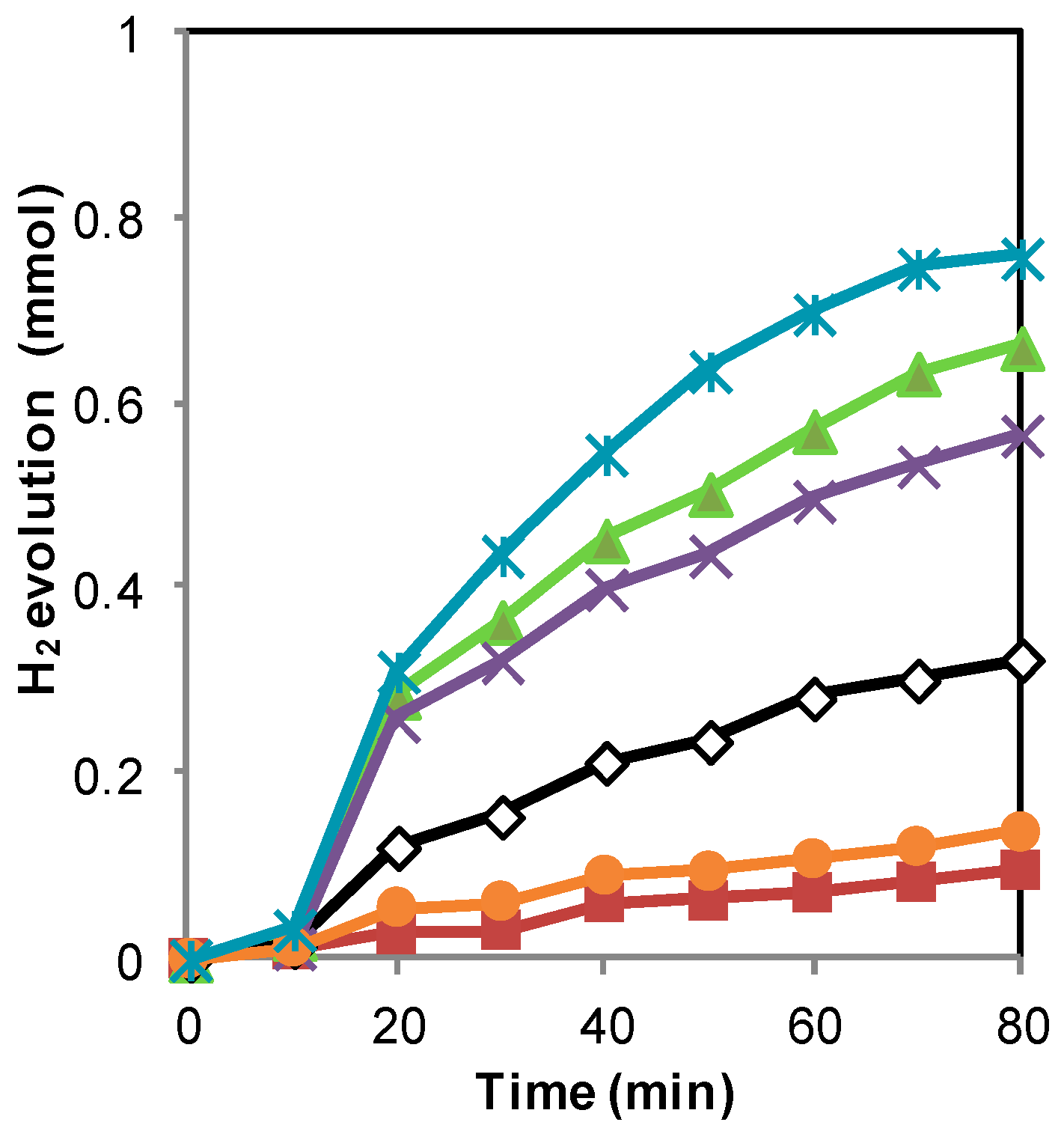
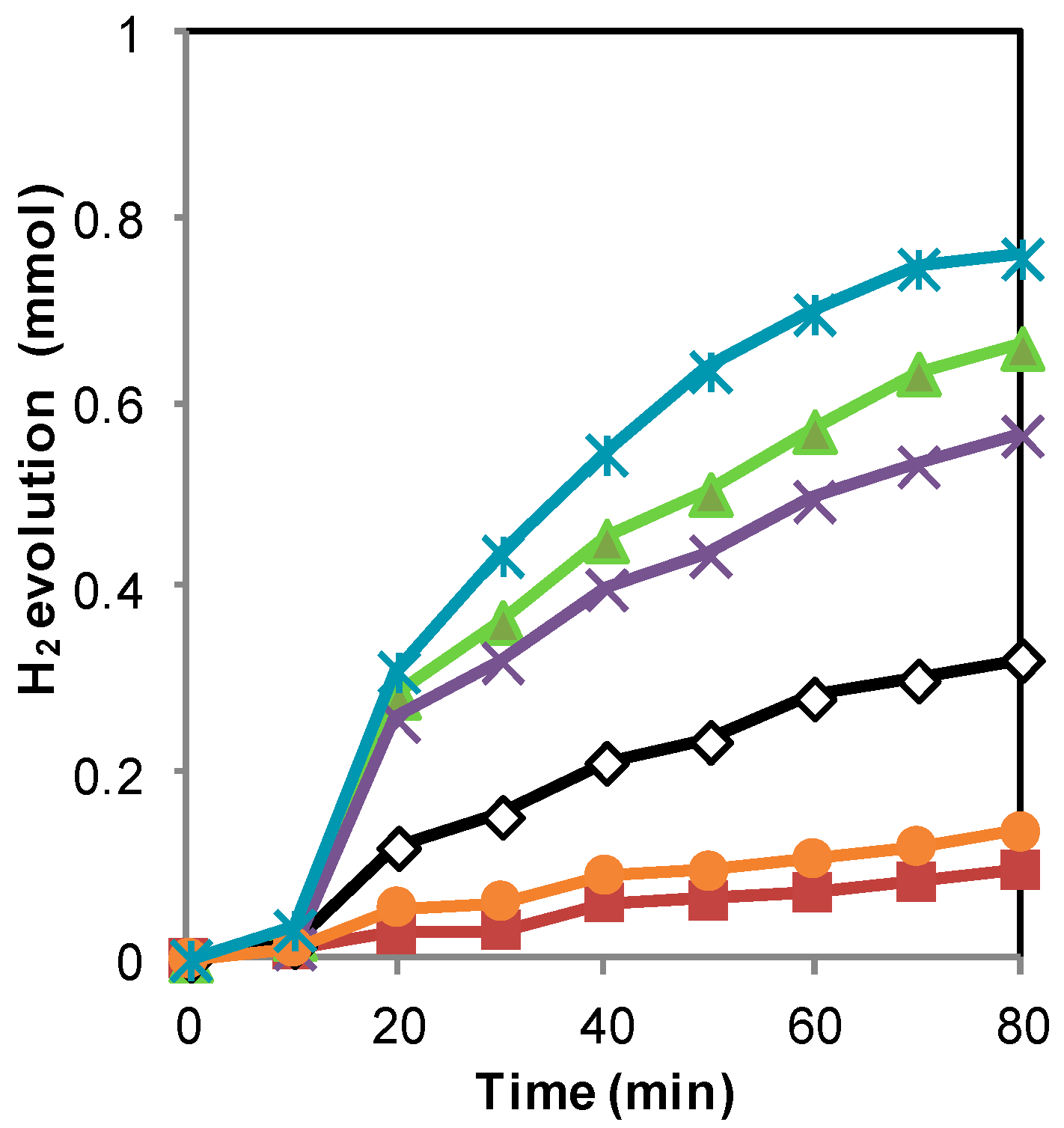
2.1. H2 Generation by Photocatalytic Water Splitting
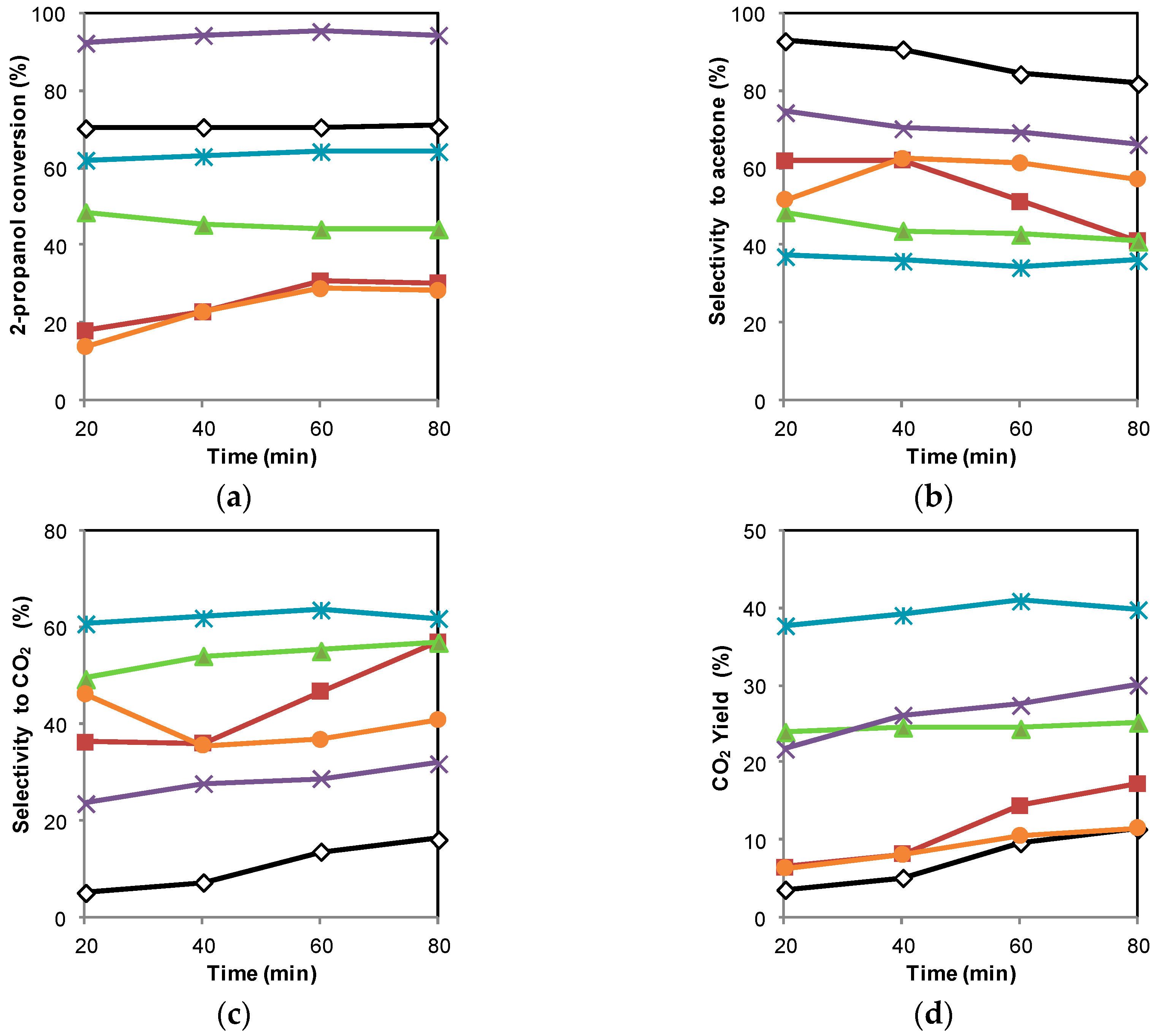
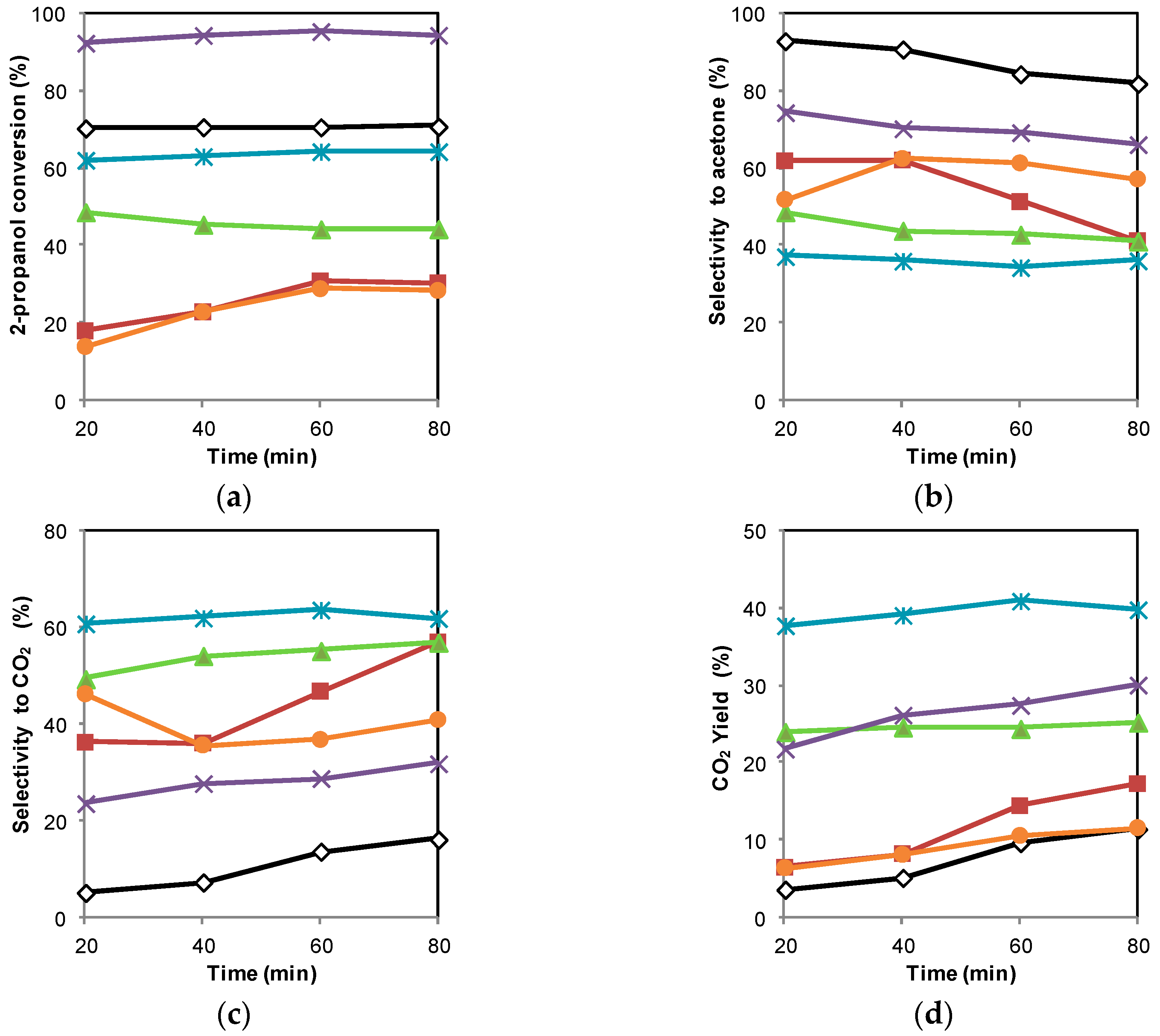
2.2. Photocatalytic Oxidation of 2-Propanol
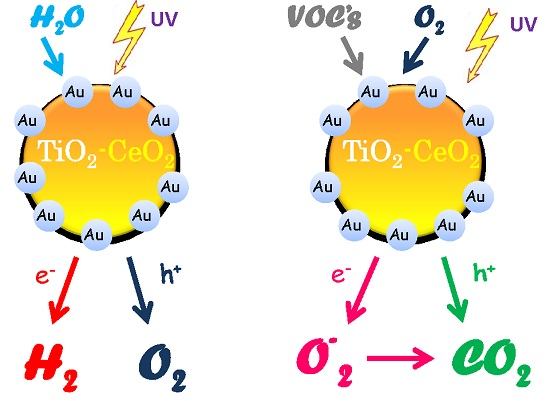
2.3. Discussion
3. Materials and Methods
3.1. Catalyst Preparation
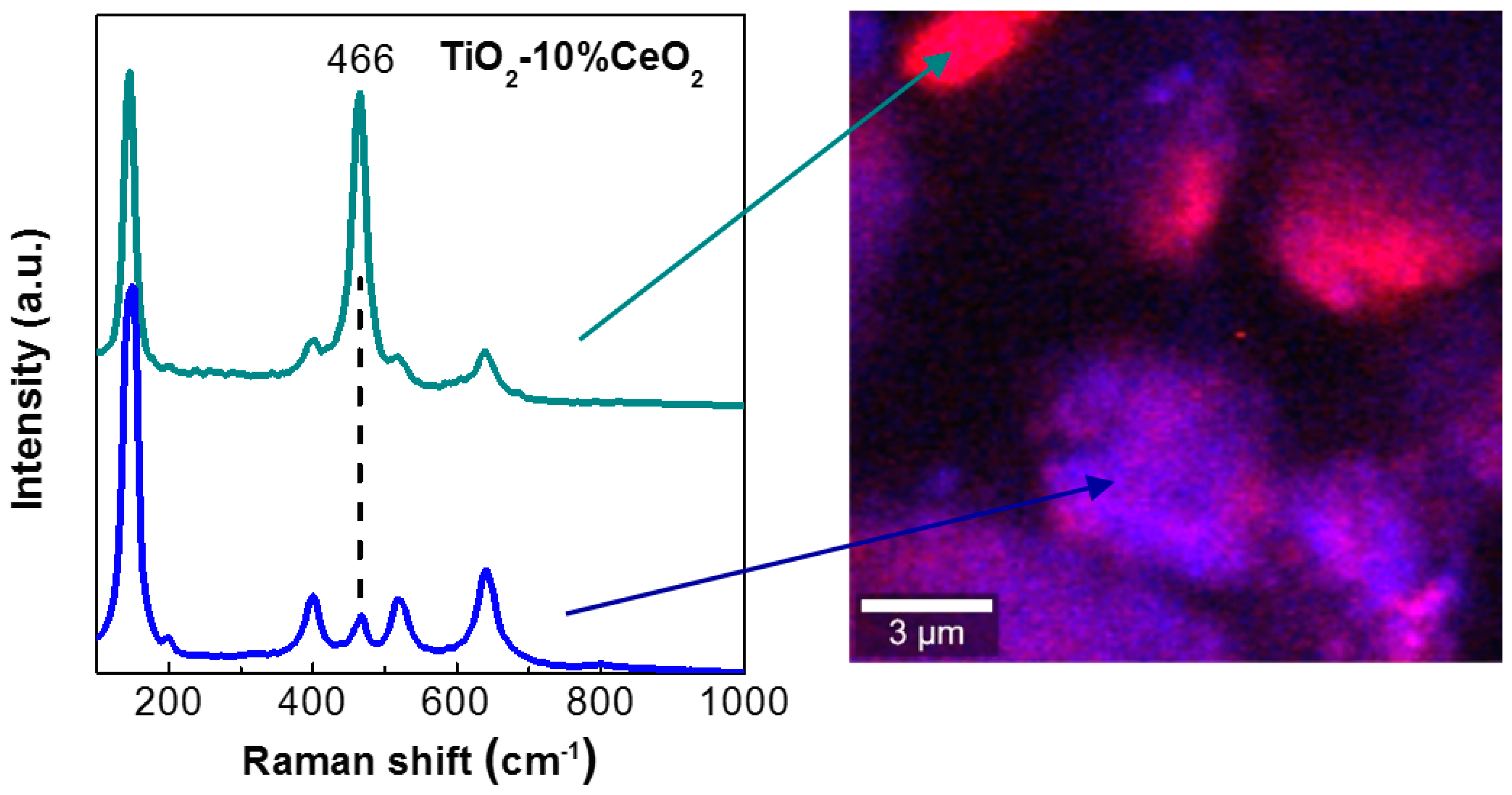
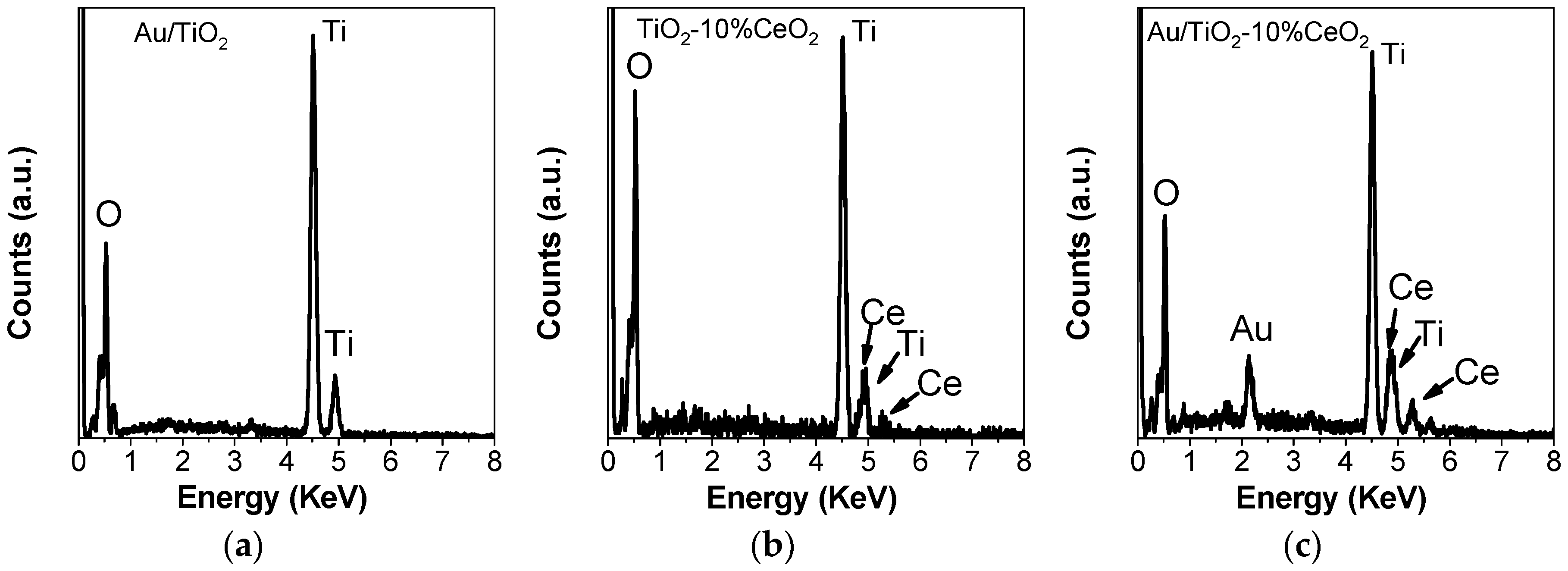
3.2. Catalyst Characterization Experiments
3.3. Photocatalytic Activity Experiments
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.H.; Huang, C.W.; Wu, J.C.S. Hydrogen Production from Semiconductor-based Photocatalysis via Water Splitting. Catalysts 2012, 2, 490–516. [Google Scholar] [CrossRef]
- Zou, Z.; Ye, J.; Sayama, K.; Arakawa, H. Direct splitting of water under visible light irradiation with an oxide semiconductor photocatalyst. Nature 2001, 414, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.M. Heterogeneous photocatalysis: Fundamentals and applications to the removal of various types of aqueous pollutants. Catal. Today 1999, 53, 115–129. [Google Scholar] [CrossRef]
- Ibhadon, A.O.; Fitzpatrick, P. Heterogeneous Photocatalysis: Recent Advances and Applications. Catalysts 2013, 3, 189–218. [Google Scholar] [CrossRef]
- Chen, X.; Mao, S.S. Titanium Dioxide Nanomaterials: Synthesis, Properties, Modifications and Applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef] [PubMed]
- Lazar, M.A.; Varghese, S.; Nair, S.S. Photocatalytic Water Treatment by Titanium Dioxide: Recent Updates. Catalysts 2012, 2, 572–601. [Google Scholar] [CrossRef]
- Gaya, U.I.; Abdullah, A.H. Heterogeneous photocatalytic degradation of organic contaminants over titanium dioxide: A review of fundamentals, progress and problems. J. Photochem. Photobiol. C 2008, 9, 1–12. [Google Scholar] [CrossRef]
- Hisanga, T.; Harada, K.; Tanaka, K. Photocatalytic degradation of organochlorine compounds in suspended TiO2. J. Photochem. Photobiol. A 1990, 54, 113–118. [Google Scholar] [CrossRef]
- Wang, C.; Ying, J. Sol-Gel Synthesis and Hydrothermal Processing of Anatase and Rutile Titania Nanocrystals. Chem. Mater. 1999, 11, 3113–3120. [Google Scholar] [CrossRef]
- Pillai, S.C.; Periyat, P.; George, R.; McCormack, D.E.; Seery, M.K.; Hayden, H.; Colreavy, J.; Corr, D.; Hinder, S.J. Synthesis of high-temperature stable anatase TiO2 photocatalyst. J. Phys. Chem. C 2007, 111, 1605–1611. [Google Scholar] [CrossRef]
- Kudo, T.; Nakamura, Y.; Ruike, A. Development of rectangular column structured titanium oxide photocatalysts anchored on silica sheets by a wet process. Res. Chem. Intermed. 2003, 29, 631–639. [Google Scholar] [CrossRef]
- Bahnemann, D. Photocatalytic water treatment: Solar energy applications. Sol. Energy 2004, 77, 445–459. [Google Scholar] [CrossRef]
- Zeltner, W.A.; Tompkin, D.T. Shedding Light on Photocatalysis. In ASHRAE Transactions, part 2; ASHRAE: Atlanta, GA, USA, 2005; Volume 111, pp. 523–532. [Google Scholar]
- Einaga, H.; Mochiduki, K.; Teraoka, Y. Photocatalytic Oxidation Processes for Toluene Oxidation over TiO2 Catalysts. Catalysts 2013, 3, 219–231. [Google Scholar] [CrossRef]
- Zhao, B.; Mele, G.; Pio, I.; Li, J.; Palmisano, L.; Vasapollo, G. Degradation of 4-nitrophenol (4-NP) using Fe-TiO2 as a heterogeneous photo-Fenton catalyst. J. Haz. Mat. 2010, 176, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Arana, J.; Dona-Rodriguez, J.M.; Gonzalez-Diaz, O.; Tello Rendon, E.; Herrera Melian, J.A.; Colon, G.; Navìo, J.A.; Perez Pena, J. Gas-phase ethanol photocatalytic degradation study with TiO2 doped with Fe, Pd and Cu. J. Mol. Catal. A 2004, 215, 153–160. [Google Scholar] [CrossRef]
- Colmenares, J.C.; Aramendia, M.A.; Marinas, A.; Marinas, J.M.; Urbano, F.J. Synthesis, characterization and photocatalytic activity of different metal-doped titania systems. Appl. Catal. A: Gen. 2006, 306, 120–127. [Google Scholar] [CrossRef]
- Bashir, S.; Wahab, A.K.; Idriss, H. Synergism and photocatalytic water splitting to hydrogen over M/TiO2 catalysts: Effect of initial particle size of TiO2. Catal. Today 2015, 240, 242–247. [Google Scholar] [CrossRef]
- Bellardita, M.; García-López, E.I.; Marcì, G.; Palmisano, L. Photocatalytic formation of H2 and value-added chemicals in aqueous glucose (Pt)-TiO2 suspension. Int. J. Hydrog. Energy 2016, 41, 5934–5947. [Google Scholar] [CrossRef]
- Gombac, V.; Sordelli, L.; Montini, T.; Delgado, J.J.; Adamski, A.; Adami, G.; Cargnello, M.; Bernal, S.; Fornasiero, P. CuOx-TiO2 Photocatalysts for H2 Production from Ethanol and Glycerol Solutions. J. Phys. Chem. A 2010, 114, 3916–3925. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, R.; Bellardita, M.; Palmisano, L.; Sciré, S. A comparison between Photocatalytic and Catalytic oxidation of 2-Propanol over Au/TiO2-CeO2 catalysts. J. Mol. Catal. A: Chem. 2016, 415, 56–64. [Google Scholar] [CrossRef]
- Zeng, M.; Li, Y.; Mao, M.; Bai, J.; Ren, L.; Zhao, X. Synergetic Effect between Photocatalysis on TiO2 and Thermocatalysis on CeO2 for Gas-Phase Oxidation of Benzene on TiO2/CeO2 Nanocomposites. ACS Catal. 2015, 5, 3278–3286. [Google Scholar] [CrossRef]
- Marcì, G.; Augugliaro, V.; Lopez-Munoz, M.J.; Martın, C.; Palmisano, L.; Rives, V.; Schiavello, M.; Tilley, R.J.D.; Venezia, A.M. Preparation Characterization and Photocatalytic Activity of Polycrystalline ZnO/TiO2. J. Phys. Chem. B 2001, 105, 1026–1032. [Google Scholar]
- Pérez-Larios, A.; Lopez, R.; Hernández-Gordillo, A.; Tzompantzi, F.; Gómez, R.; Torres-Guerra, L.M. Improved hydrogen production from water splitting using TiO2-ZnO mixed oxides. Fuel 2012, 100, 139–143. [Google Scholar] [CrossRef]
- Bellardita, M.; Addamo, M.; Di Paola, A.; Marcì, G.; Palmisano, L.; Cassar, L.; Borsa, M. Photocatalytic activity of TiO2/SiO2 systems. J. Haz. Mat. 2010, 174, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Huang, B.; Dai, Y.; Whangbo, M.H. Plasmonic photocatalysts: Harvesting visible light with noble metal nanoparticles. Phys. Chem. Chem. Phys. 2012, 149, 9813–9825. [Google Scholar] [CrossRef] [PubMed]
- Orendorff, C.J.; Sau, T.K.; Murphy, C.J. Shape-Dependent Plasmon-Resonant Gold Nanoparticles. Small 2006, 2, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, E.; Mahaney, O.O.P.; Abe, R.; Ohtani, B. Visible-light-induced photocatalysis through surface plasmon excitation of gold on titania surfaces. Phys. Chem. Chem. Phys. 2010, 12, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Naya, S.I.; Inoue, A.; Tada, H. Self-Assembled Heterosupramolecular Visible Light Photocatalyst Consisting of Gold Nanoparticle-Loaded Titanium(IV) Dioxide and Surfactant. J. Am. Chem. Soc. 2010, 132, 6292–6293. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Hung, W.H.; Pavaskar, P.; Goeppert, A.; Aykol, M.; Cronin, S.B. Photocatalytic conversion of CO2 to hydrocarbon fuels via plasmon-enhanced absorption and metallic interband transitions. ACS Catal. 2011, 1, 929–936. [Google Scholar] [CrossRef]
- Jovic, V.; Chen, W.T.; Sun-Waterhouse, D.; Blackford, M.G.; Idriss, H.; Waterhouse, G.I.N. Effect of gold loading and TiO2 support composition on the activity of Au/TiO2 photocatalysts for H2 production from ethanol-water mixtures. J. Catal. 2013, 305, 307–317. [Google Scholar] [CrossRef]
- Hinojosa-Reyes, M.; Hernández-Gordillo, A.; Zanella, R.; Rodríguez-González, V. Renewable hydrogen harvest process by hydrazine as scavenging electron donor using gold TiO2 photocatalysts. Catal. Today 2016, 266, 2–8. [Google Scholar] [CrossRef]
- Silva, C.G.; Juarez, R.; Tiziana, M.; Molinari, R.; Garcia, H. Influence of Excitation Wavelength (UV or Visible Light) on the Photocatalytic Activity of Titania Containing Gold Nanoparticles for the Generation of Hydrogen or Oxygen from Water. J. Am. Chem. Soc. 2011, 133, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Noguez, C. Plasmonic Optical Properties and Applications of Metal Nanostructures. Plasmonics 2008, 3, 127–150. [Google Scholar] [CrossRef]
- Galińska, A.; Walendziewski, J. Photocatalytic Water Splitting over Pt−TiO2 in the Presence of Sacrificial Reagents. Energy Fuels 2005, 19, 1143–1147. [Google Scholar] [CrossRef]
- Ni, M.; Leung, M.K.H.; Leung, D.Y.C.; Sumathy, K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew. Sustain. Energy Rev. 2007, 11, 401–425. [Google Scholar] [CrossRef]
- Maeda, K. Photocatalytic water splitting using semiconductor particles: History and recent developments. J. Photochem. Photobiol. C Photochem. Rev. 2011, 12, 237–268. [Google Scholar] [CrossRef]
- Shankhamala, K.; Ciston, J.; Senanayake, S.D.; Arena, D.A.; Fujita, E.; Stacchiola, D.; Barrio, L.; Navarro, R.M.; Fierro, J.L.G.; Rodriguez, J.A. Exploring the Structural and Electronic Properties of Pt/Ceria-Modified TiO2 and its Photocatalytic Activity for Water Splitting under Visible Light. J. Phys. Chem. C 2012, 116, 14062–14070. [Google Scholar]
- Guisheng, L.; Dieqing, Z.; Jimmy, C.Y. Thermally stable ordered mesoporous CeO2/TiO2 visible-light photocatalysts. Phys. Chem. Chem. Phys. 2009, 11, 3775–3782. [Google Scholar]
- Holz, M.C.; Kähler, K.; Tölle, K.; van Veen, A.C.; Muhler, M. Gas-phase oxidation of 2-propanol over Au/TiO2 catalysts to probe metal–support interactions. Phys. Status Solidi B 2013, 250, 1094–1106. [Google Scholar] [CrossRef]
- Liu, S.Y.; Yang, S.M. Complete oxidation of 2-propanol over gold-based catalysts supported on metal oxides. Appl. Catal. Gen. 2008, 334, 92–99. [Google Scholar] [CrossRef]
- Manrı́quez, M.E.; López, T.; Gómez, R.; Navarrete, J. Preparation of TiO2–ZrO2 mixed oxides with controlled acid–basic properties. J. Mol. Catal. Chem. 2004, 220, 229–237. [Google Scholar] [CrossRef]
- Haffad, D.; Chambellan, A.; Lavalley, J.C. Propan-2-ol transformation on simple metal oxides TiO2, ZrO2 and CeO2. J. Mol. Catal. A Chem. 2001, 168, 153–164. [Google Scholar] [CrossRef]
- Hirakawa, T.; Kamat, P.V. Charge Separation and Catalytic Activity of Ag@TiO2 Core−Shell Composite Clusters under UV−Irradiation. J. Am. Chem. Soc. 2005, 127, 3928–3934. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, E.; Abe, R.; Ohtani, B. Visible light-induced photocatalytic reaction of gold-modified titanium(IV) oxide particles: Action spectrum analysis. Chem. Commun. 2009, 2, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Smirniotis, P.G. Interaction of anatase and rutile TiO2 particles in aqueous photooxidation. Catal. Today 2003, 88, 49–59. [Google Scholar] [CrossRef]
- Ohno, T.; Sarukawa, K.; Tokieda, K.; Matsumura, M. Morphology of a TiO2 Photocatalyst (Degussa, P-25) Consisting of Anatase and Rutile Crystalline Phases. J. Catal. 2001, 203, 82–86. [Google Scholar] [CrossRef]
- Aramendìa, M.A.; Borau, V.; Colmenares, J.C.; Marinas, A.; Marinas, J.M.; Navìo, J.A.; Urbano, F.J. Modification of the photocatalytic activity of Pd/TiO2 and Zn/TiO2 systems through different oxidative and reductive calcination treatments. Appl. Catal. B: Environ. 2008, 80, 88–97. [Google Scholar] [CrossRef]
- Hinojosa-Reyes, M.; Rodríguez-González, V.; Zanella, R. Gold nanoparticles supported on TiO2–Ni as catalysts for hydrogen purification via water–gas shift reaction. RSC Adv. 2013, 4, 4308–4316. [Google Scholar] [CrossRef]
- Choudhury, B.; Borah, B.; Choudhury, A. Ce–Nd codoping effect on the structural and optical properties of TiO2 nanoparticles. Mater. Sci. Eng. B 2013, 178, 239–247. [Google Scholar] [CrossRef]
- Contreras-García, M.E.; García-Benjume, M.L.; Macías-Andrés, V.I.; Barajas-Ledesma, E.; Medina-Flores, A.; Espitia-Cabrera, M.I. Synergic effect of the TiO2-CeO2 nanoconjugate system on the band-gap for visible light photocatalysis. Mater. Sci. Eng. B 2014, 183, 78–85. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Gamarra, D.; Hungría, A.B.; Fernández-García, M.; Munuera, G.; Hornés, A.; Bera, P.; Conesa, J.C.; Cámara, A.L. Characterization of Active Sites/Entities and Redox/Catalytic Correlations in Copper-Ceria-Based Catalysts for Preferential Oxidation of CO in H2-Rich Streams. Catalysts 2013, 3, 378–400. [Google Scholar] [CrossRef]
- Ilieva, L.; Petrova, P.; Pantaleo, G.; Zanella, R.; Liotta, L.F.; Georgiev, V.; Boghosian, S.; Kaszkur, Z.; Sobczak, J.W.; Lisowski, W.; et al. Gold catalysts supported on Y-modified ceria for CO-free hydrogen production via PROX. Appl. Catal. B Environ. 2016, 188, 154–168. [Google Scholar] [CrossRef]
- Mamontov, E.; Egami, T.; Brezny, R.; Koranne, M.; Tyagi, S. Lattice Defects and Oxygen Storage Capacity of Nanocrystalline Ceria and Ceria-Zirconia. J. Phys. Chem. B 2000, 104, 11110–11116. [Google Scholar] [CrossRef]
- Vindigni, F.; Manzoli, M.; Damin, A.; Tabakova, T.; Zecchina, A. Surface and Inner Defects in Au/CeO2 WGS Catalysts: Relation between Raman Properties, Reactivity and Morphology. Chem. Eur. J. 2011, 17, 4356–4361. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I.; Atherton, S.J.; Brigham, E.S.; Mallouk, T.E. Sensitized layered metal oxide semiconductor particles for photochemical hydrogen evolution from non sacrificial electron donors. J. Phys. Chem. 1993, 97, 11802–11810. [Google Scholar] [CrossRef]
- Galindo, F.; Gómez, R.; Aguilar, M. Photodegradation of the herbicide 2,4-dichlorophenoxyacetic acid on nanocrystalline TiO2–CeO2 sol–gel catalysts. J. Mol. Catal. Chem. 2008, 281, 119–125. [Google Scholar] [CrossRef]
- Claus, P.; Brückner, A.; Mohr, C.; Hofmeister, H. Supported Gold Nanoparticles from Quantum Dot to Mesoscopic Size Scale: Effect of Electronic and Structural Properties on Catalytic Hydrogenation of Conjugated Functional Groups. J. Am. Chem. Soc. 2000, 122, 11430–11439. [Google Scholar] [CrossRef]
- Porter, J.F.; Li, Y.-G.; Chan, C.K. The effect of calcination on the microstructural characteristics and photoreactivity of Degussa P-25 TiO2. J. Mater. Sci. 1999, 34, 1523–1531. [Google Scholar] [CrossRef]
- Ohtani, B. Titania Photocatalysis beyond Recombination: A Critical Review. Catalysts 2013, 3, 942–953. [Google Scholar] [CrossRef]
- Watanabe, S.; Ma, X.; Song, C. Characterization of Structural and Surface Properties of Nanocrystalline TiO2−CeO2 Mixed Oxides by XRD, XPS, TPR, and TPD. J. Phys. Chem. C 2009, 113, 14249–14257. [Google Scholar] [CrossRef]
- Sun, X.; Liu, H.; Dong, J.; Wei, J.; Zhang, Y. Preparation and Characterization of Ce/N-Codoped TiO2 Particles for Production of H2 by Photocatalytic Splitting Water Under Visible Light. Catal. Lett. 2010, 135, 219–225. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Surface Area (m2/g) | Eg (eV) | Crystallite Size (nm) a | Crystal Phase a |
|---|---|---|---|---|
| TiO2 | 44.8 | 2.98 | 24 | TiO2 Anatase-Rutile |
| CeO2 | 110.2 | 2.90 | 10 | CeO2 Fluorite |
| TiO2-10%CeO2 | 47.5 | 2.93 | 22 | TiO2 Anatase-Rutile CeO2 Fluorite |
| Au/TiO2 | 46.4 | 2.97 | 21 | TiO2 Anatase-Rutile |
| Au/CeO2 | 112.7 | 2.95 | 11 | CeO2 Fluorite |
| Au/TiO2-10%CeO2 | 50.5 | 2.96 | 19 | TiO2 Anatase-Rutile, CeO2 Fluorite |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorenza, R.; Bellardita, M.; D’Urso, L.; Compagnini, G.; Palmisano, L.; Scirè, S. Au/TiO2-CeO2 Catalysts for Photocatalytic Water Splitting and VOCs Oxidation Reactions. Catalysts 2016, 6, 121. https://doi.org/10.3390/catal6080121
Fiorenza R, Bellardita M, D’Urso L, Compagnini G, Palmisano L, Scirè S. Au/TiO2-CeO2 Catalysts for Photocatalytic Water Splitting and VOCs Oxidation Reactions. Catalysts. 2016; 6(8):121. https://doi.org/10.3390/catal6080121
Chicago/Turabian StyleFiorenza, Roberto, Marianna Bellardita, Luisa D’Urso, Giuseppe Compagnini, Leonardo Palmisano, and Salvatore Scirè. 2016. "Au/TiO2-CeO2 Catalysts for Photocatalytic Water Splitting and VOCs Oxidation Reactions" Catalysts 6, no. 8: 121. https://doi.org/10.3390/catal6080121
APA StyleFiorenza, R., Bellardita, M., D’Urso, L., Compagnini, G., Palmisano, L., & Scirè, S. (2016). Au/TiO2-CeO2 Catalysts for Photocatalytic Water Splitting and VOCs Oxidation Reactions. Catalysts, 6(8), 121. https://doi.org/10.3390/catal6080121