
Biomass Derived Chemicals: Furfural Oxidative Esterification to Methyl-2-furoate over Gold Catalysts





Abstract
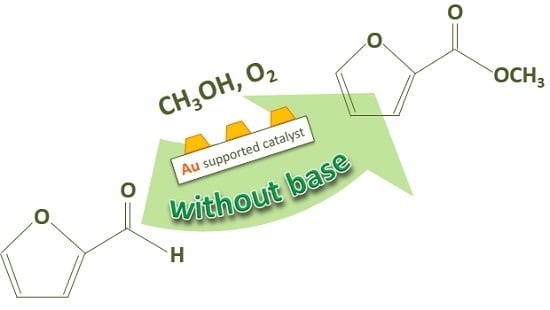
:
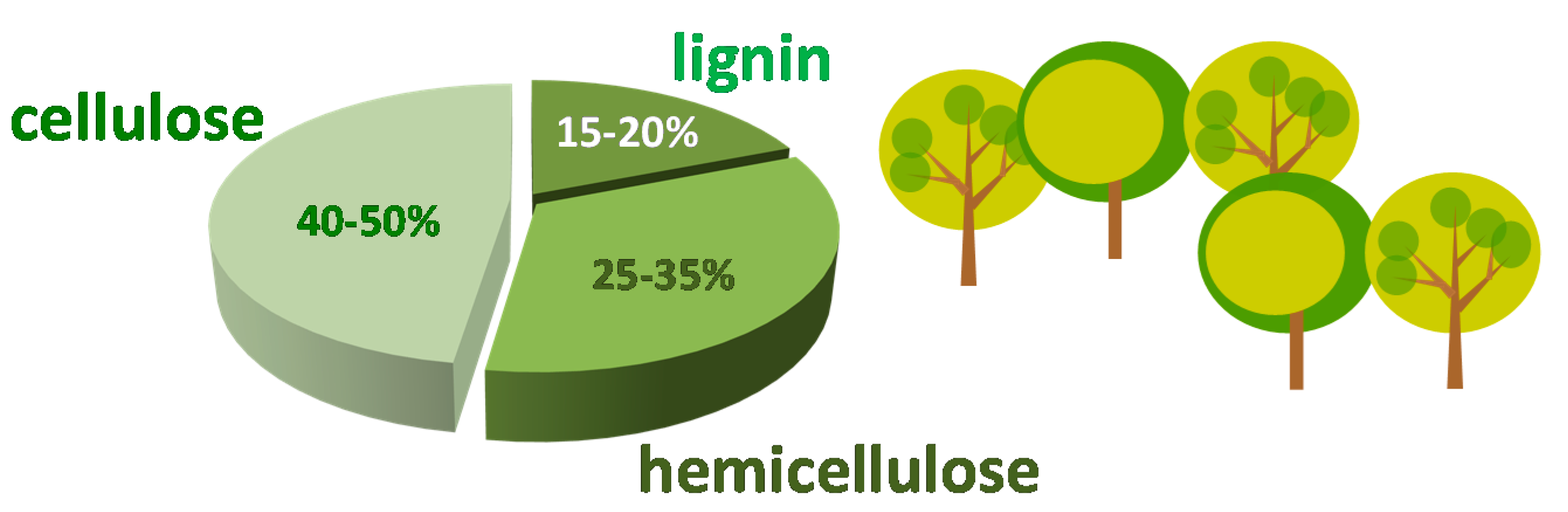
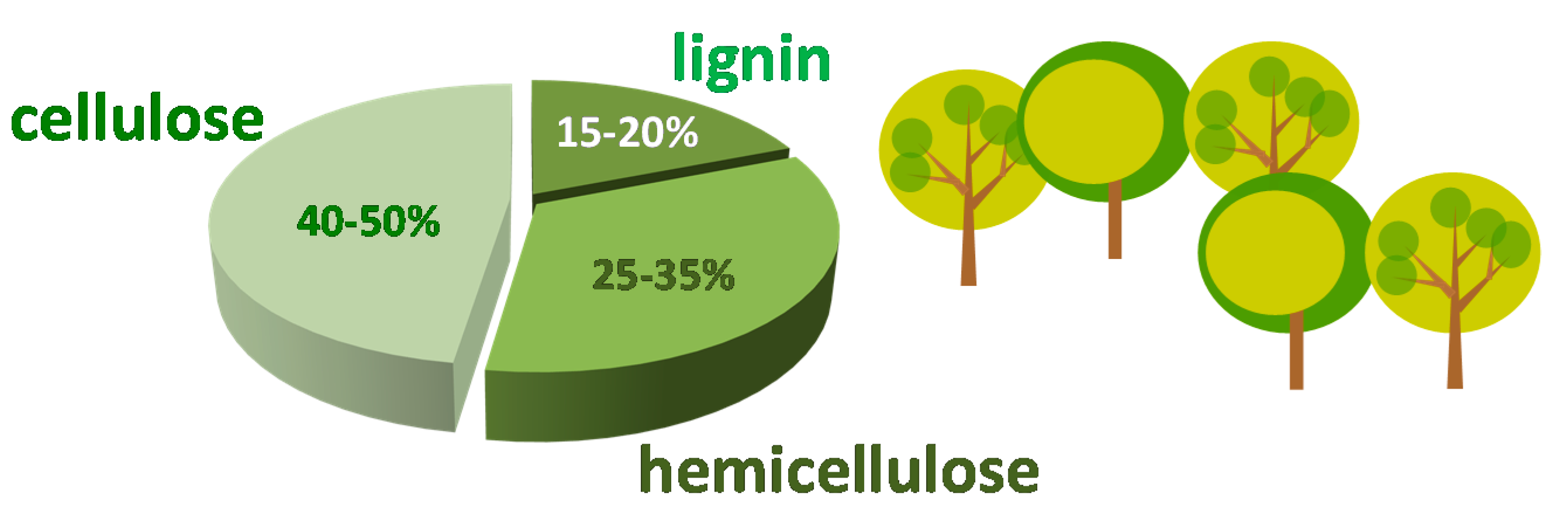
1. Furfural as Platform Molecule Coming from Biomass
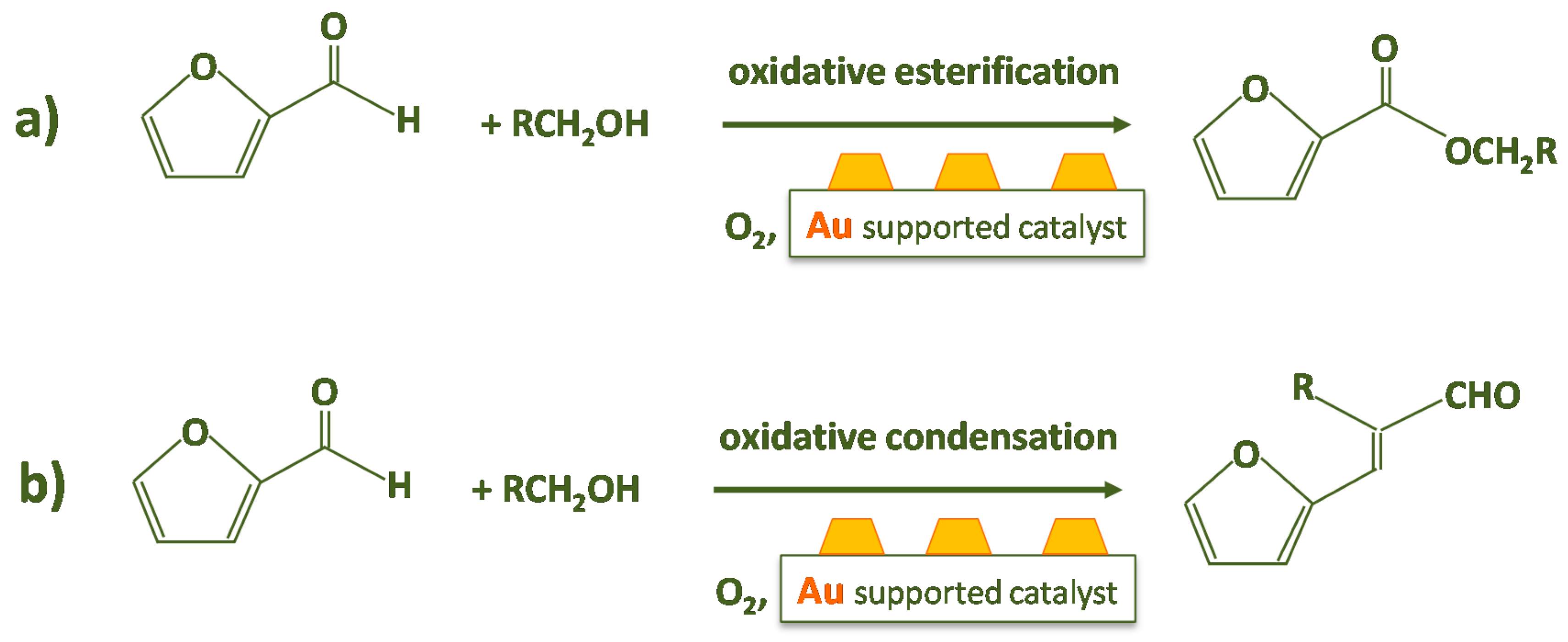
2. Furfural Oxidative Esterification to Methyl-2-furoate
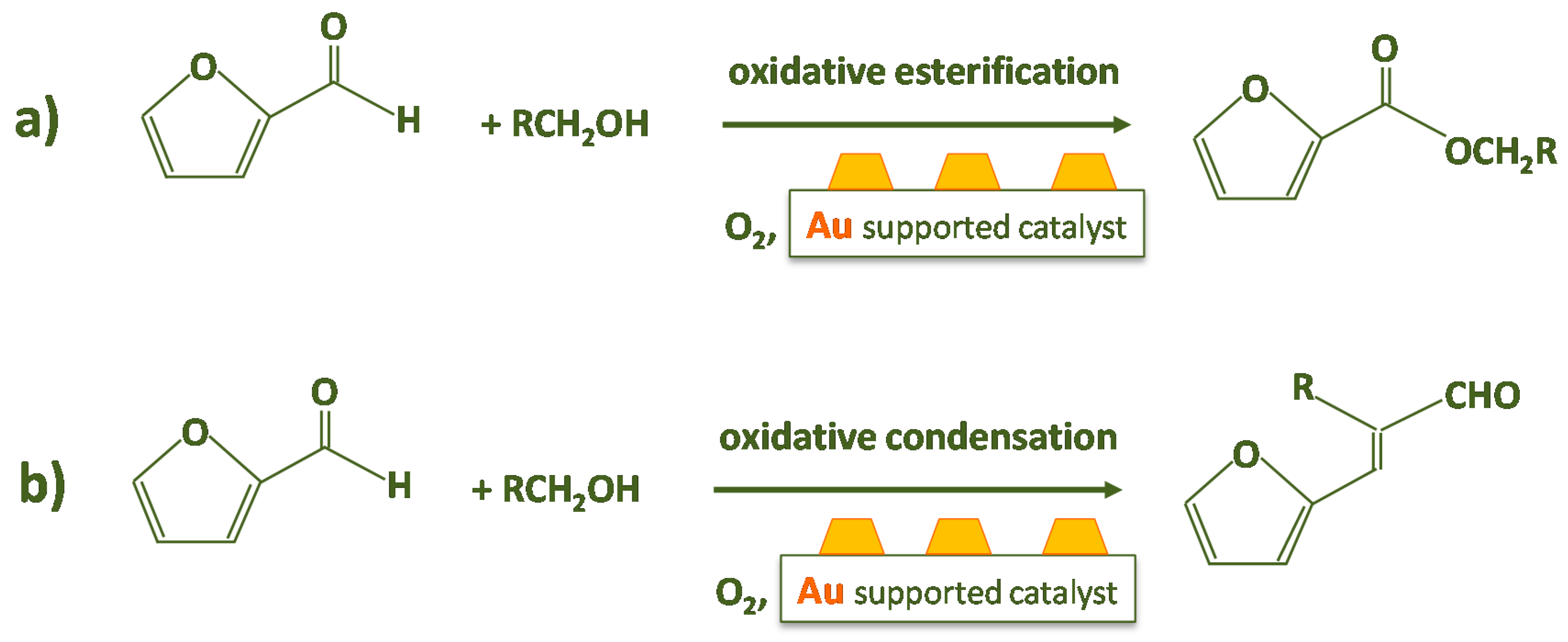
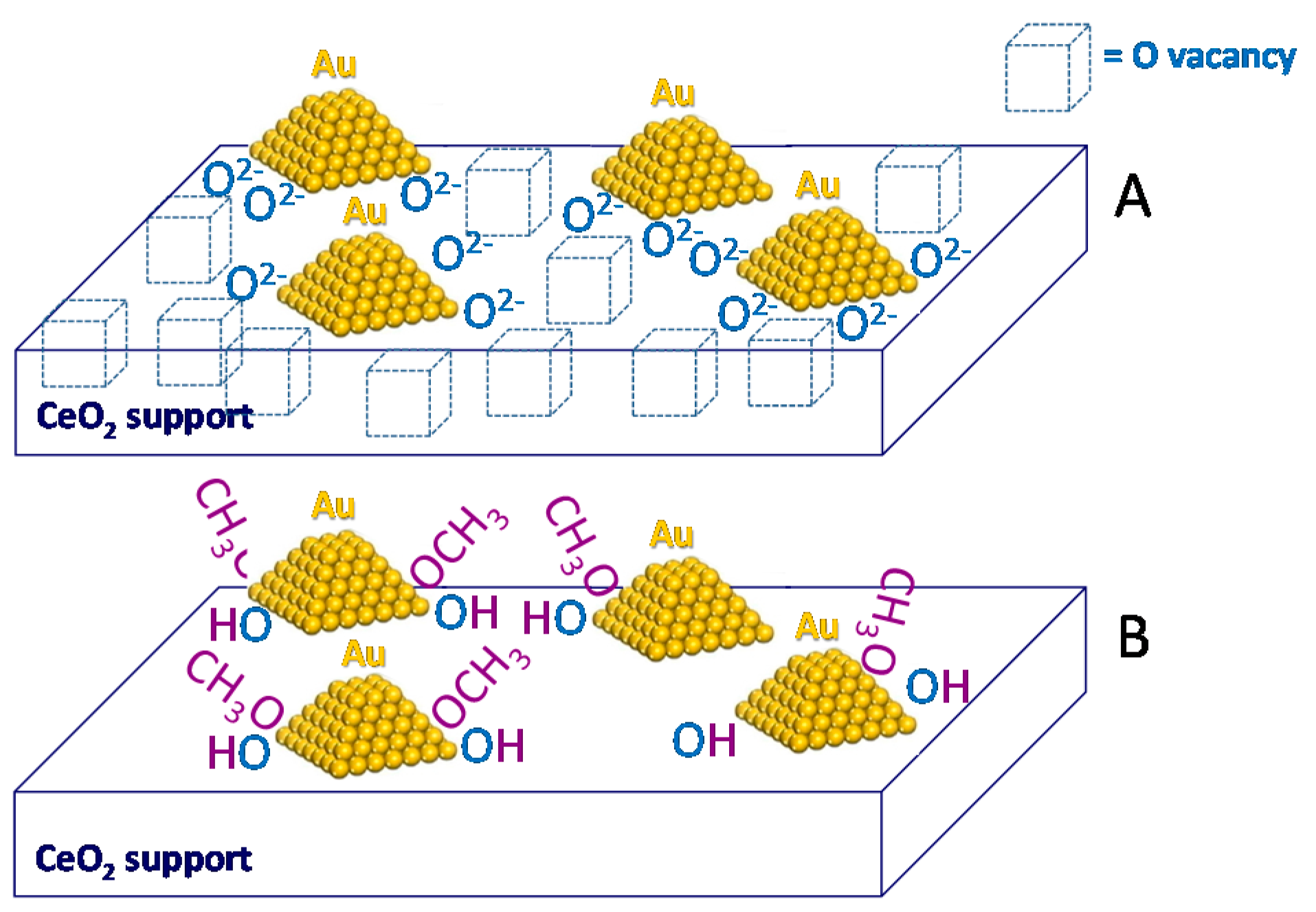
On the Reaction Mechanism
- (i)
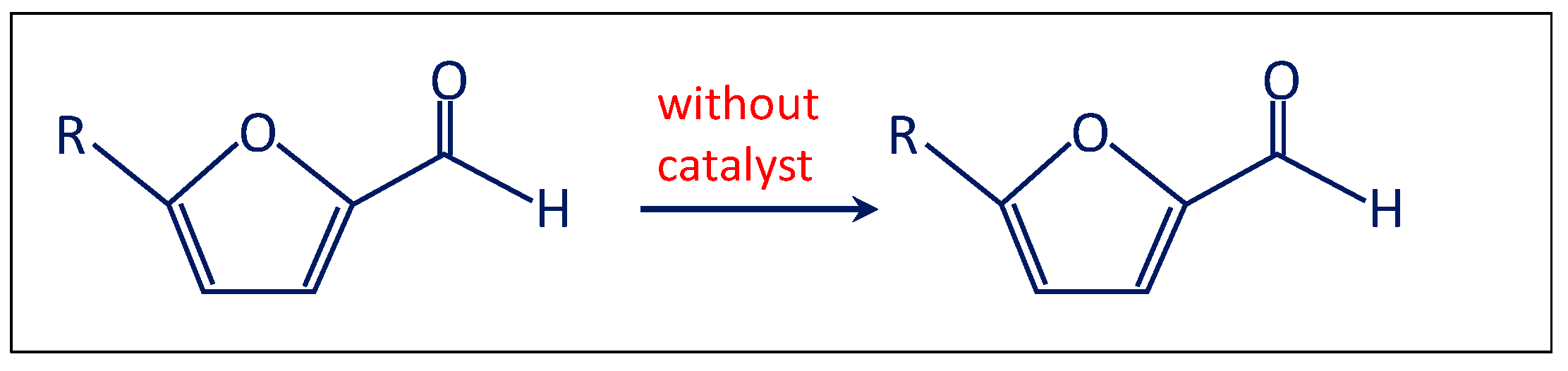
- Firstly, the reaction was performed without the catalyst (Scheme 2). Therefore, furfural, n-octane, methanol and 6 bar of O2 were charged in the autoclave. No traces of furoate, acetal or other products were detected after 3 h of reaction at 120 °C, indicating that the catalyst is needed for performing the reaction.
- (ii)
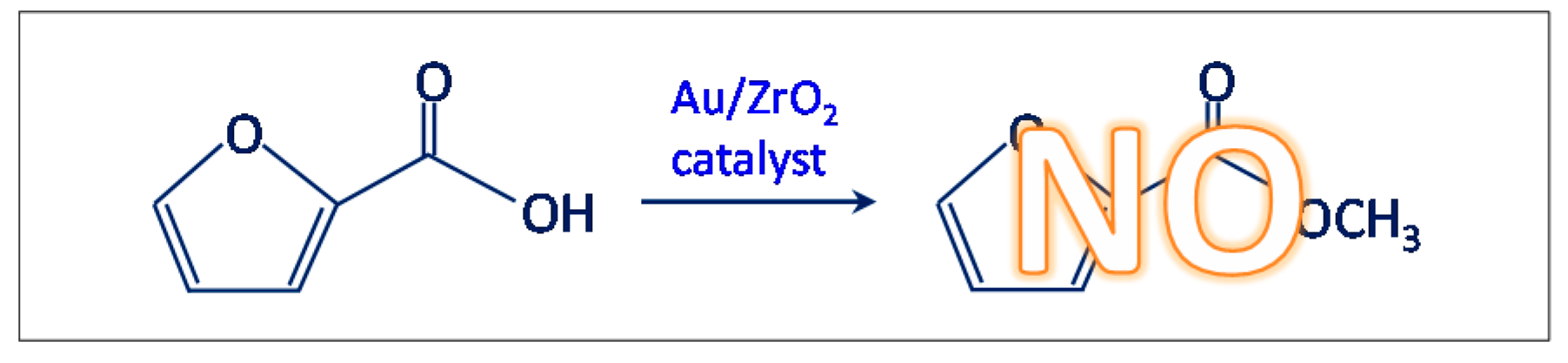
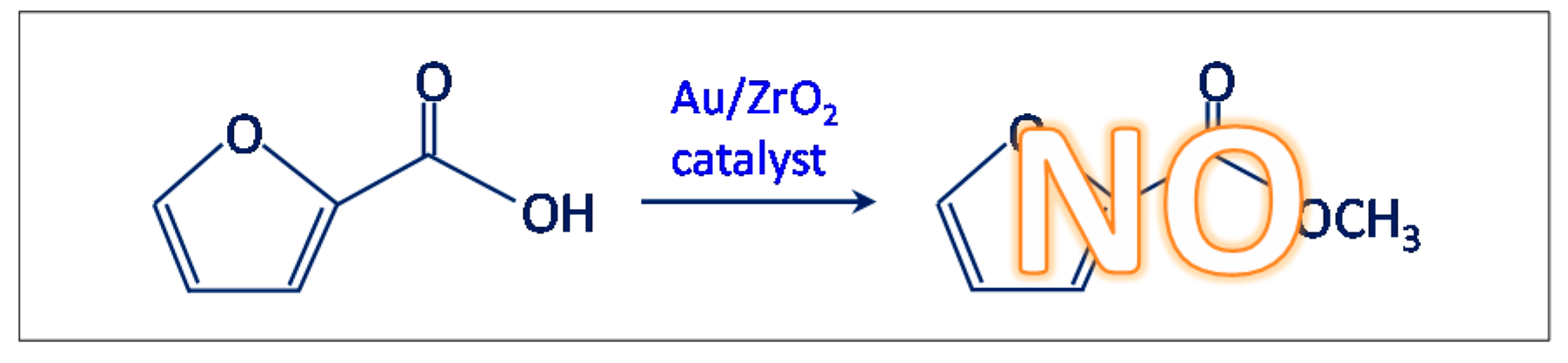
- Another test was carried out in the presence of the Au/ZrO2 catalyst by adopting the usual reaction conditions, but employing furoic acid as substrate instead of furfural (see Scheme 3). The aim was to rule out the furoate formation via furoic acid. No furoate was formed during such test.
- (iii)
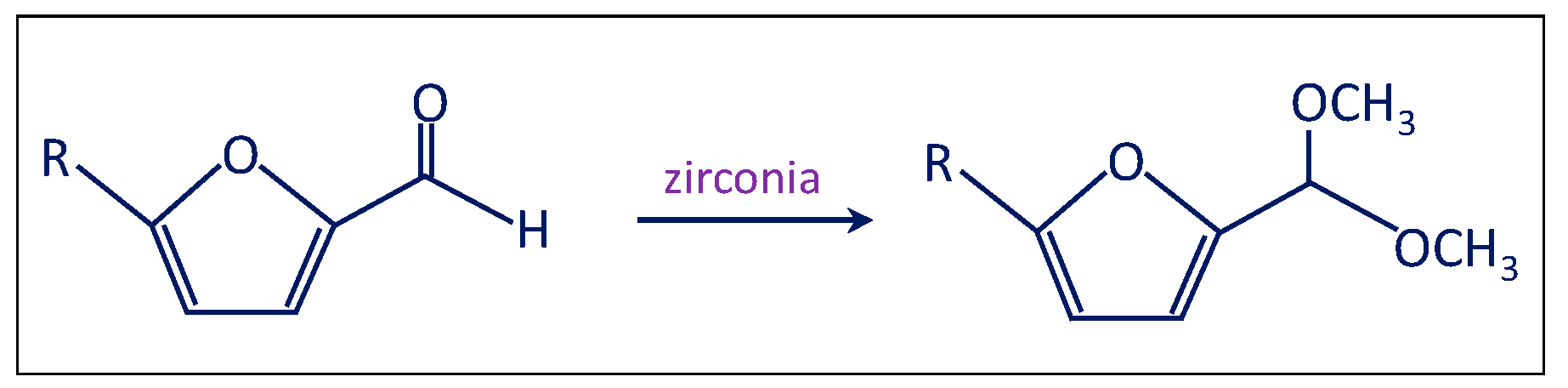
- The third test involved the use of bare zirconia (either plain zirconia or sulfated zirconia), instead of employing the Au/ZrO2 catalyst (Scheme 4). The autoclave was charged as usual and the typical reaction conditions were chosen. Furfural was totally converted (100%); nevertheless, only acetal was produced, possibly due to the Lewis acidity displayed by zirconia [40,41].
- (iv)
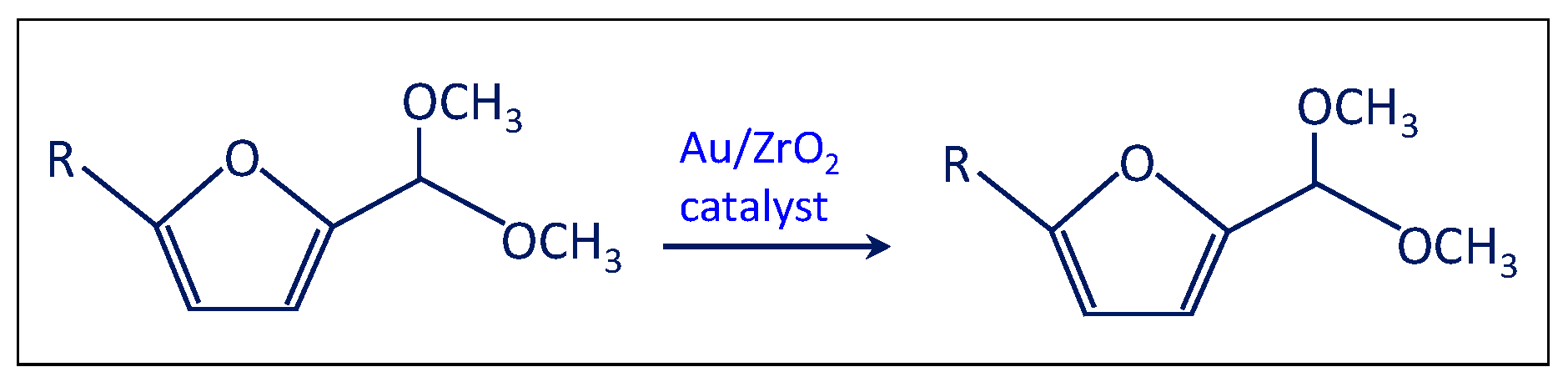
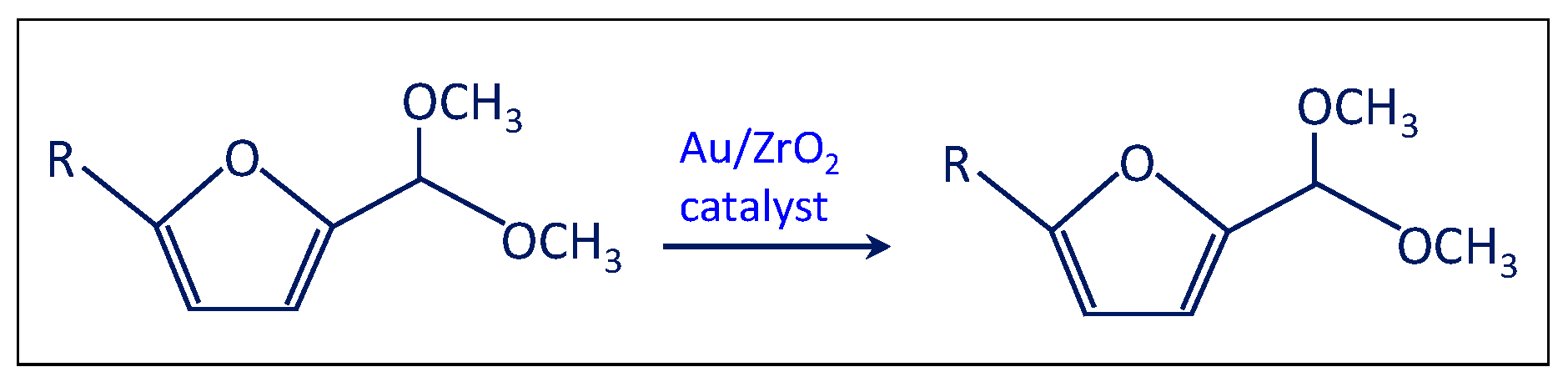
- The solution was filtered and separated from zirconia after the test at point (iii) and performed in the presence of the bare support. Then the solution of acetal in methanol was charged in the autoclave in the presence of the Au/ZrO2 catalyst in order to verify if the acetal molecule can be converted in the furoate over the catalyst under reaction conditions. According to Scheme 5, only acetal was found after the catalytic test. Hence, the results indicate that furfural can be converted into acetal in the presence of the bare support, but acetal, due to its stability, cannot be transformed into the desired furoate.
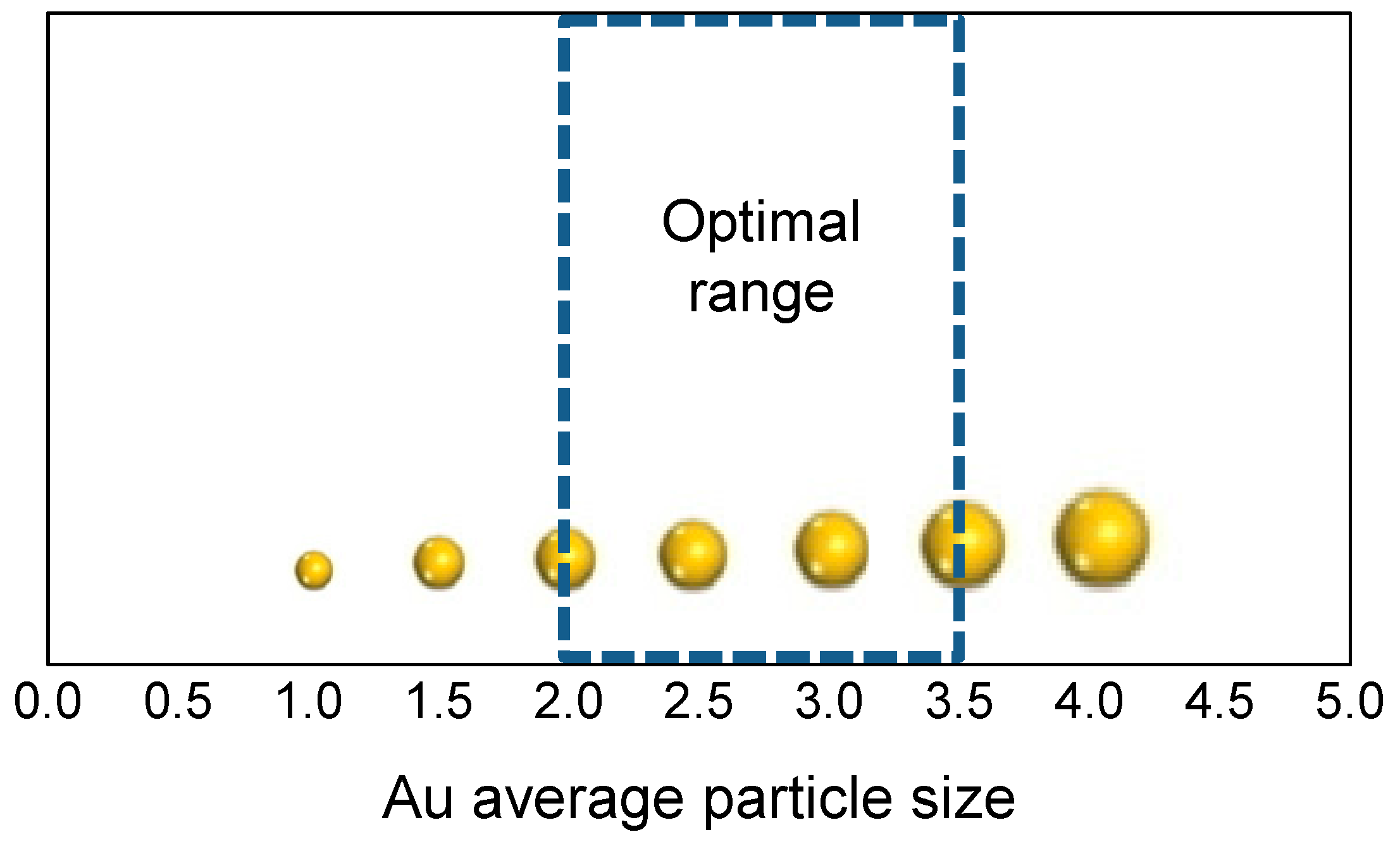
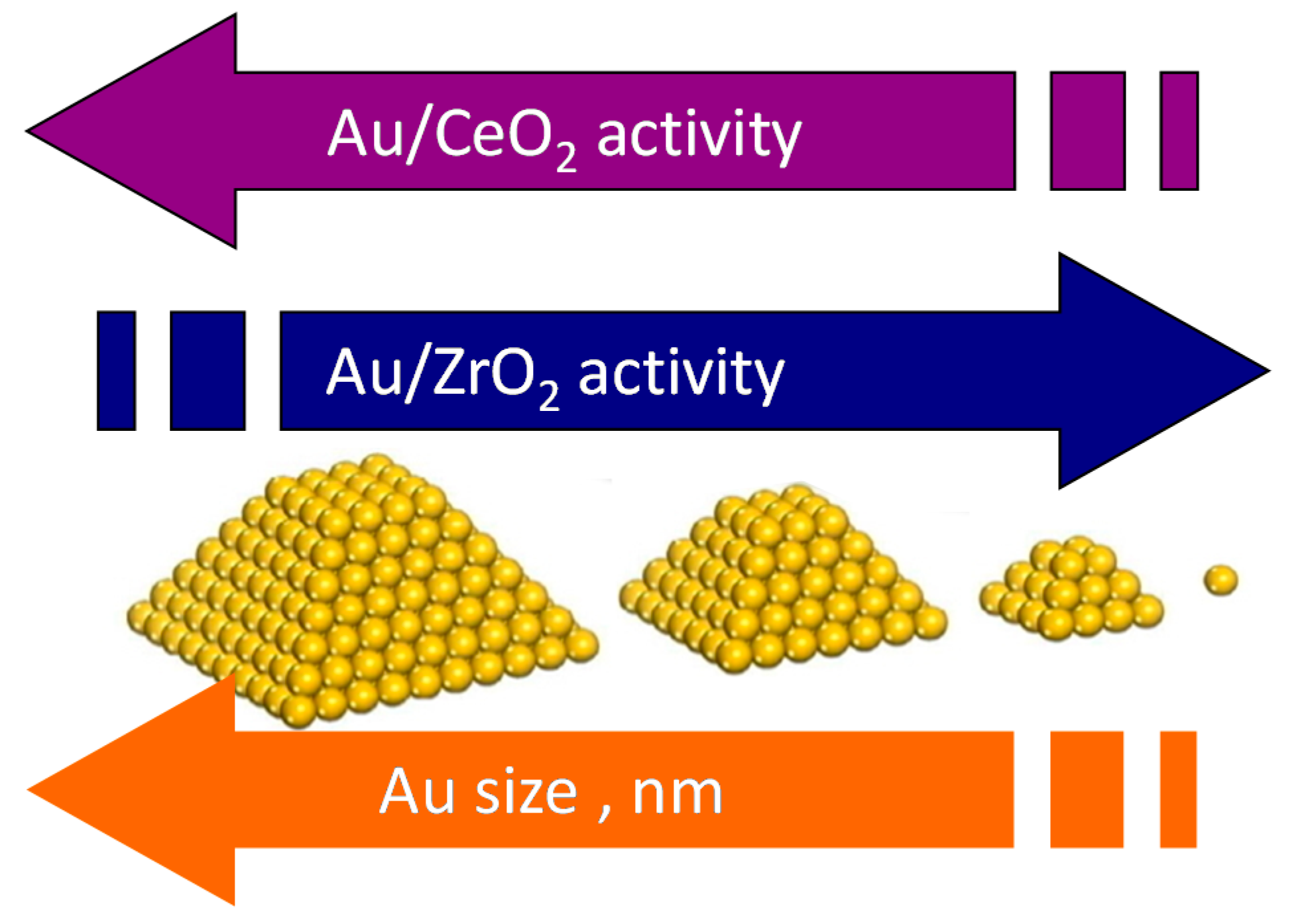
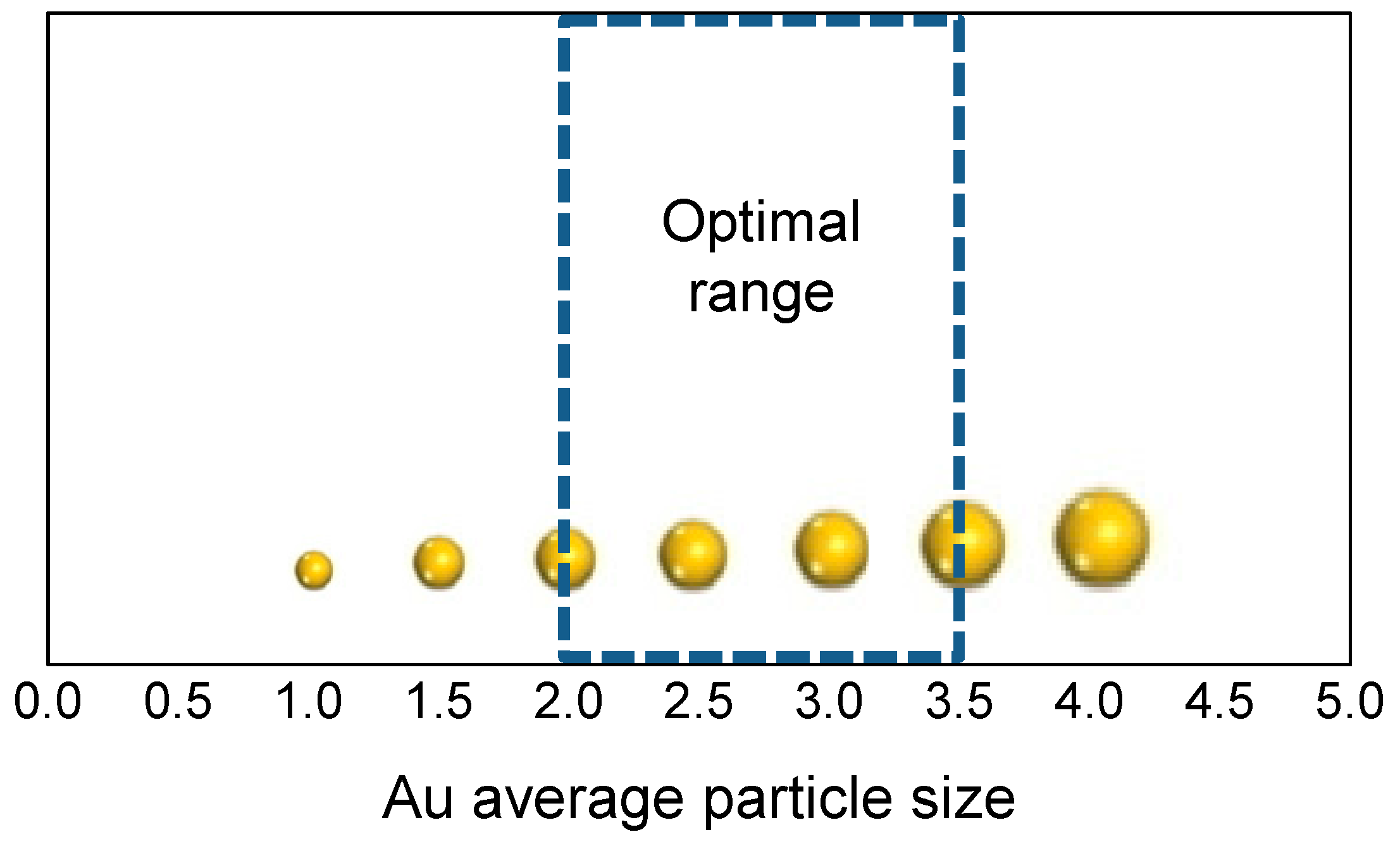
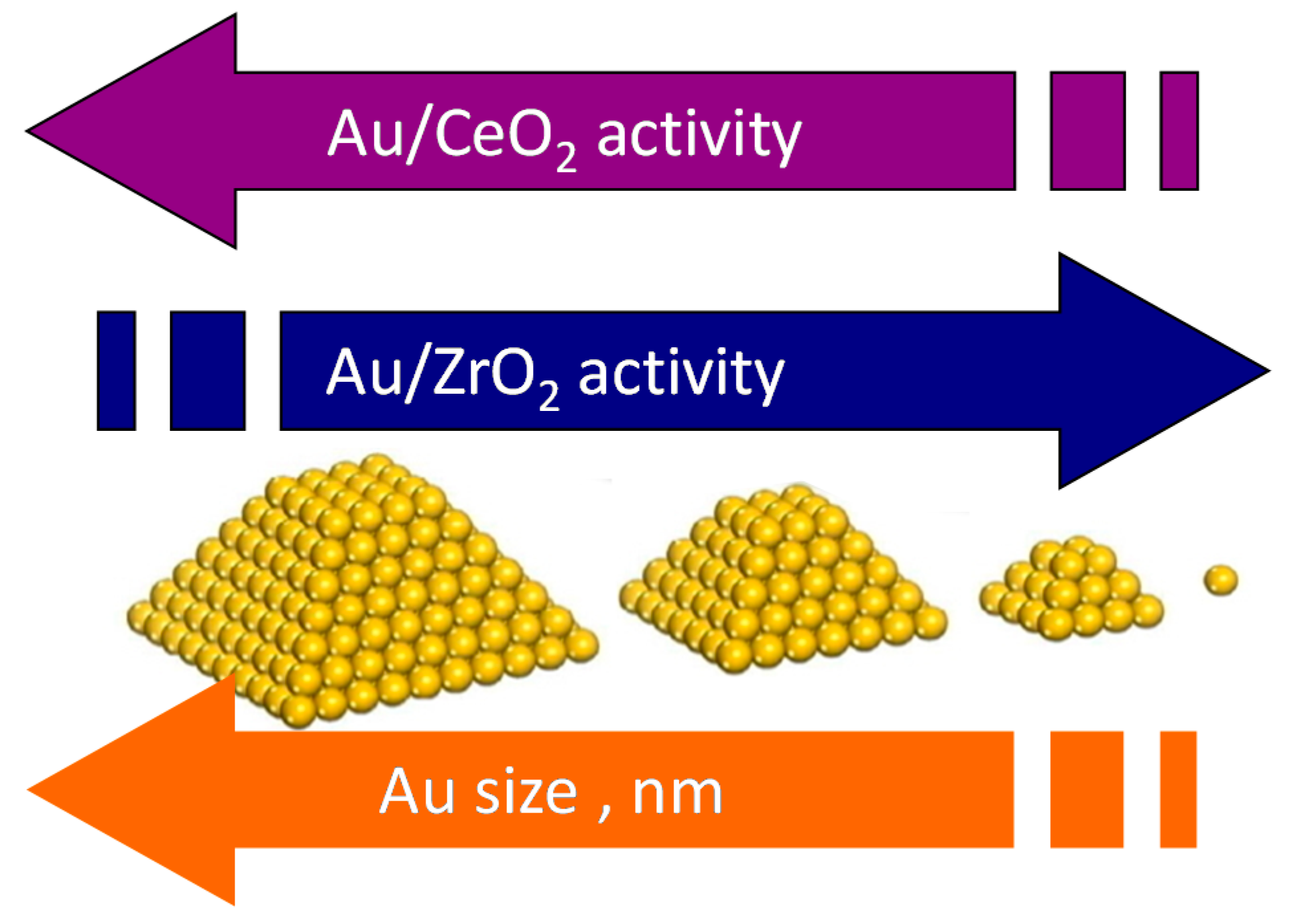
3. Role of the Gold Size
4. Effect of the Nature of the Support
4.1. ZrO2 vs. TiO2: A Comparison with the Au/TiO2 Reference Catalyst Provided by the Worl Gold Council
4.2. Which Could Be the Best Choice of the Support?
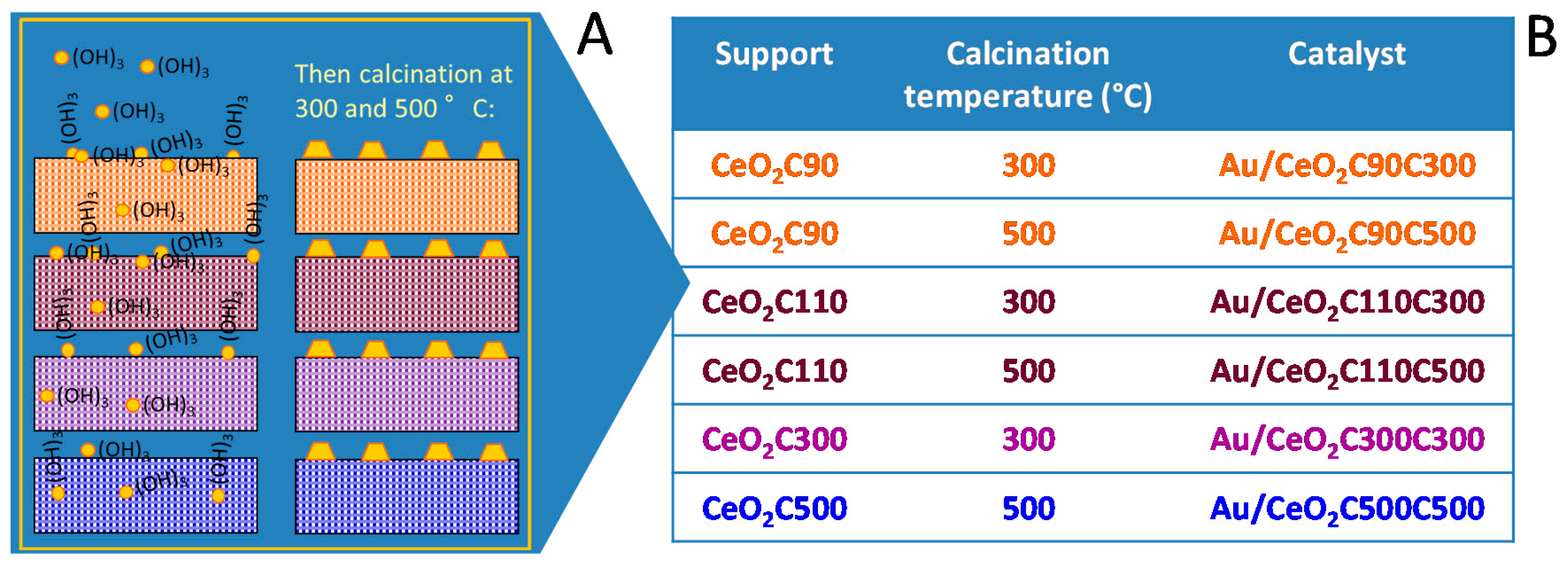
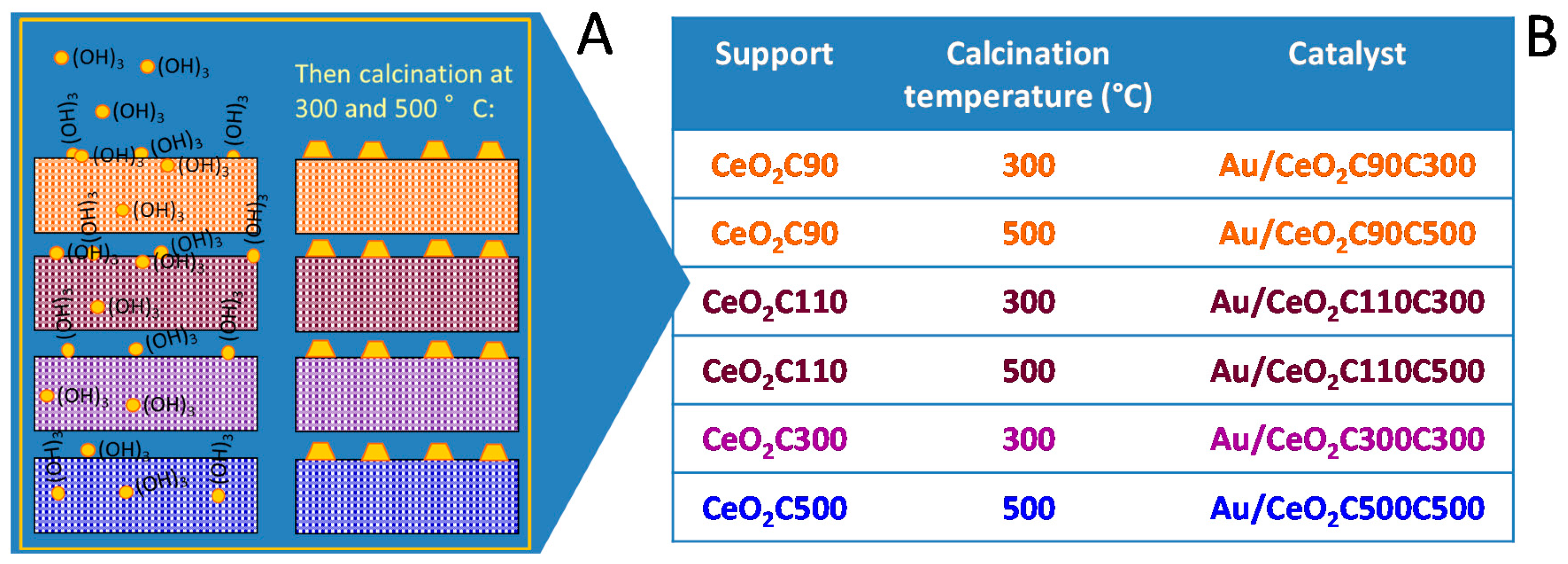
Catalyst Design. Optimization of Gold Catalysts Supported on Ceria: A Case Study
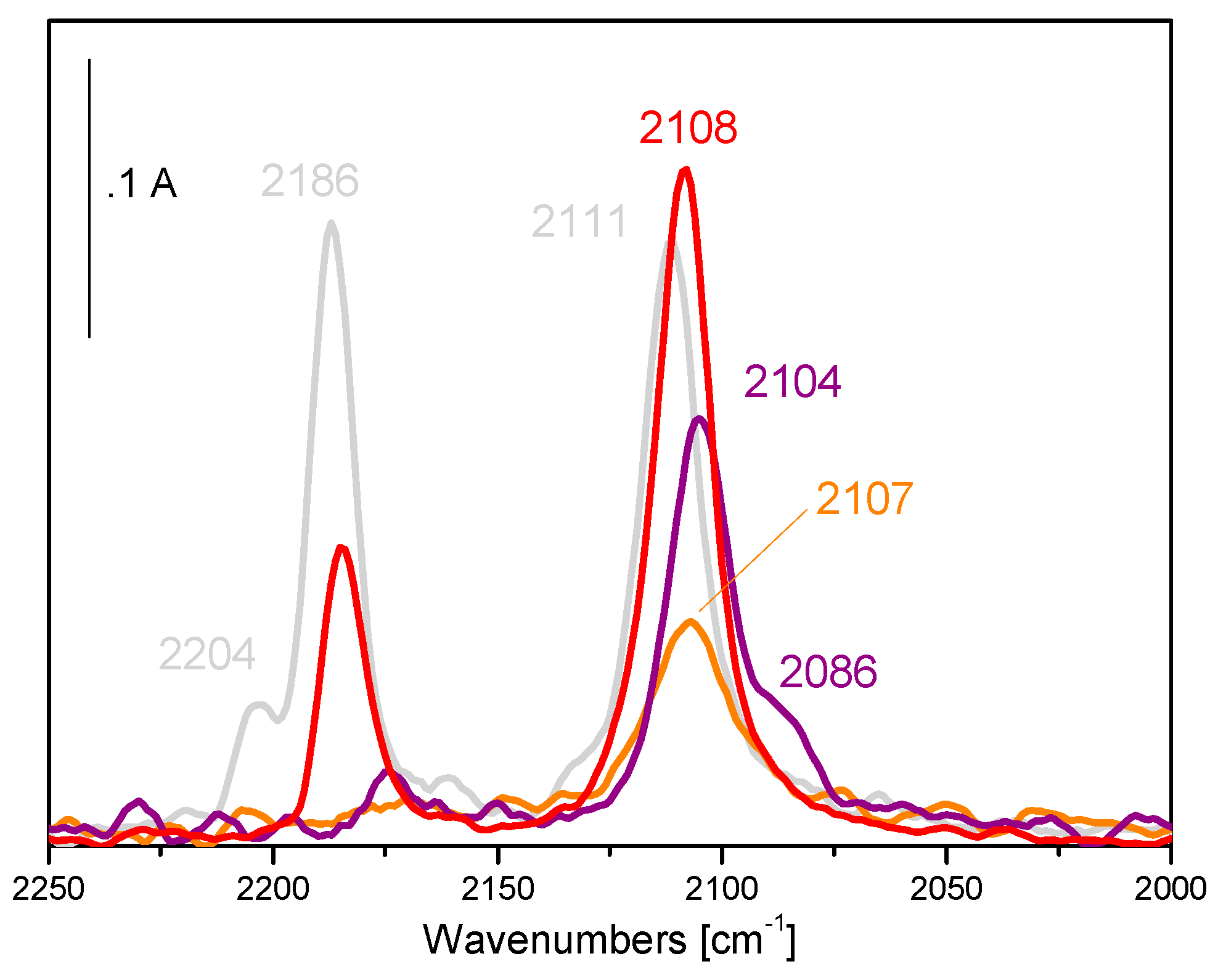
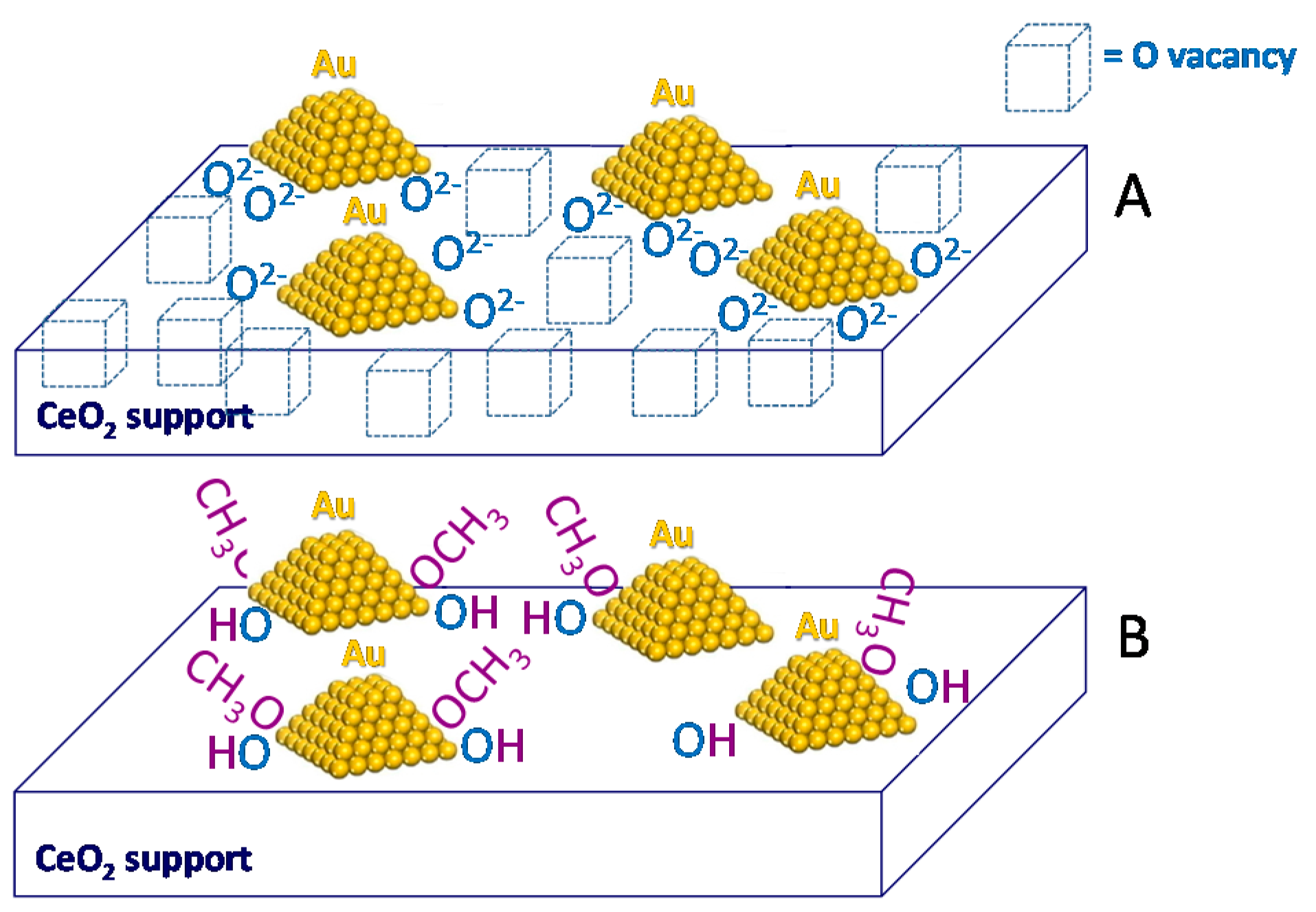
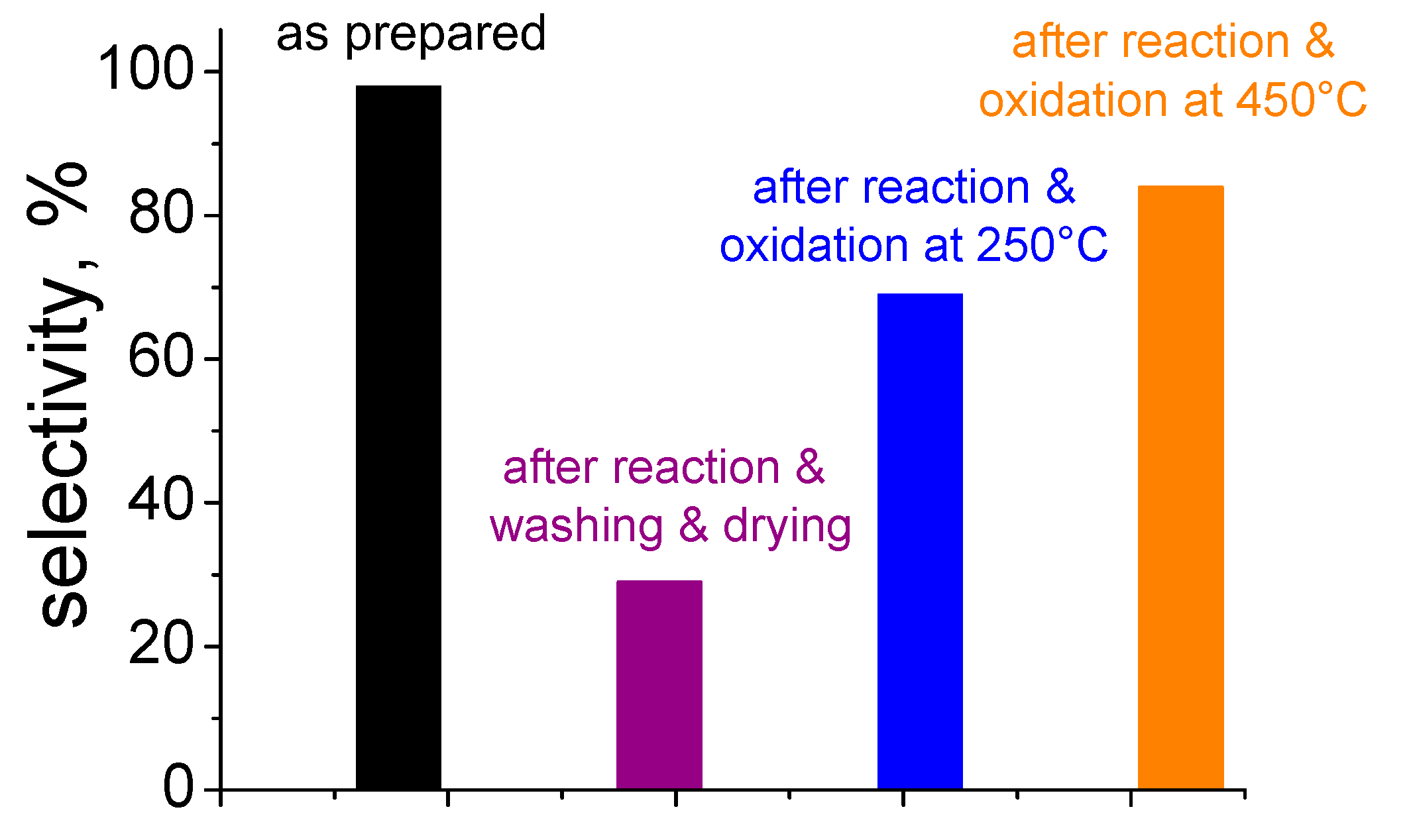
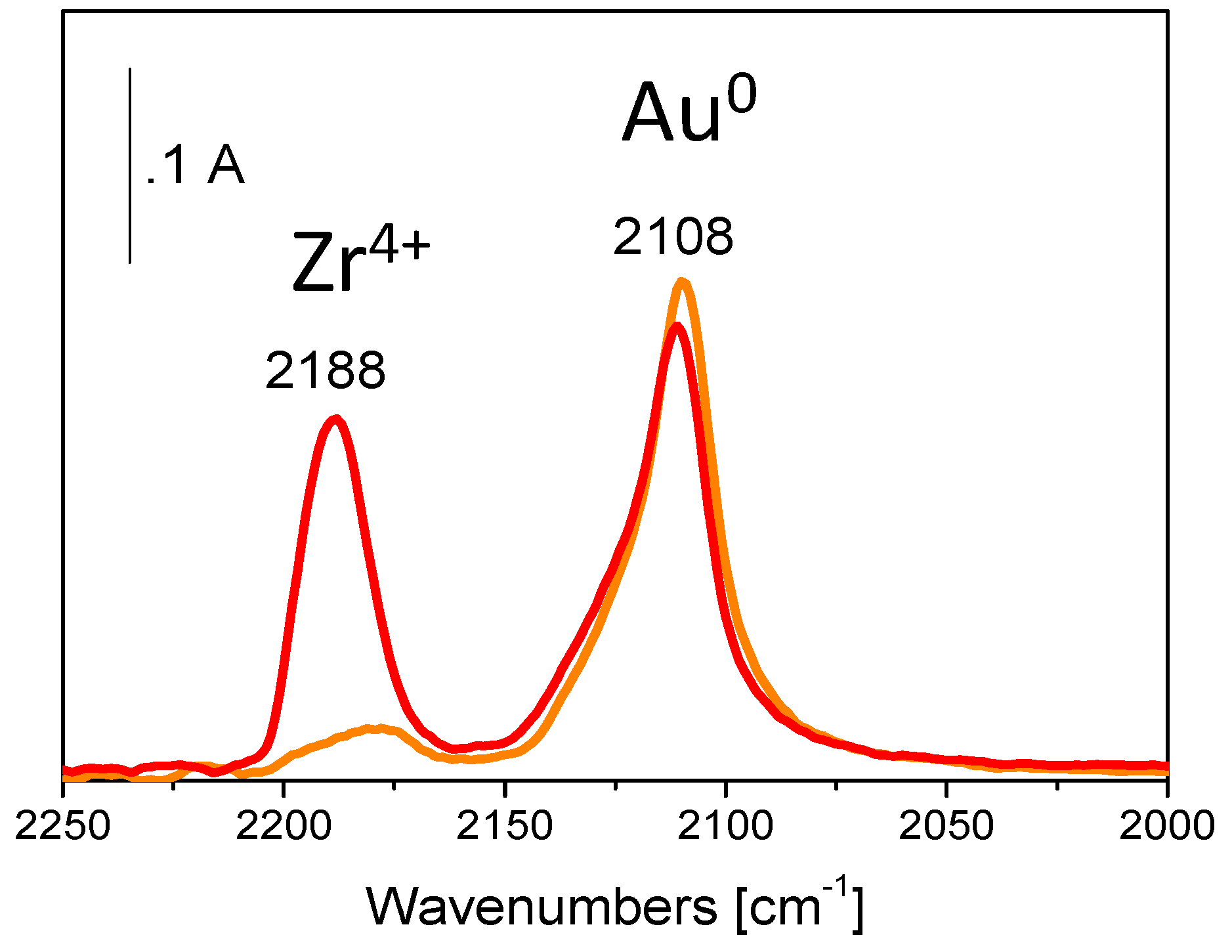
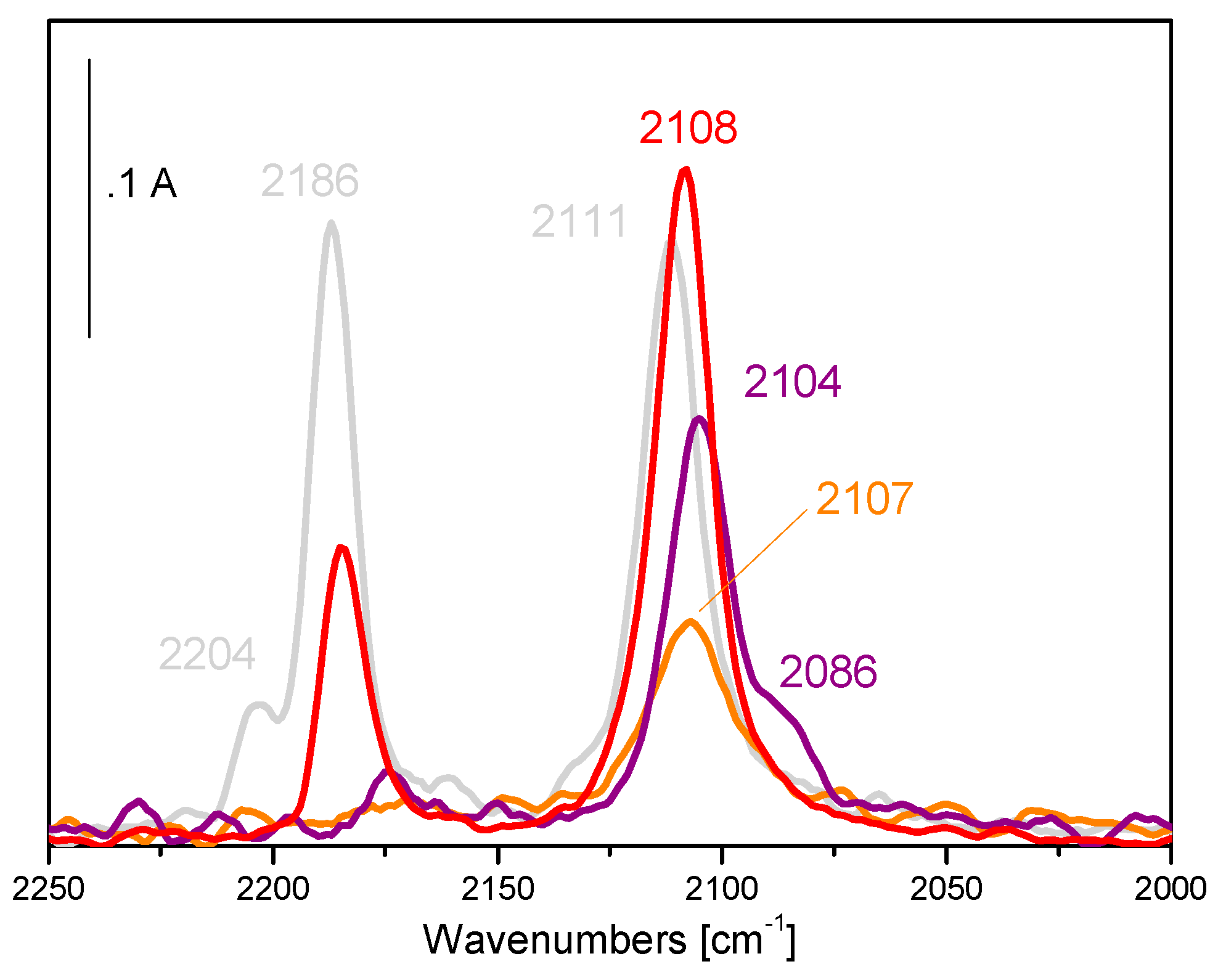
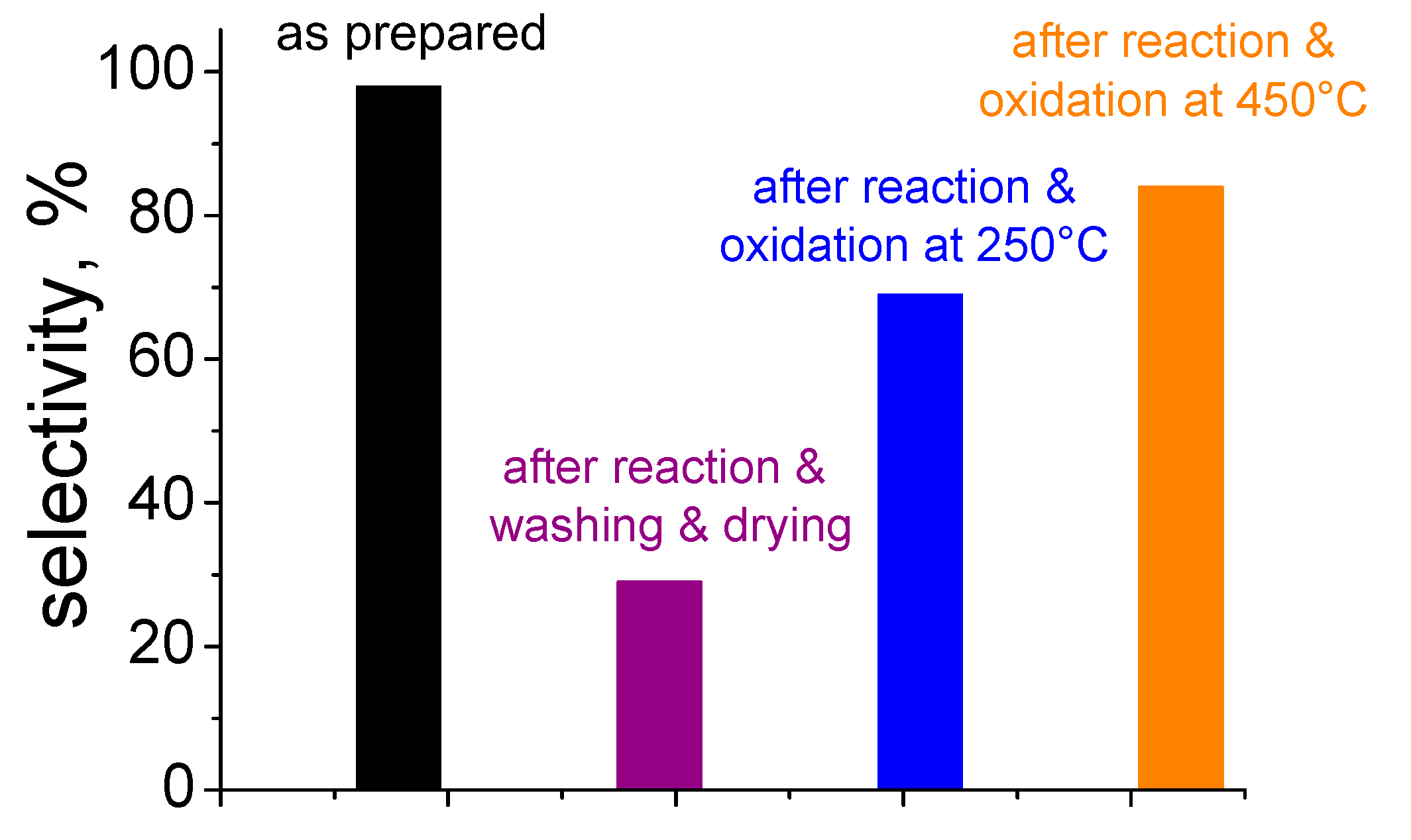
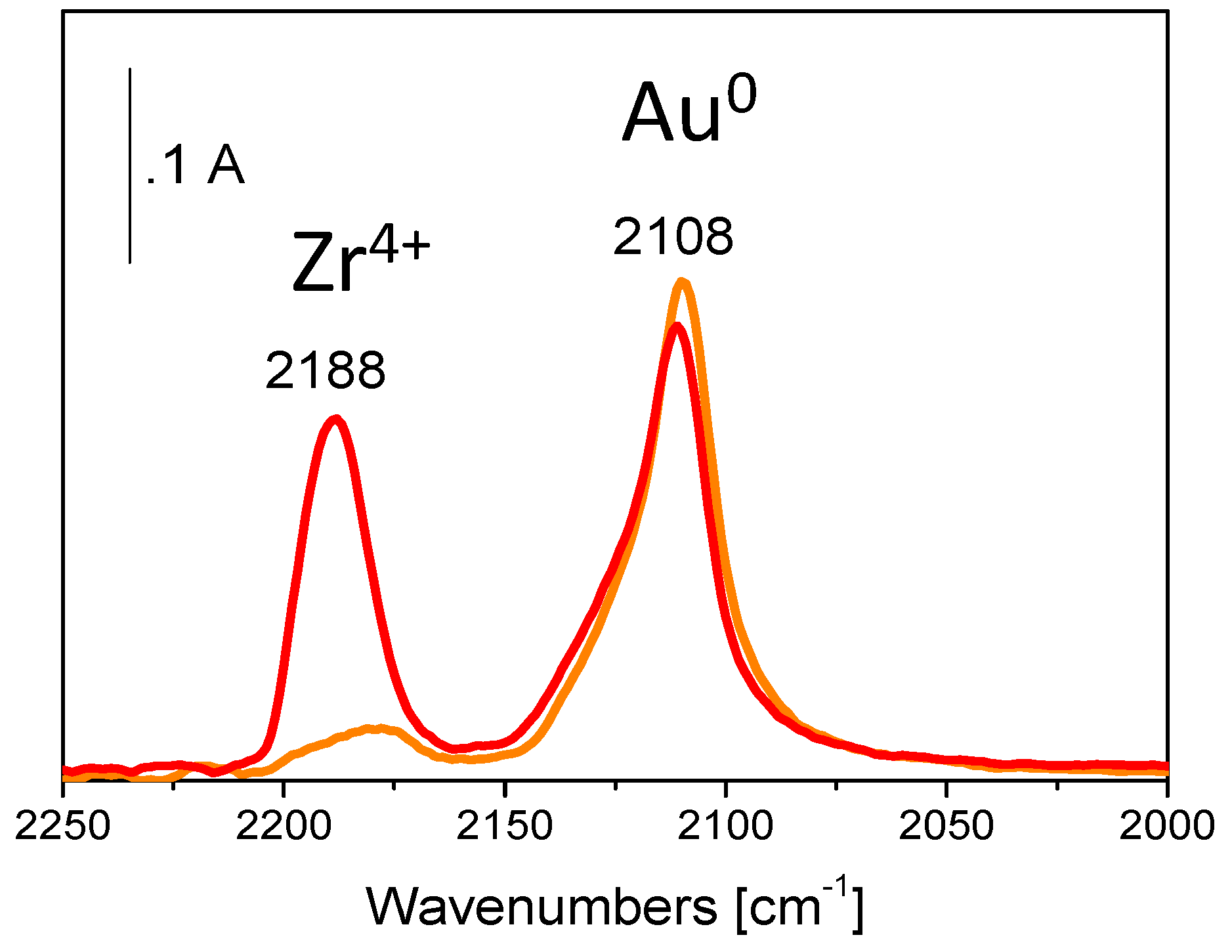
5. Stability of the Gold Species under Reaction Conditions
6. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Perlack, R.D.; Wright, L.L.; Turhollow, A.; Graham, R.L.; Stokes, B.; Erbach, D.C. Biomass as Feedstock for a Bioenergy and Bioproducts Industry: The Technical Feasibility of a Billion-Ton Annual Supply; Oak Ridge Naional Laboratory, US Department of Energy: Oak Ridge, TN, USA, 2005.
- Kudakasseril, K.J.; Raveendran, N.G.; Hussain, A.; Vijaya Raghavan, G.S. Feedstocks, logistics and pre-treatment processes for sustainable lignocellulosic bio-refineries: A comprehensive review. Renew. Sustain. Energy Rev. 2013, 25, 205–219. [Google Scholar] [CrossRef]
- Tekin, K.; Karagöz, S.; Bektaş, S. A review of hydrothermal biomass processing. Renew. Sustain. Energy Rev. 2014, 40, 673–687. [Google Scholar] [CrossRef]
- Yan, K.; Jarvis, C.; Gu, J.; Yan, Y. Production and catalytic transformation of levulinic acid: A platform for speciality chemicals and fuels. Renew. Sustain. Energy Rev. 2015, 51, 986–997. [Google Scholar] [CrossRef]
- Yan, K.; Yang, Y.; Chai, J.; Lu, Y. Catalytic reactions of gamma-valerolactone: A platform to fuels and value-added chemicals. Appl. Catal. B 2015, 179, 292–304. [Google Scholar] [CrossRef]
- Yan, K.; Wu, G.; Lafleur, T.; Jarvis, C. Production, properties and catalytic hydrogenation of furfural to fuel additives and value-added chemicals. Renew. Sustain. Energy Rev. 2014, 38, 663–676. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels. Green Chem. 2014, 16, 516–547. [Google Scholar] [CrossRef]
- Dutta, S.; De, S.; Saha, B.; Alam, M.I. Advances in conversion of hemicellulosic biomass to furfural and upgrading to biofuels. Catal. Sci. Technol. 2012, 2, 2025–2036. [Google Scholar] [CrossRef]
- Ratanakhanokchai, K.; Waeonukul, R.; Pason, P.; Tachaapaikoon, C.; Kyu, K.L.; Sakka, K.; Kosugi, A.; Mori, Y. Paenibacillus Curdlanolyticus Strain B-6 Multienzyme Complex: A Novel System for Biomass Utilization; Matovic, M.D., Ed.; InTech: Rijeka, Croatia, 2013; ISBN 978-953-51-1106-1. [Google Scholar] [CrossRef]
- Lange, J.P. Lignocellulose conversion: An introduction to chemistry, process and economics. Biofuels Bioprod. Biorefin. 2007, 1, 39–48. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G.R. Results of Screening for Potential Candidates from Sugars and Synthesis Gas. In Top Value Added Chemicals from Biomass; U.S. Department of Energy: Washington, DC, USA; Volume I, 2004. Available online: http://www.nrel.gov/docs/fy04osti/35523.pdf (accessed on 15 March 2016). [Google Scholar]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—The US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Wu, C.; Chen, W.; Zhong, L.; Peng, X.; Sun, R.; Fang, J.; Zheng, S. Conversion of xylose into furfural using lignocellulosic acid as catalyst in ionic liquid. J. Agric. Food Chem. 2014, 62, 7430–7435. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Liu, S.; Abu-Omar, M.M.; Mosier, N.S. Selective conversion of biomass hemicellulose to furfural using maleic acid with microwave heating. Energy Fuels 2012, 26, 1298–1304. [Google Scholar] [CrossRef]
- Lange, J.-P.; van der Heide, E.; van Buijtenen, J.; Price, R. Furfural—A promising platform for lignocellulosic biofuels. ChemSusChem 2012, 5, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Bohre, A.; Dutta, S.; Saha, B.; Abu-Omar, M.M. Upgrading furfurals to drop-in biofuels: An overview. ACS Sustain. Chem. Eng. 2015, 3, 1263–1277. [Google Scholar] [CrossRef]
- Stevens, J.G.; Bourne, R.A.; Twigg, M.V.; Poliakoff, M. Real-time product switching using a twin catalyst system for the hydrogenation of furfural in supercritical CO2. Angew. Chem. Int. Ed. 2010, 49, 8856–8859. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Liu, H.; Luque, R.; Li, Y. Efficient and selective hydrogenation of biomass-derived furfural to cyclopentanone using Ru catalysts. Green Chem. 2015, 17, 4183–4188. [Google Scholar] [CrossRef]
- Zhang, G.-S.; Zhu, M.-M.; Zhang, Q.; Liu, Y.-M.; He, H.-Y.; Cao, Y. Towards quantitative and scalable transformation of furfural to cyclopentanone with supported gold catalysts. Green Chem. 2016. [Google Scholar] [CrossRef]
- Guo, H.; Yin, G. Catalytic aerobic oxidation of renewable furfural with phosphomolybdic acid catalyst: An alternative route to maleic acid. J. Phys. Chem. C 2011, 115, 17516–17522. [Google Scholar] [CrossRef]
- Weingarten, R.; Cho, J.; Conner, C.; Huber, G.W. Kinetics of furfural production by dehydration of xylose in a biphasic reactor with microwave heating. Green Chem. 2010, 12, 1423–1429. [Google Scholar] [CrossRef]
- Fernando, S.; Adhikari, S.; Chandrapal, C.; Murali, N. Biorefineries: Current status, challenges, and future direction. Energy Fuels 2006, 20, 1727–1737. [Google Scholar] [CrossRef]
- Liu, Z.; Tong, X.; Liu, J.; Xue, S. A smart catalyst system for the valorization of renewable furfural in aliphatic alcohols. Catal. Sci. Technol. 2016, 6, 1214–1221. [Google Scholar] [CrossRef]
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Liu, Z.; Yu, L. A tunable process: Catalytic transformation of renewable furfural with aliphatic alcohols in the presence of molecular oxygen. Chem. Commun. 2015, 51, 3674–3677. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Lothschütz, C.; Ackermann, M.; Doepp, R.; Anantharaman, S.; Marchetti, B.; Bertagnolli, H.; Rominger, F. Gold Catalysis: In situ EXAFS Study of Homogeneous Oxidative Esterification. Chem. Eur. J. 2010, 16, 7933–8215. [Google Scholar] [CrossRef] [PubMed]
- Taaring, E.; Nielsen, I.S.; Egeblad, K.; Madsen, R.; Christensen, C.H. Chemicals from renewables: Aerobic oxidation of furfural and hydroxymethylfurfural over gold catalysts. ChemSusChem 2008, 1, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into chemicals: One pot-base free oxidative esterification of 5-hydroxymethyl-2-furfural into 2,5-dimethylfuroate with gold on nanoparticulated ceria. J. Catal. 2009, 265, 109–116. [Google Scholar] [CrossRef]
- Kegnæs, S.; Mielby, D.; Mentzel, U.V.; Jensen, T.; Fristrup, P.; Riisager, A. One-pot synthesis of amides by aerobic oxidative coupling of alcohols or aldehydes with amines using supported gold and a base as catalyst. Chem. Commun. 2012, 48, 2427–2429. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, G.J. Vapor phase hydrochlorination of acetylene: Correlation of catalytic activity of supported metal chloride catalysts. J. Catal. 1985, 96, 292–295. [Google Scholar] [CrossRef]
- Abad, A.; Concepcion, P.; Corma, A.; Garcia, H. A collaborative effect between gold and a support induces the selective oxidation of alcohols. Angew. Chem. Int. Ed. 2005, 44, 4066–4069. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Hutchings, G.J.; Brust, M.; Schmidbaur, H. Gold-an introductory perspective. Chem. Soc. Rev. 2008, 37, 1759–1765. [Google Scholar] [CrossRef] [PubMed]
- Pinna, F.; Olivo, A.; Trevisan, V.; Menegazzo, F.; Signoretto, M.; Manzoli, M.; Boccuzzi, F. The effects of gold nanosize for the exploitation of furfural by selective oxidation. Catal. Today 2013, 203, 196–201. [Google Scholar] [CrossRef]
- Jagadeesh, R.V.; Junge, H.; Pohl, M.-M.; Radnik, J.; Brükner, A.; Beller, M. Selective oxidation of alcohols to esters using heterogeneous Co3O4-N@C catalysts under mild conditions. J. Am. Chem. Soc. 2013, 135, 10776–10782. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Song, H.-J.; Cui, M.S.; Du, Y.-P.; Fu, Y. Aerobic oxidation of hydroxymethylfurfural and furfural by using heterogeneous CoxOy-N@C catalysts. ChemSusChem 2014, 7, 3334–3340. [Google Scholar] [CrossRef] [PubMed]
- Kiran, I.N.C.; Lalwani, K.; Sudalai, A. N-heterocyclic carbene catalysed esterification of aromatic aldehydes with alcohols under aerobic conditions. RSC Adv. 2013, 3, 1695–1698. [Google Scholar] [CrossRef]
- Fristrup, P.; Johansen, L.B.; Christensen, C.H. Mechanistic investigation of the gold-catalyzed aerobic oxidation of aldehydes: Added insight from Hammett studies and isotopic labelling experiments. Chem. Commun. 2008, 2750–2752. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, F.; Fantinel, T.; Signoretto, M.; Pinna, F.; Manzoli, M. On the process for furfural and HMF oxidative esterification over Au/ZrO2. J. Catal. 2014, 319, 61–70. [Google Scholar] [CrossRef]
- Morterra, C.; Cerrato, G.; Bolis, V.; Di Ciero, S.; Signoretto, M. On the strength of Lewis- and Brønsted-acid sites at the surface of sulfated zirconia catalysts. J. Chem. Soc. Faraday Trans. 1997, 93, 1179–1184. [Google Scholar] [CrossRef]
- Bensitel, M.; Saur, O.; Lavalley, J.-C.; Mabilon, G. Acidity of zirconium oxide and sulfated ZrO2 samples. Mater. Chem. Phys. 1987, 17, 249–258. [Google Scholar] [CrossRef]
- Lewis, B.; von Elbe, G. Combustion, Flames and Explosion of Gases; Academic Press: New York, NY, USA, 1961. [Google Scholar]
- Tsutsumi, K.; Yoshitake, H. Coupling reaction between α,β-unsaturated aldehyde and methanol catalyzed by gold-supported on mesostructured cerias. Appl. Catal. A Gen. 2014, 484, 64–73. [Google Scholar] [CrossRef]
- Gates, B.C. Supported gold catalysts: New properties offered by nanometer and sub-nanometers structures. Chem. Commun. 2013, 49, 7876–7877. [Google Scholar] [CrossRef] [PubMed]
- Strakakis, M.; Garcia, H. Catalysis by supported gold nanoparticles: Beyond aerobic oxidative processes. Chem. Rev. 2012, 112, 4469–4506. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M. Relevance of Metal Nanoclusters Size Control in Gold (0) Catalytic Chemistry. In Metal Nanoclusters in Catalysis and Materials Science: The Issue of Size Control; Corain, B.S., Schmid, G., Toshima, N., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 183–189. [Google Scholar]
- Hashmi, A.S.K.; Hutchings, G.J. Gold catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Schmid, G.; Corain, B. Nanoparticulated gold: Syntheses, structures, electronics, and reactivities. Eur. J. Inorg. Chem. 2003, 3081–3098. [Google Scholar] [CrossRef]
- Guzman, J.; Carretin, S.; Fierro-Gonzales, J.-C.; Hao, Y.; Gates, B.C.; Corma, A. CO oxidation catalyzed by supported gold: Cooperation between gold and nanocrystalline rare-earth supports forms reactive surface superoxide and peroxide species. Angew. Chem. Int. Ed. 2005, 44, 4778–4781. [Google Scholar] [CrossRef] [PubMed]
- Boccuzzi, F.; Chiorino, A.; Manzoli, M.; Lu, P.; Akita, T.; Ichikawa, S.; Haruta, M. Au/TiO2 nanosized samples: A catalytic, TEM, and FTIR study of the effect of calcination temperature on the CO oxidation. J. Catal. 2001, 202, 256–267. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Manzoli, M.; Boccuzzi, F.; Tabakova, T.; Papavasiliou, J.; Ioannides, T.; Idakiev, V. Catalytic performance and characterization of Au/doped-ceria catalysts for the preferential CO oxidation reaction. J. Catal. 2008, 256, 237–247. [Google Scholar] [CrossRef]
- Carretin, S.; Guzman, J.; Corma, A. Supported gold catalyzes the homocoupling of phenylboronic acid with high conversion and selectivity. Angew. Chem. Int. Ed. 2005, 44, 2242–2245. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Serna, P. Chemoselective hydrogenation of nitro compounds with supported gold catalysts. Science 2006, 313, 332–333. [Google Scholar] [CrossRef] [PubMed]
- Okumura, M.; Akita, T.; Haruta, M. Hydrogenation of 1,3-butadiene and of crotonaldehyde over highly dispersed Au catalysts. Catal. Today 2002, 74, 265–269. [Google Scholar] [CrossRef]
- Della Pina, C.; Falletta, E.; Prati, L.; Rossi, M. Selective oxidation using gold. Chem. Soc. Rev. 2008, 37, 2077–2095. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, G.J. Nanocrystalline gold and gold-palladium alloy oxidation catalysts: A personal reflection on the nature of the active sites. Dalton Trans. 2008, 5523–5536. [Google Scholar] [CrossRef] [PubMed]
- Abad, A.; Corma, A.; Gracia, H. Catalyst parameters determining activity and selectivity of supported gold nanoparticles for the aerobic oxidation of alcohols: The molecular reaction mechanism. Chem. A Eur. J. 2008, 14, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Haruta, M.; Daté, M. Advances in the catalysis of Au nanoparticles. Appl. Catal. A 2001, 222, 427–437. [Google Scholar] [CrossRef]
- Ryabenkova, Y.; Miedziak, P.J.; Dummer, N.F.; Taylor, S.H.; Dimitratos, N.; Willock, D.J.; Bethell, D.; Knight, D.W.; Hutchings, G.J. The selective oxidation of 1,2-propanediol by supported gold-based nanoparticulate catalysts. Top. Catal. 2012, 55, 1283–1288. [Google Scholar] [CrossRef]
- Dimitraos, N.; Villa, A.; Prati, L.; Hammond, C.; Chan-Thaw, C.E.; Cookson, J.; Bishop, P.T. Effect of the preparation method of supported Au nanoparticles in the liquid phase oxidation of glycerol. Appl. Catal. A 2016, 514, 267–275. [Google Scholar] [CrossRef]
- Rogers, S.M.; Catlow, C.R.A.; Chan-Thaw, C.E.; Gianolio, D.; Gibson, E.K.; Gould, A.L.; Jian, N.; Logsdail, A.J.; Palmer, R.E.; Prati, L.; et al. Tailoring gold nanoparticle characteristics and the impact on aqueous-phase oxidation of glycerol. ACS Catal. 2015, 5, 4377–4384. [Google Scholar] [CrossRef]
- Christensen, C.H.; Jørgensen, B.; Rass-Hansen, J.; Egeblad, K.; Madsen, R.; Klitgaard, S.K.; Hansen, S.M.; Hansen, M.R.; Andersen, H.C.; Riisager, A. Formation of acetic acid by aqueous-phase oxidation of ethanol with air in the presence of a heterogeneous gold catalyst. Angew. Chem. Int. Ed. 2006, 45, 4648–4651. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.; Taarning, E.; Hansen, D.; Johansen, L.; Klitgaard, S.K.; Egeblad, K.; Christensen, C.H. Aerobic oxidation of aldehydes under ambient conditions using supported gold nanoparticle catalysts. Green Chem. 2008, 10, 168–170. [Google Scholar] [CrossRef]
- Signoretto, M.; Menegazzo, F.; Contessotto, L.; Pinna, F.; Manzoli, M.; Boccuzzi, F. Au/ZrO2: An efficient and reusable catalyst for the oxidative esterification of renewable furfural. Appl. Catal. B 2013, 129, 287–293. [Google Scholar] [CrossRef]
- Manzoli, M.; Boccuzzi, F.; Trevisan, V.; Menegazzo, F.; Signoretto, M.; Pinna, F. Au/ZrO2 catalysts for LT-WGSR: Active role of sulfates during gold deposition. Appl. Catal. B 2010, 96, 28–33. [Google Scholar] [CrossRef]
- Schubert, M.M.; Hackenberg, S.; Van Veen, A.C.; Muhler, M.; Plzak, V. CO Oxidation over supported gold catalysts—“Inert” and “Active” support materials and their role for the oxygen supply during reaction. J. Catal. 2001, 197, 113–122. [Google Scholar] [CrossRef]
- Sinha, A.K.; Seelan, S.; Tsubota, S.; Haruta, M. Catalysis by gold nanoparticles: Epoxidation of propene. Top. Catal. 2004, 29, 95–102. [Google Scholar] [CrossRef]
- Turner, M.; Golovko, V.B.; Vaughan, O.P.H.; Abdulkin, P.; Berenguer-Murcia, A.; Tikhov, M.S.; Johnson, B.F.G.; Lambert, R. Selective oxidation with dioxygen by gold nanoparticle catalysts derived from 55-atom clusters. Nature 2008, 454, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, F.; Pinna, F.; Signoretto, M.; Trevisan, V.; Boccuzzi, F.; Chiorino, A.; Manzoli, M. Quantitative determination of sites able to chemisorb CO on Au/ZrO2 catalysts. Appl. Catal. A 2009, 356, 31–35. [Google Scholar] [CrossRef]
- Miyamura, H.; Yasukawa, T.; Kobayashi, S. Aerobic oxidative esterification of alcohols catalyzed by polymer-incarcerated gold nanoclusters under ambient conditions. Green Chem. 2010, 12, 776–778. [Google Scholar] [CrossRef]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, F.; Pinna, F.; Signoretto, M.; Trevisan, V.; Boccuzzi, F.; Chiorino, A.; Manzoli, M. Highly dispersed gold on zirconia: Characterization and activity in low-temperature water gas shift tests. ChemSusChem 2008, 1, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Morterra, C.; Bolis, V.; Fubini, B.; Orio, L.; Williams, T.B. A FTIR and HREM study of some morphological and adsorptive properties of monoclinic ZrO2 microcrystals. Surf. Sci. 1991, 251–252, 540–545. [Google Scholar] [CrossRef]
- Menegazzo, F.; Manzoli, M.; Chiorino, A.; Boccuzzi, F.; Tabakova, T.; Signoretto, M.; Pinna, F.; Pernicone, N. Quantitative determination of gold active sites by chemisorption and by infrared measurements of adsorbed CO. J. Catal. 2006, 237, 431–434. [Google Scholar] [CrossRef]
- Sample Number 17C. Supplied by World Gold Council. Available online: http://www.gold.org (accessed on 15 March 2016).
- Martra, G. Lewis acid and base sites at the surface of microcrystalline TiO2 anatase: Relationships between surface morphology and chemical behaviour. Appl. Catal. A 2000, 200, 275–285. [Google Scholar] [CrossRef]
- Signoretto, M.; Menegazzo, F.; Pinna, F.; Manzoli, M.; Aina, V.; Cerrato, G.; Boccuzzi, F. Oxidative esterification of renewable furfural on gold based catalysts: Which is the best support? J. Catal. 2014, 309, 241–247. [Google Scholar]
- Song, X.; Sayari, A. Sulfated zirconia-based strong solid-acid catalysts: Recent progress. Catal. Rev.-Sci. Eng. 1996, 38, 329–412. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic properties of ceria and CeO2-containing materials. Catal. Rev.-Sci. Eng. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Trevisan, V.; Signoretto, M.; Pinna, F.; Cruciani, G.; Cerrato, G. Investigation on titania synthesis for photocatalytic NOx abatement. Chem. Today 2012, 30, 25–28. [Google Scholar]
- Ishida, T.; Ogihara, Y.; Ohashi, H.; Akita, T.; Honma, T.; Oji, H.; Haruta, M. Base-free direct oxidation of 1-octanol to octanoic acid and its octyl ester over supported gold catalysts. ChemSusChem 2012, 5, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Shou, H.; Kou, Y.; Liu, H. Base-free aqueous-phase oxidation of non-activated alcohols with molecular oxygen on soluble Pt nanoparticles. Green Chem. 2009, 11, 562–568. [Google Scholar] [CrossRef]
- Su, F.Z.; Liu, Y.M.; Wang, L.-C.; Cao, Y.; He, H.-Y.; Fan, K.-N. Ga-Al mixed-oxide-supported gold nanoparticles with enhanced activity for aerobic alcohol oxidation. Angew. Chem. 2008, 120, 340–343. [Google Scholar] [CrossRef]
- Carretin, S.; Conceptión, P.; Corma, A.; Lopez Nieto, J.M.; Puntes, V.F. Nanocrystalline CeO2 Increases the Activity of Au for CO Oxidation by Two Orders of Magnitude. Angew. Chem. Int. Ed. 2004, 43, 2538–2540. [Google Scholar]
- Longo, A.; Liotta, L.F.; Pantaleo, G.; Giannici, F.; Venezia, A.M.; Martorana, A. Structure of the metal-support interface and oxidation state of gold nanoparticles supported on ceria. J. Phys. Chem. C. 2012, 116, 2960–2966. [Google Scholar] [CrossRef]
- Manzoli, M.; Menegazzo, F.; Signoretto, M.; Cruciani, G.; Pinna, F. Effects of synthetic parameters on the catalytic performance of Au/CeO2 for furfural oxidative esterification. J. Catal. 2015, 330, 465–473. [Google Scholar] [CrossRef]
- Kundakovic, L.; Flytzani-Stephanopoulos, M. Cu- and Ag-modified cerium oxide catalysts for methane oxidation. J. Catal. 1998, 179, 203–221. [Google Scholar] [CrossRef]
- Badri, A.; Binet, C.; Lavalley, J.-C. Use of methanol as an IR molecular probe to study the surface of polycrystalline ceria. J. Chem. Soc. Faraday Trans. 1997, 93, 1159–1168. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M. Methanol as an IR probe to study the reduction process in ceria-zirconia mixed compounds. Catal. Today 2001, 70, 155–167. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.-C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Boccuzzi, F.; Chiorino, A.; Manzoli, M.; Andreeva, D.; Tabakova, T. FTIR study of the low-temperature water gas shift reaction on Au/Fe2O3 and Au/TiO2 catalysts. J. Catal. 1999, 188, 176–185. [Google Scholar] [CrossRef]
- Tabakova, T.; Boccuzzi, F.; Manzoli, M.; Andreeva, D. FTIR study of low-temperature water-gas shift reaction on gold/ceria catalyst. Appl. Catal. A 2003, 252, 385–397. [Google Scholar] [CrossRef]
- Manzoli, M.; Boccuzzi, F.; Chiorino, A.; Vindigni, F.; Deng, W.; Flytzani-Stephanopoulos, M. Spectroscopic features and reactivity of CO adsorbed on different Au/CeO2 catalysts. J. Catal. 2007, 245, 308–315. [Google Scholar] [CrossRef]
- Smolentseva, E.; Costa, V.V.; Cotta, R.F.; Smakova, O.; Beloshapkin, S.; Gusevskaya, E.V.; Simakov, A. Aerobic oxidative esterification of benzyl alcohol and acetaldehyde over gold supported on nanostructured ceria-alumina mixed oxides. ChemCatChem 2015, 7, 1011–1017. [Google Scholar] [CrossRef]
- Sie, S.T. Consequences of catalyst deactivation for process design and operation. Appl. Catal. A Gen. 2001, 212, 129–151. [Google Scholar] [CrossRef]
- Sadana, A.; Doraiswamy, L.K. Effect of catalyst fouling in fixed-, moving-, and fluid-bed reactors. J. Catal. 1971, 23, 147–157. [Google Scholar] [CrossRef]
- Douglas, J.M.; Reiff, E.K., Jr.; Kittrell, J.R. Process design with catalyst deactivation. Chem. Eng. Sci. 1980, 35, 322–329. [Google Scholar] [CrossRef]
- Sá, J.; Taylor, S.F.R.; Daly, H.; Goguet, A.; Tiruvalam, R.; He, Q.; Kiely, C.J.; Hutchings, G.H.; Hardacre, C. Redispersion of gold supported on oxides. ACS Catal. 2012, 2, 552–560. [Google Scholar] [CrossRef]
- Walsh, M.J.; Yoshida, K.; Pay, M.L.; Gai, P.L.; Boyes, E.D. On the effect of atomic structure on the activity and deactivation of catalytic gold nanoparticles. ChemCatChem 2012, 4, 1638–1644. [Google Scholar] [CrossRef]
- Tebandeke, E.; Coman, C.; Guillois, K.; Canning, G.; Ataman, E.; Knudsen, J.; Wallenberg, L.R.; Ssekaalo, H.; Schnadt, J.; Wendt, O.F. Epoxidation of olefins with molecular oxygen as the oxidant using gold catalysts supported on polyoxometalates. Green Chem. 2014, 16, 1586–1593. [Google Scholar] [CrossRef]
- Signoretto, M.; Menegazzo, F.; Trevisan, V.; Pinna, F.; Manzoli, M.; Boccuzzi, F. Investigation on the stability of supported gold nanoparticles. Catalysts 2013, 3, 656–670. [Google Scholar] [CrossRef]
- Sha, J.; Zheng, E.-J.; Zhou, W.-J.; Liebens, A.; Pera-Titus, M. Selective oxidation of fatty alcohol ethoxylates with H2O2 over Au catalysts for the synthesis of alkyl ether carboxylic acids in alkaline solution. J. Catal. 2016, 337, 199–207. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Metal Loading (wt %) | O2 (bar) | Alcohol | Base | T (°C) | Time (h) | Conversion (%) | Selectivity (or Yield, Y) (%) a | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Pt/FH b | 5 | 3 | ethanol | none | 140 | 4 | 53.0 | 100 | [23] |
| Pt/FH b | 5 | 3 | ethanol | K2CO3 | 140 | 4 | 93.9 | 32.1 | [23] |
| Pt/H c | 5 | 3 | ethanol | K2CO3 | 140 | 4 | 92.5 | 31.3 | [23] |
| Pt/HTc d | 5 | 3 | ethanol | K2CO3 | 140 | 4 | 97.2 | 30.9 | [23] |
| Pt/Fe3O4 | 5 | 3 | ethanol | K2CO3 | 140 | 4 | 93.8 | 28.8 | [23] |
| Pt/Al2O34 | 5 | 3 | ethanol | K2CO3 | 140 | 4 | 97.1 | 31.4 | [23] |
| Pt/ZrO2 | 5 | 3 | ethanol | K2CO3 | 140 | 4 | 58.9 | 18.7 | [23] |
| PtOx/HTc d | 5 | 3 | ethanol | K2CO3 | 140 | 4 | 33.7 | 52.0 | [23] |
| Pt/FH b | 5 | 3 | n-propanol | none | 140 | 4 | 46.3 | 100 | [23] |
| Pt/FH b | 5 | 3 | n-propanol | K2CO3 | 140 | 4 | 90.1 | 10.0 | [23] |
| Pt/H c | 5 | 3 | n-propanol | K2CO3 | 140 | 4 | 73.0 | 9.9 | [23] |
| Pt/HTc d | 5 | 3 | n-propanol | K2CO3 | 140 | 4 | 87.8 | 11.5 | [23] |
| Pt/Fe3O4 | 5 | 3 | n-propanol | K2CO3 | 140 | 4 | 80.6 | 8.9 | [23] |
| Pt/Al2O34 | 5 | 3 | n-propanol | K2CO3 | 140 | 4 | 81.4 | 9.6 | [23] |
| Pt/ZrO2 | 5 | 3 | n-propanol | K2CO3 | 140 | 4 | 31.9 | 10.0 | [23] |
| PtOx/HTc d | 5 | 3 | n-propanol | K2CO3 | 140 | 4 | 33.7 | 46.6 | [23] |
| Au/FH b | 5 | 3 | methanol | K2CO3 | 140 | 4 | 93 | 99 | [25] |
| Au/H c | 5 | 3 | methanol | K2CO3 | 140 | 4 | 48 | 93 | [25] |
| Au/Fe3O4 | 5 | 3 | methanol | K2CO3 | 140 | 4 | 41 | 82 | [25] |
| Au/CeO2 | 5 | 3 | methanol | K2CO3 | 140 | 4 | 55 | 82 | [25] |
| Au/TiO2 | 1 | 1 | methanol | NaCH3O | 22 | 10–12 | 100 | 100 | [27] |
| Au/CeO2 e | 2.1 | 10 | methanol | none | 130 | 2 | >99 | 91 (Y) | [28] |
| Au/Fe2O3 f | 4.5 | 10 | methanol | none | 130 | 24 | 94 | 6.2 (Y) | [28] |
| Au/TiO2 f | 1 | 10 | methanol | none | 130 | 24 | >99 | 96.3 (Y) | [28] |
| Au/C f | 1.5 | 10 | methanol | none | 130 | 24 | 96 | 7.6 (Y) | [28] |
| Au/CeO2 f | 1 | 10 | methanol | none | 130 | 5 | >99 | >99 (Y) | [28] |
| Au/TiO2 e | 1 | ~1 | methanol | KCH3O | 25 | 24 | 100 | >99 (Y) | [29] |
| Au/ZrO2 | 1.51 | 6 | methanol | none | 120 | 1.5 | 100 | 100 | [34] |
| AuTiO2 | 1.56 | 6 | methanol | none | 120 | 3 | 90 | 65 | [34] |
| Co3O4-N@C e | 3 | 1 | methanol | K2CO3 | 70 | 24 | 100 | 77 (Y) | [35] |
| Co(1,10-phen)/C | 3 | 1 | methanol | K2CO3 | 60 | 12 | 100 | 95 (Y) | [36] |
| Pd(1,10-phen)/C | 3 | 1 | methanol | K2CO3 | 60 | 12 | 12 | 4 (Y) | [36] |
| Co(2,2-diPy)/C | 3 | 1 | methanol | K2CO3 | 60 | 12 | 58 | 46 (Y) | [36] |
| Co(1,10-phen)/C e | 3 | 1 | methanol | K2CO3 | 60 | 12 | 54 | 42 (Y) | [36] |
| Mn(1,10-phen)/C e | 3 | 1 | methanol | K2CO3 | 60 | 12 | 9 | - | [36] |
| Pd(1,10-phen)/C e | 3 | 1 | methanol | K2CO3 | 60 | 12 | 15 | 1 (Y) | [36] |
| NHC g | none | ~1 | methanol | DBU h | 25 | - | 24 | 63 (Y) | [37] |
| Catalyst | Au Loading (wt %) | molCO/molAu | Au Size (nm) |
|---|---|---|---|
| Au/ZrO2 | 1.56 | 0.190 | 1.68 ± 0.38 |
| Au/TiO2 | 1.51 | 0.033 | 3.80 ± 0.75 |
| Au/TiO2 | Au/ZrO2 | |||
|---|---|---|---|---|
| Time (min) | Conversion (%) | Selectivity (%) | Conversion (%) | Selectivity (%) |
| 0 | - | - | - | - |
| 30 | 35 | 79 | 35 | 98 |
| 60 | 60 | 73 | 79 | 99 |
| 90 | 72 | 73 | 96 | 99 |
| 120 | 80 | 69 | 99 | 99 |
| 150 | 82 | 68 | 99 | 99 |
| 180 | 88 | 66 | 100 | 100 |
| Catalyst | Au Loading (wt %) | molCO/molAu | Surface Area (m2/g) | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|---|
| Au/ZrO2 | 1.0 | 0.24 | 39 | 82 | 92 |
| Au/CeO2 | 2.2 | 0.06 | 105 | 66 | 69 |
| Au/TiO2 | 1.2 | 0.004 | 166 | 20 | 91 |
| Catalyst | Conversion (%) | Selectivity (%) | molCO/molAu | Reference |
|---|---|---|---|---|
| Au/ZrO2 calcined at 500 °C | 96 | 97 | 0.03 | [64] |
| Au/Ce(OH)4 | 80 | 56 | 0.04 | [this review] |
| Au/TiO2 WGC | 64 | 64 | 0.03 | [34] |
| Catalyst | Au Nominal Content (wt %) | Au Effective Content (wt %) | Surface Area (m2/g) | Conversion (%) | Selectivity (%) |
|---|---|---|---|---|---|
| Au/CeO2C90C300 | 1.5 | 2.1 | 180 | 29 | 100 |
| Au/CeO2C90C500 | 1.5 | 2.1 | 105 | 6 | 100 |
| Au/CeO2C110C300 | 1.5 | 1.3 | 121 | 29 | 100 |
| Au/CeO2C110C500 | 1.5 | 1.3 | 121 | 28 | 100 |
| Au/CeO2C300C300 | 1.5 | 1.4 | 124 | 54 | 100 |
| Au/CeO2C500C500 | 1.5 | 1.5 | 110 | 74 | 100 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manzoli, M.; Menegazzo, F.; Signoretto, M.; Marchese, D. Biomass Derived Chemicals: Furfural Oxidative Esterification to Methyl-2-furoate over Gold Catalysts. Catalysts 2016, 6, 107. https://doi.org/10.3390/catal6070107
Manzoli M, Menegazzo F, Signoretto M, Marchese D. Biomass Derived Chemicals: Furfural Oxidative Esterification to Methyl-2-furoate over Gold Catalysts. Catalysts. 2016; 6(7):107. https://doi.org/10.3390/catal6070107
Chicago/Turabian StyleManzoli, Maela, Federica Menegazzo, Michela Signoretto, and Damiano Marchese. 2016. "Biomass Derived Chemicals: Furfural Oxidative Esterification to Methyl-2-furoate over Gold Catalysts" Catalysts 6, no. 7: 107. https://doi.org/10.3390/catal6070107
APA StyleManzoli, M., Menegazzo, F., Signoretto, M., & Marchese, D. (2016). Biomass Derived Chemicals: Furfural Oxidative Esterification to Methyl-2-furoate over Gold Catalysts. Catalysts, 6(7), 107. https://doi.org/10.3390/catal6070107