Oxidation Catalysts for Elemental Mercury in Flue Gases—A Review

Abstract
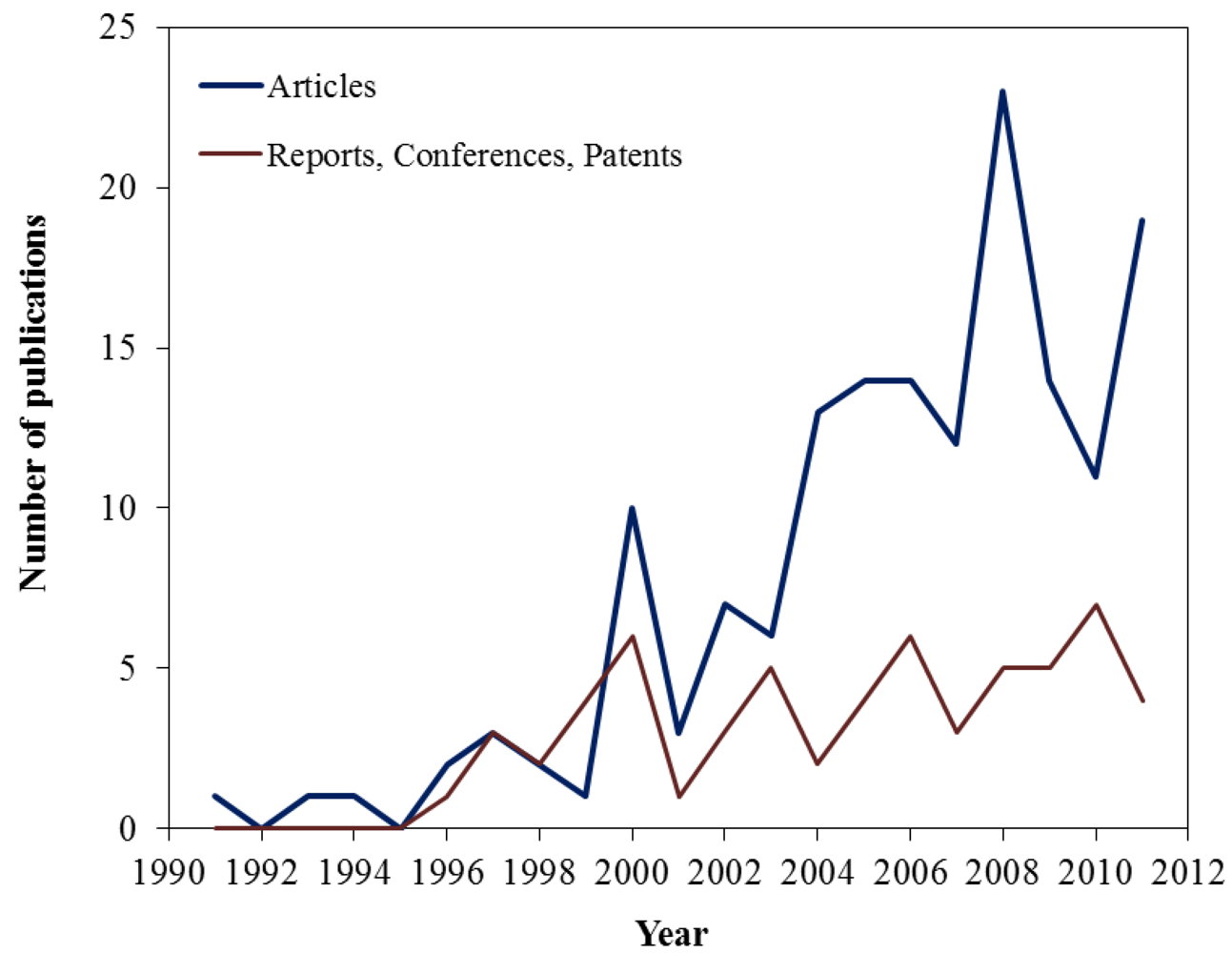
:1. Introduction

2. Operation Condition and Constraints of Hg Oxidation Catalysts in Flue Gases
3. Proposed Mechanisms for the Catalytic Oxidation of Elemental Mercury
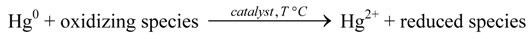
3.1. Deacon Reaction
3.2. Eley-Rideal Mechanism
3.3. Langmuir-Hinshelwood Mechanism
3.4. Mars-Maessen Mechanism
4. Noble Metal-Based Catalysts for Hgel Oxidation

{kind=link}
{kind=link}
| Catalyst type | Gas composition | T, °C | Space velocity, h−1 | Hgel oxidation, % | Reference | |||||||
| O2 | H2O | HCl | NO | NH3 | SO2 | Hgel | ||||||
| vol.% | vol.% | ppm | ppm | ppm | ppm | µg/Nm3 | ||||||
| Lab scale | Ru/TiO2●● | - | 4 | 2–12 | 30–300 | 30–260 | 500 | 50 | 150–350 | 79000 | 30–90 | [36] |
| Bench scale | Au/Al2O3 ° | - | - | 0–1000 | 6–18 | 138–160 | 8–10 ■ | ■■ 2.2 × 10−10 | [37] | |||
| Pd/Al2O3 ° | 0–5.25 | - | 0–100 | 500 | - | ■■ 1.6 × 10−10 | ||||||
| Pt/Al2O3 ° | - | - | ■■ 4.1 × 10−10 | |||||||||
| Au/Teflon ° | 6 | 0–8 | 50 | 600 | - | 2000 | 55 | 175–225 | 5–60 | [30] * | ||
| Ir/Al2O3 ° | 8 | 8 | - | <500 | - | <2000 | 12 | 138 | 7.5 ■ | 75 | [38] | |
| Au/TiO2/FF ° | 1200 | 9–65 | ||||||||||
| 4 | 10 | 50 | 100 | - | 1000 | 20–30 | 150 | [29] ● | ||||
| Pd/Al2O3/FF ° | 4800 | 4–84 | ||||||||||
| Pilot scale | Au/γ-Al2O3 ° | ~8 | 9–12 | 1–20 | - | - | 200–1200 | 10–31 | 139–149 | 3200–3600 *** | 40–99 | [39] ** |
| Pd/γ-Al2O3 ° | - | - | 41–87 | |||||||||
| Full scale | Au/γ-Al2O3 ° | 7–9 | 12 | 1.67 | - | - | 501 | 11–14 | ~150 | 21300 | 52–86 | [40] |
4.1. Activity of Platinum Group Based Catalysts for the Oxidation of Hgel
4.2. Activity of Gold Based Catalysts for the Oxidation of Hgel
4.3. Summary
5. Mercury Oxidation by Transition Metal Oxide Catalysts
- Loading and composition of the metal oxide material;
- Temperature and;
- HCl, Cl2, O2, H2O, NO and SO2 concentrations in the gaseous phase.
| Catalysts | Catalysts characteristics | Reaction temperature, | Mercury oxidation/removal, | Reference | |
| Synthesis method | Metal loading, wt.-% | °C | % | ||
| nano-CuO | commercial | 100 | 90–300 | 20–96 | [51] |
| nano-CuO | commercial | 100 | 150 | 75 | [52] |
| CuCl2/TiO2 | impregnation | 1.5–6 | 350 | 60–100 | [53] |
| CuCl2/TiO2–Al2O3 | wetness impregnation | 0.25–9 | 125–175 | 28–62 | [54] |
| CuCoO4/γ-Al2O3 | thermal decomposition | 1 *** | 100–450 | 10–92 | [55] |
| Co–oxide/TiO2 | sol-gel | 0.5–15 | 90–360 | 10->90 | [33] |
| nano-Fe2O3 | hydrothermal | 100 | 80–400 | <40 | [34] |
| Fe2O3/TiO2 | impregnation | 0.6–5 | 80 | 60–80 | [56] |
| MnOx/Al2O3 | wet impregnation | 1–8 | 100–500 | 45–90 | [14] |
| Mn/α-Al2O3 | wet impregnation | 1 | 100–250 | 30–95 | [57] |
| MnOx/TiO2 | wet impregnation | 10–20 | 175–200 | ~90 | [31] |
| MnOx–CeO2/TiO2 | impregnation | 0.18:0.82:1 ** | 120–400 | 40–>90 | [26] |
| CeO2/TiO2 | impregnation | 0.5–2 * | 120–400 | 40–95 | [58] |
| CeO2/γ-Al2O3 | thermal decomposition | 3–15 | 150–450 | 33–90 | [35] |
| V2O5/TiO2 | sol-gel | 1–10 | 100–500 | 69–100 | [59] |
| SiO2–TiO2 | sol-gel | 12 ● | 135 | 10–90 | [60] |
| SiO2–TiO2–V2O5 | sol-gel | 6–18 ■; 5 ■■ | 135–400 | 40–100 | [61] |
5.1. Copper/Cobalt Based Catalysts
5.2. Iron/Manganese Based Catalysts
5.3. Cerium Based Catalysts
5.4. Various Metal Based Catalysts
5.5. Summary
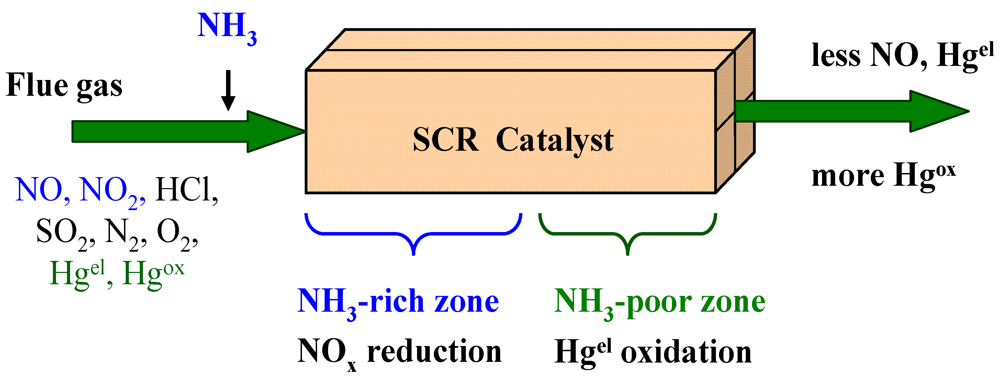
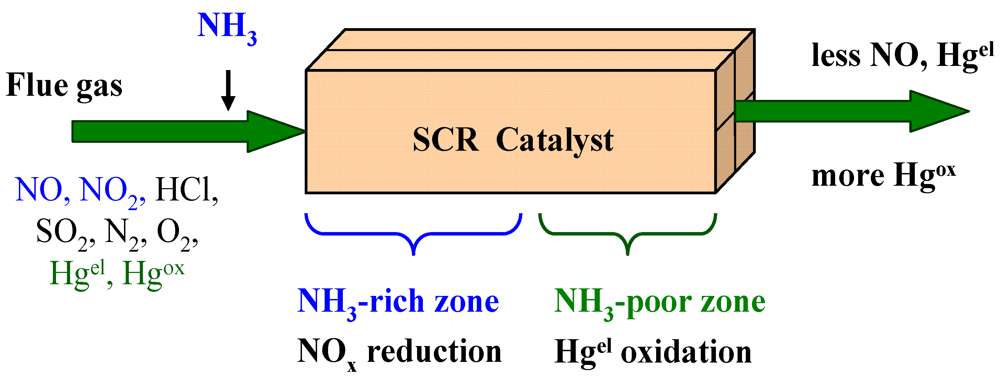
6. Mercury Oxidation on SCR Catalysts
6.1. Mercury Adsorption on SCR-Catalysts
| Gas composition | T, °C | Space velocity,h −1 | Hgel oxidation, % | Reference | ||||||||
| O2 vol.% | H2 Ovol.% | HCl ppm | NO ppm | NH3 ppm | SO2 ppm | SO3 ppm | Hgel µg/Nm3 | |||||
| Lab scale (simulated flue gases) | ||||||||||||
| 6 | - | 50 | 400 | 400 | - | - | 36–39 | 350 | 4000 | 3–91 | [25] | |
| 6 | 8 | 0–35 | 400 | 360 | 1000 | - | 10–20 | 371 | 4000 | 12–70 | [76] ■ | |
| 5 | 1.8 | 0–20 | 150 | - | 500 | - | 30 | 350 | 72 ● | 70–90 | [3] | |
| 3 | - | 10–50 | 500 | 500 | - | - | 50 | 250–350 | 120 ● | 85–98 | [78] | |
| 3 | 8 | 5–35 | 400 | 360 | - | - | ~20 | 390 | 3600 | 40–86 | [79] ■,** | |
| Bench scale (simulated flue gases) | ||||||||||||
| - | 15 | 0.3–3 | 400 | 300 | 70 | - | 160 | 260–320 | 170 ● | 50–90 | [73] | |
| 6 | 8 | 0–50 | 600 | 550 | 0–2000 | 0–50 | 13 | 343 | 20–71 | [72] | ||
| 3.5 | 5.3 | 0–204 | 350 | 315 | 280–2891 | - | 19 | 350 | 2609 | 0–>90 | [77] | |
| 7.1 | 6.8 | 0–20 | 200 | 180 | 500 | - | 20–25 | 350–400 | 20004000 | 30–88 | [80] | |
| Pilot scale (flue gases) | ||||||||||||
| 2.7–4.4 | - | 246 | 960 | 765 | 222–2921 | - | 5–10 | 300–400 | 2943 | 9–20 | [74] | |
| 3 | - | 500 | 250 | 275 | 2000 | 50 | 120 | 300–350 | 1800 | <80 | [71] * | |
6.2. Influence of Flue Gas Constituents
6.3. Influence of Catalyst Composition, Temperature and Space Velocity
6.4. Loss of Hgel Oxidation Activity
6.5. Mechanism
6.6. Optimization of Hg Oxidation Activity
6.7. Commercial Development
6.8. Summary
7. Novel Catalytic Methods for Mercury Oxidation in Flue Gases
8. Conclusions and Future Research
Acknowledgments
References
- Division of Technology, Industry and Economics (DTIE) Chemicals Branch, Study on Mercury Sources and Emissions and Analysis of the Cost and Effectiveness of Control Measures; UNEP: Geneva, Switzerland, 2010.
- Pirrone, N.; Cinnirella, S.; Feng, X.; Finkelman, B.R.; Friedli, R.H.; Leaner, J.; Mason, R.; Mukherjee, B.A.; Stracher, G.; Streets, G.D.; Telmer, K. Mercury Fate and Transport in the Global Atmosphere—Emissions, Measurements and Models; Springer: New York, NY, USA, 2009. [Google Scholar]
- Lee, B.J.; Lee, M.S.; Lee, Y.I. The characteristics of catalysts for mercury oxidation in thermal power plants. Proc. World Acad. Sci. Eng. Technol. 2008, 44, 256–257. [Google Scholar]
- United States Environmental Protection Agency (U.S. EPA). Mercury News, December 2011. Available online: http://www.epa.gov/hg/ (accessed on 2 January 2012).
- European Commission. Directive 2010/75/EU of the European Parliament and of the Council on industrial emissions (integrated pollution prevention and control). Off. J. Eur. Union 2010, L334, 17–117.
- Pavlish, J.H.; Sondreal, E.A.; Mann, D.M.; Olson, S.E.; Galbreath, C.K.; Laudal, L.; Benson, A.S. Status review of mercury control options for coal-fired power plants. Fuel Process. Technol. 2003, 82, 89–165. [Google Scholar] [CrossRef]
- Pavlish, H.J.; Hamre, L.L.; Zhuang, Y. Mercury control technologies for coal combustion and gasification systems. Fuel 2010, 89, 838–847. [Google Scholar] [CrossRef]
- Schofield, K. Fuel-mercury combustion emissions: An important heterogeneous mechanism and an overall review of its implications. Environ. Sci. Technol. 2008, 42, 9014–9030. [Google Scholar] [CrossRef]
- Presto, A.A.; Granite, E.J. Survey of catalysts for oxidation of mercury in flue gas. Environ. Sci. Technol. 2006, 40, 5601–5609. [Google Scholar] [CrossRef]
- Wilhelm, S.M. Estimate of mercury emissions to the atmosphere from petroleum. Environ. Sci. Technol. 2001, 35, 4704–4710. [Google Scholar] [CrossRef]
- Yudovich, Y.E.; Ketris, M.P. Mercury in coal: A review. Part 2 coal use and environmental problems. Int. J. Coal Geol 2005, 62, 136–165. [Google Scholar]
- Sondreal, E.A.; Benson, S.A.; Pavlish, S.A.; Ralston, N.V.C. An overview of air quality III: Mercury, trace elements, and particulate matter. Fuel Process. Technol. 2004, 85, 425–440. [Google Scholar] [CrossRef]
- Vosteen, B.W. Native Halogens in Coals from US, China and Elsewhere—Low Chlorine Coal Need Bromide Addition for Effective Mercury Capture. In Proceedings of MEC7—Mercury Emissions from Coal—International Experts Workshop, Glasgow, UK, 16–18 June 2010.
- Qiao, S.; Chen, J.; Li, J.; Qu, Z.; Liu, P.; Yan, N.; Jia, J. Adsorption and catalytic oxidation of gaseous elemental mercury in flue gas over MnOx/alumina. Ind. Eng. Chem. Res. 2009, 48, 3317–3322. [Google Scholar]
- Vosteen, W.B.; Kanefke, R.; Köser, H. Bromine-enhanced mercury abatement from combustion flue gases-recent industrial applications and laboratory research. VGB PowerTech 2006, 86, 70–75. [Google Scholar]
- Vosteen, W.B.; Straube, S.; Köser, H. Mercury Sorption and Mercury Oxidation by Chlorine and Bromine at SCR-DeNOx Catalysts—Part A: Oxidation. In Proceedings of 9th Annual EPA, DOE, EPRI, EEI Conference on Clean Air, Global Warming & Renewable Energy, Tucson, AZ, USA, 24–25 January 2006.
- Lopez, N.; Gomez-Segura, J.; Marin, R.P.; Perez-Ramirez, J.P. Mechanism of HCl oxidation (Deacon process) over RuO2. J. Catal. 2008, 255, 29. [Google Scholar]
- Bierögel, S.; Baltin, G.; Koeser, H. Important side reactions in DeNOx facilities—Investigation for halogen formation. Chem. Ing. Tech. 2003, 75, 1066–1067. [Google Scholar]
- Griffin, R.D. A new theory of dioxin formation in municipal solid waste combustion. Chemophere 1986, 15, 1987–1990. [Google Scholar] [CrossRef]
- Niksa, S.; Fujiwara, N. A predictive mechanism for mercury oxidation on selective catalytic reduction catalyst under coal-derived flue gas. J. Air Waste Manag. Assoc. 2005, 55, 1866–1875. [Google Scholar] [CrossRef]
- Kamata, H.; Uueno, S.I.; Naito, T.; Yukimura, A. Mercury oxidation over the V2O5(WO3)/TiO2 commercial SCR catalyst. Ind. Eng. Chem. Res. 2008, 47, 8136–8141. [Google Scholar] [CrossRef]
- Li, Y.; Murphy, D.P.; Wu, C.Y.; Powers, W.K.; Bonzongo, J.C. Development of silica/vanadia/titania catalysts for removal of elemental mercury from coal-combustion flue gas. Environ. Sci. Technol. 2008, 42, 5304–5309. [Google Scholar]
- He, S.; Zhou, J.; Zhu, Y.; Luo, Z.; Ni, M.; Cen, K. Mercury oxidation over a vanadia-based selective catalytic reduction catalyst. Energy Fuels 2009, 23, 253–259. [Google Scholar] [CrossRef]
- Kamata, H.; Uueno, S.I.; Naito, T.; Yamaguchi, A.; Ito, S. Mercury oxidation by hydrochloric acid over a VOx/TiO2 catalyst. Catal. Commun. 2008, 9, 2441–2444. [Google Scholar] [CrossRef]
- Eom, Y.; Jeon, H.S.; Ngo, A.T.; Kim, J.; Lee, G.T. Heterogeneous mercury reaction on a selective catalytic reduction (SCR) catalyst. Catal. Lett. 2008, 121, 219–225. [Google Scholar] [CrossRef]
- Li, H.; Wu, Y.C.; Li, Y.; Zhang, J. Superior activity of MnOx–CeO2/TiO2 catalyst for catalytic oxidation of elemental mercury at low flue gas temperatures. Appl. Catal. B Environ. 2012, 111–112, 381–388. [Google Scholar] [CrossRef]
- Hu, C.; Zhou, J.; Luo, Z.; Cen, K. Oxidative adsorption of elemental mercury by activated carbon in simulated coal-fired flue gas. Energy Fuels 2011, 25, 154–158. [Google Scholar] [CrossRef]
- Granite, J.E.; Pennline, H.W.; Hargis, A.R. Novel sorbents for mercury removal from flue gas. Ind. Eng. Chem. Res. 2000, 39, 1020–1029. [Google Scholar] [CrossRef]
- Hrdlicka, J.A.; Seames, W.S.; Mann, M.D.; Muggli, D.S.; Horabik, C.A. Mercury oxidation in flue gas using gold and palladium catalysts on fabric filters. Environ. Sci. Technol. 2008, 42, 6677–6682. [Google Scholar]
- Zhao, Y.; Mann, M.D.; Pavlish, J.H.; Mibeck, B.A.F.; Dunham, E.G.; Olson, E.S. Application of gold catalyst for mercury oxidation by chlorine. Environ. Sci. Technol. 2006, 40, 1603–1608. [Google Scholar]
- Ji, L.; Sreekanth, M.P.; Smirniotis, G.P.; Thiel, W.S.; Pinto, G.N. Manganese oxide/Titania materials for removal of NOx and elemental mercury from flue gas. Energy Fuels 2008, 22, 2299–2306. [Google Scholar] [CrossRef]
- Yang, S.; Guo, Y.; Yan, N.; Wu, D.; He, H.; Qu, Z.; Jia, J. Elemental mercury capture from flue gas by magnetic Mn-Fe spinel: Effect of chemical heterogeneity. Ind. Eng. Chem. Res. 2011, 50, 9650–9656. [Google Scholar]
- Liu, Y.; Wang, Y.; Wang, H.; Wu, Z. Catalytic oxidation of gas-phase mercury over Co/TiO2 catalysts prepared by sol-gel method. Catal. Commun. 2011, 12, 1291–1294. [Google Scholar] [CrossRef]
- Kong, F.; Qui, J.; Liu, H.; Zhao, R.; Ai, Z. Catalytic oxidation of gas-phase elemental mercury by nano-Fe2O3. J. Environ. Sci. 2011, 24, 699–704. [Google Scholar]
- Wen, X.; Li, C.; Fan, X.; Gao, H.; Zhang, W.; Chen, L.; Zeng, G.; Zhao, G. Experimental study of gaseous elemental mercury removal with CeO2/γ-Al2O3. Energy Fuels 2011, 25, 2939–2944. [Google Scholar] [CrossRef]
- Yan, N.; Chen, W.; Chen, J.; Qu, Z.; Guo, Y.; Yang, S.; Jinping, J. Significance of RuO2 modified SCR catalysts for elemental mercury oxidation in coal-fired flue gas. Environ. Sci. Technol. 2011, 45, 5725–5730. [Google Scholar]
- Presto, A.A.; Granite, E.J. Noble Metal Catalysts for mercury oxidation in utility flue gas. Platin. Met. Rev. 2008, 52, 144–154. [Google Scholar] [CrossRef]
- Granite, J.E.; Pennline, H.W. Catalysts for oxidation of mercury in flue gases. U.S. Patent 7,776,780 B1, 17 August 2010. [Google Scholar]
- Blythe, M.G.; Braman, C.; Dombrowski, K.; Machalek, T. Pilot Testing of Mercury Oxidation Catalysts for Upstream of Wet FGD Systems—Final Technical Report; Cooperative Agreement No. DE-FC26-04NT41992; DOE-NETL: Austin, TX, USA, 2010. [Google Scholar]
- Blythe, M.G.; Paradis, J. Full-Scale Testing of a Mercury Oxidation Catalyst Upstream of a Wet FGD System, Final Technical Report; Cooperative Agreement No. DE-FC26-06NT42778; DOE-NETL: Austin, TX, USA, 2010. [Google Scholar]
- Poulston, S.; Granite, J.E.; Pennline, W.H.; Myers, R.C.; Stanko, P.D.; Hamilton, H.; Rowsell, L.; Smith, J.W.A.; Ilkenhans, T.; Chu, W. Metal sorbents for high temperature mercury capture from fuel gas. Fuel 2007, 86, 2201–2203. [Google Scholar] [CrossRef]
- Granite, J.E.; Pennline, H.W. Method for high temperature mercury capture from gas streams. U.S. Patent 7,033,419 B1, 25 April 2006. [Google Scholar]
- Koutsopoulos, S.; Johannessen, T.; Eriksen, K.M.; Fehrmann, R. Titania-supported Pt and Pt–Pd nanoparticle catalysts for the oxidation of sulfur dioxide. J. Catal. 2006, 238, 206–213. [Google Scholar]
- Ji, Y.; Toops, T.J.; Graham, U.M.; Jacobs, G.; Crocker, M. A kinetic and DRIFTS study of supported Pt catalysts for NO oxidation. Catal. Lett. 2006, 110, 29–37. [Google Scholar] [CrossRef]
- Bond, G.C.; Louis, C.; Thompson, D.T. Catalysis by Gold; Imperial College Press: London, UK, 2006. [Google Scholar]
- Blythe, G.; Dombrowski, K.; Machalek, T.; Richardson, C.; Richardson, M. Pilot Testing of Mercury Oxidation Catalysts for Upstream of Wet FGD Systems, Final Report; Cooperative Agreement No. DE-FC26-01NT41185; DOE-NETL: Austin, TX, USA, 2006. [Google Scholar]
- Blythe, G.; Miller, C.; Freeman, B.; Madrid, J. Full-scale demonstration of oxidation catalyst for enhanced mercury control by wet FGD. In Proceedings of Power Plant Air Pollutant Control“Mega” Symposium, Baltimore, MD, USA, 25–28 August 2008.
- Richardson, C. Evaluation of MerCAP for Power Plant Mercury Control—Final Project Report for US Department of Energy—NETL; Cooperative Agreement No. DE-FC26-03NT41993; DOE-NETL: Austin, TX, USA, 2009. [Google Scholar]
- Tao, F.M. A new approach to the efficient basis set for accurate molecular calculations: Applications to diatomic molecules. J. Chem. Phys. 1994, 100, 3645–3650. [Google Scholar]
- Boreskov, G.K.; Slinko, M.G.; Volkova, E.I. Catalysis of the conversion of sulfur dioxide to sulfur trioxide by metals and platinum-gold alloys. Dokl. Akad. Nauk SSSR 1953, 92, 109–120. [Google Scholar]
- Yamaguchi, A.; Akiho, H.; Ito, S. Mercury oxidation by copper oxides in combustion flue gases. Powder Technol. 2008, 180, 222–226. [Google Scholar] [CrossRef]
- Kamata, H.; Mouri, S.; Uueno, S.I.; Takano, K.; Watanabe, K.; Yamaguchi, A.; Ito, S. Mercury oxidation by hydrogen chloride over the CuO based catalysts. Stud. Surf. Sci. Catal. 2007, 172, 621–622. [Google Scholar]
- Kim, H.M.; Ham, S.W.; Li, J.B. Oxidation of gaseous elemental mercury by hydrochloric acid over CuCl2/TiO2-based catalysts in SCR process. Appl. Catal. B Environ. 2010, 99, 272–278. [Google Scholar] [CrossRef]
- Miksche, J.S.; Ghorishi, B.S. Catalytic Oxidation of Elemental Mercury at Low Temperatures-Technical Paper. In Proceedings of 32nd International Technical Conference on Coal Utilization & Fuel Systems, Clearwater, FL, USA, 10–15 June 2007.
- Mei, Z.; Shen, Z.; Mei, Z.; Zhang, Y.; Xiang, F.; Chen, J.; Wang, W. The effect of N-doping and halide-doping on the activity of CuCoO4 for the oxidation of elemental mercury. Appl. Catal. B Environ. 2008, 78, 112–119. [Google Scholar] [CrossRef]
- Wu, S.; Ozaki, M.; Uddin, A.Md.; Sasaoka, E. Development of iron-based sorbents for Hg0 removal from coal derived fuel gas: Effect of hydrogen chloride. Fuel 2008, 87, 467–474. [Google Scholar] [CrossRef]
- Li, J.; Yan, N.; Qu, Z.; Qiao, S.; Yang, S.; Guo, Y.; Liu, P.; Jia, J. Catalytic oxidation of elemental mercury over the modified catalyst Mn/α-Al2O3 at lower temperatures. Environ. Sci. Technol. 2010, 44, 426–431. [Google Scholar]
- Li, H.; Wu, Y.C.; Li, Y.; Zhang, J. CeO2–TiO2 catalysts for catalytic oxidation of elemental mercury in low-rank coal combustion flue gas. Environ. Sci. Technol. 2011, 45, 7394–7400. [Google Scholar]
- Lee, W.; Bae, G.N. Removal of elemental mercury Hg0 by nanosized V2O5/TiO2 catalysts. Environ. Sci. Technol. 2009, 43, 1522–1527. [Google Scholar] [CrossRef]
- Li, Y.; Murphy, P.; Wu, Y.C. Removal of elemental mercury from simulated coal-combustion flue gas using a SiO2–TiO2 nanocomposite. Fuel Process. Technol. 2008, 89, 567–573. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Wu, Y.C.; Zhang, J. Oxidation and capture of elemental mercury over SiO2-TiO2-V2O5 catalysts in simulated low-rank coal combustion flue gas. Chem. Eng. J. 2011, 169, 186–193. [Google Scholar] [CrossRef]
- Ghorishi, S.B.; Lee, W.C.; Jozewicz, S.W.; Kilgroe, D.J. Effects of fly ash transition metal content and flue gas HCl/SO2 ration on mercury speciation on waste combustion. Environ. Eng. Sci. 2005, 22, 221–231. [Google Scholar] [CrossRef]
- Kamata, H.; Uueno, S.I.; Sato, N.; Naito, T. Mercury oxidation by hydrochloric acid over TiO2 supported metal oxide catalysts in coal combustion flue gas. Fuel Proc. Technol. 2009, 90, 947–951. [Google Scholar] [CrossRef]
- Mei, Z.; Shen, Z.; Zhao, Q.; Yuan, T.; Zhang, Y.; Xiang, F.; Wang, W. Removing and recovering gas-phase elemental mercury by CuxCo3−xO4 0.75 ≤ x ≤ 2.25, in the presence of sulphur compounds. Chemosphere 2008, 70, 1399–1404. [Google Scholar] [CrossRef]
- Yang, S.; Guo, Y.; Yan, N.; Wu, D.; He, H.; Xie, J.; Qu, Z.; Jia, J. Remarkable effect of the incorporation of titanium on the catalytic activity and SO2 poisoning resistance of magnetic Mn–Fe spinel for elemental mercury capture. Appl. Catal. B Environ. 2011, 101, 698–708. [Google Scholar] [CrossRef]
- Yang, S.; Guo, Y.; Yan, N.; Wu, D.; He, H.; Qu, Z.; Yang, C.; Zhou, Q.; Jia, J. Nanosized cation-deficient Fe–Ti spinel: A novel magnetic sorbent for elemental mercury capture from flue gas. Appl. Mater. Interfaces 2011, 3, 209–217. [Google Scholar]
- Yang, S.; Yan, N.; Guo, Y.; Wu, D.; He, H.; Qu, Z.; Li, J.; Zhou, Q.; Jia, J. Gaseous elemental mercury capture from flue gas using magnetic nanosized (Fe3−xMnx)1-δO4. Environ. Sci. Technol. 2011, 45, 1540–1546. [Google Scholar]
- Granger, P.; Parlvulescu, V.I. Past and Present in DeNOx Catalysis—From Molecular Modeling to Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Finocchio, E.; Baldi, M.; Busca, G.; Pistarino, C.; Romezzano, G.; Bregani, F.; Toledo, G.P. A study of the abatement of VOC over V2O3–WO3–TiO2 and alternative SCR catalysts. Catal. Today 2000, H59, 261–268. [Google Scholar]
- Lazar, L.; Köser, H.; Balasanian, I.; Bandrabur, F. Catalytic destruction of aromatic VOCs on SCR-DeNOx commercial catalyst. Environ. Eng. Manag. J. 2007, 6, 13–20. [Google Scholar]
- Cao, Y.; Chen, B.; Wu, J.; Cui, H.; Smith, J.; Chen, K.C.; Chu, P.; Pan, P.W. Study of mercury oxidation by a selective catalytic reduction catalyst in a pilot-scale slipstream reactor at a utility boiler burning bituminous coal. Energy Fuels 2007, 21, 145–156. [Google Scholar]
- Zhuang, Y.; Laumb, J.; Liggett, R.; Holmes, M.; Pavlish, J. Impacts of acid gases on mercury oxidation across SCR catalyst. Fuel Process. Technol. 2007, 88, 929–934. [Google Scholar] [CrossRef]
- Straube, S.; Hahn, T.; Koeser, H. Adsorption and oxidation of mercury in tail-end SCR-DeNOx plants-Bench scale investigations and speciation experiments. App. Catal. B Environ. 2008, 79, 286–295. [Google Scholar] [CrossRef]
- Lee, W.C.; Srivastava, K.R.; Ghorishi, B.S.; Kaewowski, J.; Hastings, W.T.; Hirschi, C.J. Pilot-scale study of the effect of selective catalytic reduction catalysts on mercury speciation in Illinois and Powder River Basin coal combustion flue gases. J. Air Waste Manag. Assoc. 2006, 56, 643–649. [Google Scholar] [CrossRef]
- Senior, C. Mercury oxidation across SCRs in coal-fired power plants. In Proceedings ofMercury Control Technology R&D Program Review, Pittsburgh, PA, USA, 12–14 July 2005.
- Eswaran, S.; Stenger, H.G. Understanding mercury conversion in selective catalytic reduction (SCR) catalysts. Energy Fuels 2005, 19, 2328–2334. [Google Scholar] [CrossRef]
- Lee, W.C.; Srivastava, K.R.; Ghorishi, B.S.; Hastings, W.T.; Stevens, M.F. Investigation of selective catalytic reduction impact on mercury speciation under simulated NOx emission control conditions. J. Air Waste Manag. Assoc. 2004, 54, 1560–1566. [Google Scholar] [CrossRef]
- Hong, J.H.; Ham, W.S.; Kim, H.M.; Lee, M.S.; Lee, B.J. Characteristics of commercial selective catalytic reduction catalysts for the oxidation of gaseous elemental mercury with respect to reaction conditions. Korean J. Chem. Eng. 2010, 27, 1117–1122. [Google Scholar] [CrossRef]
- Eswaran, S.; Stenger, H.G. Effect of halogens on mercury conversion in SCR catalysts. Fuel Process. Technol. 2008, 89, 1153–1159. [Google Scholar] [CrossRef]
- Lee, W.C.; Serre, D.S.; Zhao, Y.; Lee, J.S.; Hastings, W.T. Mercury oxidation promoted by a selective catalytic reduction catalyst under simulated powder river basin coal combustion conditions. J. Air Waste Manag. Assoc. 2008, 58, 484–493. [Google Scholar] [CrossRef]
- Yang, H.M.; Pan, W.P. Transformation of mercury speciation through the SCR system in power plants. J. Environ. Sci. 2007, 19, 181–184. [Google Scholar] [CrossRef]
- Cao, Y.; Gao, Z.; Zhu, J.; Wang, Q.; Huang, Y.; Chiu, C.; Parker, B.; Chu, P.; Pan, W.P. Impacts of halogen addition in mercury oxidation, in a slipstream selective catalyst reduction (SCR), reactor when burning sub-bituminous coal. Environ. Sci. Technol. 2008, 42, 256–261. [Google Scholar]
- Senior, C. Oxidation of Mercury Across SCR Catalysts in Coal-Fired Power Plants Burning Low Rank Fuels. Final Report to U.S. DOE/NETL; U.S. Department of Energy Agreement No. DE-FC26-03NT41728; Reaction Engineering International: Salt Lake City, UT, USA, 2004. [Google Scholar]
- Sibley, F.A.; Dene, C.; Jimenez, A.; Hinton, W.S. Pilot scale studies on mercury oxidation by SCR catalyst. In Proceedings of Power Plant Air Pollutant Control “Mega” Symposium Symposium, Baltimore, MD, USA, 25–28 August 2008.
- Crocker, R.C.; Benson, A.S.; Laumb, D.J. SCR catalyst blinding due to sodium and calcium sulphate formation. Prepr. Pap. Am. Chem. Soc. Div. Fuel Chem. 2004, 49, 169–172. [Google Scholar]
- Wan, Q.; Duan, L.; Li, J.; Chen, L.; He, K.; Hao, J. Deactivation performance and mechanism of alkali (earth) metals on V2O5–WO3/TiO2 catalyst for oxidation of gaseous elemental mercury in simulated coal-fired flue gas. Catal. Today 2011, 175, 189–195. [Google Scholar]
- Guo, X.; Beutler, J.; Anderson, C.; Nackos, A.; Ashton, J.; Bartholomew, C.; Hecker, W.; Baxter, L. Poisoning/Deactivation Study of V2O5/TiO2 SCR Catalysts. In Proceedings of ACERC Annual Conference, Provo, UT, USA, 26 February 2008.
- Staudt, E.J.; Engelmeyer, T.; Weston, H.W.; Sigling, R. The impact of arsenic on coal fired power plants equipped with SCR. In Presented at ICAC Forum, Houston, TX, USA, 12–13 February 2002.
- Thowarth, H. Trace Element Behavior in Pulverised Fuel Fired Power Plants—Impact of Fuels and Emission Control Technologies; Cuvillier Verlag: Göttingen, Germany, 2007. [Google Scholar]
- Senior, C.; Lignell, O.D.; Sarofim, F.A.; Mehta, A. Modelling arsenic partitioning in coal-fired power plants. Combust. Flame 2006, 1473, 209–221. [Google Scholar]
- Senior, C. Oxidation of mercury across selective catalytic reduction catalysts in coal fired power plants. J. Air Waste Manag. Assoc. 2006, 56, 23–31. [Google Scholar] [CrossRef]
- Liu, J.; He, M.; Zheng, C.; Chang, M. Density functional theory study of mercury adsorption on V2O5(001) surface with implications for oxidation. Proc. Combust. Inst. 2011, 33, 2771–2777. [Google Scholar] [CrossRef]
- Madsen, K.; Jensen, D.A.; Frandsen, J.F.; Thogersen, R.J. A mechanistic Study on the Inhibition of the DeNOx Reaction on the Mercury Oxidation over SCR catalysts. In Proceedings of Air Quality VIII Conference, Arlington, VA, USA, 24–27 October 2011.
- Stolle, R.; Köser, H.; Gutberlet, H. Determination of Hg-oxidation-activity of SCR-DeNOx-catalysts. In Presented at VGB Workshop “Flue Gas Cleaning 2008”, Vilnius, Lithuania, 3–4 June 2008.
- Zeng, K.; Stolle, R.; Köser, H. Quecksilberoxidation an metalleoxiddotierten SCR-DeNOx-Katalysatoren. In Proceedings of ProzessNet Conference, Mannheim, Germany, 8–10 September 2009.
- Kai, K.; Kato, Y.; Kikkawa, H.; Imada, N.; Nagai, Y. New SCR Catalysts with Improved Mercury Oxidation for Bituminous Coal-Fired Boilers. In Proceedings of Air Quality Conference, Arlington, VA, USA, 24–27 September 2007.
- Kai, K.; Kato, Y. Catalysts for oxidation of metal mercury. WIPO Patent Application WO/2008/035773, 27 March 2008. [Google Scholar]
- Nochi, K.; Yonemura, M.; Kiyosawa, M. Mercury oxidation catalyst and method for producing the same. U.S. Patent Application 2011/0082028A1, 7 April 2011. [Google Scholar]
- Kai, K.; Kato, Y. Oxidation treatment of metallic mercury in flue gas using oxidation catalysts. JP Patent 2010/088984A, 22 April 2010. [Google Scholar]
- Kai, K.; Kato, Y.; Imada, N. Method and catalyst for oxidation treatment of mercury metal in flue gas. JP Patent 2010/088983A, 22 April 2010. [Google Scholar]
- Kai, K.; Kato, Y. Method for waste gas treatment with a catalyst. JP Patent 2009226238A, 8 October 2009. [Google Scholar]
- Nagayasu, T.; Nakashoji, H.; Imai, N. Multi-pollutant control capabilities of double contact flow scrubber (DCFS). VGB PowerTech 2011, 91, 68–73. [Google Scholar]
- Zhuang, Y.; Thompson, S.J.; Zxgarlicke, J.C.; Pavlish, H.J. Impact of calcium chloride addition on mercury transformations and control in coal flue gas. Fuel 2007, 86, 2351–2359. [Google Scholar] [CrossRef]
- Niksa, S.; Padak, B.; Krishnakumar, B.; Naik, V.C. Process Chemistry of Br addition to utility flue gas for Hg emissions control. Energy Fuels 2010, 24, 1020–1029. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, S.; Xiao, L.; Wang, Y.; Liu, Y.; Wu, Z. Titania nanotubes—A unique photocatalyst and adsorbent for elemental mercury removal. Catal. Today 2011, 175, 202–208. [Google Scholar]
- Snider, G.; Ariya, P. Photo-catalytic oxidation reaction of gaseous mercury over titanium dioxide nanoparticle surfaces. Chem. Phys. Lett. 2010, 491, 23–28. [Google Scholar] [CrossRef]
- Jeon, H.S.; Eom, Y.; Lee, G.T. Photocatalytic oxidation of gas-phase elemental mercury by nanotitanosilicate fibers. Chemosphere 2008, 71, 969–974. [Google Scholar] [CrossRef]
- Guo, Y.; Yan, N.; Yang, S.; Qu, Z.; Wu, Z.; Liu, Y.; Liu, P.; Jia, J. Conversion of elemental mercury with a novel membrane delivery catalytic oxidation system (MDCOs). Environ. Sci. Technol. 2011, 45, 706–711. [Google Scholar]
- Granite, J.E.; Freeman, C.M.; Hargis, A.R.; O’Dowd, J.W.; Pennline, H.W. The thief process for mercury removal from flue gas. J. Environ. Manag. 2006, 84, 628–634. [Google Scholar]
- Dunham, E.G.; DeWall, A.R.; Senior, L.C. Fixed-bed studies of the interactions between mercury and coal combustion fly ash. Fuel Process. Technol. 2003, 82, 197–213. [Google Scholar] [CrossRef]
- Claudino, A.; Soares, J.L.; Moreira, M.P.F.R.; José, J.H. Adsorption equilibrium and breakthrough analysis for NO adsorption on activated carbons at low temperatures. Carbon 2004, 42, 1483–1490. [Google Scholar] [CrossRef]
- Ochiai, R.; Uddin, A.Md.; Sasaoka, E.; Wu, S. Effects of HCl and SO2 concentration on mercury removal by activated carbon sorbents in coal-derived flue gas. Energy Fuels 2009, 23, 4734–4739. [Google Scholar] [CrossRef]
- Presto, A.A.; Granite, E.J. Impact of sulfur oxides on mercury capture by activated carbon. Environ. Sci. Technol. 2007, 41, 6579–6584. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dranga, B.-A.; Lazar, L.; Koeser, H. Oxidation Catalysts for Elemental Mercury in Flue Gases—A Review. Catalysts 2012, 2, 139-170. https://doi.org/10.3390/catal2010139
Dranga B-A, Lazar L, Koeser H. Oxidation Catalysts for Elemental Mercury in Flue Gases—A Review. Catalysts. 2012; 2(1):139-170. https://doi.org/10.3390/catal2010139
Chicago/Turabian StyleDranga, Beatrice-Andreea, Liliana Lazar, and Heinz Koeser. 2012. "Oxidation Catalysts for Elemental Mercury in Flue Gases—A Review" Catalysts 2, no. 1: 139-170. https://doi.org/10.3390/catal2010139
APA StyleDranga, B.-A., Lazar, L., & Koeser, H. (2012). Oxidation Catalysts for Elemental Mercury in Flue Gases—A Review. Catalysts, 2(1), 139-170. https://doi.org/10.3390/catal2010139