
Gold Catalysts on Y-Doped Ceria Supports for Complete Benzene Oxidation


 ,
,  ,
, 
Abstract
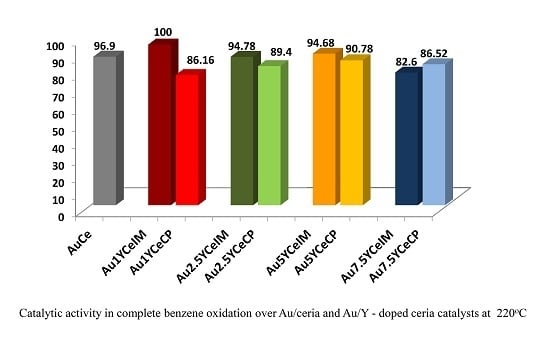
:
1. Introduction
2. Results and Discussion
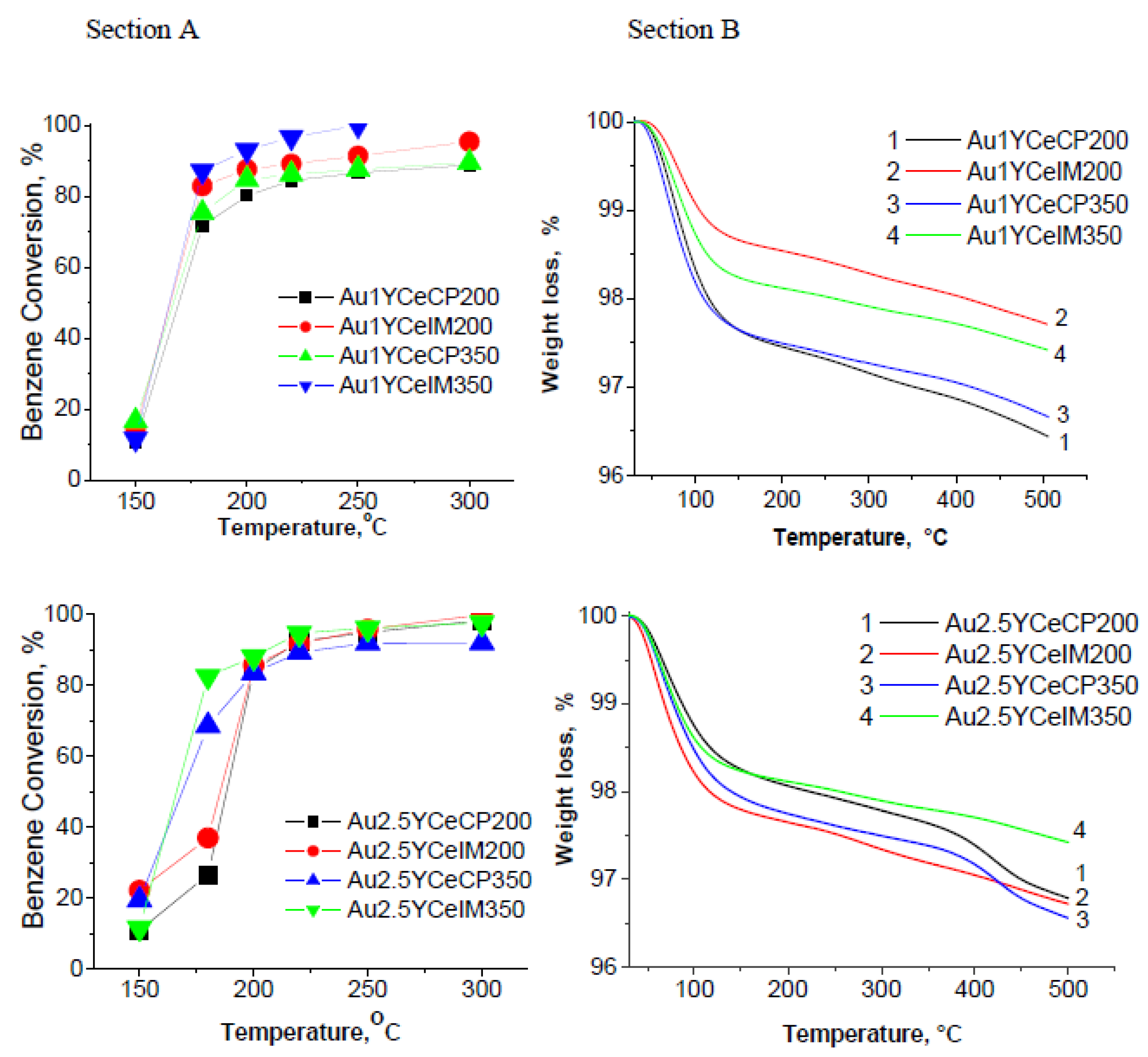
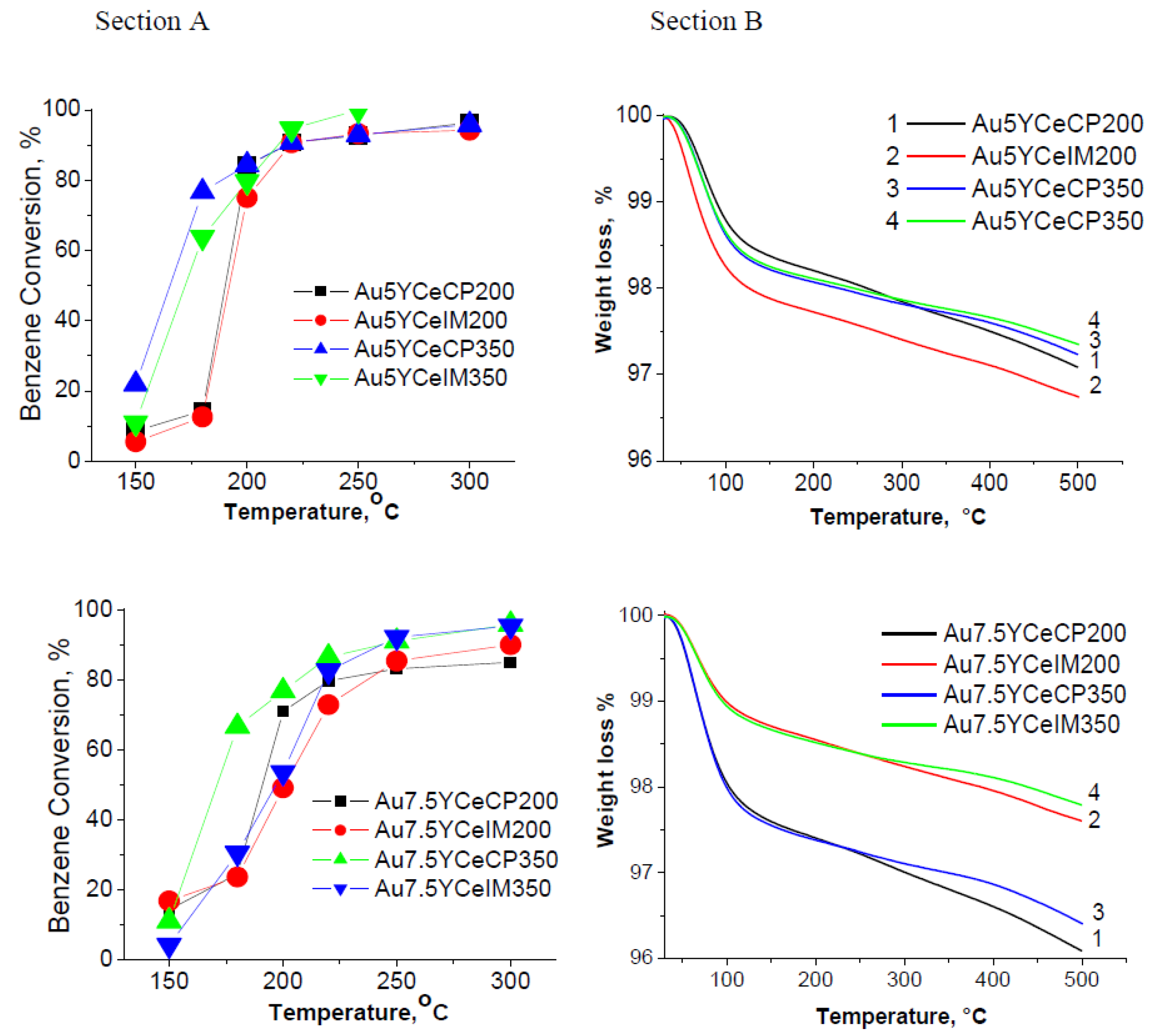
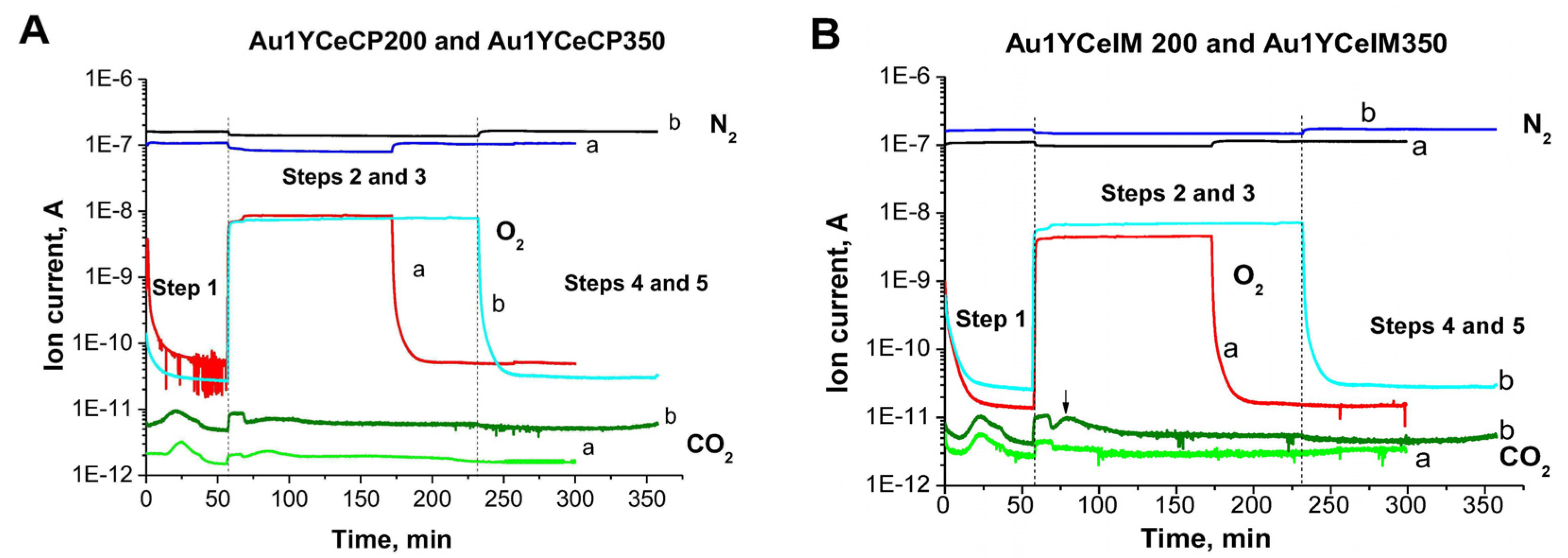
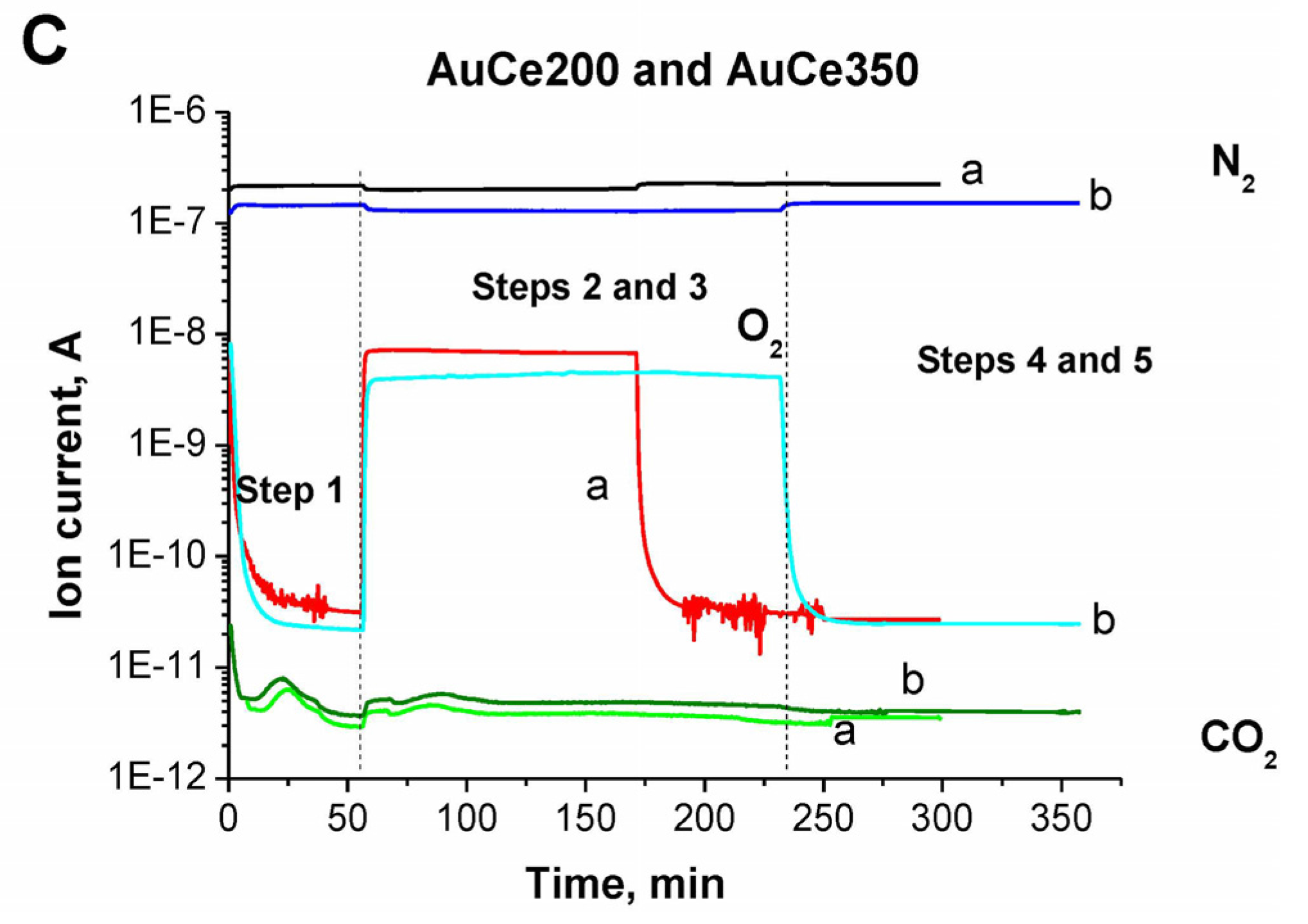
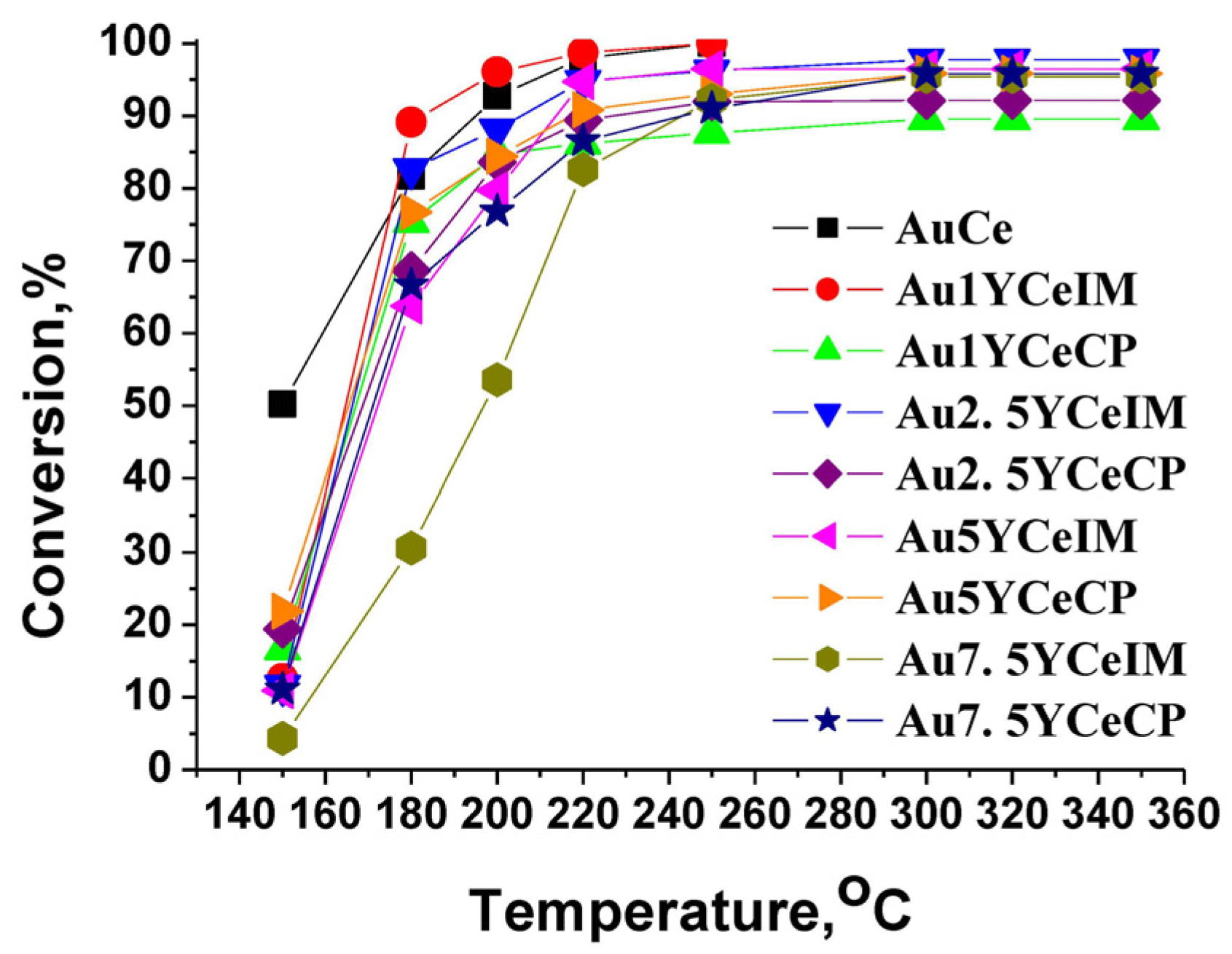
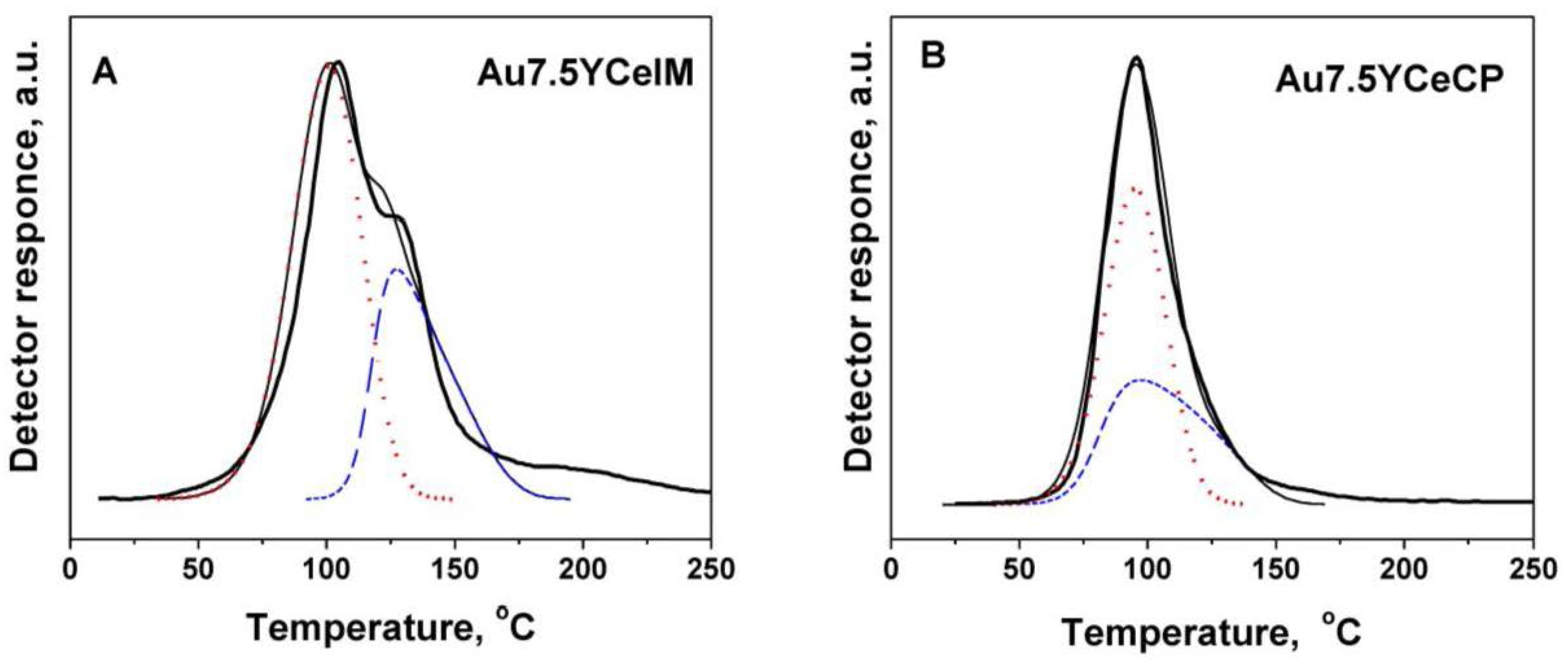
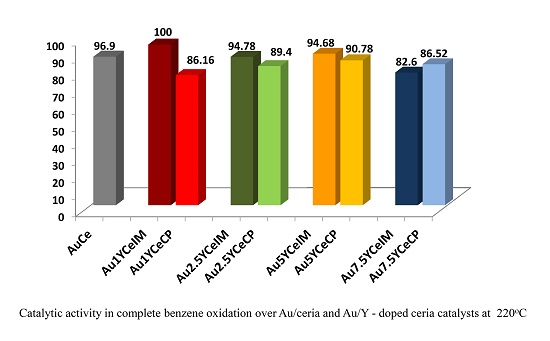
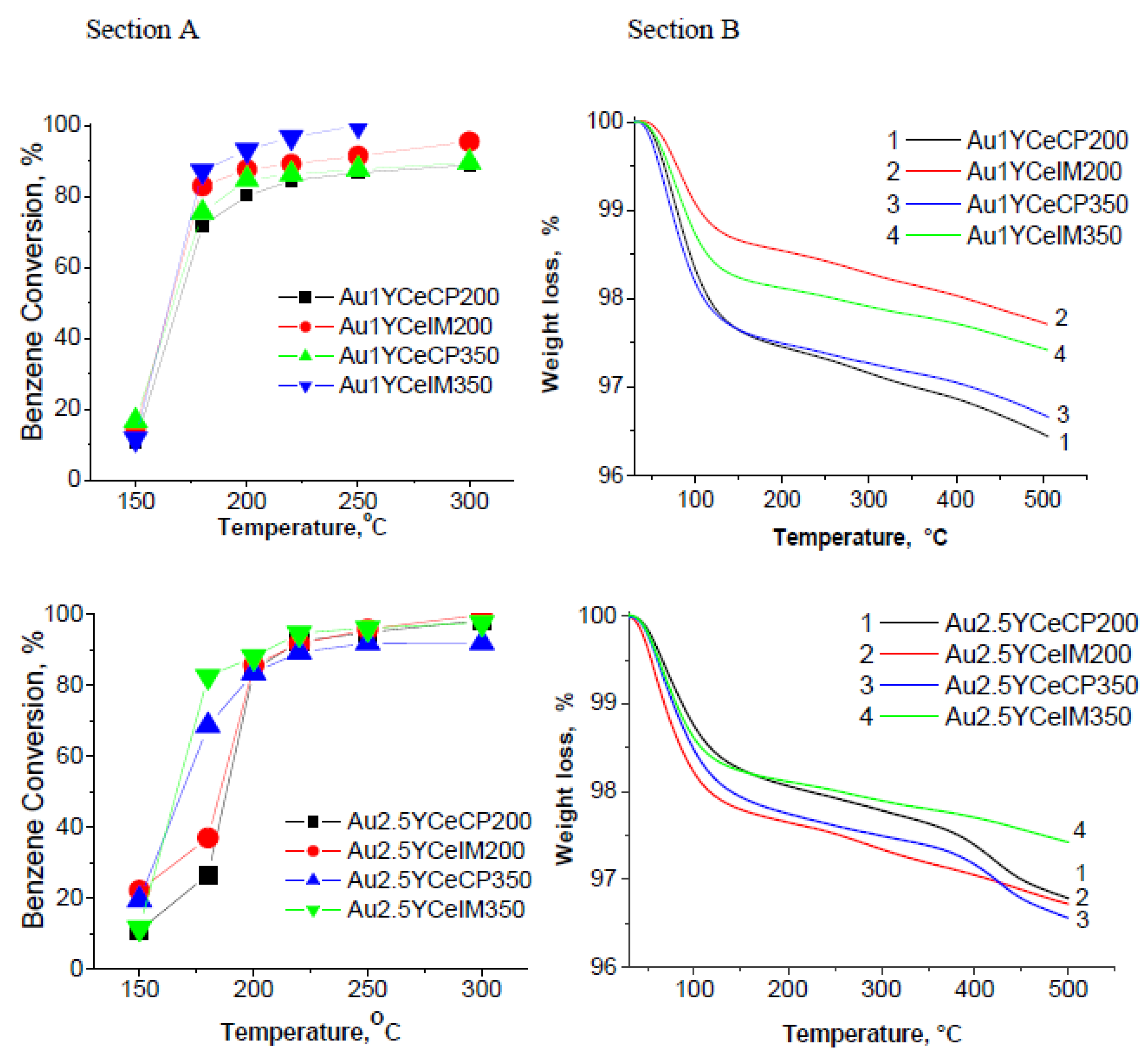
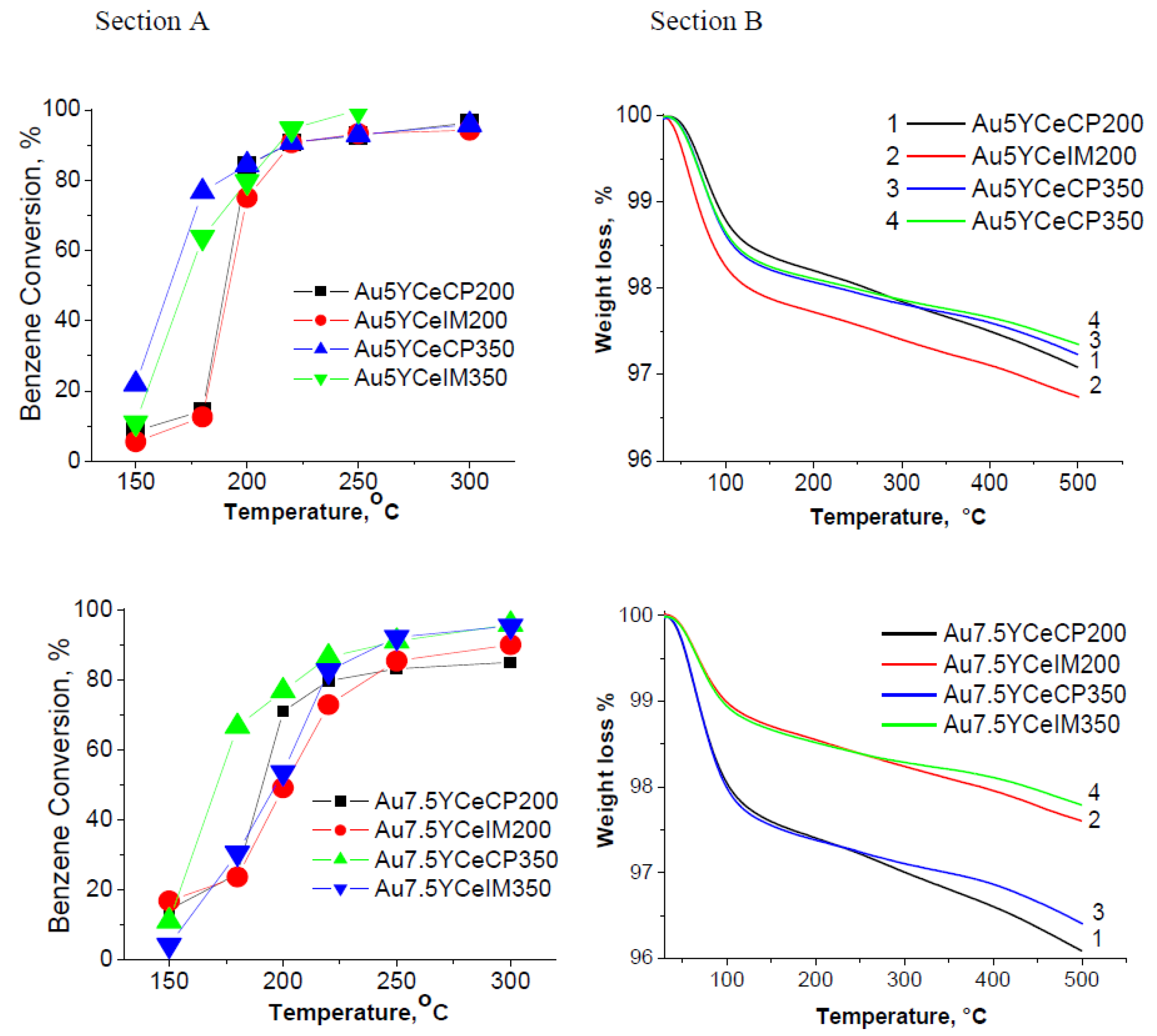
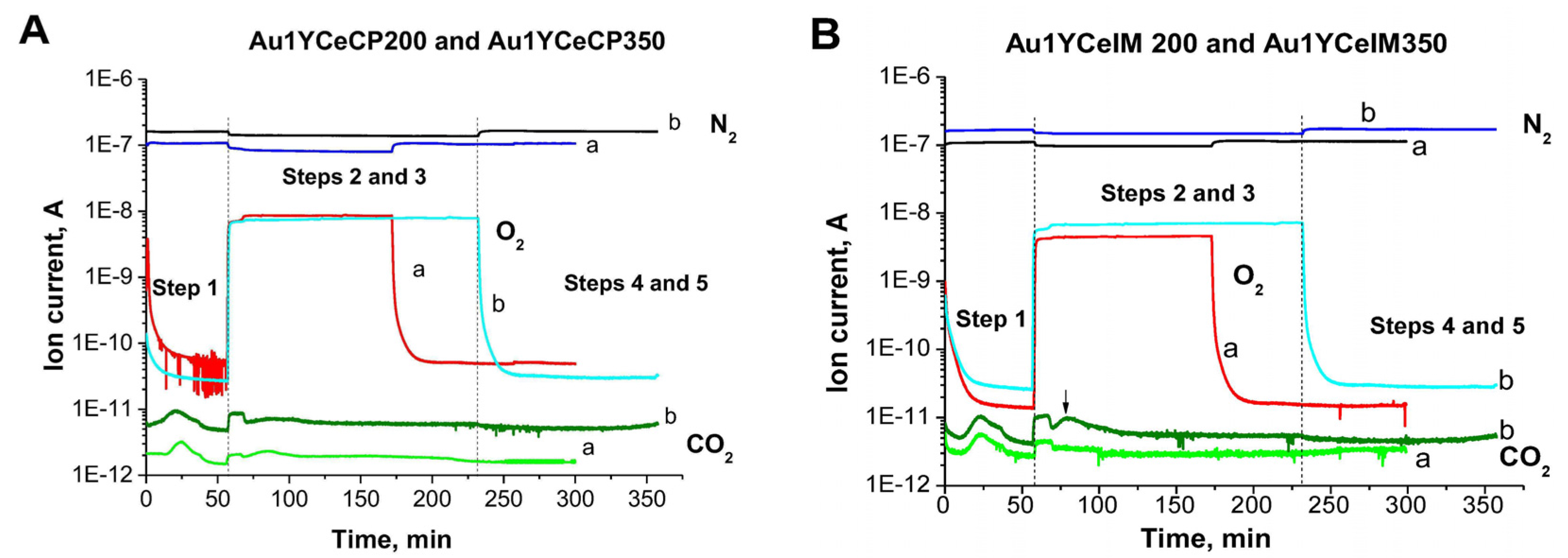
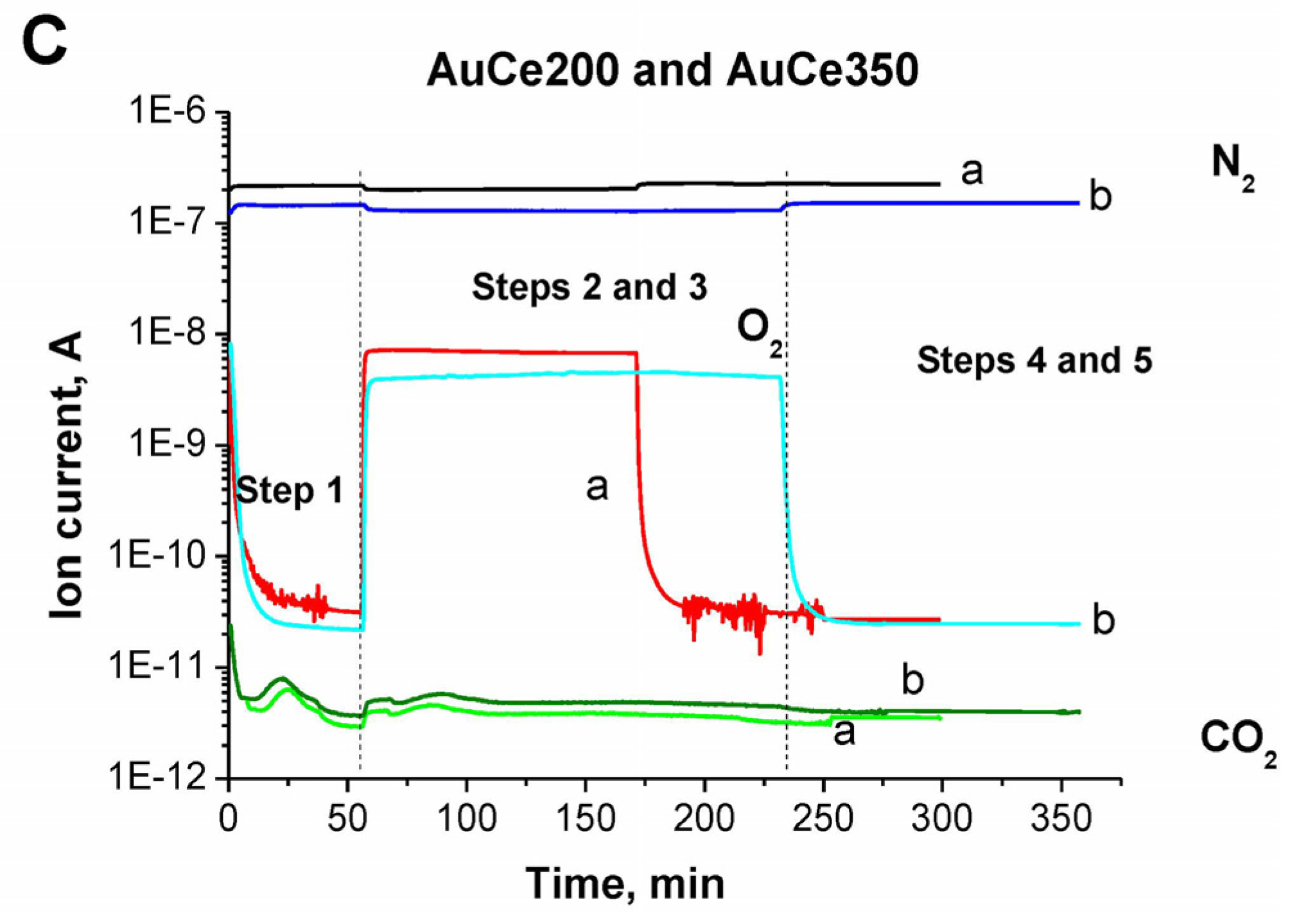
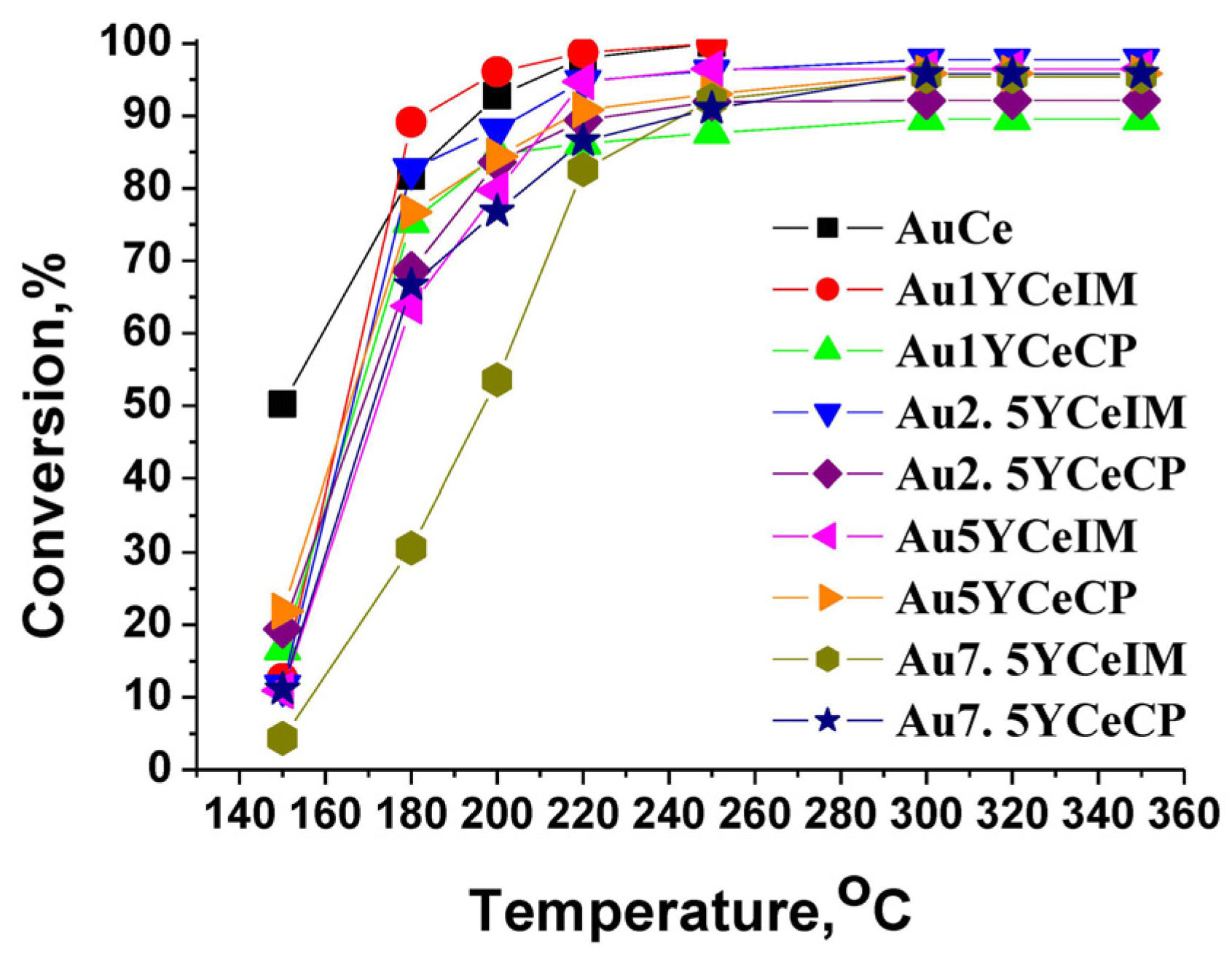
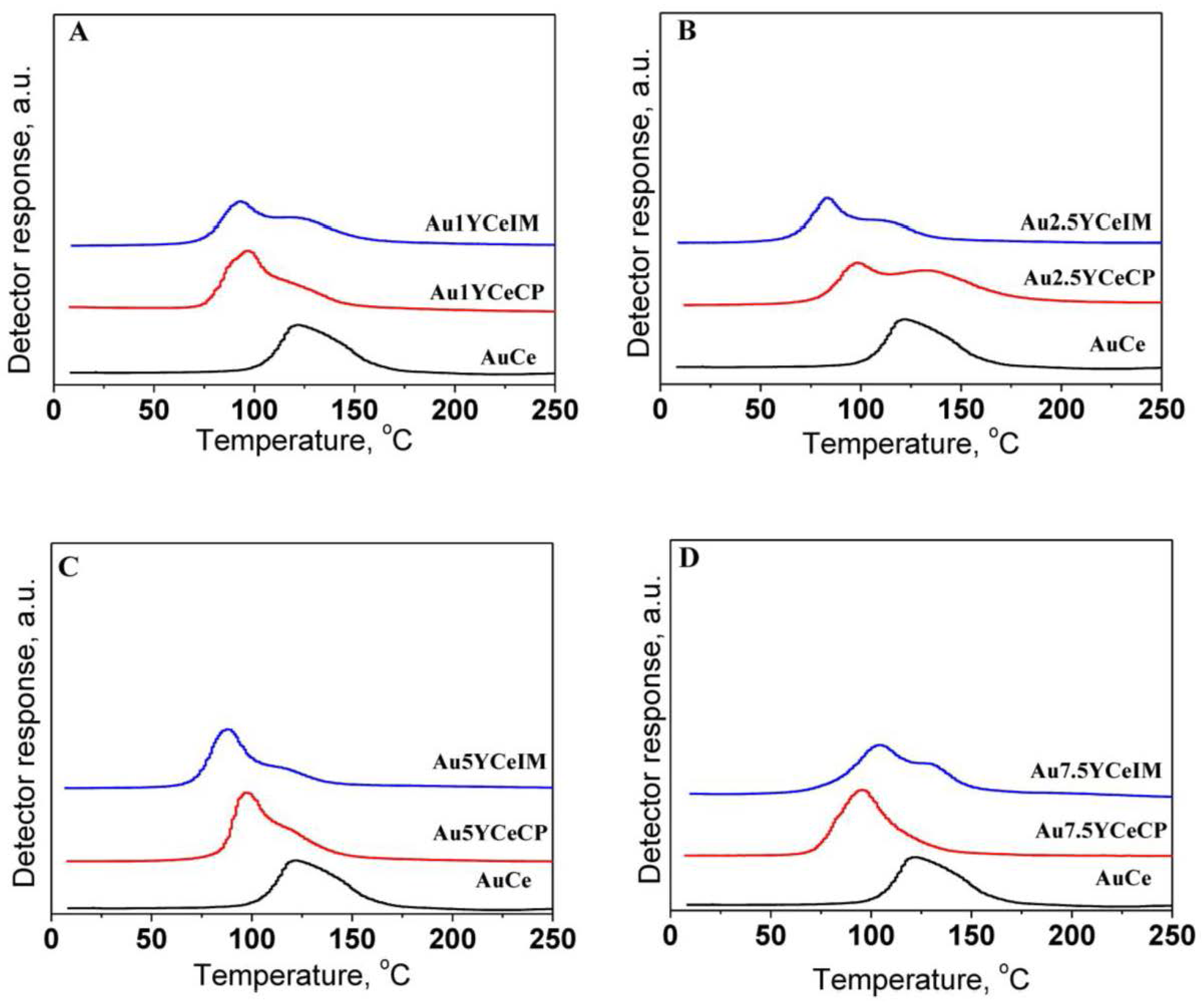
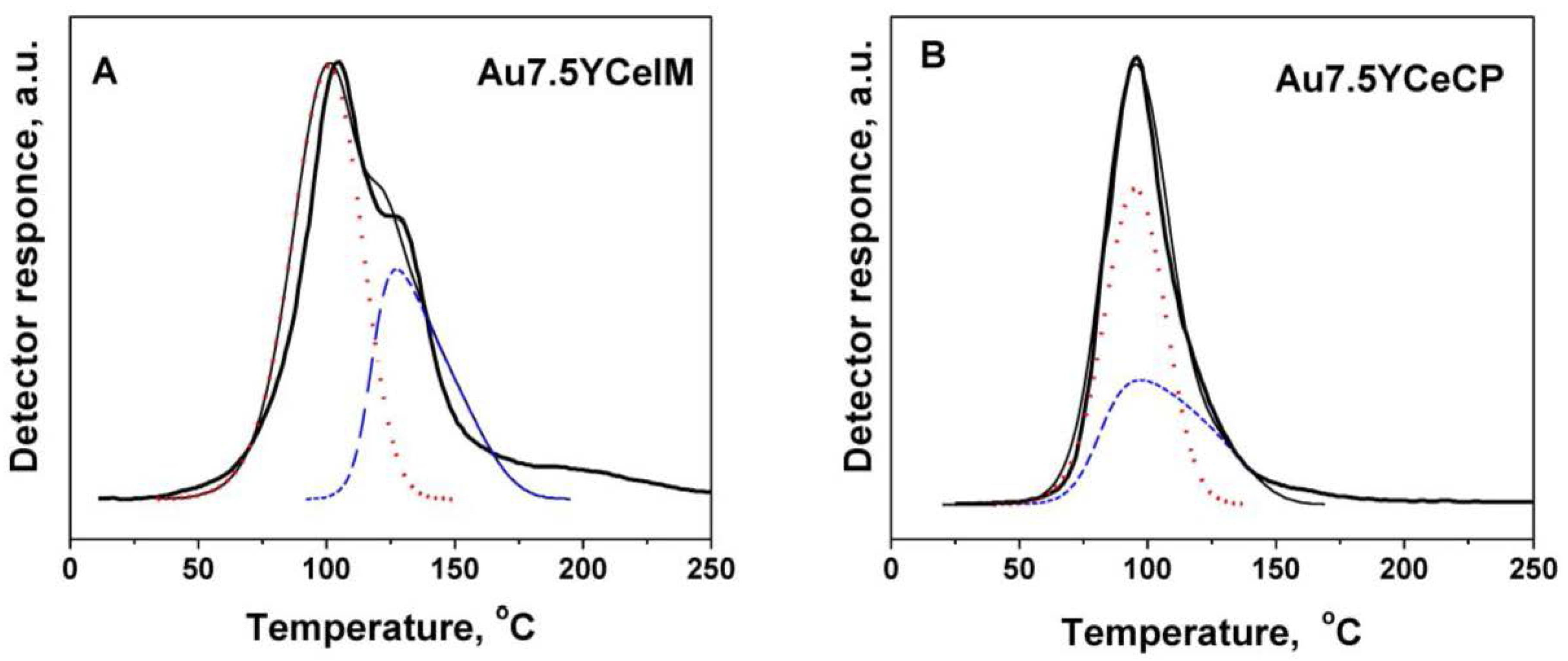
2.1. Catalytic Activity Measurements and TGA/QM Analysis
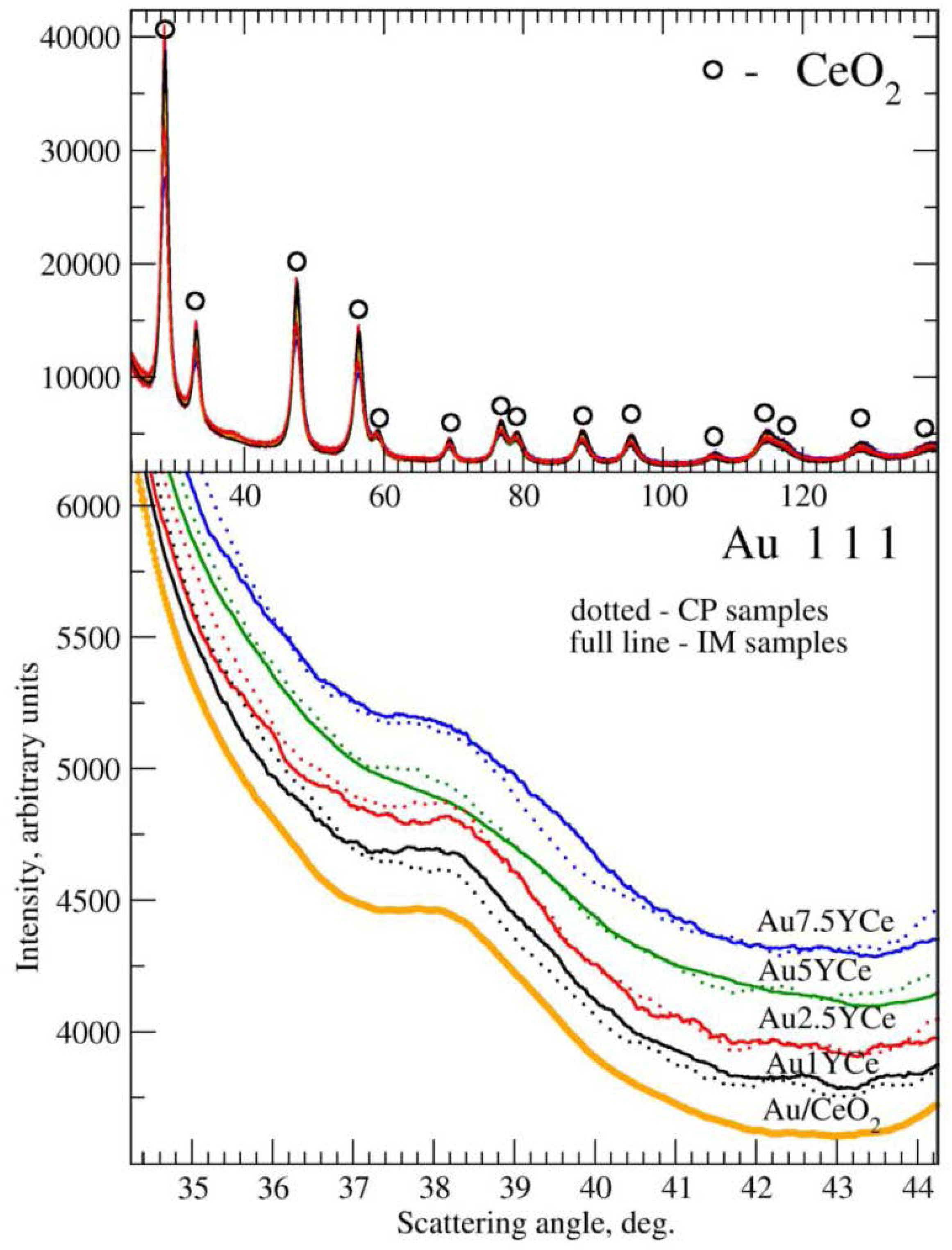
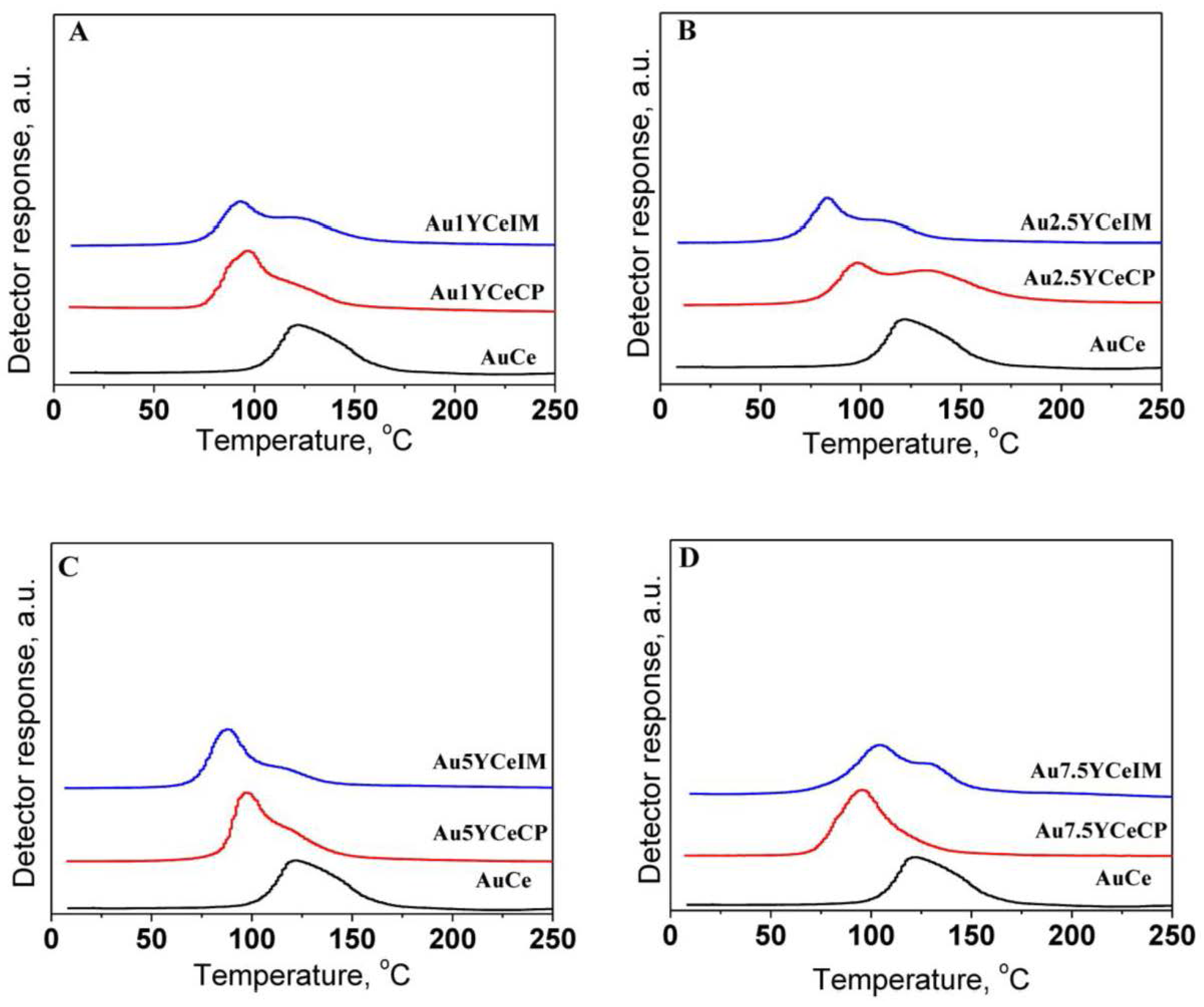
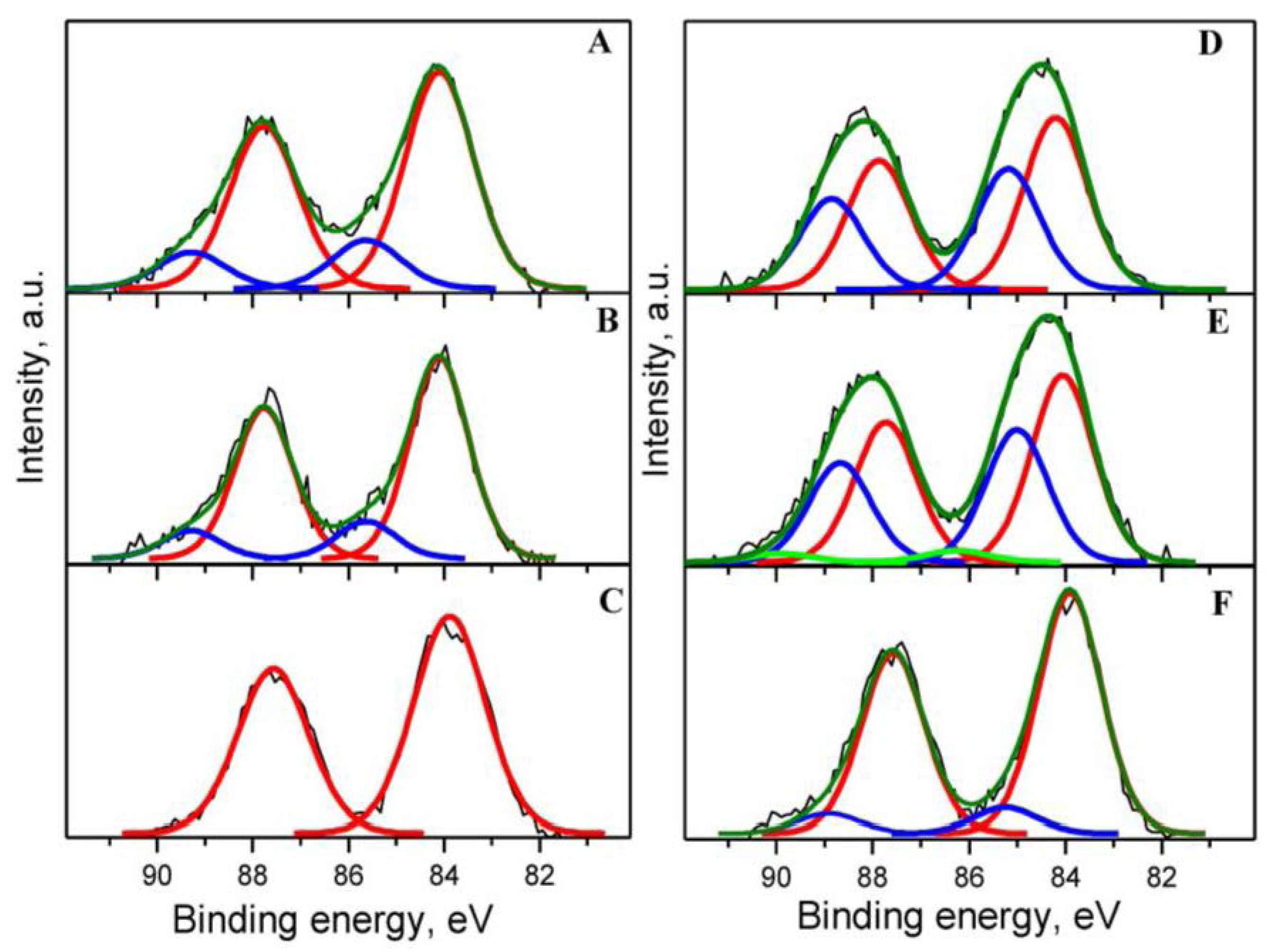
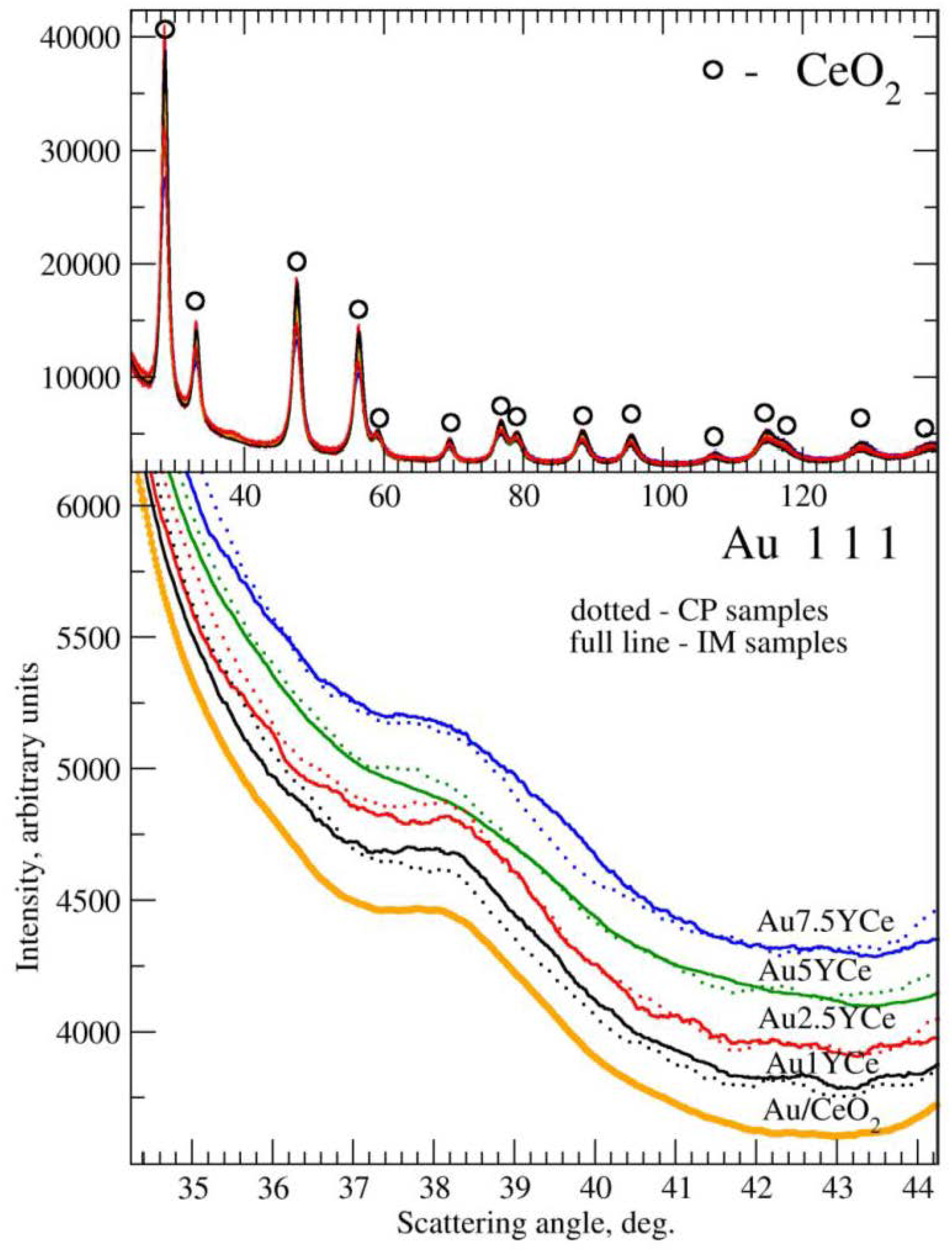
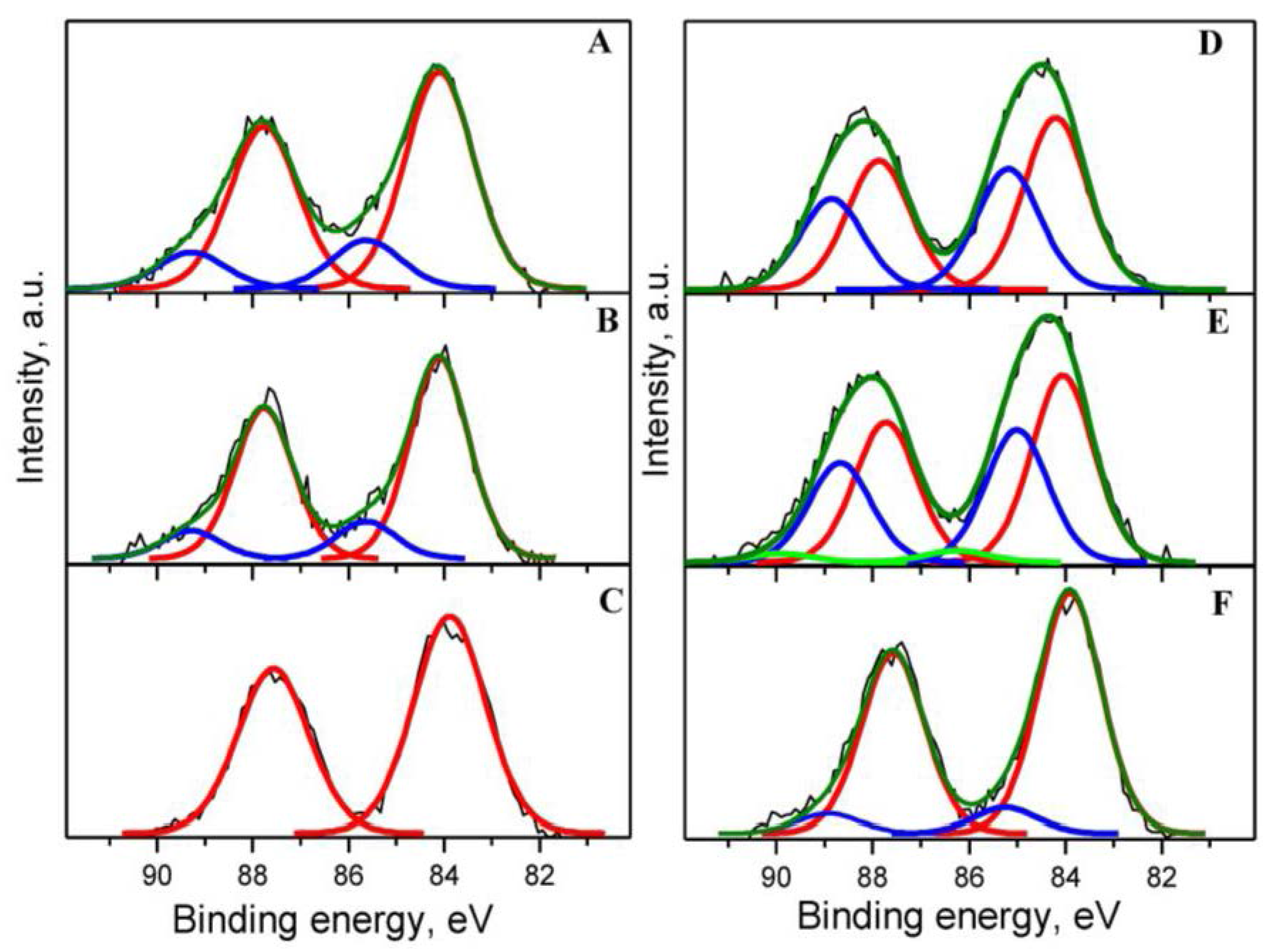
2.2. Sample Characterization
3. Experimental Section
3.1. Sample Preparation
3.2. Sample Characterization
3.3. Catalytic Test in CBO
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scire, S.; Liotta, L.F. Supported gold catalysts for the total oxidation of volatile organic compounds. Appl. Catal. B 2012, 125, 222–246. [Google Scholar] [CrossRef]
- Scirè, S.; Minicò, S.; Crisafulli, C.; Satriano, C.; Pistone, A. Catalytic combustion of volatile organic compounds on gold/cerium oxide catalysts. Appl. Catal. B 2003, 40, 43–49. [Google Scholar] [CrossRef]
- Centeno, M.A.; Paulis, M.; Montes, M.; Odriozola, J.A. Catalytic combustion of volatile organic compounds on Au/CeO2/Al2O3 and Au/Al2O3 catalysts. Appl. Catal. A 2002, 234, 65–78. [Google Scholar] [CrossRef]
- Lai, S.Y.; Qiu, Y.F.; Wang, S.J. Effects of the structure of ceria on the activity of gold/ceria catalysts for the oxidation of carbon monoxide and benzene. J. Catal. 2006, 237, 303–313. [Google Scholar] [CrossRef]
- Ousmane, M.; Liotta, L.F.; Di Carlo, G.; Pantaleo, G.; Venezia, A.M.; Deganello, G.; Retailleau, L.; Boreave, A.; Giroir-Fendler, A. Supported Au catalysts for low temperature abatement of propene and toluene, as model VOCs: Support effect. Appl. Catal. B 2011, 101, 629–637. [Google Scholar] [CrossRef]
- Jiang, X.; Hua, J.; Deng, H.; Wu, Z. Influence of pre-added NaOH on the microstructure of Au–CeO2 catalyst and its activity for benzene oxidation. J. Mol. Catal. A: Chem. 2014, 383–384, 188–193. [Google Scholar] [CrossRef]
- Andreeva, D.; Nedyalkova, R.; Ilieva, L.; Abrashev, M.V. Nanosize gold–ceria catalysts promoted by vanadia for complete benzene oxidation. Appl. Catal. A 2003, 246, 2938. [Google Scholar] [CrossRef]
- Andreeva, D.; Nedyalkova, R.; Ilieva, L.; Abrashev, M.V. Gold–vanadia catalysts supported on ceria–alumina for complete benzene oxidation. Appl. Catal. B 2004, 52, 157–165. [Google Scholar] [CrossRef]
- Andreeva, D.; Petrova, P.; Ilieva, L.; Abrashev, M.V. Gold supported on ceria and ceria–alumina promoted by molybdena for complete benzene oxidation. Appl. Catal. B 2006, 67, 237–245. [Google Scholar] [CrossRef]
- Andreeva, D.; Petrova, P.; Ilieva, L.; Sobczak, J.W.; Abrashev, M.V. Design of new gold catalysts supported on mechanochemically activated ceria–alumina, promoted by molybdena for complete benzene oxidation. Appl. Catal. B 2008, 77, 364–372. [Google Scholar] [CrossRef]
- Yang, S.M.; Liu, D.M.; Liu, S.Y. Catalytic Combustion of Benzene over Au Supported on Ceria and Vanadia Promoted Ceria. Top. Catal. 2008, 47, 101–108. [Google Scholar] [CrossRef]
- Ilieva, L.; Petrova, P.; Tabakova, T.; Zanella, R.; Abrashev, M.V.; Sobczak, J.W.; Lisowski, W.; Kaszkur, Z.; Andreeva, D. Relationship between structural properties and activity in complete benzene oxidation over Au/CeO2–CoOx catalysts. Catal. Today 2012, 187, 30–38. [Google Scholar] [CrossRef]
- Petrova, P.; Tabakova, T.; Munteanu, G.; Zanella, R.; Tsvetkov, M.; Ilieva, L. Gold catalysts on Co-doped ceria for complete benzene oxidation: Relationship between reducibility and catalytic activity. Catal. Commun. 2013, 36, 84–88. [Google Scholar] [CrossRef]
- Ilieva, L.; Petrova, P.; Tabakova, T.; Zanella, R.; Kaszkur, Z. Gold catalysts on ceria doped with MeOx (Me = Fe, Mn, Co and Sn) for complete benzene oxidation: Effect of composition and structure of the mixed supports. React. Kinet. Mech. Catal. 2012, 105, 23–37. [Google Scholar] [CrossRef]
- Ilieva, L.; Petrova, P.; Pantaleo, G.; Zanella, R.; Liotta, L.F.; Georgiev, V.; Boghosian, S.; Kaszkur, Z.; Sobczak, J.W.; Lisowski, W.; et al. Gold catalysts supported on Y-modified ceria for CO-free hydrogen production via PROX. Appl. Catal. B 2016, 188, 154–168. [Google Scholar] [CrossRef]
- Ou, D.R.; Mori, T.; Ye, F.; Takahashi, M.; Zou, J.; Drennan, J. Microstructures and electrolytic properties of yttrium-doped ceria electrolytes: Dopant concentration and grain size dependences. Acta Mater. 2006, 54, 3737–3746. [Google Scholar] [CrossRef]
- Burbano, M.; Norberg, S.T.; Hull, S.; Eriksson, S.G.; Marroccelli, D.; Madden, P.A.; Watson, G.W. Oxygen vacancy ordering and the conductivity maximum in Y2O3-doped CeO2. Chem. Mater. 2011, 24, 222–229. [Google Scholar] [CrossRef]
- Yan, P.F.; Mori, T.; Suzuki, A.; Wu, Y.Y.; Auchterlonie, G.J.; Zou, J.; Drennan, J. Grain boundary’s conductivity in heavily yttrium doped ceria. Solid State Ion. 2012, 222–223, 31–37. [Google Scholar] [CrossRef]
- Vayssilov, G.N.; Mihaylov, M.; Petkov, P.S.; Hadjiivanov, K.I.; Neyman, K.M. Reassignment of the Vibrational Spectra of Carbonates, Formates, and Related Surface Species on Ceria: A Combined Density Functional and Infrared Spectroscopy Investigation. J. Phys. Chem. C 2011, 115, 23435–23454. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev. Sci. Eng. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Ying, F.; Wang, S.; Au, C.-T.; Lai, S.-Y. Highly active and stable mesoporous Au/CeO2 catalysts prepared from MCM-48 hard-template. Microporous Mesoporous Mater. 2011, 142, 308–315. [Google Scholar] [CrossRef]
- Ilieva, L.; Petrova, P.; Ivanov, I.; Munteanu, G.; Boutonnet, M.; Sobczac, J.W.; Lisowski, W.; Kaszkur, Z.; Markov, P.; Venezia, A.M.; et al. Nanosized gold catalysts on Pr-modified ceria for pure hydrogen production via WGS reaction. Mat. Chem. Phys. 2015, 157, 138–146. [Google Scholar] [CrossRef]
- Yao, H.C.; Yao, Y.F.Y. Ceria in automotive exhaust catalysts: I. Oxygen storage. J. Catal. 1984, 86, 254–265. [Google Scholar] [CrossRef]
- Fu, Q.; Weber, A.; Flytzani-Stephanopoulos, M. Nanostructured Au–CeO2 catalysts for low-temperature water–gas shift. Catal. Lett. 2001, 77, 87–95. [Google Scholar] [CrossRef]
- Andreeva, D.; Idakiev, V.; Tabakova, T.; Ilieva, L.; Falaras, P.; Bourlinos, A.; Travlos, A. Low-temperature water-gas shift reaction over Au/CeO2 catalysts. Catal. Today 2002, 72, 51–57. [Google Scholar] [CrossRef]
- Sanchez, M.G.; Gazquez, J.L. Oxygen vacancy model in strong metal-support interaction. J. Catal. 1987, 104, 120–135. [Google Scholar] [CrossRef]
- Laachir, A.; Perrichon, V.; Bardi, A.; Lamotte, J.; Catherine, E.; Lavalley, J.C.; El Faallah, J.; Hilaire, L.; Le Normand, F.; Quemere, E.; et al. Reduction of CeO2 by hydrogen. Magnetic susceptibility and Fourier-transform infrared, ultraviolet and X-ray photoelectron spectroscopy measurements. J. Chem. Soc. Faraday Trans. 1991, 87, 1601–1609. [Google Scholar] [CrossRef]
- Munteanu, G.; Ilieva, L.; Nedyalkova, R.; Andreeva, D. Influence of gold on the reduction behaviour of Au–V2O5/CeO2 catalytic systems: TPR and kinetic parameters of reduction. Appl. Catal. A 2004, 277, 31–40. [Google Scholar] [CrossRef]
- Guzman, J.; Corma, A. Nanocrystalline and mesostructured Y2O3 as supports for gold catalysts. Chem. Commun. 2005, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Carabineiro, S.A.C.; Chen, X.; Martynyuk, O.; Bogdanchikova, N.; Avalos-Borja, M.; Pestryakov, A.; Tavares, P.B.; Órfão, J.J.M.; Pereira, M.F.R.; Figueiredo, J.L. Gold supported on metal oxides for volatile organic compounds total oxidation. Catal. Today 2015, 244, 103–114. [Google Scholar] [CrossRef]
- Monte, M.; Munuera, G.; Costa, D.; Conesa, J.C.; Martínez-Arias, A. Near-ambient XPS characterization of interfacial copper species in ceria-supported copper catalysts. Phys. Chem. Chem. Phys. 2015, 17, 29995–30004. [Google Scholar] [CrossRef] [PubMed]
- Óvári, L.; Calderon, S.K.; Lykhach, Y.; Libuda, J.; Erdőhelyi, A.; Papp, C.; Kiss, J.; Steinrück, H.-P. Near ambient pressure XPS investigation of the interaction of ethanol with Co/CeO2(1 1 1). J. Catal. 2013, 307, 132–139. [Google Scholar] [CrossRef]
- Gamarra, D.; López Cámara, A.; Monte, M.; Rasmussen, S.B.; Chinchilla, L.E.; Hungría, A.B.; Munuera, G.; Gyorffy, N.; Schay, Z.; Corberán, V.C.; et al. Preferential oxidation of CO in excess H2 over CuO/CeO2 catalysts: Characterization and performance as a function of the exposed face present in the CeO2 support. Appl. Catal. B 2013, 130–131, 224–238. [Google Scholar] [CrossRef]
- Tidahy, V.; Siffert, S.; Lamonier, J.-F.; Cousin, R.; Zhilinskaya, E.A.; Aboukais, A.; Su, B.-L.; Canet, X.; De Weireld, G.; Frere, M.; et al. Influence of the exchanged cation in Pd/BEA and Pd/FAU zeolites for catalytic oxidation of VOCs. Appl. Catal. B 2007, 70, 377–383. [Google Scholar] [CrossRef]
- Williamson, G.K.; Hall, W.H. X-ray line broadening from filed aluminium and wolfram. Acta Metall. 1953, 1, 22–31. [Google Scholar] [CrossRef]
- Mote, V.D.; Purushotham, Y.; Dole, B.N. Williamson-Hall analysis in estimation of lattice strain in nanometer-sized ZnO particles. J. Theor. Appl. Phys. 2012, 6, 6–28. [Google Scholar] [CrossRef]
- Barr, L.; Fries, G.; Cariati, F.; Bart, C.J.; Giordano, N. A spectroscopic investigation of cerium molybdenum oxides. J. Chem. Soc. Dalton Trans. 1983, 9, 1825–1829. [Google Scholar] [CrossRef]
- Paparazzo, E.; Ingo, G.M.; Zaccheti, N. X-ray induced reduction effects at CeO2 surfaces: An X-ray photoelectron spectroscopy study. J. Vac. Sci. Technol. A 1991, 9, 1416–1420. [Google Scholar] [CrossRef]
- Trudeau, M.L.; Tschope, A.; Ying, J.Y. XPS investigation of surface oxidation and reduction in nanocrystalline CexLa1−xO2−y. Surf. Interface Anal. 1995, 23, 219–226. [Google Scholar] [CrossRef]
- Sundaram, K.B.; Wahid, P.F.; Melendez, O. Deposition and X-ray photoelectron spectroscopy studies on sputtered cerium dioxide thin films. J. Vac. Sci. Technol. A 1997, 15, 52–56. [Google Scholar] [CrossRef]
- Nelson, A.E.; Graves-Brook, M.K.; Schulz, K.H. Analysis of Cerium-Zirconium Mixed Metal Oxides by X-Ray Photoelectron Spectroscopy. Surf. Sci. Spectra 2001, 8, 126. [Google Scholar] [CrossRef]
- He, H.; Dai, H.X.; Wong, K.W.; Au, C.T. RE0.6Zr0.4−xYxO2 (RE = Ce, Pr; x = 0, 0.05) solid solutions: An investigation on defective structure, oxygen mobility, oxygen storage capacity, and redox properties. Appl. Catal. A 2003, 251, 61–74. [Google Scholar] [CrossRef]
- Borchert, H.; Frolova, Y.V.; Kaichev, V.V.; Prosvirin, I.P.; Alikina, G.M.; Lukashevich, A.I.; Zaikovskii, V.I.; Moroz, E.M.; Trukhan, S.N.; Ivanov, V.P.; et al. Electronic and Chemical Properties of Nanostructured Cerium Dioxide Doped with Praseodymium. J. Phys. Chem. B 2005, 109, 5728–5738. [Google Scholar] [CrossRef] [PubMed]
- Bêche, E.; Charvin, P.; Perarnau, D.; Abanades, S.; Flamant, G. Ce 3d XPS investigation of cerium oxides and mixed cerium oxide (CexTiyOz). Surf. Interface Anal. 2008, 40, 264–267. [Google Scholar] [CrossRef]
- Fang, J.; Bi, X.; Si, D.; Jiang, Z.; Huang, W. Spectroscopic studies of interfacial structures of CeO2-TiO2 mixed oxides. Appl. Surf. Sci. 2007, 253, 8952–8961. [Google Scholar] [CrossRef]
- Rebellato, J.; Natile, M.; Glisenti, A. Influence of the synthesis procedure on the properties and reactivity of nanostructured ceria powders. Appl. Catal. A 2008, 339, 108–120. [Google Scholar] [CrossRef]
- Xi, L.; Yan, S.; Zhu, L.; Li, L.; Wenhui, S. Synthesis and Properties of the Solid Solution Ce1−xPrxO2−δ (x = 0.05~0.30). Acta Chim. Sinica 2009, 67, 1389–1394. [Google Scholar]
- Reddy, B.M.; Thrimurthulu, G.; Katta, L.; Yamada, Y.; Park, S.E. Structural Characteristics and Catalytic Activity of Nanocrystalline Ceria-Praseodymia Solid Solutions. J. Phys. Chem. C 2009, 113, 15882–15890. [Google Scholar] [CrossRef]
- Yu, Q.; Li, Y.; Zou, X.; Zhuo, H.; Yao, Y.; Suo, Z. Effect of Alkali Metal Promoters on Water-Gas Shift Activity over Au-Pt/CeO2 Catalyst. Chin. J. Catal. 2010, 31, 671–676. [Google Scholar]
- Reddy, B.M.; Lakshmi, K.; Thrimurthulu, G. Novel Nanocrystalline Ce1−xLaxO2−δ (x = 0.2) Solid Solutions: Structural Characteristics and Catalytic Performance. Chem. Mater. 2010, 22, 467–475. [Google Scholar] [CrossRef]
- Cai, W.; Chen, F.; Shen, X.; Chen, L.; Zhang, J. Enhanced catalytic degradation of AO7 in the CeO2-H2O2 system with Fe3+ doping. Appl. Catal. B 2010, 101, 160–168. [Google Scholar] [CrossRef]
- Kim, W.-H.; Kim, M.-K.; Maeng, W.J.; Gatineau, J.; Pallem, V.; Dussarrat, C.; Noori, A.; Thompson, D.; Chu, S.; Kim, H. Growth Characteristics and Film Properties of Cerium Dioxide Prepared by Plasma-Enhanced Atomic Layer Deposition. J. Electrochem. Soc. 2011, 158, G169. [Google Scholar] [CrossRef]
- Paunović, N.; Dohčević-Mitrović, Z.; Scurtu, R.; Aškrabić, S.; Prekajski, M.; Matović, B.; Popović, Z.V. Suppression of inherent ferromagnetism in Pr-doped CeO2 nanocrystals. Nanoscale 2012, 4, 5469–5476. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; Gazquez, J.; Palau, A.; Varela, M.; Obradors, X.; Puig, T. Low Temperature Epitaxial Oxide Ultrathin Films and Nanostructures by Atomic Layer Deposition. Chem. Mater. 2012, 24, 3732–3737. [Google Scholar] [CrossRef]
- Gong, L.; Luo, L.-T.; Wang, R.; Zhang, N. Effect of preparation method of CeO2-MnOx mixed oxides n preferential oxidation of CO in H2-rich gases over CuO-based catalysts. J. Chil. Chem. Soc. 2012, 57, 1048–1053. [Google Scholar] [CrossRef]
- Beche, E.; Peraudeau, G.; Flaud, V.; Perarnau, D. An XPS investigation of (La2O3)1−x (CeO2)2x (ZrO2)2 compounds. Surf. Interface Anal. 2012, 44, 1045–1050. [Google Scholar] [CrossRef]
- Tan, J.; Zhang, W.; Lv, Y.-H.; Xia, A.-L. Facile Preparation of Mn-doped CeO2 Submicrorods by Composite-Hydroxide-Salt-Mediated Approach and Their Magnetic Property. Mater. Res. 2013, 16, 689–694. [Google Scholar] [CrossRef]
- Konysheva, E.Y.; Francis, S.M. Identification of surface composition and chemical states in composites comprised of phases with fluorite and perovskite structures by X-ray photoelectron spectroscopy. Appl. Surf. Sci. 2013, 268, 278–287. [Google Scholar] [CrossRef]
- Santra, C.; Rahman, S.; Bojja, S.; James, O.; Sen, D.; Maity, S.; Mohanty, A.K.; Mazumder, S.; Chowdhury, B. Barium, calcium and magnesium doped mesoporous ceria supported gold nanoparticle for benzyl alcohol oxidation using molecular O2. Catal. Sci. Technol. 2013, 3, 360–370. [Google Scholar] [CrossRef]
- Alshankiti, I.; Al-Otaibi, F.; Alsalik, Y.; Idriss, H. Solar Thermal Hydrogen Production from Water over Modified CeO2 Materials. Top. Catal. 2013, 56, 1129–1138. [Google Scholar] [CrossRef]
- Kotzev, N.; Shopov, D. A thermodesorption study of the system olefin-NiO. J. Catal. 1971, 22, 297–301. [Google Scholar] [CrossRef]
- Monti, D.A.M.; Baiker, A. Temperature-programmed reduction. Parametric sensitivity and estimation of kinetic parameters. J. Catal. 1983, 83, 323–335. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2·g−1) | DXRD(CeO2) (nm) | DXRD(Au) (nm) |
|---|---|---|---|
| AuCe | 102.4 | 5.7(1) * | 4.1 |
| Au1YCeIM | 87.2 | 7.1(6)/6.7(1) a | 4.0/2.8; 5.1 a,b |
| Au2.5YCeIM | 83.2 | 5.3(8) | 3.7 |
| Au5YCeIM | 76.1 | 7.2(6) | 4.5 |
| Au7.5YCeIM | 55.9 | 6.7(2) | 3.2 |
| Au1YCeCP | 90.0 | 8.1(2)/7.3(1) a | 5.8/5.7 a |
| Au2.5YCeCP | 88.7 | 5.1(8) | 5.3 |
| Au5YCeCP | 82.4 | 4.3(8) | 5.4 |
| Au7.5YCeCP | 60.1 | 4.7(9) | 5.2 |
| Catalysts | H2 Consumption (mmol·g−1) | |
|---|---|---|
| CP | IM | |
| AuCe | 0.54 | |
| Au1YCe | 0.69 | 0.62 |
| Au2.5YCe | 0.87 | 0.59 |
| Au5YCe | 0.80 | 0.61 |
| Au7.5YCe | 0.78 | 0.63 |
| Catalyst | Au 4f7/2 | Ce 3d5/2 (Ce3+) | O 1s | C 1s | Au/Ce | Y/Ce | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| BE (eV) | Area (%) | BE (eV) | (at. %) | BE (eV) | (at. %) | BE (eV) | (at. %) | |||
| Au1YCeIM as received | 84.07 (Au0) 85.59 (Auδ+) | 81.8 18.2 | 880.73 884.53 | 35.7 | 529.46(O2−) 531.89(-OH) | 45.6 15.2 | 285.22 (C-C,C-H) 286.72 (C-OH) 288.22 (C=O) | 12.6 2.2 1.6 | 0.022 | 0.041 |
| Σ = 16.4 | ||||||||||
| Au1YCeIM oxidized at 350 °C | 84.10 (Au0) 85.63 (Auδ+) | 84.4 15.6 | 880.68 884.11 | 28.4 | 529.49 (O2−) 531.64 (-OH) | 49.0 12.0 | 285.27 (C-C,C-H) 286.77 (C-OH) 288.27 (C=O) | 10.1 1.0 2.9 | 0.022 | 0.042 |
| Σ = 14.2 | ||||||||||
| Au1YCeIM used in CBO | 83.88 (Au0) | 100 | 880.78 885.05 | 33.6 | 529.48 (O2−) 531.82 (-OH) | 51.4 13.1 | 285.15 (C-C,C-H) 286.50 (C-OH) | 8.9 0.7 | 0.019 | 0.035 |
| Σ = 9.6 | ||||||||||
| Au1YCeCP as received | 84.20 (Au0) 85.18 (Auδ+) | 58.7 41.3 | 880.65 885.13 | 30.9 | 529.54 (O2−) 531.85 (-OH) 533.31(C-O-C) | 47.0 13.9 3.1 | 285.00 (C-C,C-H) 286.35 (C-OH) 287.76 (C=O) | 8.8 3.7 0.4 | 0.023 | 0.029 |
| Σ = 12.9 | ||||||||||
| Au1YCeCP oxidized at 350 °C | 84.05 (Au0) 85.01 (Auδ+) 86.25 (Au3+) | 56.5 40.0 3.5 | 880.50 884.64 | 27.0 | 529.47 (O2−) 531.71 (-OH) | 53.2 11.9 | 285.01 (C-C,C-H) 285.70 (C-OH) 287.77 (C=O) | 5.0 4.6 0.2 | 0.024 | 0.026 |
| Σ = 9.8 | ||||||||||
| Au1YCeCP used in CBO | 83.90 (Au0) 85.25 (Auδ+) | 90.3 9.7 | 880.24 885.09 | 28.8 | 529.45 (O2−) 532.64 (-OH) | 30.0 29.0 | 285.01 (C-C,C-H) 286.51 (C-OH) 289.51 (COOH) | 12.4 8.9 4.2 | 0.022 | 0.076 |
| Σ = 25.4 | ||||||||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilieva, L.; Petrova, P.; Liotta, L.F.; Sobczak, J.W.; Lisowski, W.; Kaszkur, Z.; Munteanu, G.; Tabakova, T. Gold Catalysts on Y-Doped Ceria Supports for Complete Benzene Oxidation. Catalysts 2016, 6, 99. https://doi.org/10.3390/catal6070099
Ilieva L, Petrova P, Liotta LF, Sobczak JW, Lisowski W, Kaszkur Z, Munteanu G, Tabakova T. Gold Catalysts on Y-Doped Ceria Supports for Complete Benzene Oxidation. Catalysts. 2016; 6(7):99. https://doi.org/10.3390/catal6070099
Chicago/Turabian StyleIlieva, Lyuba, Petya Petrova, Leonarda F. Liotta, Janusz W. Sobczak, Wojciech Lisowski, Zbigniew Kaszkur, Gabriel Munteanu, and Tatyana Tabakova. 2016. "Gold Catalysts on Y-Doped Ceria Supports for Complete Benzene Oxidation" Catalysts 6, no. 7: 99. https://doi.org/10.3390/catal6070099
APA StyleIlieva, L., Petrova, P., Liotta, L. F., Sobczak, J. W., Lisowski, W., Kaszkur, Z., Munteanu, G., & Tabakova, T. (2016). Gold Catalysts on Y-Doped Ceria Supports for Complete Benzene Oxidation. Catalysts, 6(7), 99. https://doi.org/10.3390/catal6070099