
Mechanistic and Structural Insight to an Evolved Benzoylformate Decarboxylase with Enhanced Pyruvate Decarboxylase Activity


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Substrate Profile of T377L/A460Y
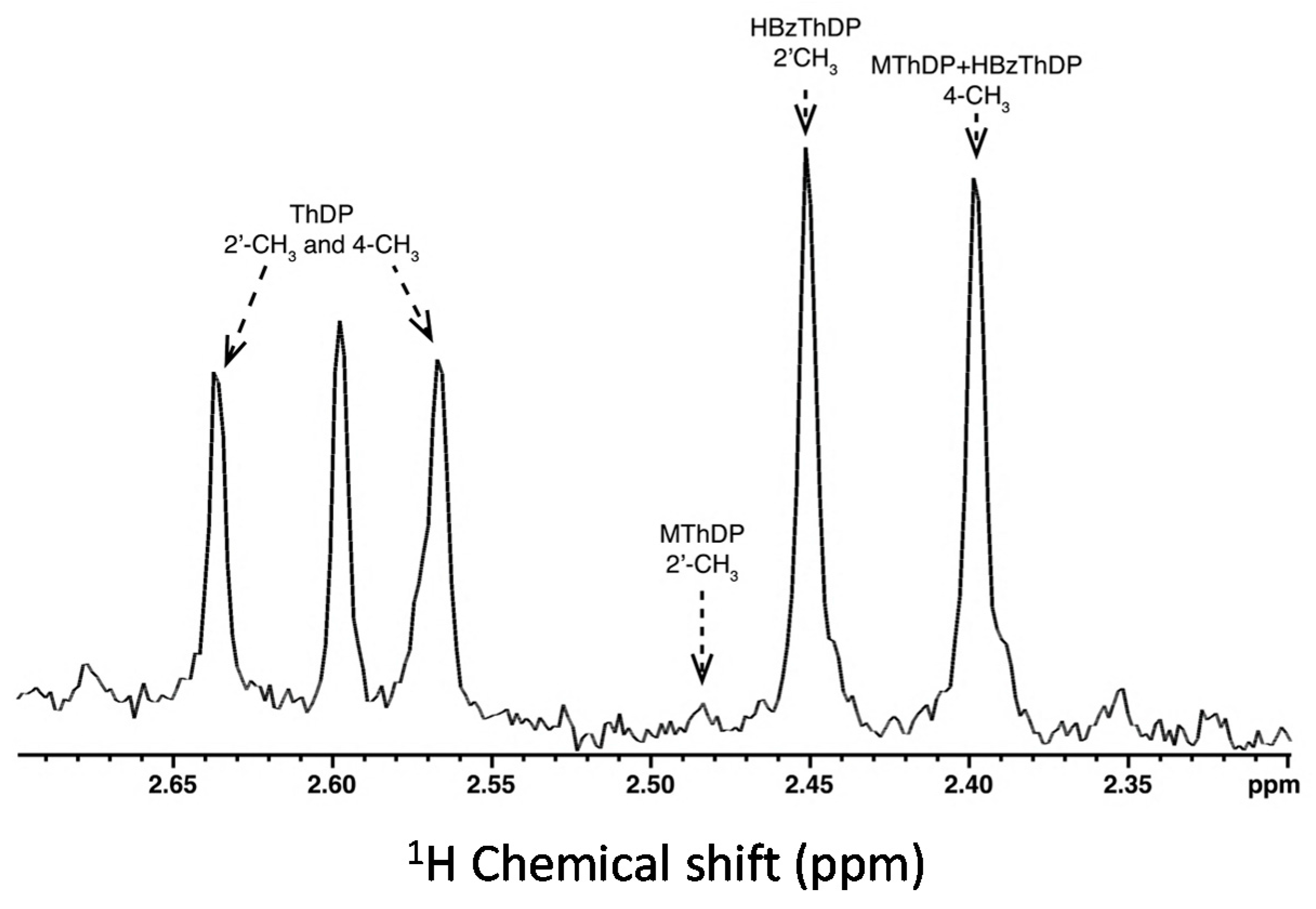
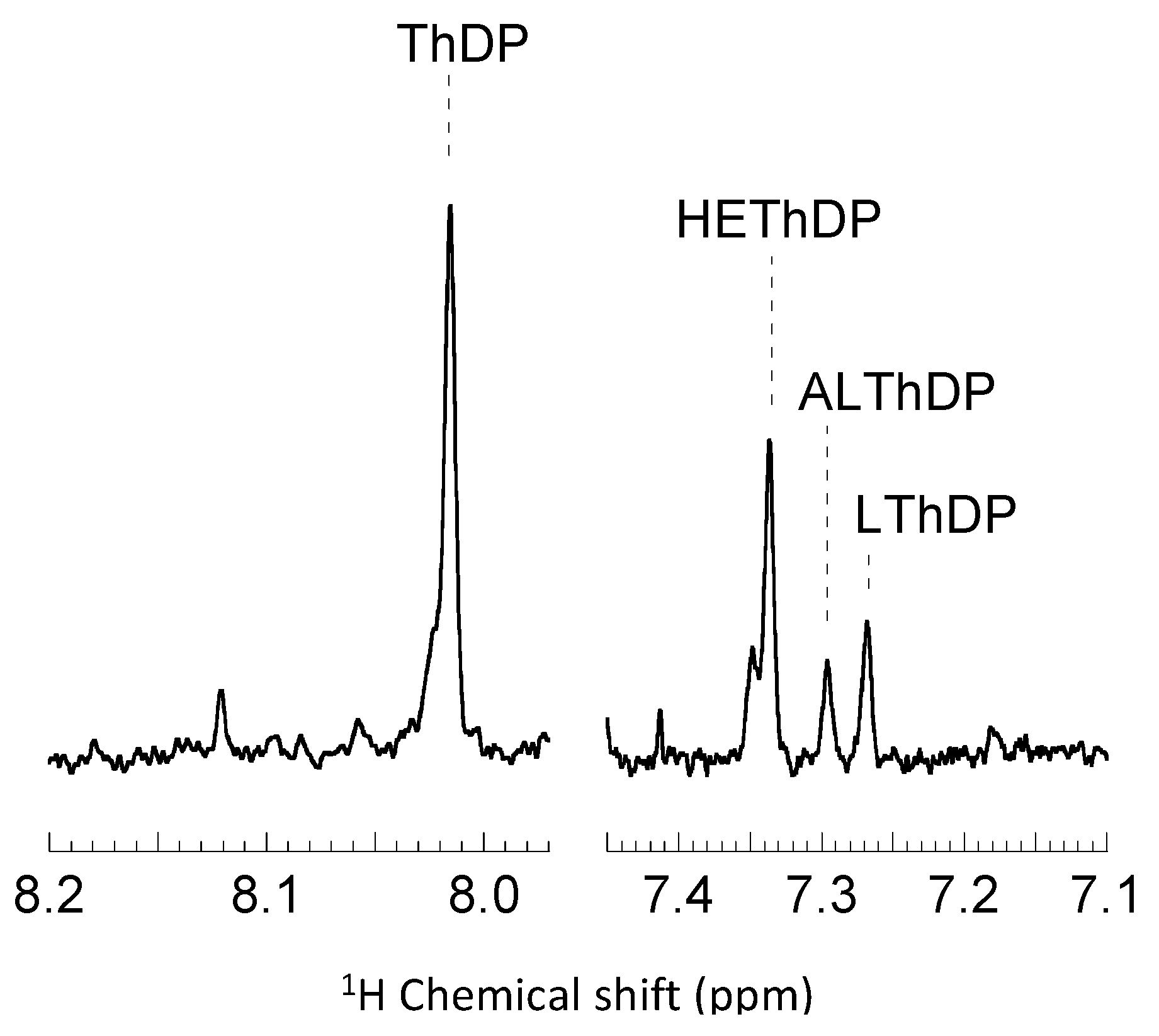
2.2. Intermediate Distribution Analysis for Reaction of T377L/A460Y with Benzoylformate
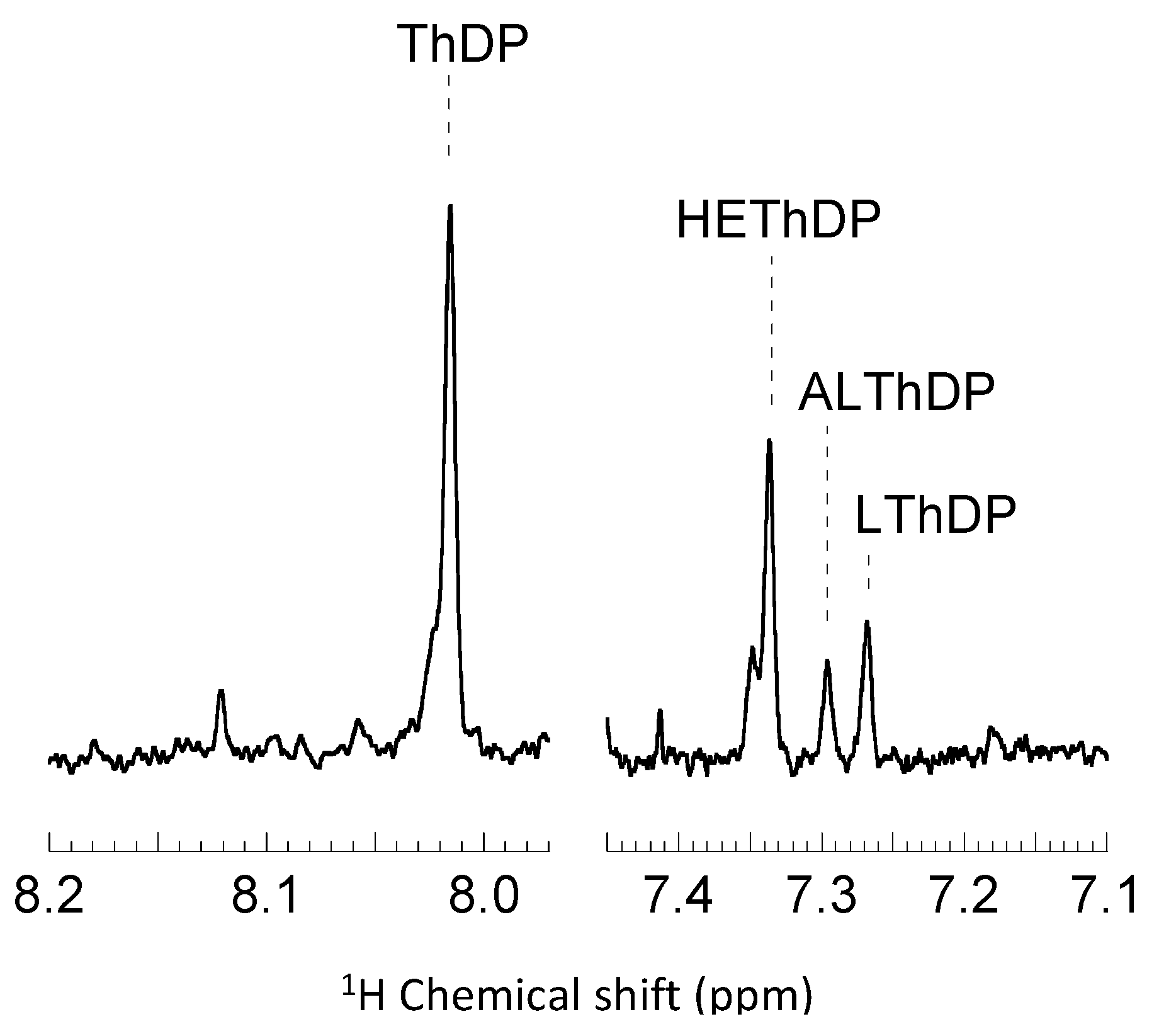
2.3. Intermediate Distribution Analysis for Reaction of T377L/A460Y with Pyruvate
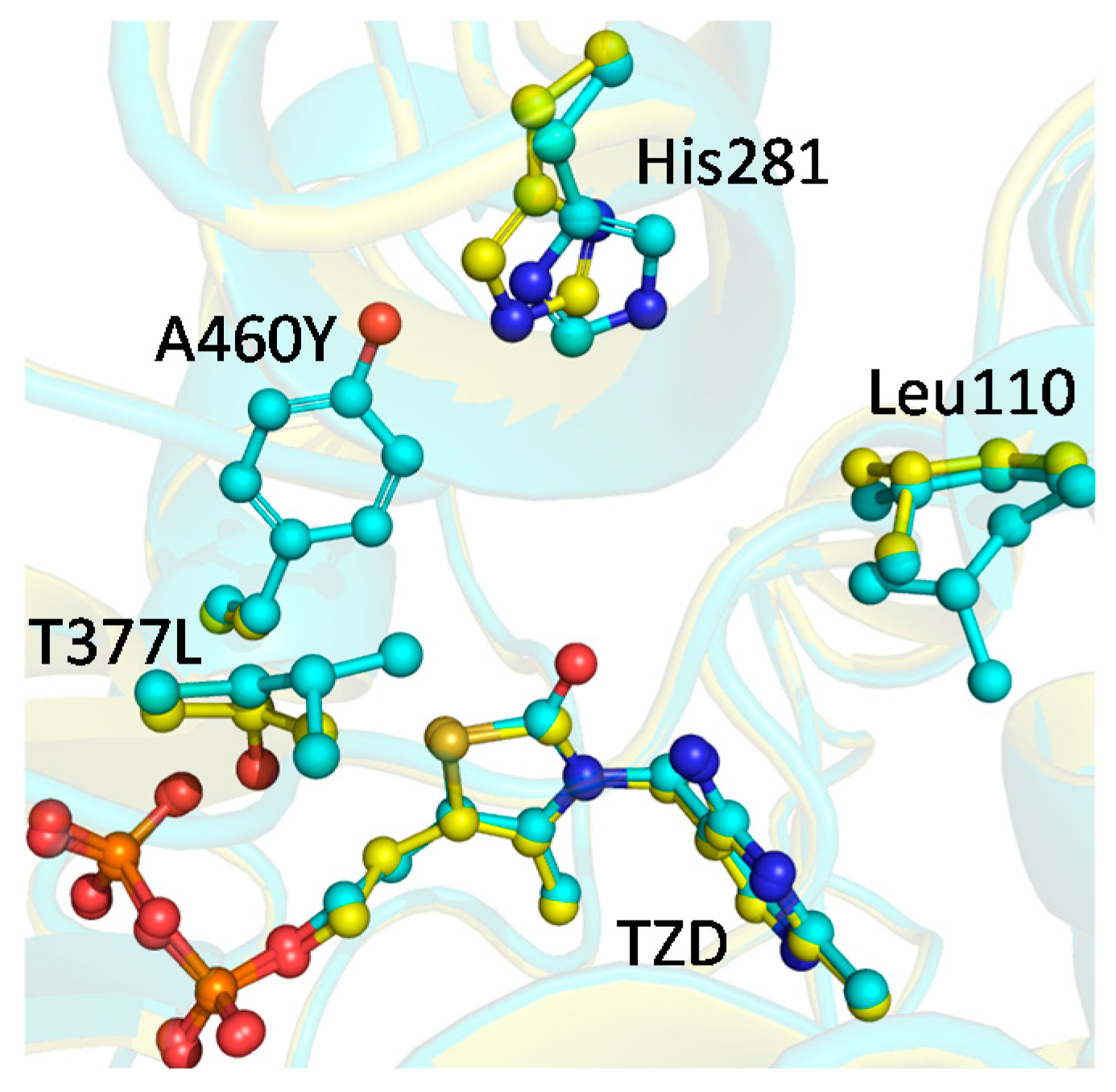
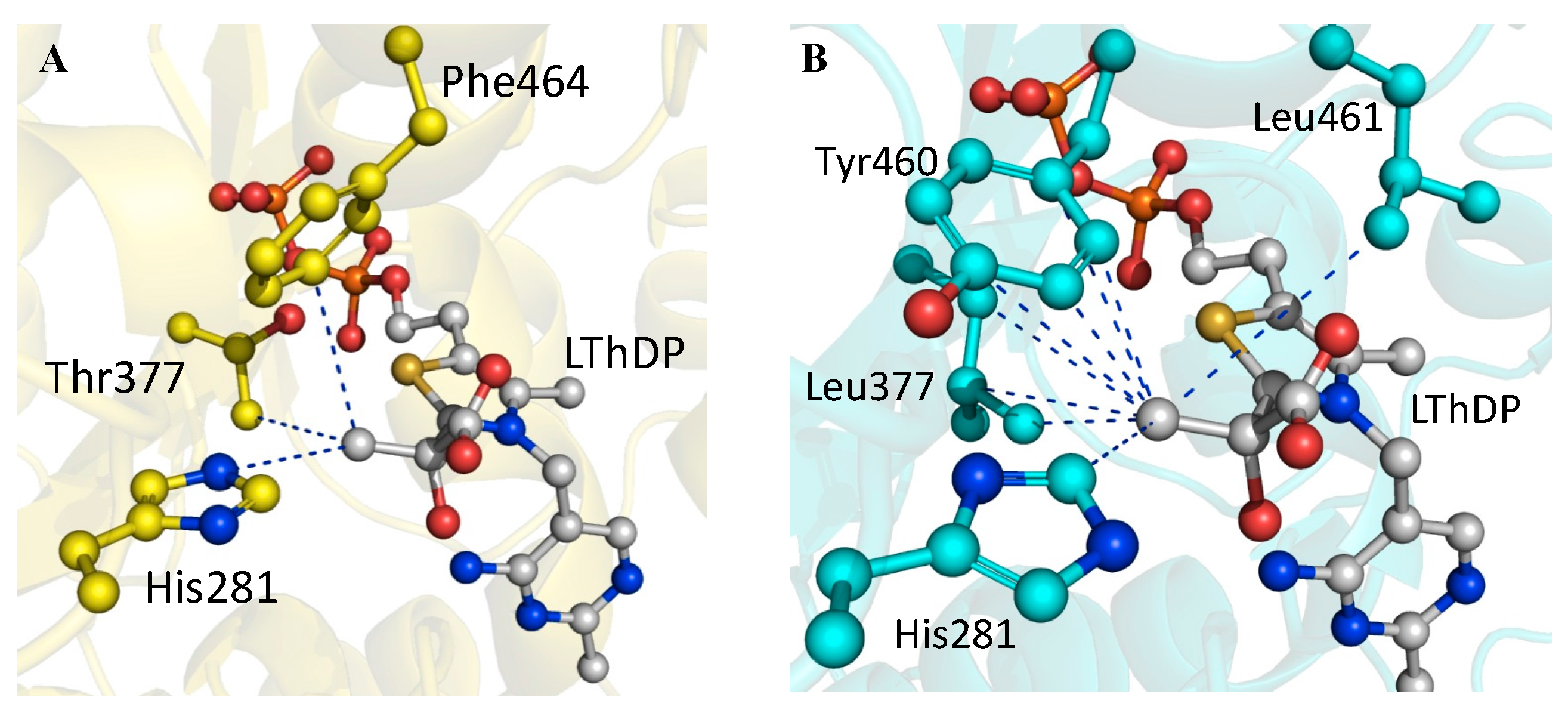
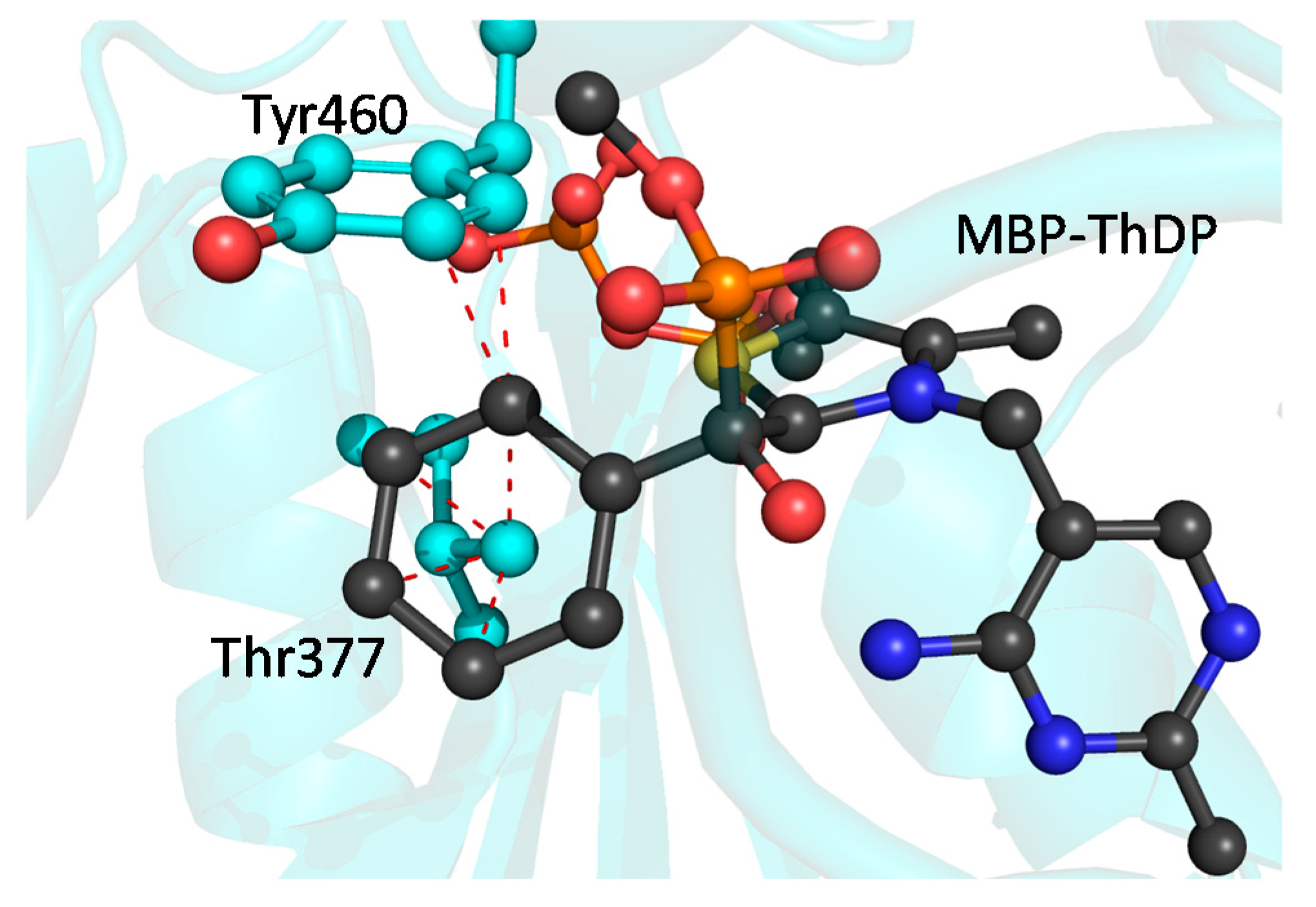
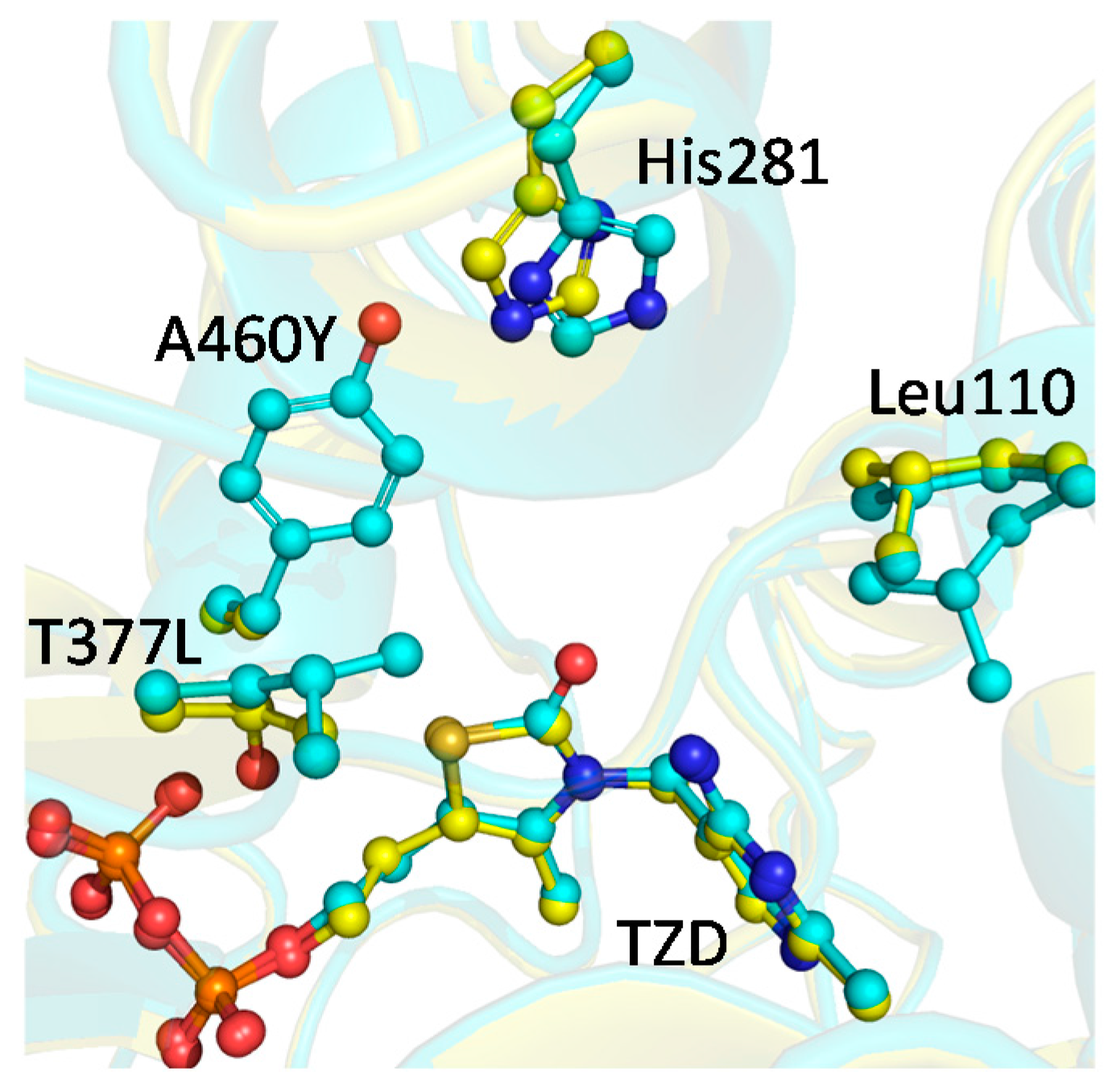
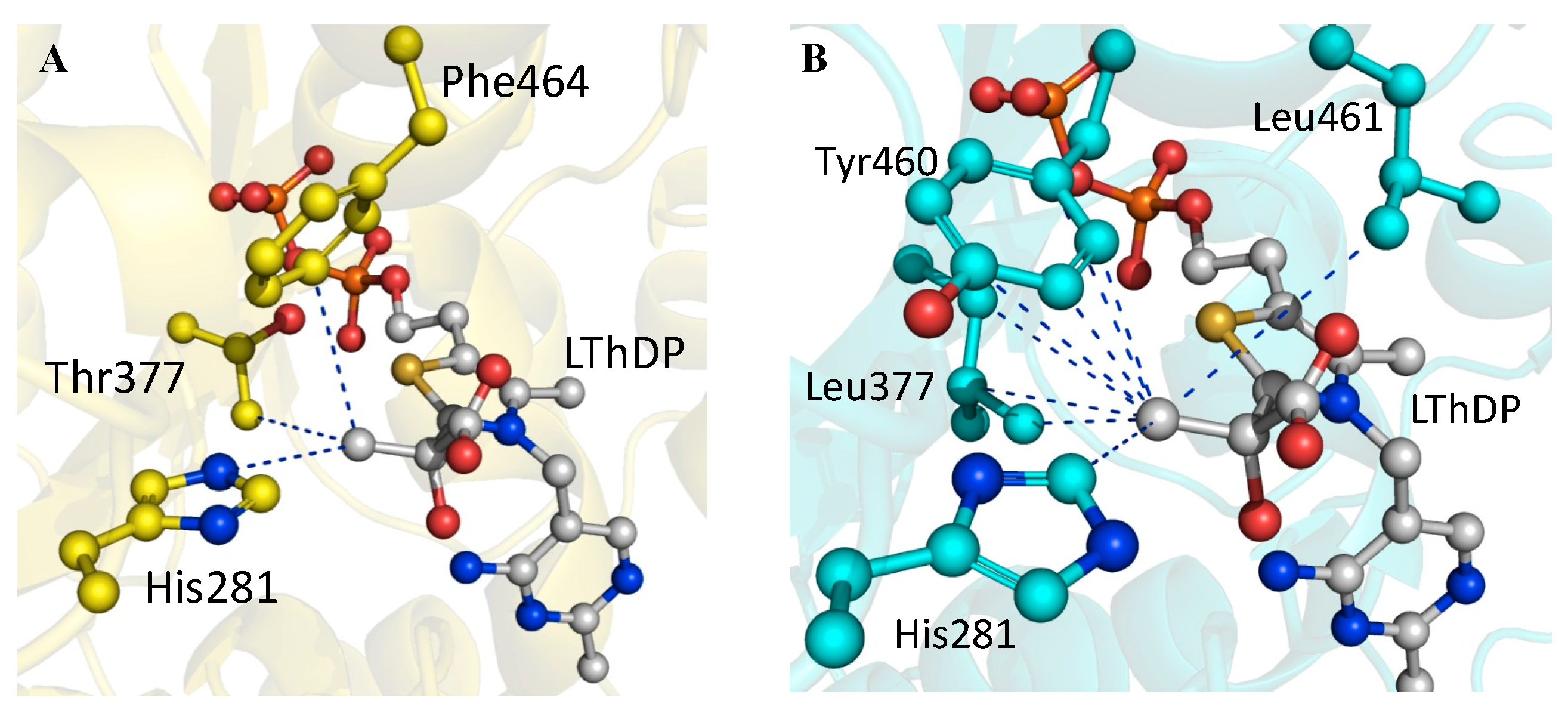
2.4. X-ray Structure of BFDC T377L/A460Y
3. Materials and Methods
3.1. Materials
3.2. Protein Expression and Purification
3.3. Analysis of Substrate Spectrum
3.4. Steady State Analysis of Reaction Intermediates by Acid Quench/NMR Spectroscopy
3.5. Crystallization of BFDC T377L/A460Y
3.6. X-ray Data Collection
3.7. Structure Solutions and Refinements
3.8. Modeling of Reaction Analogues and Intermediates
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Andrews, F.H.; McLeish, M.J. Substrate specificity in thiamin diphosphate-dependent decarboxylases. Bioorg. Chem. 2012, 43, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Nichols, N.; Dien, B.; Bothast, R. Engineering lactic acid bacteria with pyruvate decarboxylase and alcohol dehydrogenase genes for ethanol production from Zymomonas mobilis. J. Ind. Microbiol. Biotechnol. 2003, 30, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Dyda, F.; Furey, W.; Swaminathan, S.; Sax, M.; Farrenkopf, B.; Jordan, F. Catalytic centers in the thiamin diphosphate dependent enzyme pyruvate decarboxylase at 2.4 Å resolution. Biochemistry 1993, 32, 6165–6170. [Google Scholar] [CrossRef] [PubMed]
- Dobritzsch, D.; König, S.; Schneider, G.; Lu, G. High resolution crystal structure of pyruvate decarboxylase from Zymomonas mobilis. Implications for substrate activation in pyruvate decarboxylases. J. Biol. Chem. 1998, 273, 20196–20204. [Google Scholar] [CrossRef] [PubMed]
- Kutter, S.; Wille, G.; Relle, S.; Weiss, M.S.; Hübner, G.; König, S. The crystal structure of pyruvate decarboxylase from Kluyveromyces lactis. FEBS J. 2006, 273, 4199–4209. [Google Scholar] [CrossRef] [PubMed]
- Rother, D.; Kolter, G.; Gerhards, T.; Berthold, C.L.; Gauchenova, E.; Knoll, M.; Pleiss, J.; Müller, M.; Schneider, G.; Pohl, M. S-selective mixed carboligation by structure-based design of the pyruvate decarboxylase from Acetobacter pasteurianus. ChemCatChem 2011, 3, 1587–1596. [Google Scholar] [CrossRef]
- Liu, M.; Sergienko, E.A.; Guo, F.; Wang, J.; Tittmann, K.; Hübner, G.; Furey, W.; Jordan, F. Catalytic acid-base groups in yeast pyruvate decarboxylase. 1. Site-directed mutagenesis and steady-state kinetic studies on the enzyme with the D28A, H114F, H115F, and E477Q substitutions. Biochemistry 2001, 40, 7355–7368. [Google Scholar] [CrossRef] [PubMed]
- Sergienko, E.A.; Jordan, F. Catalytic acid-base groups in yeast pyruvate decarboxylase. 2. Insights into the specific roles of D28 and E477 from the rates and stereospecificity of formation of carboligase side products. Biochemistry 2001, 40, 7369–7381. [Google Scholar] [CrossRef] [PubMed]
- Sergienko, E.A.; Jordan, F. Catalytic acid-base groups in yeast pyruvate decarboxylase. 3. A steady-state kinetic model consistent with the behavior of both wild-type and variant enzymes at all relevant pH values. Biochemistry 2001, 40, 7382–7403. [Google Scholar] [CrossRef] [PubMed]
- Candy, J.M.; Duggleby, R.G. Structure and properties of pyruvate decarboxylase and site-directed mutagenesis of the Zymomonas mobilis enzyme. BBA Protein Struct. Mol. Enzymol. 1998, 1385, 323–338. [Google Scholar] [CrossRef]
- Schütz, A.; Sandalova, T.; Ricagno, S.; Hübner, G.; König, S.; Schneider, G. Crystal structure of thiamindiphosphate-dependent indolepyruvate decarboxylase from Enterobacter cloacae, an enzyme involved in the biosynthesis of the plant hormone indole-3-acetic acid. Eur. J. Biochem. 2003, 270, 2312–2321. [Google Scholar] [CrossRef] [PubMed]
- Berthold, C.L.; Gocke, D.; Wood, M.D.; Leeper, F.J.; Pohl, M.; Schneider, G. Structure of the branched-chain keto acid decarboxylase (KdcA) from Lactococcus lactis provides insights into the structural basis for the chemoselective and enantioselective carboligation reaction. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Versees, W.; Spaepen, S.; Vanderleyden, J.; Steyaert, J. The crystal structure of phenylpyruvate decarboxylase from Azospirillum brasilense at 1.5 Å resolution. Implications for its catalytic and regulatory mechanism. FEBS J. 2007, 274, 2363–2375. [Google Scholar] [CrossRef] [PubMed]
- Hegeman, G.D. Benzoylformate decarboxylase (Pseudomonas putida). Methods Enzymol. 1970, 17, 674–678. [Google Scholar]
- Hegeman, G.D. Synthesis of the enzymes of the mandelate pathway by Pseudomonas putida I. Synthesis of enzymes by the wild type. J. Bacteriol. 1966, 91, 1140–1154. [Google Scholar] [PubMed]
- Kluger, R.; Tittmann, K. Thiamin diphosphate catalysis: Enzymic and nonenzymic covalent intermediates. Chem. Rev. 2008, 108, 1797–1833. [Google Scholar] [CrossRef] [PubMed]
- Hasson, M.S.; Muscate, A.; McLeish, M.J.; Polovnikova, L.S.; Gerlt, J.A.; Kenyon, G.L.; Petsko, G.A.; Ringe, D. The crystal structure of benzoylformate decarboxylase at 1.6 Å resolution: Diversity of catalytic residues in thiamin diphosphate-dependent enzymes. Biochemistry 1998, 37, 9918–9930. [Google Scholar] [CrossRef] [PubMed]
- Polovnikova, E.S.; McLeish, M.J.; Sergienko, E.A.; Burgner, J.T.; Anderson, N.L.; Bera, A.K.; Jordan, F.; Kenyon, G.L.; Hasson, M.S. Structural and kinetic analysis of catalysis by a thiamin diphosphate-dependent enzyme, benzoylformate decarboxylase. Biochemistry 2003, 42, 1820–1830. [Google Scholar] [CrossRef] [PubMed]
- Sergienko, E.A.; Wang, J.; Polovnikova, L.; Hasson, M.S.; McLeish, M.J.; Kenyon, G.L.; Jordan, F. Spectroscopic detection of transient thiamin diphosphate-bound intermediates on benzoylformate decarboxylase. Biochemistry 2000, 39, 13862–13869. [Google Scholar] [CrossRef] [PubMed]
- Schütz, A.; Golbik, R.; König, S.; Hübner, G.; Tittmann, K. Intermediates and transition states in thiamin diphosphate-dependent decarboxylases. A kinetic and NMR study on wild-type indolepyruvate decarboxylase and variants using indolepyruvate, benzoylformate, and pyruvate as substrates. Biochemistry 2005, 44, 6164–6179. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.; Kern, G.; Neef, H.; Tittmann, K.; Killenberg-Jabs, M.; Wikner, C.; Schneider, G.; Hübner, G. How thiamin diphosphate is activated in enzymes. Science 1997, 275, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.; Leeper, F.; Luisi, B. Structure, mechanism and catalytic duality of thiamine-dependent enzymes. Cell Mol. Life Sci. 2007, 64, 892–905. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Neumann, P.; Ficner, R.; Tittmann, K. Observation of a stable carbene at the active site of a thiamin enzyme. Nat. Chem. Biol. 2013, 9, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Tittmann, K.; Golbik, R.; Uhlemann, K.; Khailova, L.; Schneider, G.; Patel, M.; Jordan, F.; Chipman, D.M.; Duggleby, R.G.; Hübner, G. NMR analysis of covalent intermediates in thiamin diphosphate enzymes. Biochemistry 2003, 42, 7885–7891. [Google Scholar] [CrossRef] [PubMed]
- Bruning, M.; Berheide, M.; Meyer, D.; Golbik, R.; Bartunik, H.; Liese, A.; Tittmann, K. Structural and kinetic studies on native intermediates and an intermediate analogue in benzoylformate decarboxylase reveal a least motion mechanism with an unprecedented short-lived predecarboxylation intermediate. Biochemistry 2009, 48, 3258–3268. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.; Lingen, B.; Müller, M. Thiamin-diphosphate-dependent enzymes: New aspects of asymmetric C–C bond formation. Chem. Eur. J. 2002, 8, 5288–5295. [Google Scholar] [CrossRef]
- Fuganti, C.; Grasselli, P. Synthesis of the C14 chromanyl moiety of natural α-tocopherol (vitamin E). J. Chem. Soc. Chem. Commun. 1982, 4, 205–206. [Google Scholar] [CrossRef]
- Gala, D.; DiBenedetto, D.J.; Clark, J.E.; Murphy, B.L.; Schumacher, D.P.; Steinman, M. Preparations of antifungal Sch 42427/SM 9164: Preparative chromatographic resolution, and total asymmetric synthesis via enzymic preparation of chiral α-hydroxy arylketones. Tetrahedron Lett. 1996, 37, 611–614. [Google Scholar] [CrossRef]
- Siegert, P.; McLeish, M.J.; Baumann, M.; Iding, H.; Kneen, M.M.; Kenyon, G.L.; Pohl, M. Exchanging the substrate specificities of pyruvate decarboxylase from Zymomonas mobilis and benzoylformate decarboxylase from Pseudomonas putida. Protein Eng. Des. Sel. 2005, 18, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Yep, A.; McLeish, M.J. Engineering the substrate binding site of benzoylformate decarboxylase. Biochemistry 2009, 48, 8387–8395. [Google Scholar] [CrossRef] [PubMed]
- Tittmann, K.; Vyazmensky, M.; Hübner, G.; Barak, Z.; Chipman, D.M. The carboligation reaction of acetohydroxyacid synthase ii: Steady-state intermediate distributions in wild type and mutants by nmr. Proc Natl Acad Sci U S A 2005, 102, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Belenky, I.; Steinmetz, A.; Vyazmensky, M.; Barak, Z.; Tittmann, K.; Chipman, D.M. Many of the functional differences between acetohydroxyacid synthase (AHAS) isozyme I and other AHASs are a result of the rapid formation and breakdown of the covalent acetolactate-thiamin diphosphate adduct in AHAS I. FEBS J. 2012, 279, 1967–1979. [Google Scholar] [CrossRef] [PubMed]
- Joseph, E.; Wei, W.; Tittmann, K.; Jordan, F. Function of a conserved loop of the β-domain, not involved in thiamin diphosphate binding, in catalysis and substrate activation in yeast pyruvate decarboxylase. Biochemistry 2006, 45, 13517–13527. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, M.; Kaplun, A.; Vyazmensky, M.; Engel, S.; Golbik, R.; Tittmann, K.; Uhlemann, K.; Meshalkina, L.; Barak, Z.; Hübner, G.; et al. Monitoring the acetohydroxy acid synthase reaction and related carboligations by circular dichroism spectroscopy. Anal. Biochem. 2005, 342, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, A.; Vyazmensky, M.; Meyer, D.; Barak, Z.; Golbik, R.; Chipman, D.M.; Tittmann, K. Valine 375 and phenylalanine 109 confer affinity and specificity for pyruvate as donor substrate in acetohydroxy acid synthase isozyme II from Escherichia coli. Biochemistry 2010, 49, 5188–5199. [Google Scholar] [CrossRef] [PubMed]
- Vyazmensky, M.; Steinmetz, A.; Meyer, D.; Golbik, R.; Barak, Z.; Tittmann, K.; Chipman, D.M. Significant catalytic roles for Glu47 and Gln110 in all four of the C–C bond-making and -breaking steps of the reactions of acetohydroxyacid synthase II. Biochemistry 2011, 50, 3250–3260. [Google Scholar] [CrossRef] [PubMed]
- Beigi, M.; Loschonsky, S.; Lehwald, P.; Brecht, V.; Andrade, S.L.A.; Leeper, F.J.; Hummel, W.; Muller, M. α-hydroxy-β-keto acid rearrangement-decarboxylation: Impact on thiamine diphosphate-dependent enzymatic transformations. Org. Biomol. Chem. 2013, 11, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Andrews, F.H.; Tom, A.R.; Gunderman, P.R.; Novak, W.R.P.; McLeish, M.J. A bulky hydrophobic residue is not required to maintain the V-conformation of enzyme-bound thiamin diphosphate. Biochemistry 2013, 52, 3028–3030. [Google Scholar] [CrossRef] [PubMed]
- Berthold, C.L.; Moussatche, P.; Richards, N.G.; Lindqvist, Y. Structural basis for activation of the thiamin diphosphate-dependent enzyme oxalyl-coA decarboxylase by adenosine diphosphate. J. Biol. Chem. 2005, 280, 41645–41654. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.K.; Polovnikova, L.S.; Roestamadji, J.; Widlanski, T.S.; Kenyon, G.L.; McLeish, M.J.; Hasson, M.S. Mechanism-based inactivation of benzoylformate decarboxylase, a thiamin diphosphate-dependent enzyme. J. Am. Chem. Soc. 2007, 129, 4120–4121. [Google Scholar] [CrossRef] [PubMed]
- Brandt, G.S.; Kneen, M.M.; Chakraborty, S.; Baykal, A.T.; Nemeria, N.; Yep, A.; Ruby, D.I.; Petsko, G.A.; Kenyon, G.L.; McLeish, M.J.; et al. Snapshot of a reaction intermediate: Analysis of benzoylformate decarboxylase in complex with a benzoylphosphonate inhibitor. Biochemistry 2009, 48, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Neumann, P.; Parthier, C.; Friedemann, R.; Nemeria, N.; Jordan, F.; Tittmann, K. Double duty for a conserved glutamate in pyruvate decarboxylase: Evidence of the participation in stereoelectronically controlled decarboxylation and in protonation of the nascent carbanion/enamine intermediate. Biochemistry 2010, 49, 8197–8212. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Nemeria, N.S.; Balakrishnan, A.; Brandt, G.S.; Kneen, M.M.; Yep, A.; McLeish, M.J.; Kenyon, G.L.; Petsko, G.A.; Ringe, D.; et al. Detection and time course of formation of major thiamin diphosphate-bound covalent intermediates derived from a chromophoric substrate analogue on benzoylformate decarboxylase. Biochemistry 2009, 48, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Andrews, F.H.; Rogers, M.P.; Paul, L.N.; McLeish, M.J. Perturbation of the monomer-monomer interfaces of the benzoylformate decarboxylase tetramer. Biochemistry 2014, 53, 4358–4367. [Google Scholar] [CrossRef] [PubMed]
- Turano, A.; Furey, W.; Pletcher, J.; Sax, M.; Pike, D.; Kluger, R. Synthesis and crystal structure of an analog of 2-(alpha-lactyl)thiamin, racemic methyl 2-hydroxy-2-(2-thiamin)ethylphosphonate chloride trihydrate. A conformation for a least-motion, maximum-overlap mechanism for thiamin catalysis. J. Am. Chem. Soc. 1982, 104, 3089–3095. [Google Scholar] [CrossRef]
- Wille, G.; Meyer, D.; Steinmetz, A.; Hinze, E.; Golbik, R.; Tittmann, K. The catalytic cycle of a thiamin diphosphate enzyme examined by cryocrystallography. Nat. Chem. Biol. 2006, 2, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.; Arjunan, P.; Furey, W.; Jordan, F. A dynamic loop at the active center of the Escherichia coli pyruvate dehydrogenase complex e1 component modulates substrate utilization and chemical communication with the E2 component. J. Biol. Chem. 2007, 282, 28106–28116. [Google Scholar] [CrossRef] [PubMed]
- Arjunan, P.; Sax, M.; Brunskill, A.; Chandrasekhar, K.; Nemeria, N.; Zhang, S.; Jordan, F.; Furey, W. A thiamin-bound, pre-decarboxylation reaction intermediate analogue in the pyruvate dehydrogenase E1 subunit induces large scale disorder-to-order transformations in the enzyme and reveals novel structural features in the covalently bound adduct. J. Biol. Chem. 2006, 281, 15296–15303. [Google Scholar] [CrossRef] [PubMed]
- Demir, A.S.; Dünnwald, T.; Iding, H.; Pohl, M.; Müller, M. Asymmetric benzoin reaction catalyzed by benzoylformate decarboxylase. Tetrahedron Asymmetry 1999, 10, 4769–4774. [Google Scholar] [CrossRef]
- Dünkelmann, P.; Kolter-Jung, D.; Nitsche, A.; Demir, A.S.; Siegert, P.; Lingen, B.; Baumann, M.; Pohl, M.; Müller, M. Development of a donor-acceptor concept for enzymatic cross-coupling reactions of aldehydes: The first asymmetric cross-benzoin condensation. J. Am. Chem. Soc. 2002, 124, 12084–12085. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.; Sprenger, G.A.; Müller, M. A new perspective on thiamine catalysis. Curr. Opin. Biotechnol. 2004, 15, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Walter, L.; Kolter, G.; Pohl, M.; Müller, M.; Tittmann, K. Conversion of pyruvate decarboxylase into an enantioselective carboligase with biosynthetic potential. J. Am. Chem. Soc. 2011, 133, 3609–3616. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Bailey, S. The CCP4 suite—Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., 3rd; Snoeyink, J.; Richardson, J.S.; et al. Molprobity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. Molprobity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | wtBFDC | BFDC T377L/A460Y | ScPDC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Km (mM) | kcat (s−1) | kcat/Km (mM−1·s−1) | Km (mM) | kcat (s−1) | kcat/Km (mM−1·s−1) | S0.5 (mM) | kcat (s−1) | kcat/S0.5 (mM−1·s−1) | |
| Benzoylformate | 0.27 ± 0.02 | 320 ± 4 | 1180 | 0.45 ± 0.04 | 15 ± 1 | 34 | NAD b | NAD b | NAD b |
| Pyruvate | - | - | 0.01 | 4.6 ± 0.3 | 3.3 ± 0.1 | 0.72 | 1.3 ± 0.1 | 60 ± 2 | 45 |
| 4-Methylthio-2-ketobutanoate | 1.3 ± 0.2 | 4.5 ± 0.3 | 3.5 | 0.14 ± 0.01 | 4.6 ± 0.1 | 33 | 0.80 ± 0.05 | 5.2 ± 0.3 | 6.6 |
| 2-Ketohexanoate | 5.3 ± 0.3 | 4.1 ± 0.1 | 1.3 | 0.14 ± 0.01 | 5.9 ± 0.2 | 42 | 1.5 ± 0.1 | 15 ± 1 | 9.8 |
| 2-Ketopentanoate | 11 ± 1 | 6.0 ± 0.5 | 1.8 | 0.15 ± 0.01 | 1.1 ± 0.1 | 7.5 | 1.1 ± 0.1 | 27 ± 2 | 24 |
| 4-Methyl-2-ketopentanoate | 9.6 c | 11 c | 1.1 c | 0.14 ± 0.02 | 2.7 ± 0.2 | 20 | 5.0 ± 0.2 | 10 ± 1 | 2.0 |
| 3-Methyl-2-ketopentanoate | - | - | 1.6 b | 1.2 ± 0.1 | 16 ± 1 | 13 | 8.7 ± 0.5 | 22 ± 1 | 2.5 |
| 2-Ketobutanoate | 4.1 ± 0.1 | 7.5 ± 0.6 | 0.5 | 0.39 ± 0.02 | 6.0 ± 0.1 | 16 | 0.49 ± 0.02 | 25 ± 1 | 49 |
| 3-Methyl-2-ketobutanoate | - | - | 2.6 c,d | 0.77 ± 0.06 | 12 ± 1 | 15 | 0.73 ± 0.03 | 6.3 ± 0.2 | 8.6 |
| Phenylpyruvate | 0.1 c,d | 0.82 ± 0.06 | 2.0 ± 0.1 | 2.5 | 0.23 ± 0.01 | 0.87 ± 0.02 | 3.7 | ||
| Enzyme Variant | Substrate | kcat | C–C (k2′) | Decarboxylation (k3′) | Release (k4′) |
|---|---|---|---|---|---|
| wtBFDC a | BF | 450 | 500 | 16,000 | 2400 |
| Pyruvate | <0.05 b | NDc | ND c | ND c | |
| BFDC T377L/A460Y | BF | 15 | 40 | 500 | 24 |
| Pyruvate | 3.3 | 6 | 24 | 10 | |
| ScPDC d | Pyruvate | 38 | 294 | 105 | 105 |
| ZmPDC e | Pyruvate | 150 | 2650 | 397 | 265 |
| Data Collection Statistics BFDC T377L/A460Y (PDB 4MZX) | |
| Beam line | APS, GM/CA-CAT, 23 ID-B |
| Wavelength | 1.03 Å |
| Space group | I222 |
| Cell constants | a = 80.98 Å; b = 95.56 Å; c = 137.2 Å; α = β = γ = 90˚ |
| Total reflections | 75885 |
| Unique reflections | 75885 |
| Resolution limit (Å) | 1.56 |
| Completeness (%) | 99.8 (100) |
| Redundancy | 7.3 (7.2) |
| I/σI | 27.5 (7.3) |
| Rmerge (%) | 5.8 (29) |
| Refinement statistics | |
| Resolution range (Å) | 1.56–19.71 |
| Rfree test set size | 2000 |
| Rcryst (%) | 11.7 |
| Rfree (%) | 15.0 |
| No. Atoms | |
| Total | 4547 |
| Protein | 4050 |
| Ligand | 41 |
| Water | 456 |
| Ramachandran | |
| Favored | 98 |
| Allowed | 2 |
| Outliers | 0 |
| B-factors | |
| Protein | 15.1 |
| Ligand | 18.4 |
| Solvent | 26.4 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.017 |
| Bond angles (˚) | 1.50 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrews, F.H.; Wechsler, C.; Rogers, M.P.; Meyer, D.; Tittmann, K.; McLeish, M.J. Mechanistic and Structural Insight to an Evolved Benzoylformate Decarboxylase with Enhanced Pyruvate Decarboxylase Activity. Catalysts 2016, 6, 190. https://doi.org/10.3390/catal6120190
Andrews FH, Wechsler C, Rogers MP, Meyer D, Tittmann K, McLeish MJ. Mechanistic and Structural Insight to an Evolved Benzoylformate Decarboxylase with Enhanced Pyruvate Decarboxylase Activity. Catalysts. 2016; 6(12):190. https://doi.org/10.3390/catal6120190
Chicago/Turabian StyleAndrews, Forest H., Cindy Wechsler, Megan P. Rogers, Danilo Meyer, Kai Tittmann, and Michael J. McLeish. 2016. "Mechanistic and Structural Insight to an Evolved Benzoylformate Decarboxylase with Enhanced Pyruvate Decarboxylase Activity" Catalysts 6, no. 12: 190. https://doi.org/10.3390/catal6120190
APA StyleAndrews, F. H., Wechsler, C., Rogers, M. P., Meyer, D., Tittmann, K., & McLeish, M. J. (2016). Mechanistic and Structural Insight to an Evolved Benzoylformate Decarboxylase with Enhanced Pyruvate Decarboxylase Activity. Catalysts, 6(12), 190. https://doi.org/10.3390/catal6120190