
CNT-TiO2−δ Composites for Improved Co-Catalyst Dispersion and Stabilized Photocatalytic Hydrogen Production


Abstract
:1. Introduction
2. Results and Discussion
2.1. Physicochemical Properties
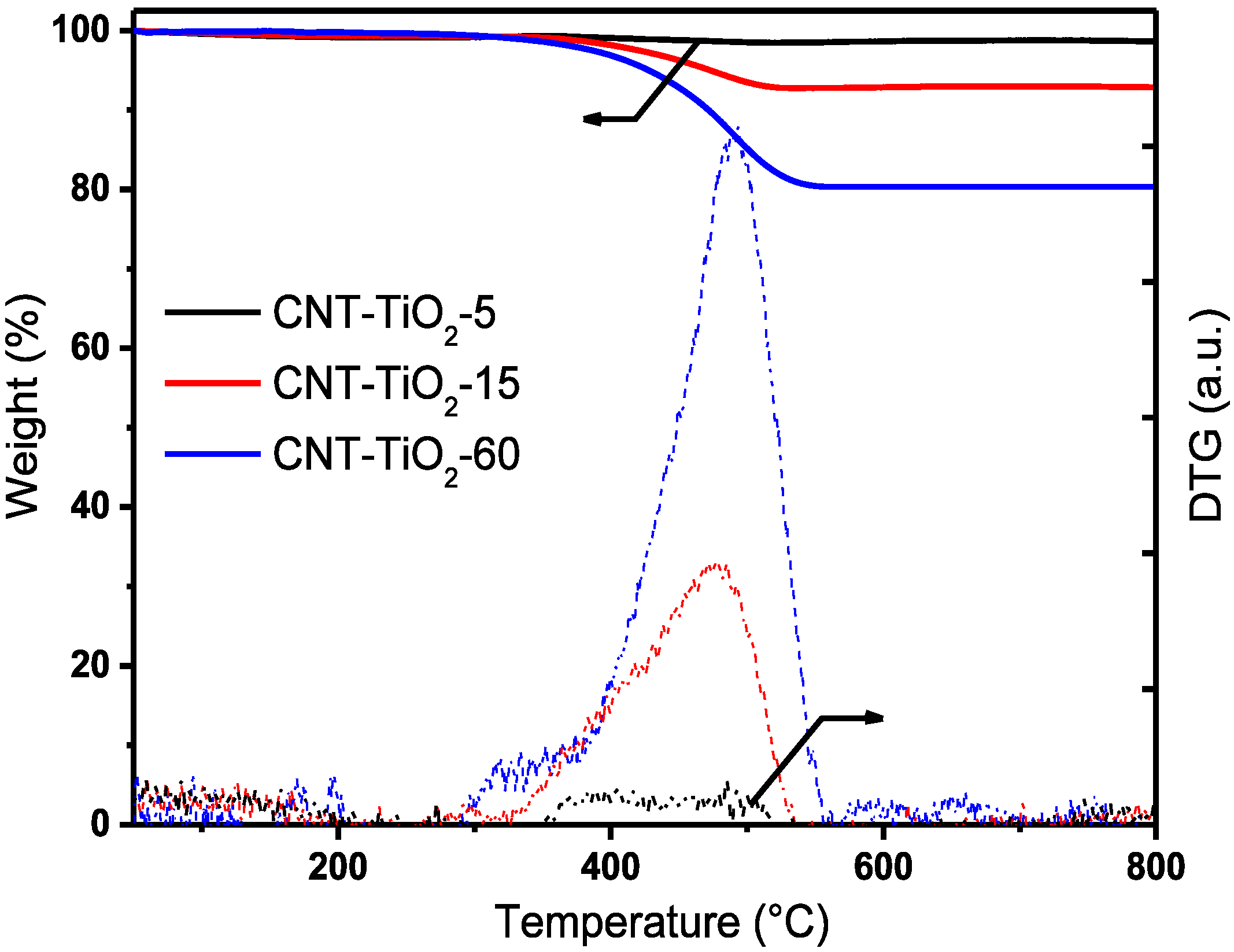

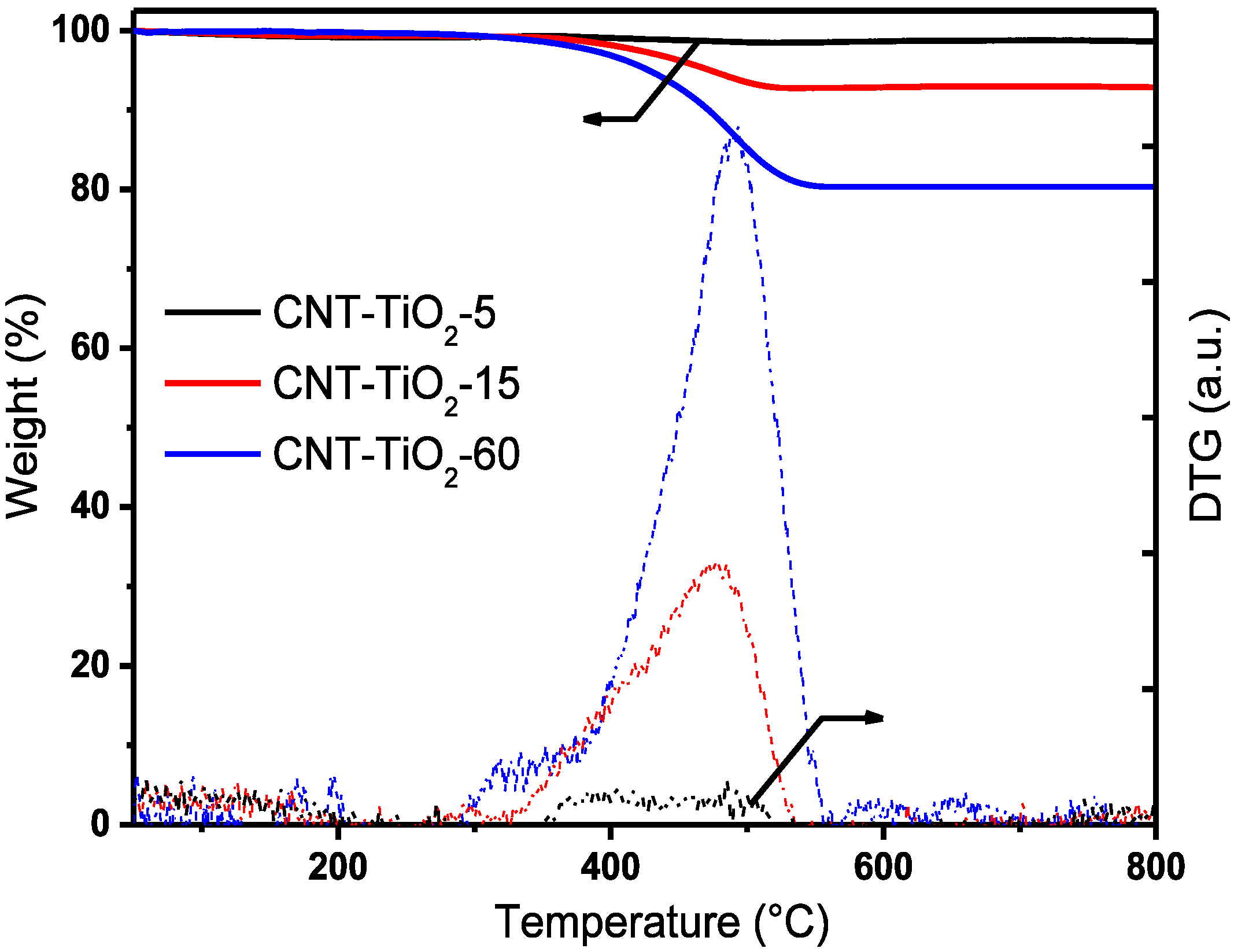{kind=link}
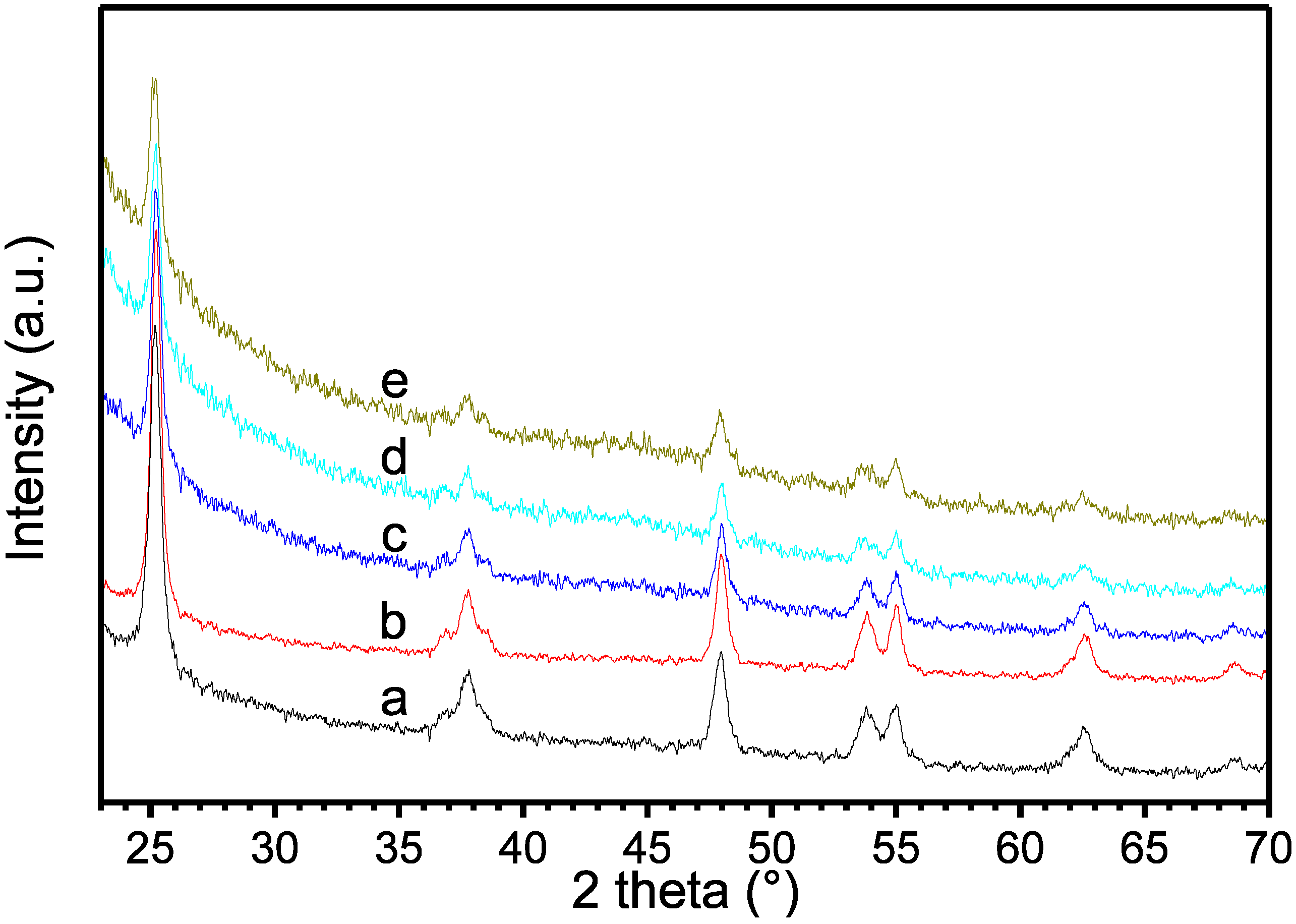
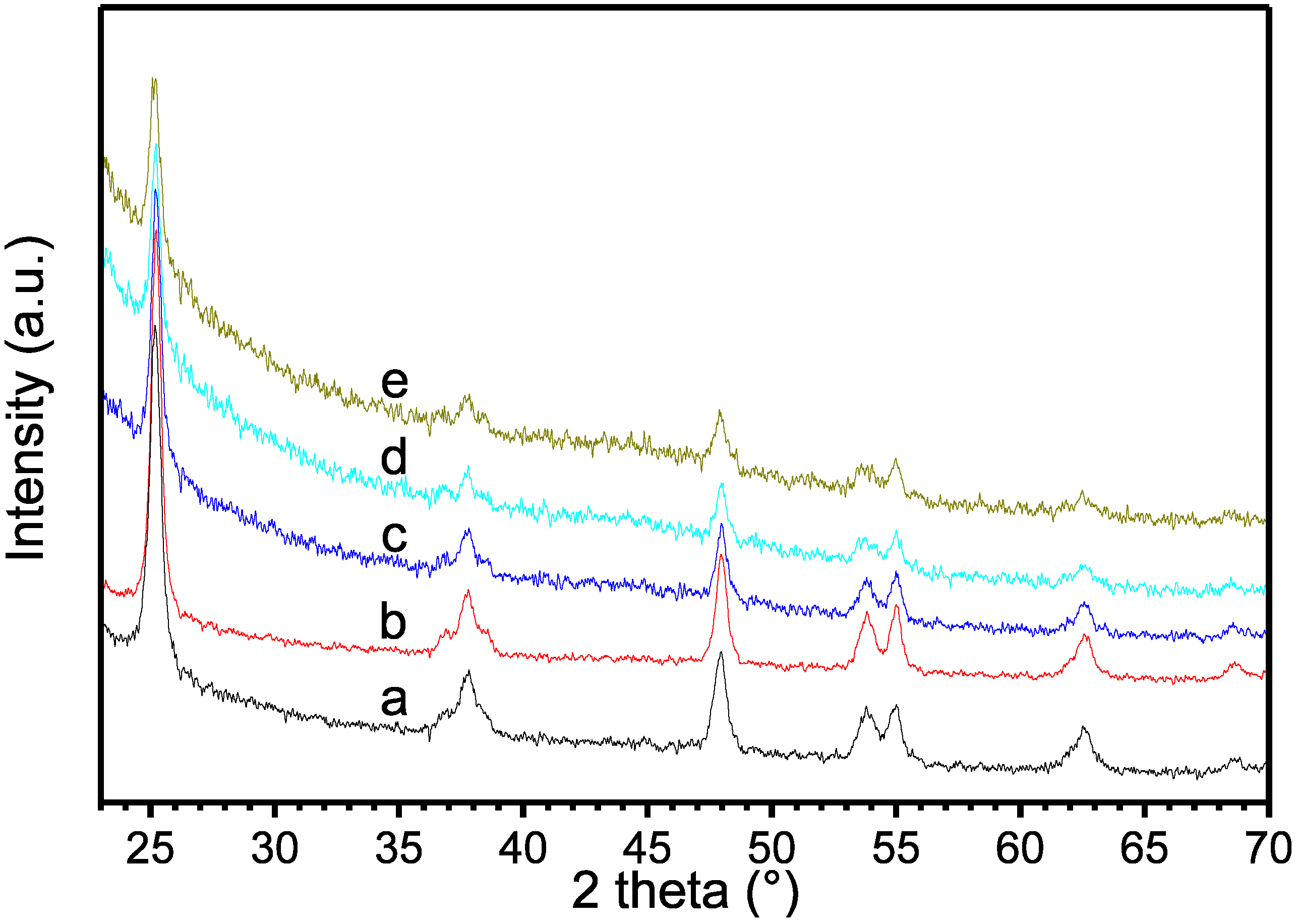{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | C (wt%) | BET (m2 g−1) | r1 (mmol h−1) | r2 (mmol h−1) |
|---|---|---|---|---|
| Pristine TiO2 | - | 104 | 13.4 | 4.2 |
| TiO2−δ | - | 92 | 18.2 | 6.2 |
| CNT-TiO2−δ-5 | 1.4 | 73 | 8.2 | 2.6 |
| CNT-TiO2−δ-15 | 7.2 | 79 | 9.7 | 8.1 |
| CNT-TiO2−δ-60 | 19.3 | 71 | 6.8 | 6.5 |

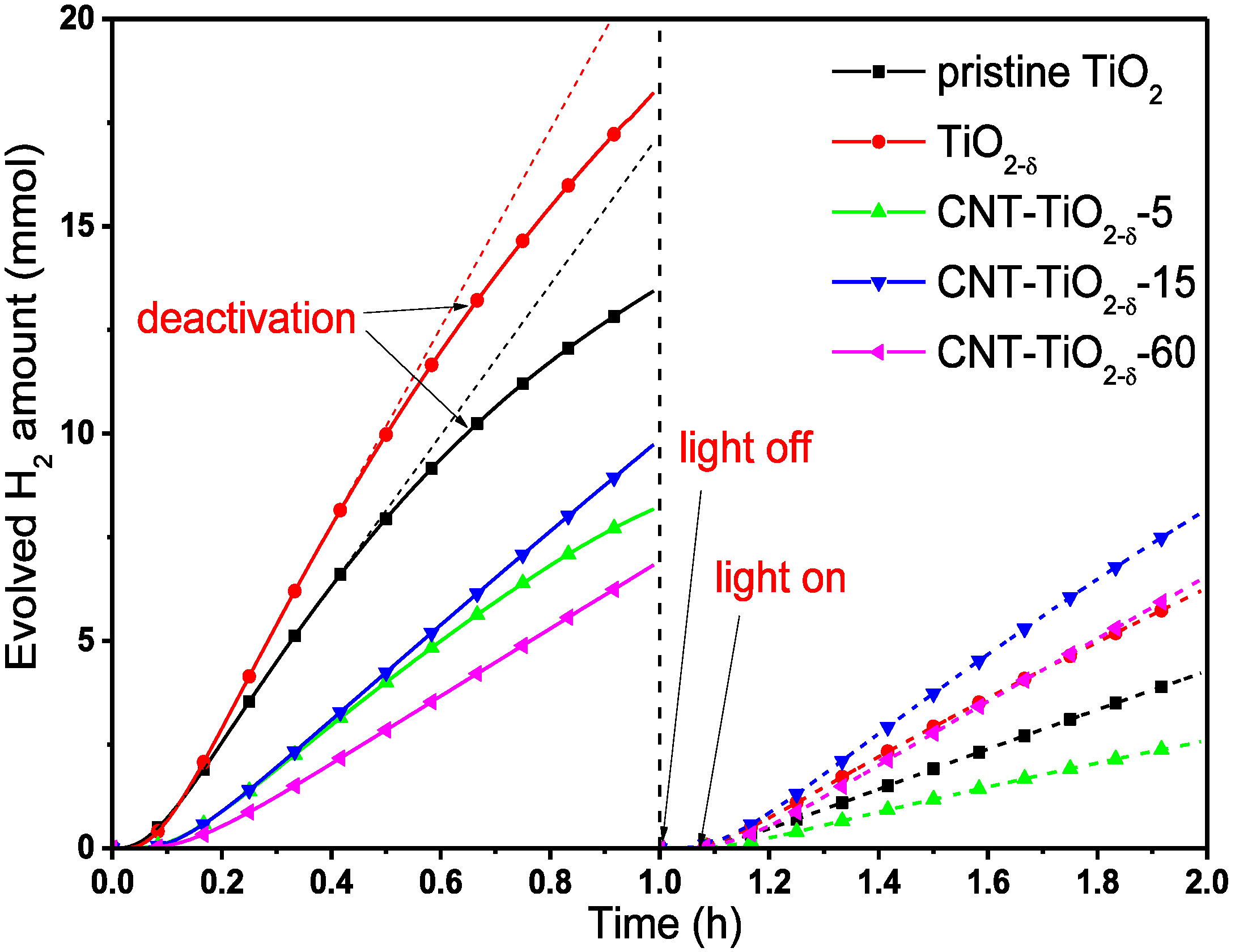
2.2. Photocatalytic Performance
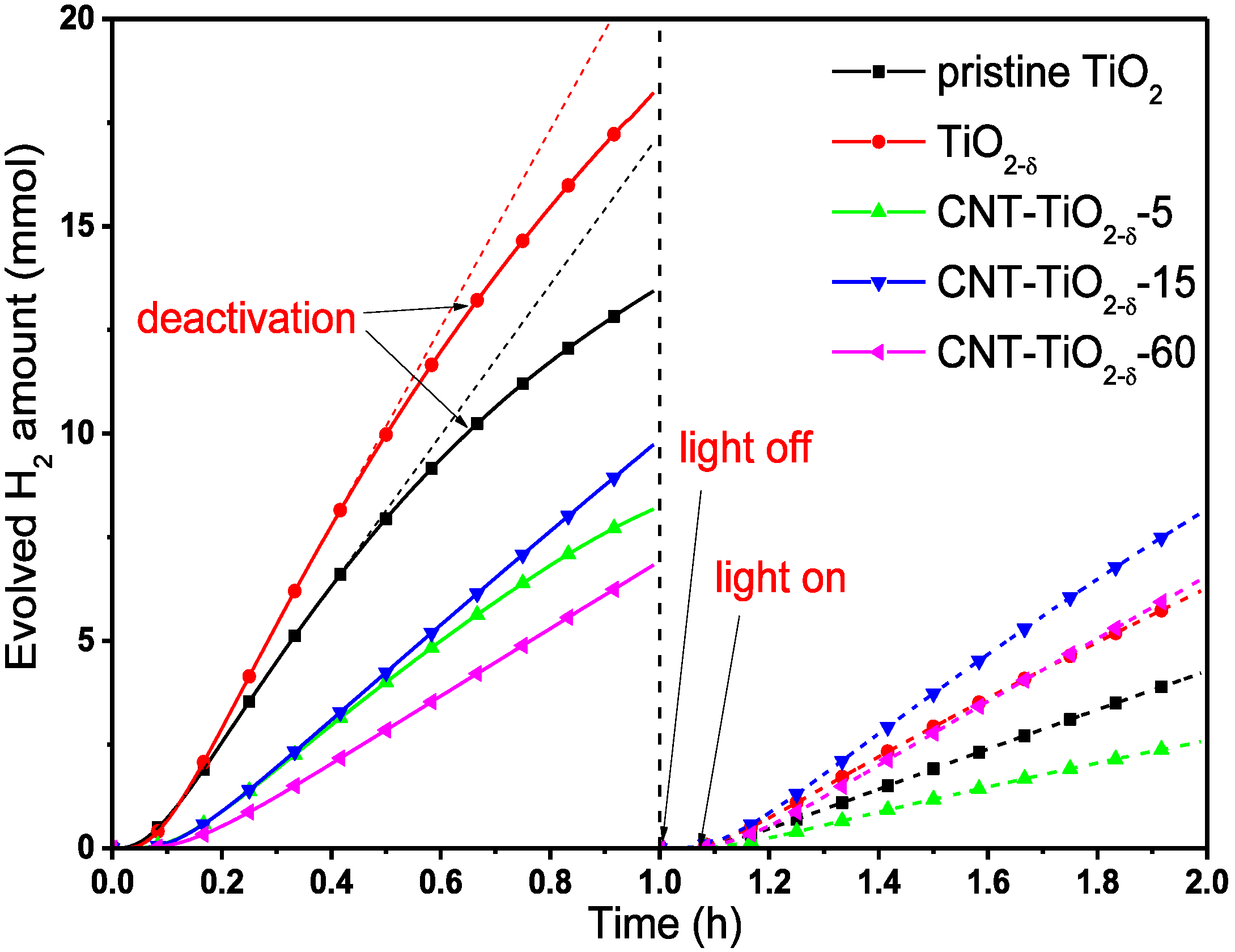
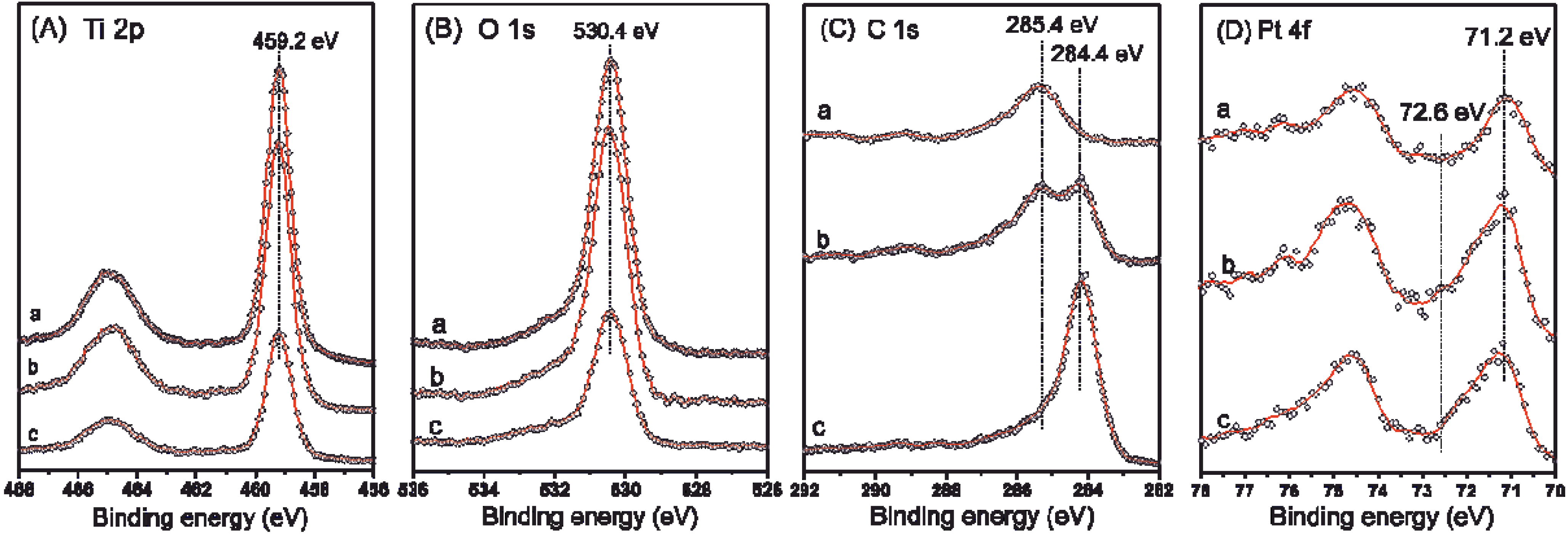
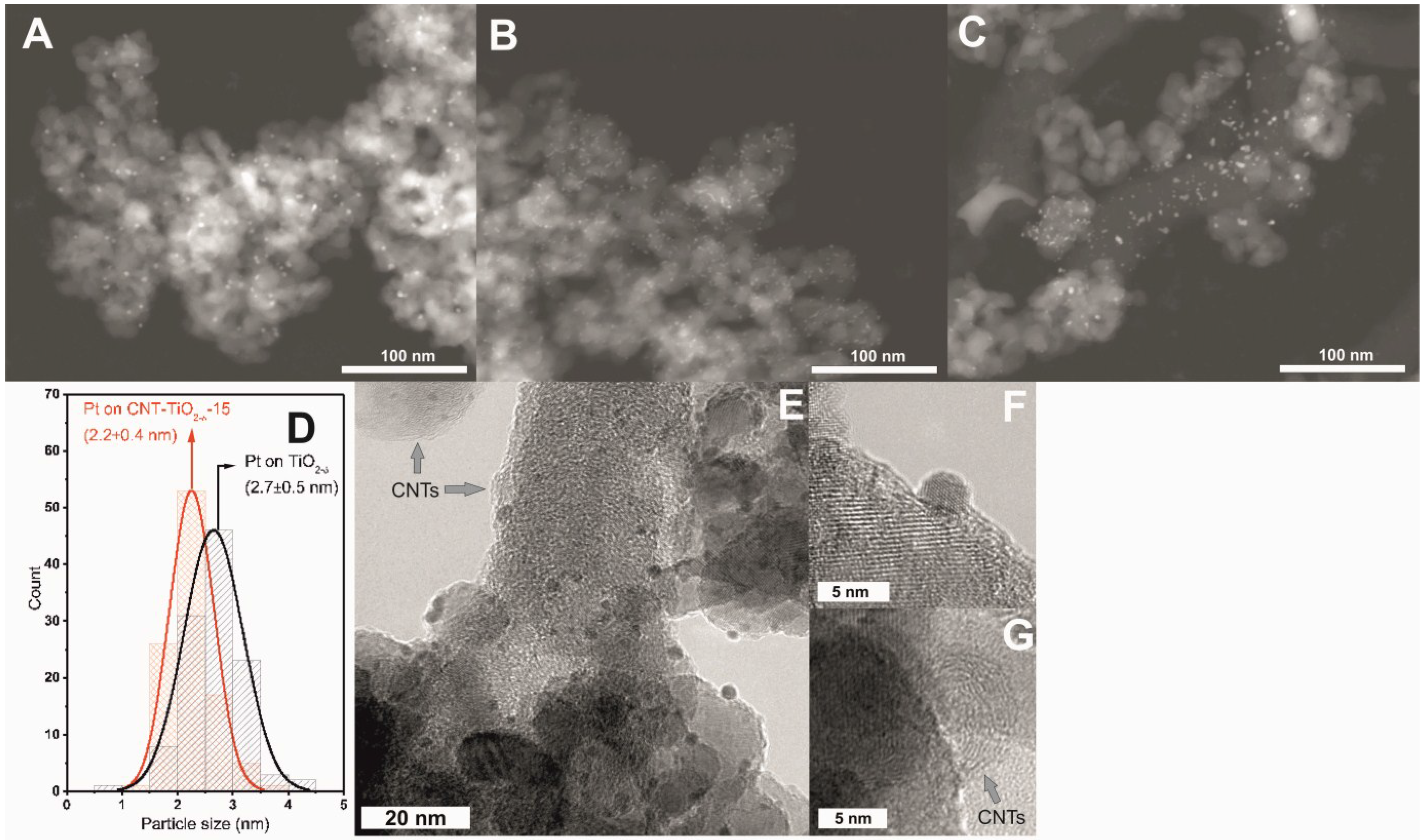
2.3. XPS and (S)TEM of the Photocatalysts after Reactions
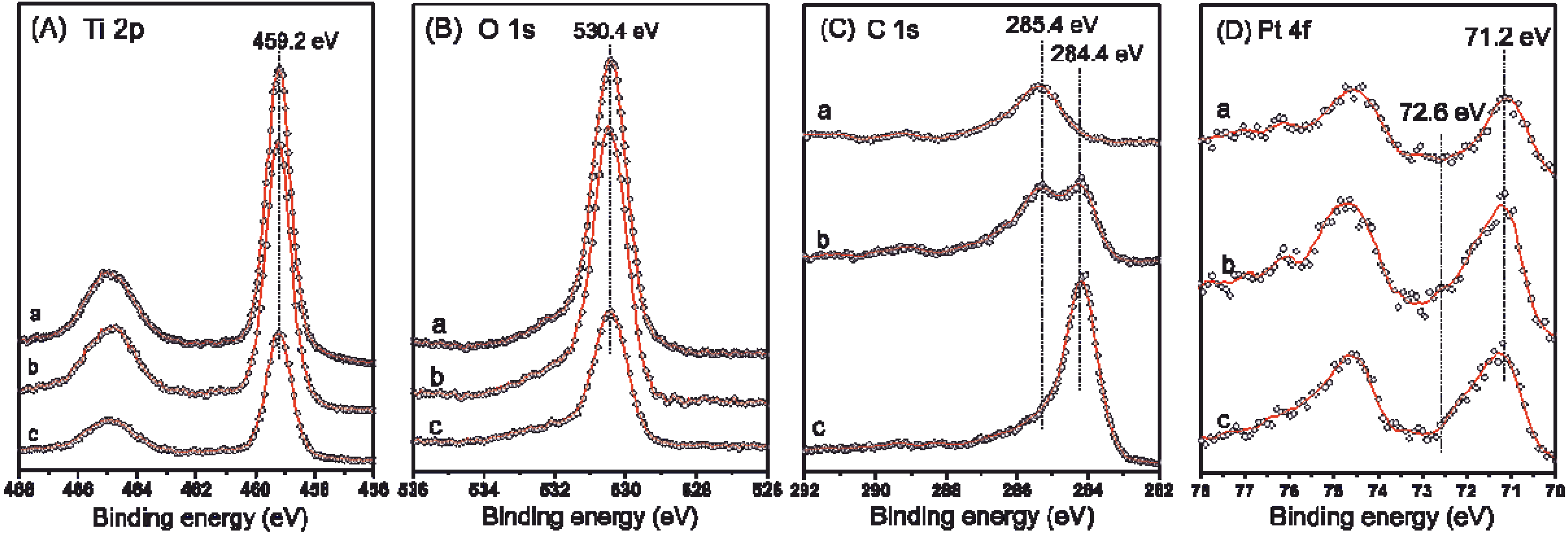
| Catalyst | C | O | Ti | Pt |
|---|---|---|---|---|
| TiO2−δ | 20.34 | 55.03 | 24.47 | 0.16 |
| CNT-TiO2−δ-15 | 31.94 | 47.22 | 20.52 | 0.32 |
| CNT-TiO2−δ-60 | 54.81 | 32.08 | 12.76 | 0.35 |
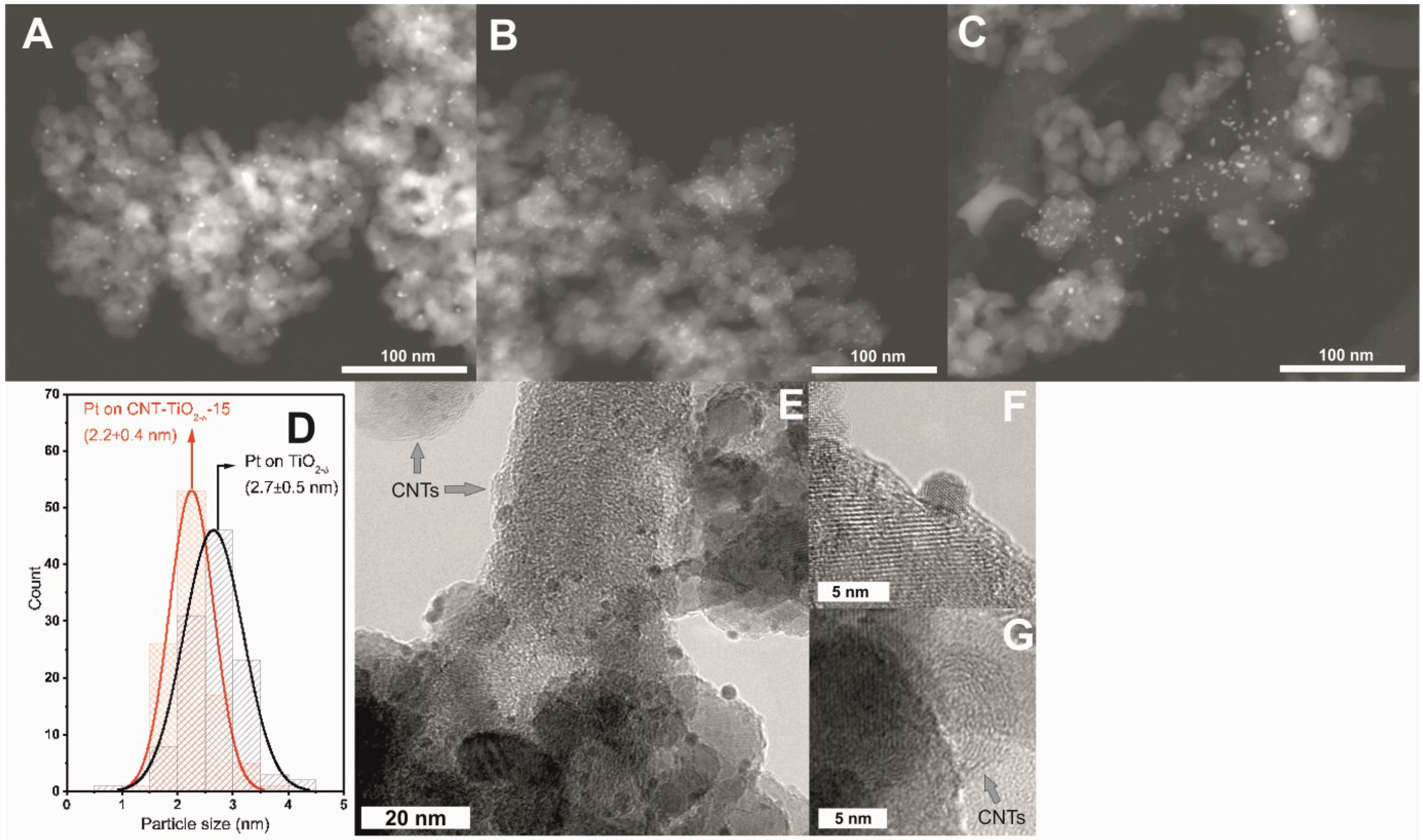
3. Experimental Section
3.1. Materials and Sample Preparation
3.2. Physicochemical Characterization
3.3. Photocatalytic and Photoelectrochemical Measurements
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ibhadon, A.O.; Fitzpatrick, P. Heterogeneous photocatalysis: Recent advances and applications. Catalysts 2013, 3, 189–218. [Google Scholar] [CrossRef]
- Martin, D.J.; Reardon, P.J.; Moniz, S.J.; Tang, J. Visible light-driven pure water splitting by a nature-inspired organic semiconductor-based system. J. Am. Chem. Soc. 2014, 136, 12568–12571. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, S.; Bard, A.J. Novel carbon-doped TiO2 nanotube arrays with high aspect ratios for efficient solar water splitting. Nano. Lett. 2006, 6, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-H.; Gopal, N.O.; Ke, S.-C. Origin of photoactivity of oxygen-deficient TiO2 under visible light. Appl. Phys. Lett. 2009, 95, 83126–83128. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, X.; Lin, B.; Gao, B. Origin of the visible-light photoactivity of NH3-treated TiO2: Effect of nitrogen doping and oxygen vacancies. Appl. Surf. Sci. 2013, 264, 845–852. [Google Scholar] [CrossRef]
- Leshuk, T.; Parviz, R.; Everett, P.; Krishnakumar, H.; Varin, R.A.; Gu, F. Photocatalytic activity of hydrogenated TiO2. ACS Appl. Mater. Interfaces 2013, 5, 1892–1895. [Google Scholar] [CrossRef] [PubMed]
- Dozzi, M.V.; Selli, E. Specific facets-dominated anatase TiO2: Fluorine-mediated synthesis and photoactivity. Catalysts 2013, 3, 455–485. [Google Scholar] [CrossRef]
- Wei, W.; Yaru, N.; Chunhua, L.; Zhongzi, X. Hydrogenation of TiO2 nanosheets with exposed {001} facets for enhanced photocatalytc activity. RSC Adv. 2012, 2, 8286. [Google Scholar] [CrossRef]
- Mei, B.; Sánchez, M.D.; Reinecke, T.; Kaluza, S.; Xia, W.; Muhler, M. The synthesis of Nb-doped TiO2 nanoparticles by spray drying: An efficient and scalable method. J. Mater. Chem. 2011, 21, 11781–11790. [Google Scholar] [CrossRef]
- Jin, Q.L.; Arimoto, H.; Fujishima, M.; Tada, H. Manganese oxide-surface modified titanium(IV) dioxide as environmental catalyst. Catalysts 2013, 3, 444–454. [Google Scholar] [CrossRef]
- Choi, J.; Park, H.; Hoffmann, M.R. Effects of single metal-ion doping on the visible-light photoreactivity of TiO2. J. Phys. Chem. C 2010, 114, 783–792. [Google Scholar] [CrossRef]
- Liu, G.; Wang, L.; Yang, H.G.; Cheng, H.-M.; Lu, G.Q. Titania-based photocatalysts—Crystal growth, doping and heterostructuring. J. Mater. Chem. 2010, 20, 831–843. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Yu, P.Y.; Mao, S.S. Increasing solar absorption for photocatalysis with black hydrogenated titanium dioxide nanocrystals. Science 2011, 331, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Zuo, F.; Wang, L.; Wu, T.; Zhang, Z.; Borchardt, D.; Feng, P. Self-doped Ti3+ enhanced photocatalyst for hydrogen production under visible light. J. Amer. Chem. Soc. 2010, 132, 11856–11857. [Google Scholar] [CrossRef]
- Leary, R.; Westwood, A. Carbonaceous nanomaterials for the enhancement of TiO2 photocatalysis. Carbon 2011, 49, 741–772. [Google Scholar] [CrossRef]
- Wong, T.J.; Lim, F.J.; Gao, M.; Lee, G.H.; Ho, G.W. Photocatalytic H2 production of composite one-dimensional TiO2 nanostructures of different morphological structures and crystal phases with graphene. Catal. Sci. Technol. 2013, 3, 1086–1093. [Google Scholar] [CrossRef]
- Tsubota, T.; Ono, A.; Murakami, N.; Ohno, T. Characterization and photocatalytic performance of carbon nanotube material-modified TiO2 synthesized by using the hot CVD process. Appl. Catal. B 2009, 91, 533–538. [Google Scholar] [CrossRef]
- Huang, Q.; Tian, S.; Zeng, D.; Wang, X.; Song, W.; Li, Y.; Xiao, W.; Xie, C. Enhanced photocatalytic activity of chemically bonded TiO2/graphene composites based on the effective interfacial charge transfer through the C–Ti bond. ACS Catal. 2013, 3, 1477–1485. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Li, C.; Chen, W.; Zeng, M. Carbon nanotube/titania composites prepared by a micro-emulsion method exhibiting improved photocatalytic activity. Appl. Catal. A 2012, 427–428, 1–7. [Google Scholar] [CrossRef]
- Ahmmad, B.; Kusumoto, Y.; Somekawa, S.; Ikeda, M. Carbon nanotubes synergistically enhance photocatalytic activity of TiO2. Catal. Commun. 2008, 9, 1410–1413. [Google Scholar] [CrossRef]
- Ventosa, E.; Chen, P.; Schuhmann, W.; Xia, W. CNTs grown on oxygen-deficient anatase TiO2−δ as high-rate composite electrode material for lithium ion batteries. Electrochem. Commun. 2012, 25, 132–135. [Google Scholar] [CrossRef]
- Chen, P.R.; Chew, L.M.; Xia, W. The influence of the residual growth catalyst in functionalized carbon nanotubes on supported Pt nanoparticles applied in selective olefin hydrogenation. J. Catal. 2013, 307, 84–93. [Google Scholar] [CrossRef]
- Becker, M.J.; Xia, W.; Tessonnier, J.-P.; Blume, R.; Yao, L.; Schlögl, R.; Muhler, M. Optimizing the synthesis of cobalt-based catalysts for the selective growth of multiwalled carbon nanotubes under industrially relevant conditions. Carbon 2011, 49, 5253–5264. [Google Scholar] [CrossRef]
- Ventosa, E.; Xia, W.; Klink, S.; La Mantia, F.; Mei, B.; Muhler, M.; Schuhmann, W. Ammonia-annealed TiO2 as a negative electrode material in Li-ion batteries: N doping or oxygen deficiency? Chem. Eur. J. 2013, 19, 14194–14199. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Su, D.; Birkner, A.; Ruppel, L.; Wang, Y.; Wöll, C.; Qian, J.; Liang, C.; Marginean, G.; Brandl, W.; Muhler, M. Chemical vapor deposition and synthesis on carbon nanofibers: sintering of ferrocene-derived supported iron nanoparticles and the catalytic growth of secondary carbon nanofibers. Chem. Mater. 2005, 17, 5737–5742. [Google Scholar] [CrossRef]
- Kundu, S.; Nagaiah, T.C.; Chen, X.X.; Xia, W.; Bron, M.; Schuhmann, W.; Muhler, M. Synthesis of an improved hierarchical carbon-fiber composite as a catalyst support for platinum and its application in electrocatalysis. Carbon 2012, 50, 4534–4542. [Google Scholar] [CrossRef]
- Sánchez, M.D.; Chen, P.; Reinecke, T.; Muhler, M.; Xia, W. The role of oxygen- and nitrogen-containing surface groups on the sintering of iron nanoparticles on carbon nanotubes in different atmospheres. ChemCatChem 2012, 4, 1997–2004. [Google Scholar] [CrossRef]
- Santato, C.; Ulmann, M.; Augustynski, J. Photoelectrochemical properties of nanostructured tungsten trioxide films. J. Phys. Chem. B 2001, 105, 936–940. [Google Scholar] [CrossRef]
- Busser, G.W.; Mei, B.; Muhler, M. Optimizing the deposition of hydrogen evolution sites on suspended semiconductor particles using on-line photocatalytic reforming of aqueous methanol solutions. ChemSusChem 2012, 5, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, B.; Iwai, K.; Nishimoto, S.; Sato, S. Role of Platinum Deposits on Titanium(IV) Oxide Particles: Structural and Kinetic Analyses of Photocatalytic Reaction in Aqueous Alcohol and Amino Acid Solutions. J. Phys. Chem. B 1997, 101, 3349–3359. [Google Scholar] [CrossRef]
- Kowalska, E.; Mahaney, O.O.; Abe, R.; Ohtani, B. Visible-light-induced photocatalysis through surface plasmon excitation of gold on titania surfaces. Phys. Chem. Chem. Phys 2010, 12, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Woan, K.; Pyrgiotakis, G.; Sigmund, W. Photocatalytic carbon-nanotube-TiO2 composites. Adv. Mater. 2009, 21, 2233–2239. [Google Scholar] [CrossRef]
- Chen, T.; Feng, Z.; Wu, G.; Shi, J.; Ma, G.; Ying, P.; Li, C. Mechanistic studies of photocatalytic reaction of methanol for hydrogen production on Pt/TiO2 by in situ Fourier transform IR and time-Resolved IR spectroscopy. J. Phys. Chem. C 2007, 111, 8005–8014. [Google Scholar] [CrossRef]
- Yu, J.G.; Xiang, Q.J.; Zhou, M.H. Preparation, characterization and visible-light-driven photocatalytic activity of Fe-doped titania nanorods and first-principles study for electronic structures. Appl. Catal. B 2009, 90, 595–602. [Google Scholar] [CrossRef]
- Dholam, R.; Patel, N.; Adami, M.; Miotello, A. Hydrogen production by photocatalytic water-splitting using Cr- or Fe-doped TiO2 composite thin films photocatalyst. Int. J. Hydrogen Energy 2009, 34, 5337–5346. [Google Scholar] [CrossRef]
- Tatarchuk, B. Physical characterization of Fe/TiO2 model supported catalysts I. Electron microscopic studies of reduction behavior. J. Catal. 1981, 70, 308–322. [Google Scholar] [CrossRef]
- Pumera, M. Carbon nanotubes contain residual metal catalyst nanoparticles even after washing with nitric acid at elevated temperature because these metal nanoparticles are sheathed by several graphene sheets. Langmuir 2007, 23, 6453–6458. [Google Scholar] [CrossRef] [PubMed]
- Tessonnier, J.P.; Rosenthal, D.; Hansen, T.W.; Hess, C.; Schuster, M.E.; Blume, R.; Girgsdies, F.; Pfander, N.; Timpe, O.; Su, D.S.; Schlögl, R. Analysis of the structure and chemical properties of some commercial carbon nanostructures. Carbon 2009, 47, 1779–1798. [Google Scholar] [CrossRef]
- Lascovich, J.C.; Giorgi, R.; Scaglione, S. Evaluation of the sp2/sp3 ratio in amorphous carbon structure by XPS and XAES. Appl. Surf. Sci. 1991, 47, 17–21. [Google Scholar] [CrossRef]
- Veziri, C.M.; Pilatos, G.; Karanikolos, G.N.; Labropoulos, A.; Kordatos, K.; Kasselouri-Rigopoulou, V.; Kanellopouios, N.K. Growth and optimization of carbon nanotubes in activated carbon by catalytic chemical vapor deposition. Micro. Mesopor. Mater. 2008, 110, 41–50. [Google Scholar] [CrossRef]
- Yang, J.; Wang, D.; Han, H.; Li, C. Roles of cocatalysts in photocatalysis and photoelectrocatalysis. Acc. Chem. Res. 2013, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Bledowski, M.; Wang, L.; Ramakrishnan, A.; Betard, A.; Khavryuchenko, O.V.; Beranek, R. Visible-light photooxidation of water to oxygen at hybrid TiO2-polyheptazine photoanodes with photodeposited Co-Pi (CoOx) cocatalyst. Chemphyschem 2012, 13, 3018–3024. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, P.; Wang, L.; Wang, P.; Kostka, A.; Wark, M.; Muhler, M.; Beranek, R. CNT-TiO2−δ Composites for Improved Co-Catalyst Dispersion and Stabilized Photocatalytic Hydrogen Production. Catalysts 2015, 5, 270-285. https://doi.org/10.3390/catal5010270
Chen P, Wang L, Wang P, Kostka A, Wark M, Muhler M, Beranek R. CNT-TiO2−δ Composites for Improved Co-Catalyst Dispersion and Stabilized Photocatalytic Hydrogen Production. Catalysts. 2015; 5(1):270-285. https://doi.org/10.3390/catal5010270
Chicago/Turabian StyleChen, Peirong, Lidong Wang, Ping Wang, Aleksander Kostka, Michael Wark, Martin Muhler, and Radim Beranek. 2015. "CNT-TiO2−δ Composites for Improved Co-Catalyst Dispersion and Stabilized Photocatalytic Hydrogen Production" Catalysts 5, no. 1: 270-285. https://doi.org/10.3390/catal5010270
APA StyleChen, P., Wang, L., Wang, P., Kostka, A., Wark, M., Muhler, M., & Beranek, R. (2015). CNT-TiO2−δ Composites for Improved Co-Catalyst Dispersion and Stabilized Photocatalytic Hydrogen Production. Catalysts, 5(1), 270-285. https://doi.org/10.3390/catal5010270