
Electrocatalytic Activity and Durability of Pt-Decorated Non-Covalently Functionalized Graphitic Structures



Abstract
:
1. Introduction
2. Results and Discussion
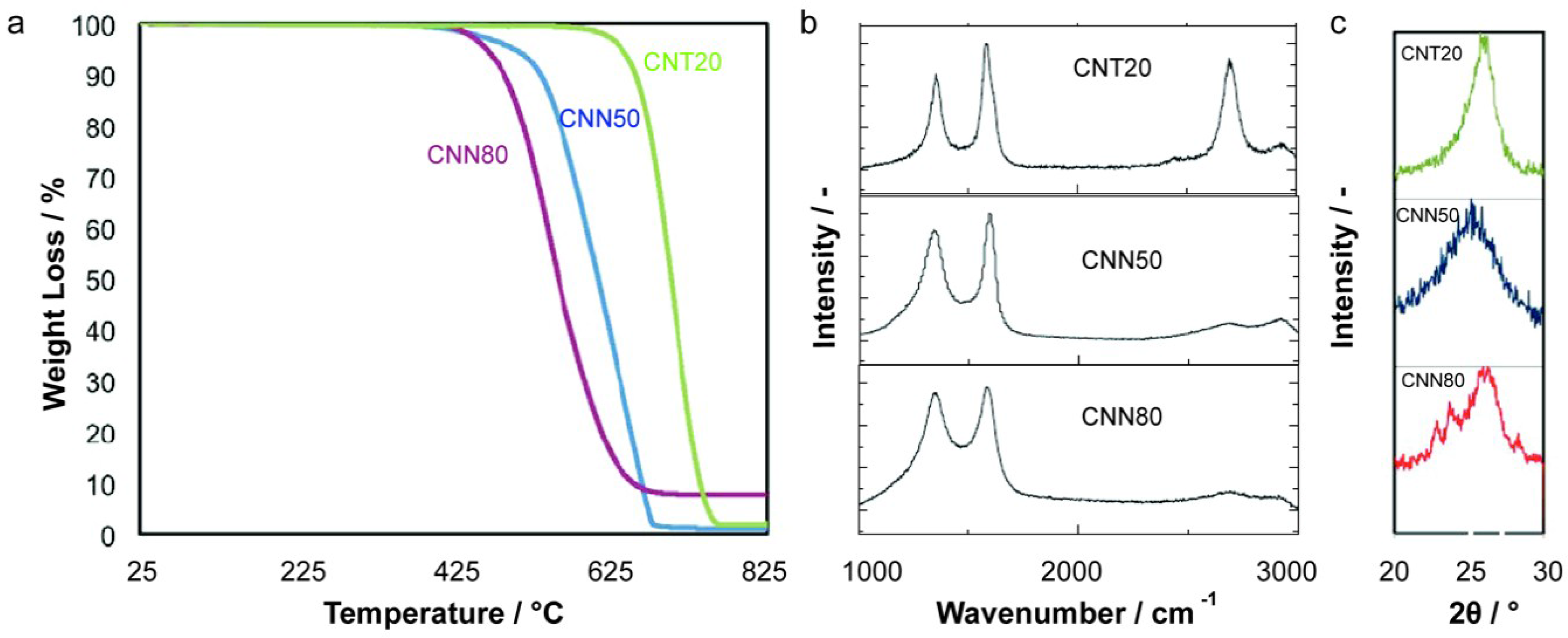
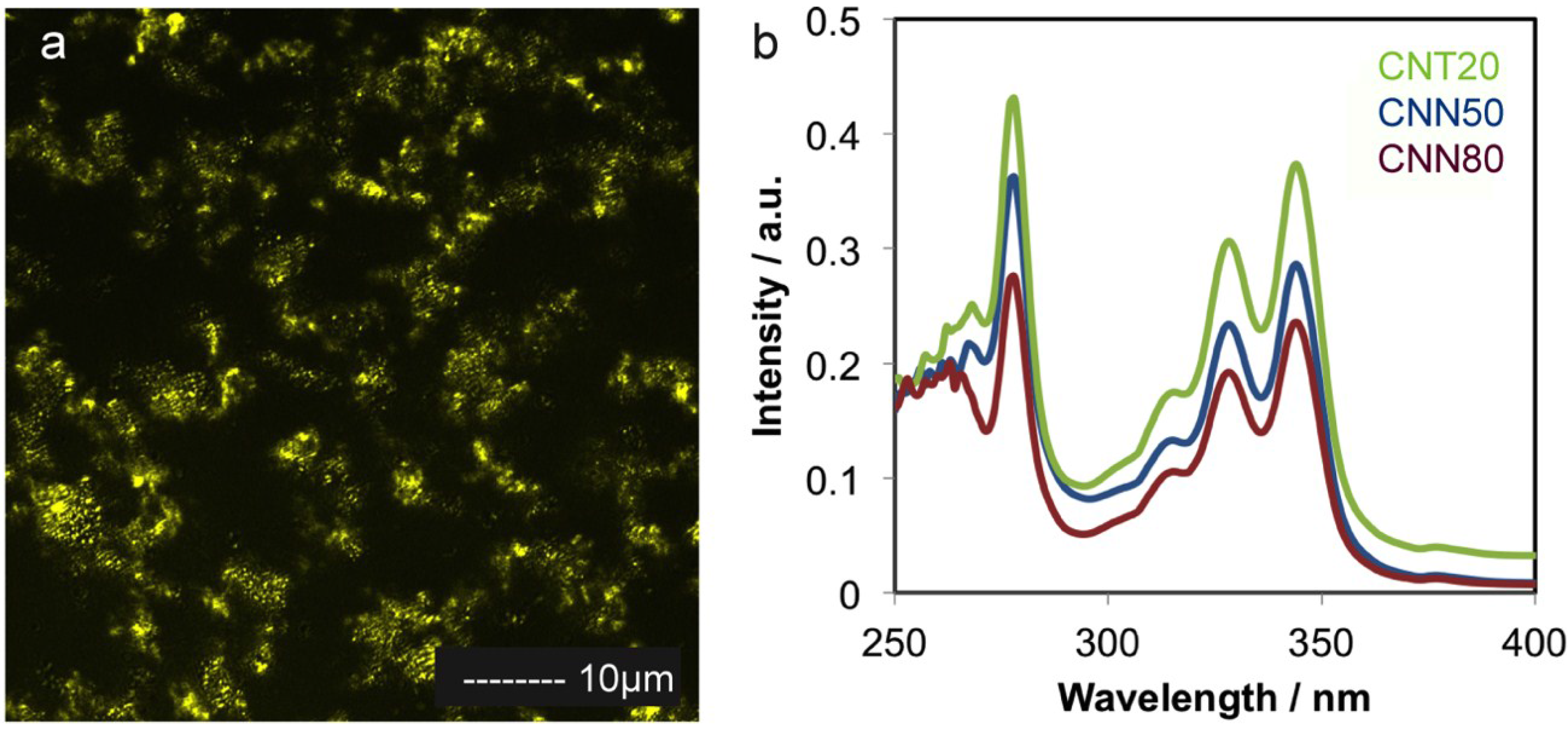
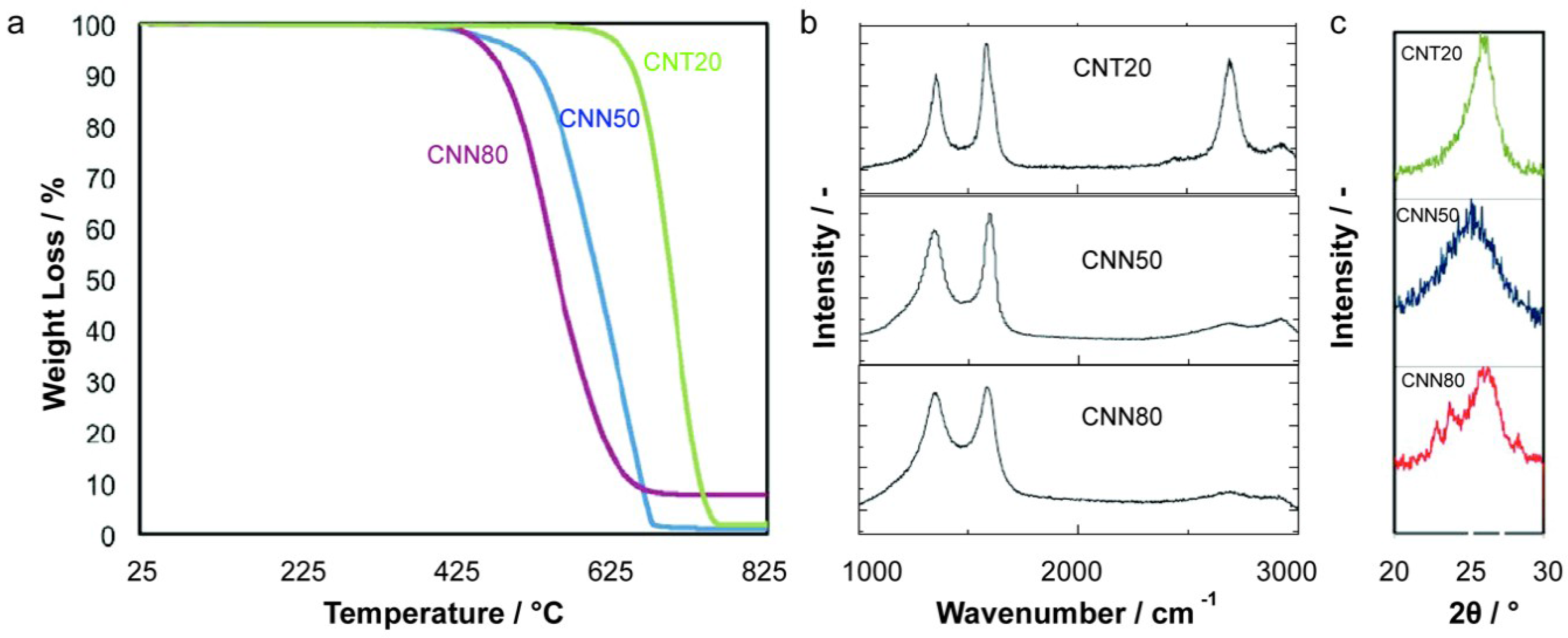
2.1. Carbon Support Synthesis, Functionalization and Characterization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Average Diameter/nm | Specific Surface Area/m2 g−1 | Tox/°C | ID/IG | PCA/% |
|---|---|---|---|---|---|
| CNT20 | 20 | 110 | 700 | 0.7 | 7.5 |
| CNN50 | 50 | 63 | 630 | 0.9 | 8.3 |
| CNN80 | 80 | 39 | 610 | 1 | 9 |
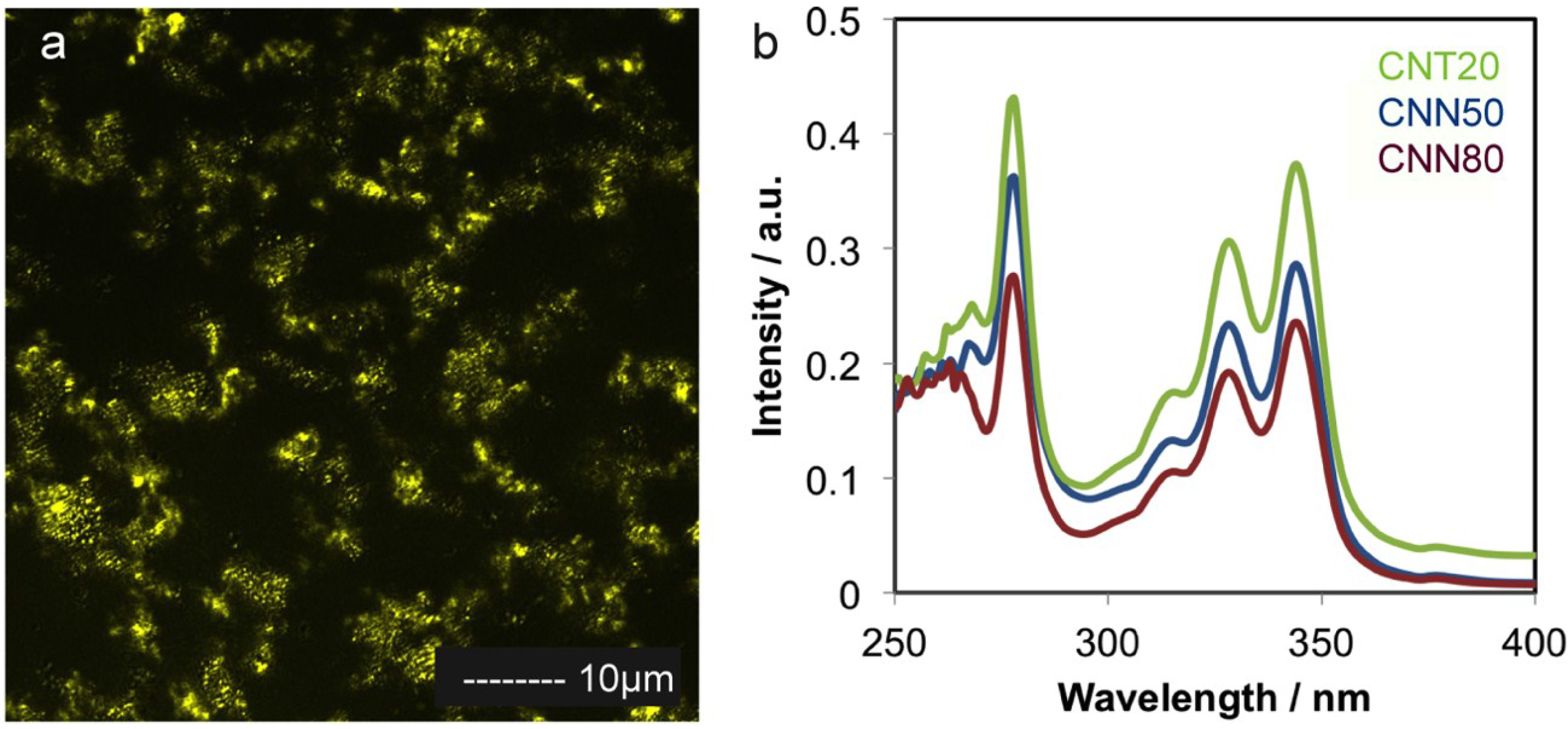
2.2. Platinum Deposition
| Sample | Pt Loading/% | Particle Size TEM/nm | Particle Size XRD/nm |
|---|---|---|---|
| CNT20 | 7 | 2.3 ± 0.2 | 3.9 |
| CNN50 | 9 | 2.3 ± 0.2 | 3.1 |
| CNN80 | 11 | 2.3 ± 0.2 | 2.4 |

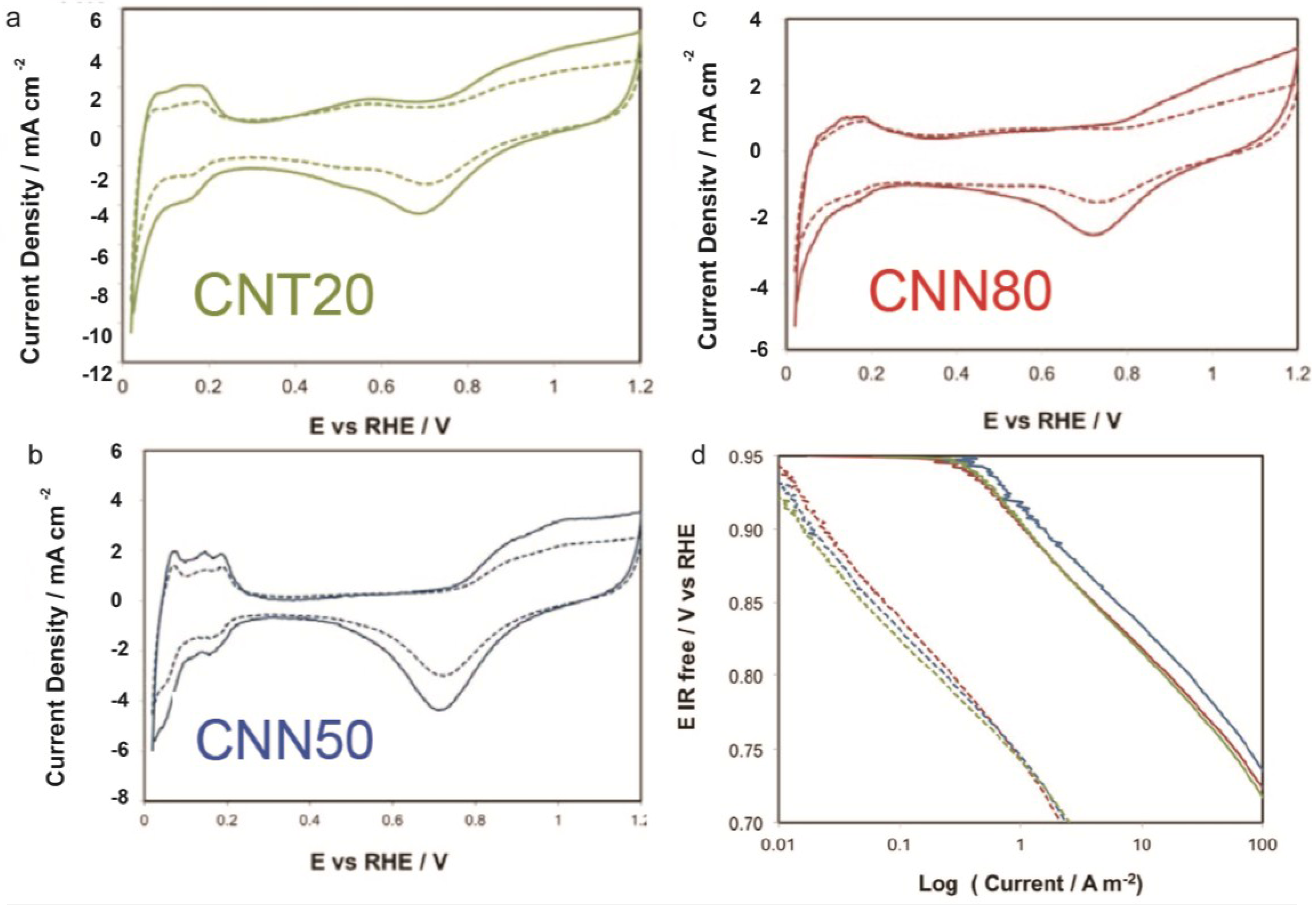
2.3. Electrochemical Characterization
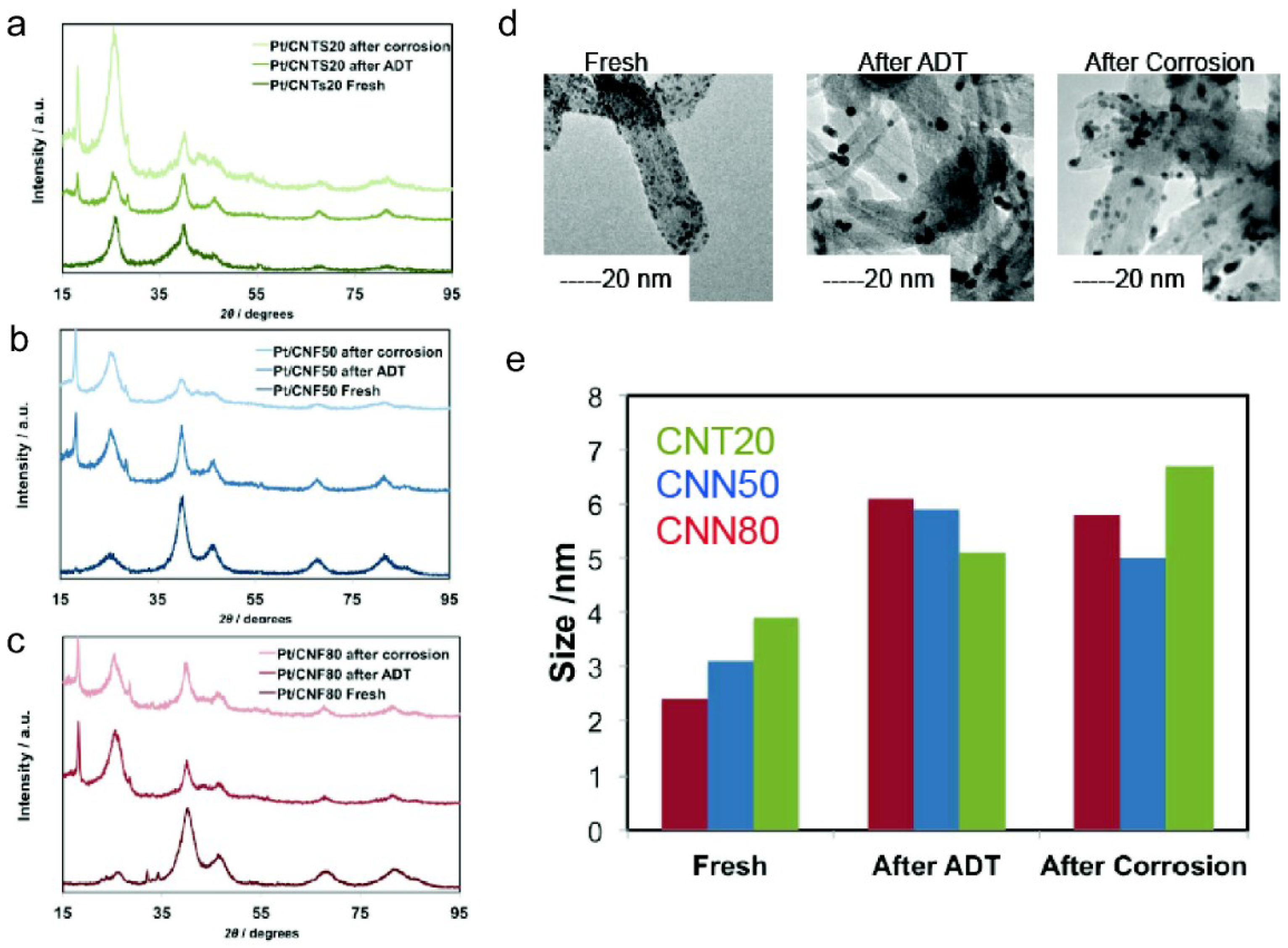
2.3.1. ECSA and ORR Activity before and after ADT
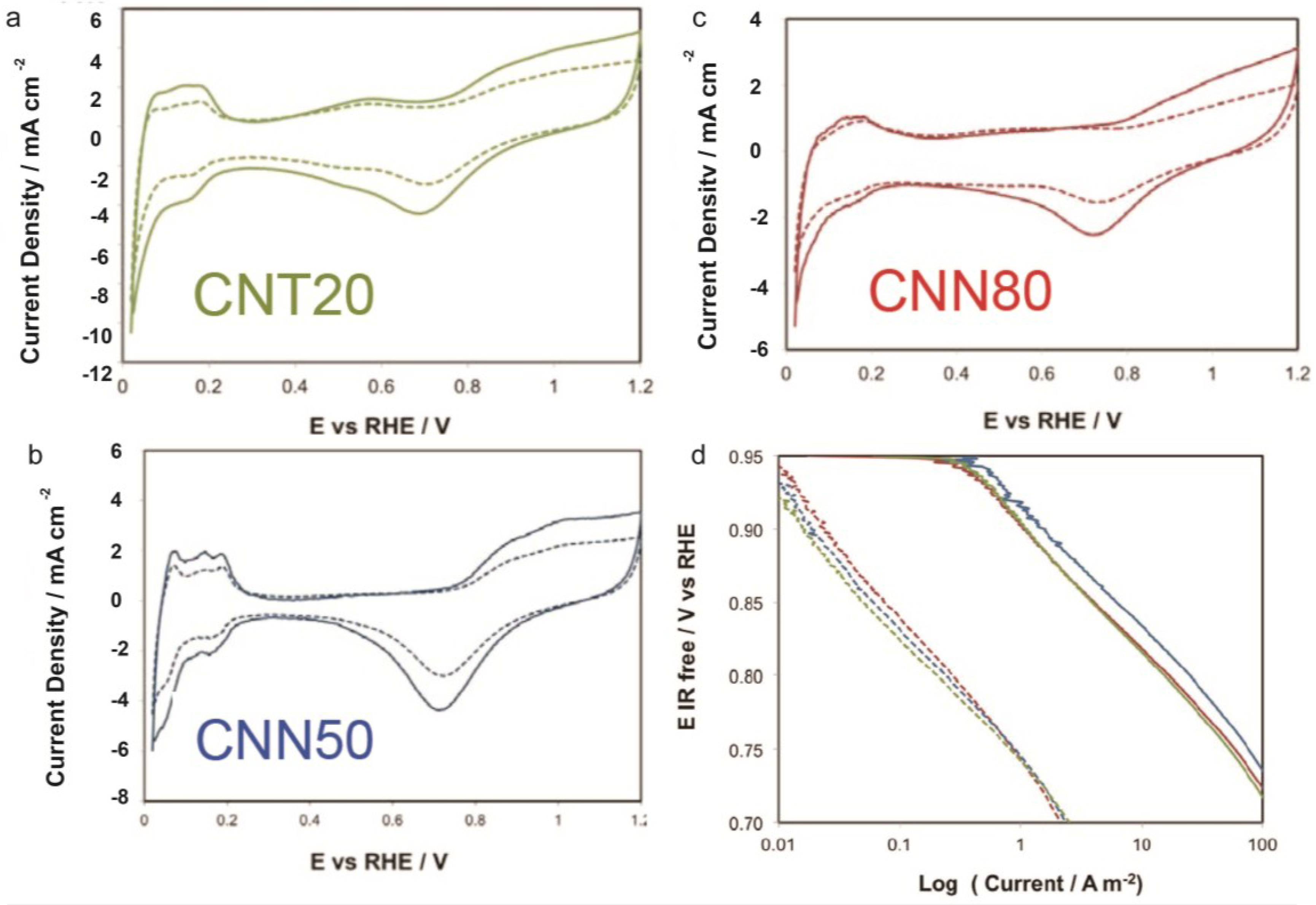
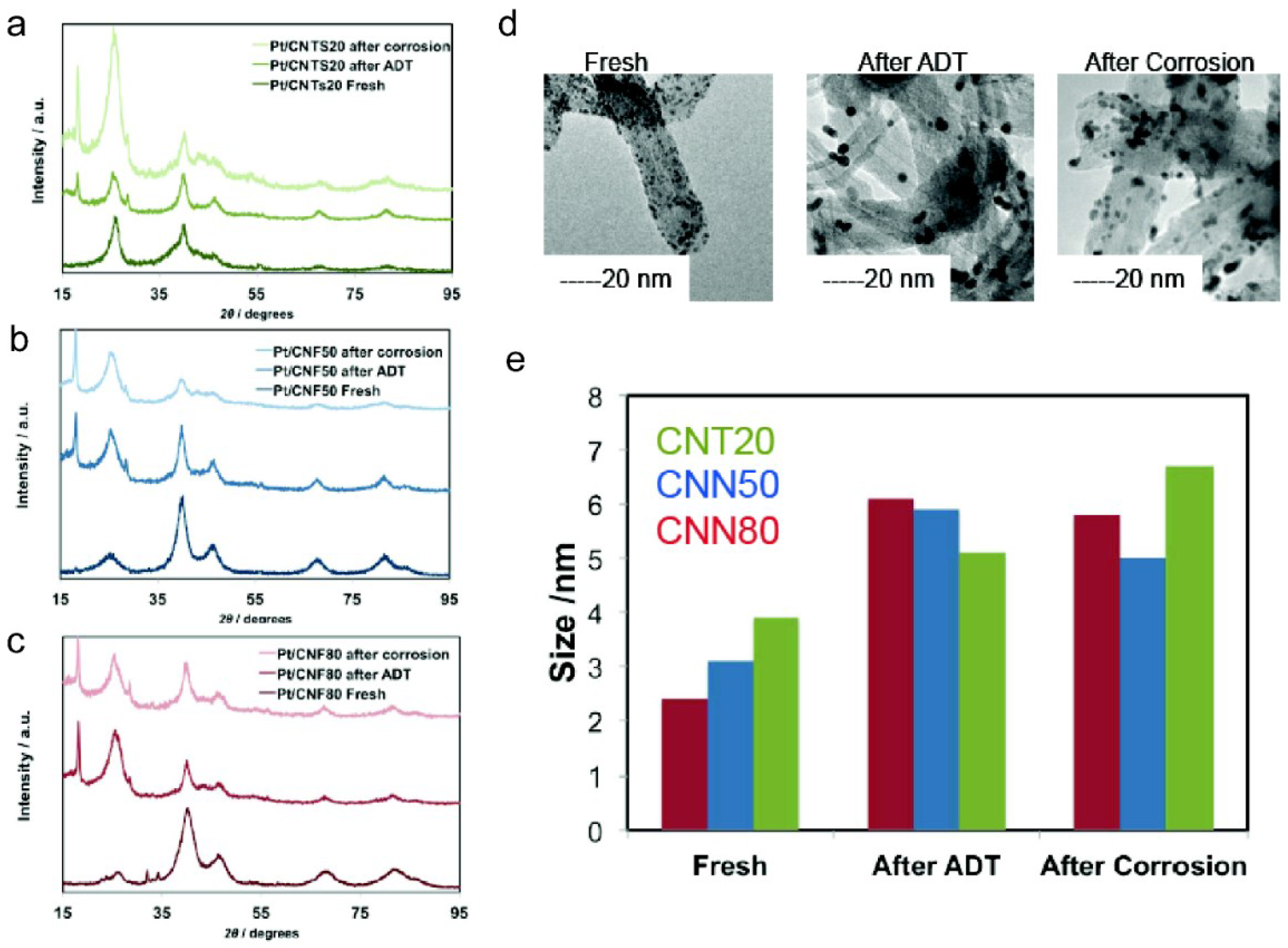
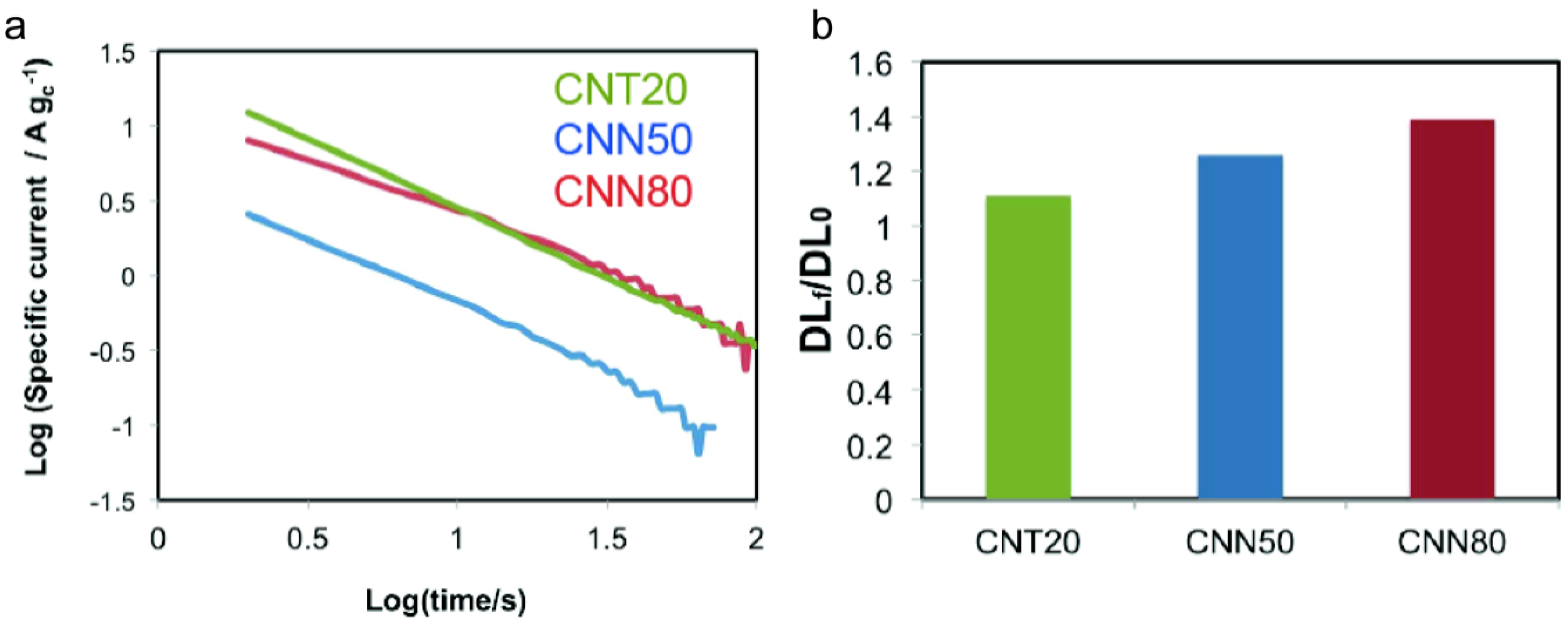
| Sample | ECSA/m2 g−1 | ECSA after ADT/m2 g−1 | ECSA Loss with ADT/% | Particle Size XRD after ADT/nm | DLC0/F g−1 | DLCf/DLC0 | k | n | Particle Size XRD after Corrosion/nm |
|---|---|---|---|---|---|---|---|---|---|
| CNT20 | 44.2 | 22.4 | −49 | 5.1 | 99 | 1.11 | 30.97 | 0.88 | 6.7 |
| CNN50 | 52.1 | 30.2 | −42 | 5.9 | 66 | 1.26 | 6.46 | 0.98 | 5 |
| CNN80 | 58.3 | 32.5 | −44 | 6.1 | 77 | 1.39 | 30.7 | 0.92 | 5.8 |
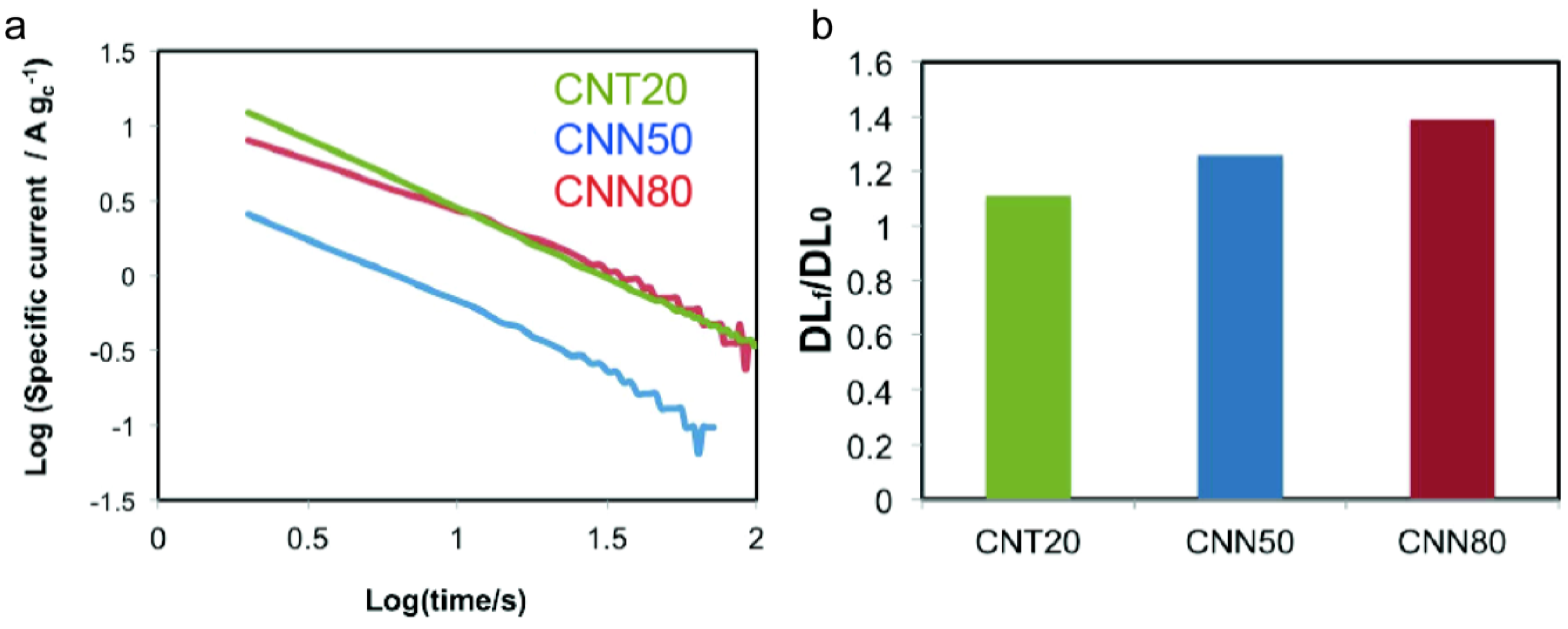
2.3.2. Corrosion Tests
3. Experimental Section
3.1. Chemicals
3.2. Carbon Support Synthesis and Functionalization
3.2.1. Preparation of CNNs
3.2.2. PCA Functionalization of Graphitic Carbon Supports
3.2.3. Catalyst Deposition
3.3. Supports and Electrocatalyst Characterization
3.4. Electrochemical Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dillon, R.; Srinivasan, S.; Aricò, A.S.; Antonucci, V. International activities in DMFC R&D: Status of technologies and potential applications. J. Power Sources 2004, 127, 112–126. [Google Scholar]
- Specchia, S.; Francia, C.; Spinelli, P. Polymer electrolyte membrane fuel cells. In Electrochemical Technologies for Energy Storage and Conversion; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 601–670. [Google Scholar]
- Aricò, A.S.; Baglio, V.; Antonucci, V. Direct Methanol Fuel Cells; Nova Science Publishers: Hauppauge, NY, USA, 2010. [Google Scholar]
- De Bruijn, F.A.; Dam, V.A.T.; Janssen, G.J.M. Review: Durability and degradation issues of PEM fuel cell components. Fuel Cells 2008, 8, 3–22. [Google Scholar] [CrossRef]
- Zeng, J.; Francia, C.; Dumitrescu, M.A.; Videla, A.H.A.M.; Ijeri, V.S.; Specchia, S.; Spinelli, P. Electrochemical performance of Pt-based catalysts supported on different ordered mesoporous carbons (Pt/OMCs) for oxygen reduction reaction. Ind. Eng. Chem. Res. 2011, 51, 7500–7509. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Galvez, M.E.; Calvillo, L.; Alegre, C.; Sebastian, D.; Suelves, I.; Perez-Rodriguez, S.; Celorrio, V.; Pastor, E.; Pardo, J.I.; Moliner, R.; et al. Nanostructured carbon materials as supports in the preparation of direct methanol fuel cell electrocatalysts. Catalysts 2013, 3, 671–682. [Google Scholar] [CrossRef]
- Ferrandon, M.; Wang, X.; Kropf, A.J.; Myers, D.J.; Wu, G.; Johnston, C.M.; Zelenay, P. Stability of iron species in heat-treated polyaniline-iron-carbon polymer electrolyte fuel cell cathode catalysts. Electrochim. Acta 2013, 110, 282–291. [Google Scholar] [CrossRef]
- Borup, R.; Meyers, J.; Pivovar, B.; Kim, Y.S.; Mukundan, R.; Garland, N.; Myers, D.; Wilson, M.; Garzon, F.; Wood, D.; et al. Scientific aspects of polymer electrolyte fuel cell durability and degradation. Chem. Rev. 2007, 107, 3904–3951. [Google Scholar] [CrossRef] [PubMed]
- Siroma, Z.; Ishii, K.; Yasuda, K.; Miyazaki, Y.; Inaba, M.; Tasaka, A. Imaging of highly oriented pyrolytic graphite corrosion accelerated by Pt particles. Electrochem. Commun. 2005, 7, 1153–1156. [Google Scholar] [CrossRef]
- Negro, E.; Dieci, M.; Sordi, D.; Kowlgi, K.; Makkee, M.; Koper, G.J.M. High yield, controlled synthesis of graphitic networks from dense micro emulsions. Chem. Commun. 2014, 50, 11848–11851. [Google Scholar] [CrossRef] [PubMed]
- Negro, E.; Latsuzbaia, R.; Dieci, M.; Boshuizen, I.; Koper, G.J.M. Pt electrodeposited over carbon nano-networks grown on carbon paper as durable catalyst for PEM fuel cells. Appl. Catal. B 2015, 166–167, 155–165. [Google Scholar] [CrossRef]
- Negro, E.; Videla, A.H.A.M.; Baglio, V.; Aricò, A.S.; Specchia, S.; Koper, G.J.M. Fe-N supported on graphitic carbon nano-networks grown from cobalt as oxygen reduction catalysts for low-temperature fuel cells. Appl. Catal. B 2015, 166–167, 75–83. [Google Scholar] [CrossRef]
- Negro, E.; Vries, M.A.D.; Latsuzbaia, R.; Koper, G.J.M. Networked graphitic structures as durable catalyst support for PEM electrodes. Fuel Cells 2014, 14, 350–356. [Google Scholar] [CrossRef]
- Sebastián, D.; Lázaro, M.J.; Suelves, I.; Moliner, R.; Baglio, V.; Stassi, A.; Aricò, A.S. The influence of carbon nanofiber support properties on the oxygen reduction behavior in proton conducting electrolyte-based direct methanol fuel cells. Int. J. Hydrogen Energy 2012, 37, 6253–6260. [Google Scholar] [CrossRef]
- Sebastián, D.; Ruíz, A.G.; Suelves, I.; Moliner, R.; Lázaro, M.J.; Baglio, V.; Stassi, A.; Aricò, A.S. Enhanced oxygen reduction activity and durability of Pt catalysts supported on carbon nanofibers. Appl. Catal. B 2012, 115–116, 269–275. [Google Scholar] [CrossRef]
- Sebastián, D.; Suelves, I.; Moliner, R.; Lázaro, M.J.; Stassi, A.; Baglio, V.; Aricò, A.S. Optimizing the synthesis of carbon nanofiber based electrocatalysts for fuel cells. Appl. Catal. B 2013, 132–133, 22–27. [Google Scholar] [CrossRef]
- Carrera-Cerritos, R.; Baglio, V.; Aricò, A.S.; Ledesma-García, J.; Sgroi, M.F.; Pullini, D.; Pruna, A.J.; Mataix, D.B.; Fuentes-Ramírez, R.; Arriaga, L.G. Improved Pd electro-catalysis for oxygen reduction reaction in direct methanol fuel cell by reduced graphene oxide. Appl. Catal. B 2014, 144, 554–560. [Google Scholar] [CrossRef]
- Kowlgi, K.N.K.; Koper, G.J.M.; van Raalten, R.A.D. Carbon nanostructures and networks produced by chemical vapor deposition. US Patent 20130244023 A1, 19 September 2013. [Google Scholar]
- Kowlgi, K.; Lafont, U.; Rappolt, M.; Koper, G. Uniform metal nanoparticles produced at high yield in dense microemulsions. J. Colloid Interf. Sci. 2012, 372, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Latsuzbaia, R.; Negro, E.; Koper, G. Bicontinuous microemulsions for high yield, wet synthesis of ultrafine nanoparticles: A general approach. Faraday Discuss. 2015, 181, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Negro, E.; Latsuzbaia, R.; Koper, G.J.M. Bicontinuous microemulsions for high yield wet synthesis of ultrafine platinum nanoparticles: Effect of precursors and kinetics. Langmuir 2014, 30, 8300–8307. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.S.; Kundu, A. Functionalizing carbon nanotubes for proton exchange membrane fuel cells electrode. J. Power Sources 2010, 195, 6255–6261. [Google Scholar] [CrossRef]
- Wu, H.; Wexler, D.; Wang, G.; Liu, H. Pt/C catalysts using different carbon supports for the cathode of PEM fuel cells. Adv. Sci. Lett. 2011, 4, 115–120. [Google Scholar] [CrossRef]
- Xu, C.; Chen, J.; Cui, Y.; Han, Q.; Choo, H.; Liaw, P.K.; Wu, D. Influence of the surface treatment on the deposition of platinum nanoparticles on the carbon nanotubes. Adv. Eng. Mater. 2006, 8, 73–77. [Google Scholar] [CrossRef]
- Xing, Y. Synthesis and electrochemical characterization of uniformly-dispersed high loading Pt nanoparticles on sonochemically-treated carbon nanotubes. J. Phys. Chem. B 2004, 108, 19255–19259. [Google Scholar] [CrossRef]
- Latsuzbaia, R.; Negro, E.; Koper, G.J.M. Environmentally friendly carbon-preserving recovery of noble metals from supported fuel cell catalysts. ChemSusChem 2015, 8, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.-S.; Kim, H. Efficient synthesis of Pt nanoparticles supported on hydrophobic graphitized carbon nanofibers for electrocatalysts using noncovalent functionalization. Adv. Funct. Mater. 2011, 21, 3954–3960. [Google Scholar] [CrossRef]
- Esmaeilifar, A.; Rowshanzamir, S.; Eikani, M.H.; Ghazanfari, E. Synthesis methods of low-Pt-loading electrocatalysts for proton exchange membrane fuel cell systems. Energy 2010, 35, 3941–3957. [Google Scholar] [CrossRef]
- Zou, J.; Zeng, X.; Xiong, X.; Tang, H.; Li, L.; Liu, Q.; Li, Z. Preparation of vapor grown carbon fibers by microwave pyrolysis chemical vapor deposition. Carbon 2007, 45, 828–832. [Google Scholar] [CrossRef]
- Stassi, A.; Modica, E.; Antonucci, V.; Aricò, A.S. A half cell study of performance and degradation of oxygen reduction catalysts for application in low temperature fuel cells. Fuel Cells 2009, 9, 201–208. [Google Scholar] [CrossRef]
- Curnick, O.J.; Mendes, P.M.; Pollet, B.G. Enhanced durability of a Pt/C electrocatalyst derived from nafion-stabilised colloidal platinum nanoparticles. Electrochem. Commun. 2010, 12, 1017–1020. [Google Scholar] [CrossRef]
- Stamatin, S.N.; Borghei, M.; Dhiman, R.; Andersen, S.M.; Ruiz, V.; Kauppinen, E.; Skou, E.M. Activity and stability studies of platinized multi-walled carbon nanotubes as fuel cell electrocatalysts. Appl. Catal. B 2015, 162, 289–299. [Google Scholar] [CrossRef]
- Oh, H.-S.; Oh, J.-G.; Kim, H. Modification of polyol process for synthesis of highly platinum loaded platinum-carbon catalysts for fuel cells. J. Power Sources 2008, 183, 600–603. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negro, E.; Stassi, A.; Baglio, V.; Aricò, A.S.; Koper, G.J.M. Electrocatalytic Activity and Durability of Pt-Decorated Non-Covalently Functionalized Graphitic Structures. Catalysts 2015, 5, 1622-1635. https://doi.org/10.3390/catal5031622
Negro E, Stassi A, Baglio V, Aricò AS, Koper GJM. Electrocatalytic Activity and Durability of Pt-Decorated Non-Covalently Functionalized Graphitic Structures. Catalysts. 2015; 5(3):1622-1635. https://doi.org/10.3390/catal5031622
Chicago/Turabian StyleNegro, Emanuela, Alessandro Stassi, Vincenzo Baglio, Antonino S. Aricò, and Ger J.M. Koper. 2015. "Electrocatalytic Activity and Durability of Pt-Decorated Non-Covalently Functionalized Graphitic Structures" Catalysts 5, no. 3: 1622-1635. https://doi.org/10.3390/catal5031622
APA StyleNegro, E., Stassi, A., Baglio, V., Aricò, A. S., & Koper, G. J. M. (2015). Electrocatalytic Activity and Durability of Pt-Decorated Non-Covalently Functionalized Graphitic Structures. Catalysts, 5(3), 1622-1635. https://doi.org/10.3390/catal5031622