Abstract
Bilirubin is a key component in the preparation of two traditional Chinese medicines: Calculus bovis sativus and Calculus bovis artifactus. Currently, industrial-scale production of bilirubin is limited to extraction from pig bile in a very low yield and its market price is very high, so it is important to develop an alternative method for producing bilirubin. This study developed a potential process for bilirubin production through biotransformation of biliverdin. The codon-optimized gene for biliverdin reductase (BVR) from Synechocystis PCC6803 was recombinantly expressed in Komagataella phaffii GS115, resulting in the genetically modified strain GS115-bvdR, which successfully expressed BVR with intracellular activity. Whole cells of GS115-bvdR were capable of transforming biliverdin to bilirubin in vitro. The overexpression conditions were optimized to enhance BVR production by GS115-bvdR, and the optimal conditions for the biotransformation of biliverdin into bilirubin using resting GS115-bvdR cells were established (pH 5.0 buffer, at 30 °C for 24 h, with 200 mg/L biliverdin). Under these conditions, a bilirubin concentration of 153 mg/L was achieved, with a conversion of 76.2% from biliverdin. These findings provide valuable insights for future studies on the biosynthesis of bilirubin through metabolic engineering.
1. Introduction
Bilirubin is an important mammalian metabolic product, mainly derived from the breakdown of hemoglobin in aging red blood cells. As red blood cells age, their membranes rupture and hemoglobin is engulfed and digested by macrophages, releasing heme. The porphyrin ring of heme is cleaved to form biliverdin IXα by heme oxygenase (HO, EC 1.14.99.3), which exists in multiple isoforms, including the well-characterized HO-1 and HO-2, as well as the more controversial HO-3, whose functional significance remains under investigation [1,2]. Among these, HO-1 is the predominant isoform. The term “biliverdin” typically refers specifically to this isomer. Biliverdin is subsequently reduced by biliverdin reductase (BVR, EC 1.3.1.24) to produce bilirubin [3]. The transformation of biliverdin into bilirubin by BVR is illustrated in Figure 1.
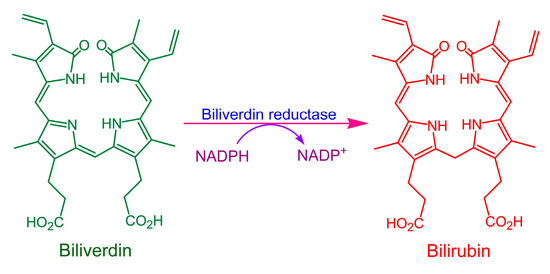
Figure 1.
Scheme of biliverdin reduction to bilirubin by biliverdin reductase (BVR).
Bilirubin was, until recently, considered a useless metabolic waste product, or even harmful to the human nervous system. If blood bilirubin concentrations become excessively high, it can accumulate in the brain, potentially causing diseases such as schizophrenia [4]; unconjugated bilirubin can also cause encephalopathy and kernicterus [5]. However, recent research has greatly improved our understanding of the beneficial effects of bilirubin on human health; bilirubin is a potent lipid-soluble antioxidant, capable of neutralizing various reactive oxygen species (ROS) [6]. When bilirubin levels in serum are normal or slightly elevated, it exerts several important physiological effects, including antioxidant [7], anti-inflammatory [8,9], immune regulatory [10], antiviral [11] and antiproliferative activities [12], so it is now regarded as the essential “yellow hormone” [13].
Bilirubin is a major component of Calculus bovis (bovine gallstones), with its content ranging from 50.4% to 56.8% [14]. C. bovis, also known as ox bezoar or Bezoar bovis, and Niuhuang in Chinese, is an important component of nearly one hundred traditional Chinese medicines documented in the 2020 edition of the Chinese Pharmacopoeia, including Angong Niuhuang Wan, Niuhuang Jiedu Wan, Niuhuang Shangqing Pian, and Niuhuang Qianjin San [15]. The Chinese Pharmacopoeia stipulates that the bilirubin content of natural C. bovis should be at least 25.0%. C. bovis typically occurs only in the gallbladders of aged and unhealthy oxen, but the mechanization of modern agriculture has led to a decline in the population of farmed oxen, resulting in a scarcity of natural C. bovis resources. Two alternatives have been developed as substitutes for natural C. bovis, Calculus bovis sativus (cultured in vitro) and Calculus bovis artifactus (artificially prepared), with bilirubin being the key component in both preparations. The bilirubin content of Calculus bovis sativus must be at least 35%, according to the Chinese Pharmacopoeia [15].
Currently, bilirubin is produced industrially by extraction from pig bile, but the bilirubin content of pig bile is relatively low, at approximately 0.06% (w/w). The shortage of raw materials and low extraction yields have caused ongoing supply shortages for the Chinese bilirubin market, causing the price to soar as high as RMB 60,000 (~USD 8500) per kilogram. Therefore, the development of cost-effective, alternative methods for bilirubin production could provide significant economic benefits.
The chemical synthesis of bilirubin has been extensively studied, but this method is not only low-yielding but also produces a product consisting of a mixture of four bilirubin isomers: IXα, IXβ, IXγ, and IXδ. An effective method for separating these isomers is not currently available, which has rendered the application of chemical synthesis for industrial bilirubin production both impractical and uneconomic. However, biosynthesis has emerged as a very effective production method for the pharmaceutical industry [16], suggesting that the biosynthetic production of bilirubin could be a viable alternative to current bilirubin production methods.
Heme is a natural precursor to bilirubin, which is produced in large quantities from animal blood, such as that of pigs and cows, and is available at a relatively low market price. Enzymic biocatalysis is a promising method for producing bilirubin from heme, but this approach faces several challenges. Recombinant Escherichia coli, which expresses HO-1 and BVR, is not a practical whole-cell biocatalyst; heme cannot easily enter the cells to interact with the enzymes because the E. coli cell membrane lacks heme transporters [17,18], resulting in low biocatalytic efficiency. Using free HO-1 and BVR for the biotransformation of heme into bilirubin is severely hampered by the low solubility of bilirubin in water and the requirement for NADPH as a cofactor for BVR, which must be added in large quantities at considerable costs or regenerated enzymically, also resulting in low biocatalytic efficiency.
Biliverdin, the immediate precursor of bilirubin, is available from natural sources, such as birds’ eggshells and the bile of birds [19,20] and fish [21]. Although the biliverdin concentration is relatively low (e.g., ~0.04% in chicken bile), this abundant natural resource has not yet been fully utilized. Biliverdin can serve as a substrate for bilirubin production, but the cost of extracting biliverdin from animal tissues may be high, making bilirubin produced from biliverdin less cost-competitive than that extracted from pig bile. Therefore, this method is unlikely to replace the extraction of bilirubin from pig bile. However, it could serve as an alternative method for producing bilirubin and increase overall bilirubin production, thereby reducing its market price. Biliverdin can be converted into bilirubin by chemical or biological processes, but the biological method has advantages, including high efficiency, excellent selectivity, mild reaction conditions, and much greater environmental sustainability.
Our previous study reported the biocatalytic production of bilirubin from biliverdin using recombinant E. coli expressing BVR; a starting biliverdin concentration of 450 mg/L resulted in a conversion of 72.3%, with a final bilirubin content of 20.8 mg/g in the dried cells [22]. However, the bilirubin yield from this method is not high enough for it to be a viable industrial process. The use of yeast cells appears to offer advantages, such as larger biomass and greater availability of endogenously produced NADPH compared to E. coli cells, suggesting that using BVR-expressing yeast cells to transform biliverdin into bilirubin would be more efficient. In this study, a recombinant Komagataella phaffii (previously known as Pichia pastoris) strain expressing intracellularly active BVR was constructed, which can produce bilirubin through the biotransformation of biliverdin.
2. Results and Discussion
2.1. Identification of the Recombinant K. phaffii
The genomic DNA of K. phaffii strains GS115 (the starting strain), GS115-pPICZB (GS115 transformed with the empty pPICZB vector), and GS115-bvdR (GS115 transformed with Synechocystis PCC6803 bvdR) was extracted for use as PCR templates. The sequences of 5′-AOX and 3′-AOX were used as PCR primers, and the resulting PCR products were analyzed by agarose gel electrophoresis (Figure 2).
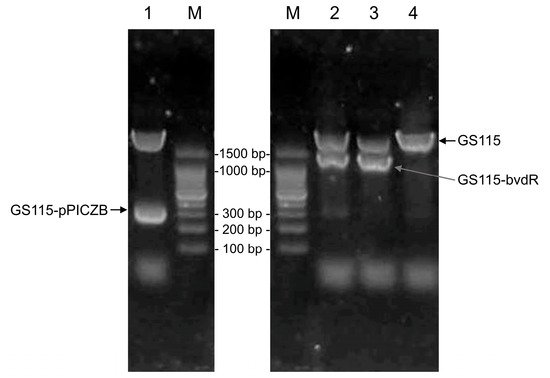
Figure 2.
Electrophoresis of PCR products from the genomic DNA of the strains GS115, GS115-pPICZB, and GS115-bvdR. Lane 1: GS115-pPICZB; Lanes 2 and 3: GS115-bvdR; Lane 4: GS115; Lane M: DNA marker.
The band above 1500 bp in Lanes 1–4 of Figure 2 represents a PCR product (~2200 bp) derived from the genomic DNA of K. phaffii GS115. In Lane 1, the band of ~300 bp matches the predicted size of the PCR product from the empty pPICZB transformants (325 bp); this band was absent in Lane 4, as GS115 does not contain pPICZB. In Lanes 2 and 3, the band between 1000 and 1500 bp corresponds to the predicted size of the GS115-bvdR PCR product (1212 bp). The DNA from this band was recovered, sequenced, and analyzed using BLAST on the NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The sequence was completely consistent with that of the target gene (Gene ID: 14615643), indicating the successful construction of GS115-bvdR.
2.2. Expression of BVR in Recombinant K. phaffii
After culturing in buffered methanol-complex medium (BMMY) and methanol induction, the cells of GS115-pPICZB and GS115-bvdR were lysed and analyzed using SDS-PAGE (Figure 3).
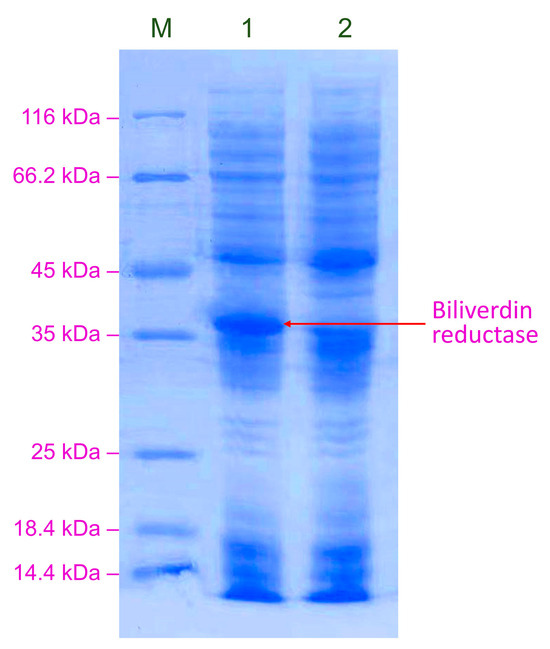
Figure 3.
SDS-PAGE analysis of BVR expressed in GS115-bvdR. Lane M: protein marker; Lane 1: GS115-bvdR; Lane 2: GS115-pPICZB (control).
GS115-bvdR expressed a protein with a molecular weight of between 35 and 40 kDa, consistent with the predicted molecular weight of BVR (36.3 kDa) (Figure 3, Lane 1), which was absent from GS115-pPICZB (Lane 2), indicating the successful construction of the recombinant strain GS115-bvdR, expressing BVR.
The BVR activity was 0.783 ± 0.021 U/mL in the cell lysate, which is equivalent to 0.157 ± 0.004 U/mL in the BMMY culture cell suspension (the cell lysate was concentrated × 5 compared with the cell suspension).
2.3. Biotransformation of Biliverdin by Recombinant K. phaffii
GS115-pPICZB and GS115-bvdR cells harvested from a 25 mL culture were resuspended in sodium citrate buffer (pH 6.0), containing 50 mg/L biliverdin, and then biotransformed at 30 °C for 16 h (Figure 4).

Figure 4.
Reaction mixtures after treatment of biliverdin with GS115-pPICZB (a) and GS115-bvdR (b) resting cells.
After the treatment of biliverdin (green) with GS115-pPICZB cells, the reaction mixture remained green, indicating that no biotransformation occurred (Figure 4). After treatment with GS115-bvdR, however, the reaction mixture turned yellow, indicating that the expressed BVR can transform biliverdin into a yellow substance, presumed to be bilirubin, which accumulated in the cells.
The clear yellow extract of the GS115-bvdR cells was analyzed by HPLC (Figure 5); the retention time of the yellow substance in the extract was consistent with that of the bilirubin standard. LC-ESI-MS analysis in negative ion mode (Figure 6) found the m/z value of the molecular ion of the yellow substance, [M-H]−, to be 583.9, consistent with the molecular mass of bilirubin (C33H36N4O6, molecular weight: 584.7). Therefore, biliverdin was successfully biotransformed into bilirubin by GS115-bvdR resting cells.
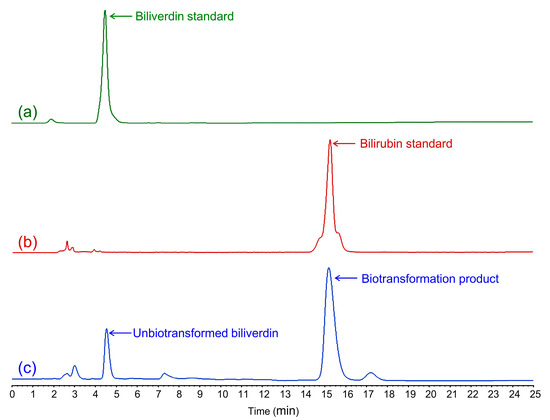
Figure 5.
HPLC analysis of cell extracts after GS115-bvdR biotransformation of biliverdin: (a) biliverdin standard; (b) bilirubin standard; (c) GS115-bvdR cell extracts.
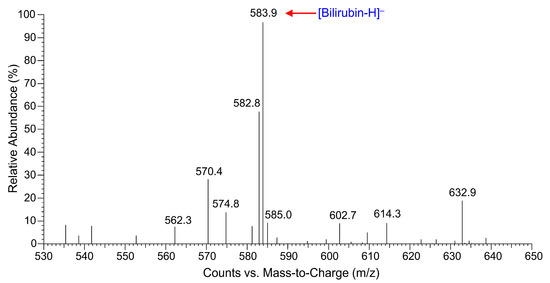
Figure 6.
LC-ESI-MS spectrum of biliverdin biotransformation product, confirming its identity as bilirubin.
2.4. Optimization of Overexpression Conditions for BVR
The concentration of the inducer (methanol), the induction culture time, the pH of the BMMY, and the concentration of sorbitol supplied for induction were optimized for the overexpression of BVR in K. phaffii GS115-bvdR. The optimal conditions for BVR expression were found to be BMMY at pH 6.0, with the addition of 1% v/v methanol and a 1.25% molar ratio of sorbitol to methanol every 24 h during the induction culture. The BVR overexpression process was then performed under these conditions. After three days of induction culture, the activity of BVR in the cell lysate was 0.906 ± 0.013 U/mL, equivalent to the 0.181 ± 0.003 U/mL activity in the GS115-bvdR culture and a 15.7% increase compared with the unoptimized conditions. SDS-PAGE analysis of BVR produced by GS115-bvdR, cultured under the optimized conditions (Figure 7), shows a markedly more intense BVR band than that shown in Figure 3.
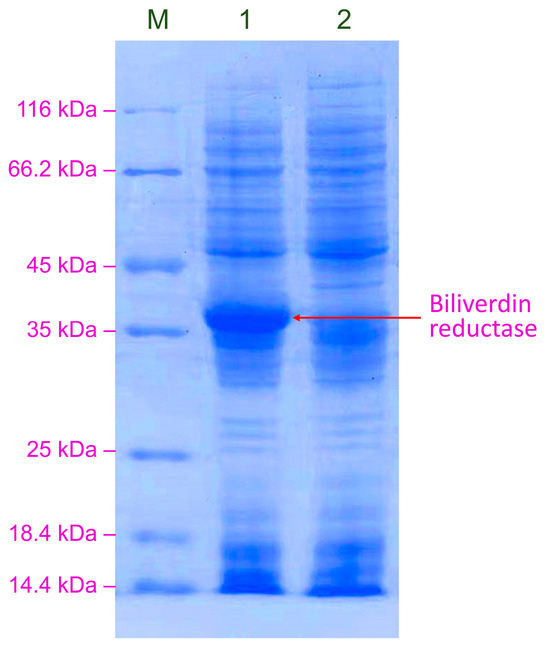
Figure 7.
SDS-PAGE analysis of BVR expressed by GS115-bvdR under optimized overexpression conditions. Lane M: protein marker; Lane 1: GS115-bvdR; Lane 2: GS115-pPICZB.
The intensity of the BVR band in the SDS-PAGE gel indicates that BVR is efficiently expressed in yeast cells and would be expected to have higher activity. However, the activity of BVR did not exceed that observed in E. coli, which achieved an activity of 0.183 ± 0.007 U/mL in culture [22]. This unexpectedly low activity may be related to the biliverdin BVR gene being derived from the prokaryotic organism Synechocystis. The recombinant protein may undergo post-translational modifications when expressed in the eukaryotic organism K. phaffii, which could interfere with its correct folding into the conformation necessary for optimal activity. Therefore, it is essential to test additional BVR genes from alternative cellular sources for expression in yeast cells in future studies.
2.5. Optimization of Biliverdin-to-Bilirubin Biotransformation Conditions
First, a suitable water-miscible organic co-solvent for facilitating the dissolution of water-insoluble biliverdin in the biotransformation cell suspension was selected. An excessive concentration of organic solvent would inhibit the BVR activity, limiting the practical biotransformation efficiency. Therefore, 20% v/v methanol and dimethyl sulfoxide (DMSO) in the resting GS115-bvdR cell suspension, with different biliverdin concentrations (50, 100, 150, 200 and 300 mg/L), were compared as solvents for biliverdin. The biotransformation was conducted at 30 °C in a shaking incubator for 24 h. The final bilirubin concentration and the conversion of biliverdin were determined (Figure 8a).
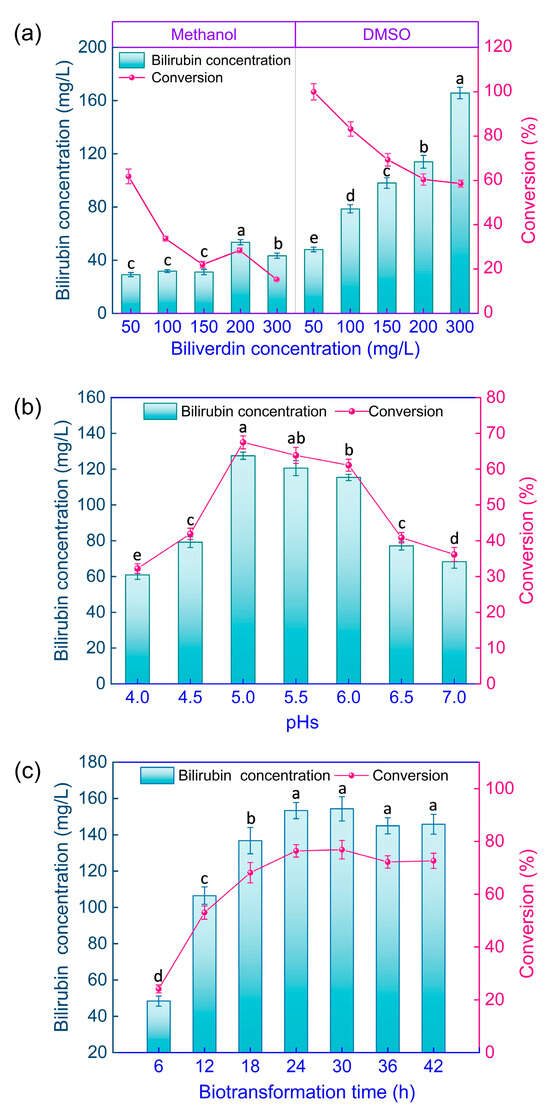
Figure 8.
Optimization of conditions for biotransformation of biliverdin to bilirubin by GS115-bvdR resting cells. Differences in final bilirubin concentration and conversion (a) with methanol or DMSO as co-solvent; (b) at different medium pH values; and (c) after different biotransformation times. Vertical bars represent the mean ± standard error. Different letters on the bars indicate that the values are significantly different at the p < 0.05 level.
At a biliverdin concentration of 50 mg/L, the conversion with methanol as a co-solvent was 61.8% (Figure 8a), whereas that with DMSO was ~100%; at 300 mg/L biliverdin, the conversion was 15.3% and 58.5%, respectively, so DMSO is clearly a superior co-solvent.
The time course for bilirubin production from biliverdin using GS115-bvdR, in the presence of 200 mg/L biliverdin and 20% v/v DMSO and at pH 5.0, was determined (Figure 8c). The final bilirubin concentration and the conversion increased with time; both parameters peaked and then plateaued after 24 h, with the conversion being 76.2% at this time. Therefore, the optimal biotransformation time was 24 h; at this point, the final bilirubin concentration was 153 mg/L and the bilirubin content of the dry cells was 2.92 mg/g.
Previously, recombinant E. coli expressing BVR derived from Synechocystis PCC6803 was used to biotransform biliverdin; the conversion of bilirubin from 450 mg/L biliverdin was 72.3% and the bilirubin content in dried E. coli cells was 20.2 mg/g [22]. Although a similar conversion was achieved, the recombinant K. phaffii expressing BVR used here did not outperform the recombinant E. coli.
Future research should aim to increase the biocatalytic efficiency of recombinant K. phaffii cells expressing BVR. One potential approach is to express BVR from various eukaryotic sources in K. phaffii, which may improve the enzyme activity of the recombinant yeast. Another approach is to conduct the biotransformation tests in a stirred batch fermenter. Culturing recombinant yeast in fermenters allows for improved oxygenation, a continuous supply of methanol, and consistent medium feeding. Both the enzyme activity and biomass of the recombinant yeast should be increased, which could enhance the conversion of bilirubin from biliverdin.
3. Materials and Methods
3.1. Strains and Plasmids
The K. phaffii strain GS115 and plasmid pPICZB were obtained from Yinyuan Biotech Co., Ltd. (Hangzhou, China). E. coli DH5α was obtained from Beijing Tsingke Biotech Co., Ltd. (Beijing, China).
3.2. Reagents, Enzymes, and Test Kits
The Rapid Yeast Genomic DNA Isolation Kit, Plasmid DNA Mini-prep Kit, SDS-PAGE Preparation Kit, DNA Gel Extraction Kit, primers of 5′-AOX and 3′-AOX (sequences shown in Supplementary Data: Table S1), DNA, and protein markers were obtained from Sangon Biotech Co., Ltd. (Shanghai, China). DNA restriction enzymes (EcoR I, Sal I., and Sac I) were obtained from Takara Biomedical Technology Co., Ltd. (Beijing, China). TS GelRed nucleic acid dye and T5 super PCR mix were obtained from Beijing Tsingke Biotech Co., Ltd. (Beijing, China). Standard biliverdin hydrochloride (≥95%) and bilirubin (≥98%) were obtained from Aladdin Reagent Co., Ltd. (Shanghai, China). Biliverdin substrate was prepared from chicken bile in our laboratory, with a purity of 33.7%. Zeocin was obtained from Thermo Fisher Scientific Inc. (Carlsbad, CA, USA), and all other reagents were commercially available at analytical or biological grades.
3.3. Culture Media
The culture media used were as follows: Luria–Bertani medium (LB): 10 g/L peptone, 5 g/L yeast extract, 10 g/L NaCl, and pH 7.0; yeast extract peptone dextrose medium (YPD): 10 g/L yeast extract, 20 g/L peptone, 20 g/L dextrose, and pH 7.2; buffered glycerol-complex medium (BMGY): 20 g/L peptone, 10 g/L yeast extract, and 10 g/L glycerol; yeast nitrogen base (YNB): total volume of 13.4 g/L, with 0.4 mg/L biotin, prepared at pH 5.0, and 0.1 mol/L sodium citrate buffer; buffered methanol-complex medium (BMMY): 20 g/L peptone, 10 g/L yeast extract, 10 mL/L methanol, 13.4 g/L YNB, and 0.4 mg/L biotin, prepared at pH 5.0, and 0.1 mol/L sodium citrate buffer.
3.4. Recombinant Plasmid Constructs
The DNA sequence encoding BvdR (bvdR, Gene ID: 14615643 (sequence shown in Supplementary Material: Table S1) from Synechocystis 6803, retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov, accessed on 15 April 2022), was optimized using K. phaffii-preferred codons (GenSmartTM, https://www.genscript.com.cn/tools/gensmart-codon-optimization, accessed on 18 April 2022; the sequence is shown in Supplementary Material: Table S1). The EcoR I and Sal I digestion sites were introduced upstream (5′-GAATTC-3′) and downstream (5′-GTCGAC-3′), respectively. The synthesized (by Beijing Qingke Biotech Co., Ltd., Beijing, China) bvdR sequence and vector pPICZB were digested with EcoR I and Sal I, respectively. They were then ligated using the T4 DNA ligase and transformed into E. coli DH5α-competent cells. The recombinant E. coli was cultured on LB plates containing zeocin (50 μg/mL). The colonies were then picked to verify the positive transformants by PCR. The recombinant vector pPICZB-bvdR (plasmid map in Figure 9) was extracted from the positive transformants after being cultured in LB medium containing zeocin (50 μg/mL).
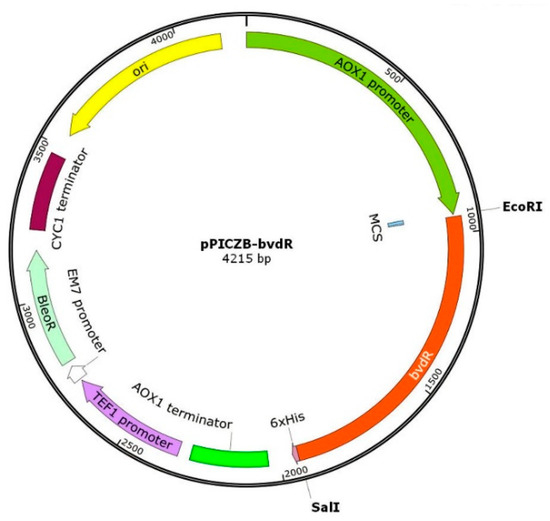
Figure 9.
Map of the recombinant plasmid pPICZB-bvdR.
3.5. K. phaffii Transformation
The plasmid pPICZB-bvdR was linearized using the Sac I restriction enzyme. A mixture of pPICZB-bvdR (20 μL) and a GS115-competent cell suspension (80 μL) was transferred into a pre-cooled electroporation cuvette (0.2 cm type). After cooling on ice for 5 min, the cells were subjected to an electric pulse of 4 ms in the “Fungi Pichia” mode using an electroporation device (MicroPulser 1652100, Bio-Rad, Philadelphia, PA, USA). Following electroporation, pre-cooled sorbitol solution (1 mL, 1 mol/L) was immediately added to the cuvette, and the cell suspension was transferred to a 1.5 mL centrifuge tube, revived in a water bath at 30 °C for 2 h, and then plated on YPD agar plates containing 50 mg/L zeocin. The plates were incubated at 30 °C for 48 h. Single colonies on the YPD agar plates were picked and inoculated individually in YPD liquid medium (5 mL, containing 50 mg/L zeocin). The cultures were incubated at 30 °C for 24 h, after which their genomic DNA was extracted. The target genes were amplified by PCR using the 5′-AOX and 3′-AOX primers, and then the PCR products were recovered and sequenced for verification. The recombinant K. phaffii GS115 strain carrying bvdR was designated as GS115-bvdR. A control strain containing the empty vector, GS115-pPICZB, was produced similarly.
3.6. Expression and SDS-PAGE Analysis of BVR Protein
The recombinant GS115-bvdR cell suspension (1 mL), which had been stored in a 20% glycerol solution at −80 °C, was inoculated in BMGY (50 mL) and then cultured at 30 °C and 180 r/min for 16 h until the OD600 reached ~6.0. The culture was then transferred into a 100 mL sterile centrifuge tube and centrifuged at 5000× g for 5 min. The supernatant was discarded, and BMMY (25 mL) was added to resuspend the cells. The suspension was transferred into a 250 mL sterile conical flask and cultured at 30 °C and 200 r/min for 3–5 d to reach an OD600 of 10–12. Methanol was added every 24 h to a final concentration of 1% v/v.
An aliquot (100 μL) of the GS115-bvdR culture was centrifuged at 8500× g for 5 min. The supernatant was discarded, and the yeast cells were resuspended in 50 μL of sodium citrate buffer (1 mL, pH 6.0, 0.1 mol/L). The cells were lysed using a freeze–thaw method, which involved boiling the cell suspension for 5 min, freezing it in liquid nitrogen for 10 min, and then boiling it again for 5 min. This process was repeated three times. Lysate (16 μL) and 5 × protein loading buffer (4 μL) were mixed, heated in boiling water for 10 min, and analyzed by SDS-PAGE [23].
3.7. BVR Enzymic Activity Determination
The GS115-bvdR culture (5 mL) was centrifuged at 8500× g for 5 min, the supernatant was discarded, and sodium citrate buffer (1 mL, pH 6.0, 0.1 mol/L) was added to resuspend the cells. The yeast cells were ultrasonically disrupted and then centrifuged at 8500× g for 10 min at 4 °C; then the supernatant was used for BVR activity assays, as described previously, with some modifications [24].
The BVR assay solution consisted of biliverdin (40 μL of a 0.15 mmol/L methanolic solution), sodium citrate buffer at pH 6.0 (100 μL, 0.1 mol/L), BSA solution (40 μL, 10 g/L), NADPH solution (120 μL, 20 mmol/L), and GS115-bvdR cell lysate (700 μL) in a 1.5 mL centrifuge tube, incubated at 30 °C for 30 min. The A450 of the reaction mixture was measured and the bilirubin concentration was calculated using a bilirubin standard curve at 450 nm. BVR activity is expressed as the amount of enzyme (in mL) required to produce 1 μg of bilirubin per minute at 30 °C and pH 6.0.
3.8. Biotransformation of Biliverdin to Bilirubin by Recombinant Yeast Cells
A GS115-bvdR resting culture (25 mL) was centrifuged at 5000× g for 5 min to collect the cells, which were then resuspended in sodium citrate buffer (pH 6.0, 4.0 mL, 0.1 mol/L). Biliverdin solution in methanol or DMSO (1.0 mL) was added to the resting cell suspension and then shaken at 30 °C for an appropriate time.
3.9. Analysis of Bilirubin
The biotransformation mixture was centrifuged at 8500× g for 10 min, the cell pellet was retained, and the cells were washed twice with deionized water and resuspended in methanol/dichloromethane (10 mL, 2:1 v/v). The extraction was performed by ultrasonication at 0 °C and 100 W for 30 min, and then the extract was centrifuged at 8500× g for 5 min, retaining the supernatant. The supernatant was analyzed by high-performance liquid chromatography (HPLC) after being filtered through a 0.22 μm membrane.
Bilirubin was quantified by HPLC using a Phenomenex C18 column (Torrance, CA, USA, 5 µm, 150 mm × 4.6 mm) fitted to a Shimadzu LC-20A System (Shimadzu, Kyoto, Japan). The mobile phase consisted of acetonitrile/water (95:5 v/v) containing 0.1% acetic acid at a flow rate of 1 mL/min. The column temperature was 30 °C and the detection wavelength was 450 nm.
The biotransformation product was confirmed as bilirubin by liquid chromatography–electrospray ionization–mass spectrometry (LC-ESI-MS) using an LCQ Deca XP Plus mass spectrometer (Thermo-Fisher, Waltham, MA, USA). The scanning range was m/z 530–650, the capillary temperature was 300 °C, and the capillary voltage was 4.0 kV.
The conversion of bilirubin from biliverdin was calculated as shown in Equation (1):
where 584.7 Da and 582.6 Da are the molecular weights of bilirubin and biliverdin, respectively.
3.10. Statistical Analysis
All experiments were conducted with three independent replicates, and the results were presented as the mean ± standard deviation (SD). The data were analyzed using analysis of variance (ANOVA). A significant difference was considered to occur at p < 0.05.
4. Conclusions
The BVR gene from Synechocystis PCC6803 was codon-optimized and successfully cloned into K. phaffii, resulting in the expression of the enzyme with intracellular activity. Following the optimization of expression conditions, enzyme activity increased from 0.157 ± 0.003 U/mL to 0.181 ± 0.003 U/mL. Utilizing whole cells of the genetically modified yeast as biocatalysts, biliverdin was effectively converted into the valuable compound bilirubin. During the biotransformation process, bilirubin accumulated in the cells, reaching up to 2.92 mg/g of dry cell weight, which is approximately 6.5 times greater than in the traditional source, pig bile (~0.6 mg/mL). This method presents a promising alternative for producing bilirubin instead of extracting it from animal bile. The findings of this study provide valuable insights for future research on the biosynthesis of bilirubin through metabolic engineering.
5. Patents
Chinese Patent CN 2023107666370 resulted from the work reported in this manuscript.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/catal15080766/s1: Table S1: DNA sequences used in this study.
Author Contributions
Conceptualization, investigation, writing—original draft, H.C.; methodology, investigation, visualization, S.Z.; validation, investigation, Y.H.; resources, data curation, W.K.; writing—review and editing, supervision, project administration, funding acquisition, J.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Key Research and Development Program of Zhejiang Province (Grant No. 2021C03088-2).
Data Availability Statement
The data within the article are available from the corresponding author upon reasonable request.
Acknowledgments
The authors gratefully acknowledge the financial support from Shanghai Hebutong Technology Co., Ltd.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
References
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Nogchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [CrossRef]
- Pink, R.C.; Wicks, K.; Caley, D.P.; Punch, E.K.; Jacobs, L.; Carter, D.R. Pseudogenes: Pseudofunctional or key regulators in health and disease? RNA 2011, 17, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.; Blanco, G. Chapter 17–Heme metabolism. In Medical Biochemistry, 2nd ed.; Blanco, A., Blanco, G., Eds.; Academic Press: New York, NY, USA, 2022; pp. 437–448. [Google Scholar] [CrossRef]
- Coradduzza, D.; di Lorenzo, B.; Sedda, S.; Nivoli, A.M.; Carru, C.; Mangoni, A.A.; Zinellu, A. Investigating bilirubin concentrations in schizophrenia: A systematic review and meta-analysis. Schizophr. Res. 2024, 271, 228–236. [Google Scholar] [CrossRef]
- Das, S.; van Landeghem, F.K.H. Clinicopathological spectrum of bilirubin encephalopathy/kernicterus. Diagnostics 2019, 9, 24. [Google Scholar] [CrossRef]
- Bin-Nun, A.; Mimouni, F.B.; Kasirer, Y.; Schors, I.; Schimmel, M.S.; Kaplan, M.; Hammerman, C. Might bilirubin serve as a natural antioxidant in response to neonatal encephalopathy? Am. J. Perinatol. 2018, 35, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Ziberna, L.; Martelanc, M.; Franko, M.; Passamonti, S. Bilirubin is an endogenous antioxidant in human vascular endothelial cells. Sci. Rep. 2016, 6, 29240. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, H.; Kang, S.; Lee, J.; Park, J.; Jon, S. Bilirubin nanoparticles as a nanomedicine for anti-inflammation therapy. Angew. Chem. Int. Ed. Engl. 2016, 55, 7460–7463. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Jeong, Y.; Kim, J.; Bae, H.; Son, S.; Kwak, S. The anti-inflammatory role of bilirubin on “two-hit” sepsis animal model. Int. J. Mol. Sci. 2020, 21, 8650. [Google Scholar] [CrossRef] [PubMed]
- Spetzler, V.N.; Goldaracena, N.; Kaths, J.M.; Marquez, M.; Selzner, N.; Cattral, M.S.; Greig, P.D.; Lilly, L.; McGilvray, I.D.; Levy, G.A.; et al. High preoperative bilirubin values protect against reperfusion injury after live donor liver transplantation. Transplant Int. 2015, 28, 1317–1325. [Google Scholar] [CrossRef]
- Shankaran, P.; Madlenakova, M.; Hajkova, V.; Jilich, D.; Svobodova, I.; Horinek, A.; Fujikura, Y.; Melkova, Z. Effects of heme degradation products on reactivation of latent HIV-1. Acta Virol. 2017, 61, 86–96. [Google Scholar] [CrossRef][Green Version]
- Ollinger, R.; Bilban, M.; Erat, A.; Froio, A.; McDaid, J.; Tyagi, S.; Csizmadia, E.; Graça-Souza, A.V.; Liloia, A.; Soares, M.P.; et al. Bilirubin: A natural inhibitor of vascular smooth muscle cell proliferation. Circulation 2005, 112, 1030–1039. [Google Scholar] [CrossRef]
- Vítek, L.; Tiribelli, C. Bilirubin: The yellow hormone? J. Hepatol. 2021, 75, 1485–1490. [Google Scholar] [CrossRef]
- Yu, Z.J.; Xu, Y.; Peng, W.; Liu, Y.J.; Zhang, J.M.; Li, J.S.; Sun, T.; Wang, P. Calculus bovis: A review of the traditional usages, origin, chemistry, pharmacological activities and toxicology. J. Ethnopharmacol. 2020, 254, 112649. [Google Scholar] [CrossRef]
- China Pharmacopoeia Committee. Pharmacopoeia of the People’s Republic of China (Part I); China Medical Science Press: Beijing, China, 2020. [Google Scholar]
- Wang, J.-T.; Shi, T.-T.; Ding, L.; Xie, J.; Zhao, P.-J. Multifunctional enzymes in microbial secondary metabolic processes. Catalysts 2023, 13, 581. [Google Scholar] [CrossRef]
- Fiege, K.; Frankenberg-Dinkel, N. Construction of a new T7 promoter compatible Escherichia coli Nissle 1917 strain for recombinant production of heme-dependent proteins. Microb. Cell Fact. 2020, 19, 190. [Google Scholar] [CrossRef]
- Huang, W.; Wilks, A. Extracellular heme uptake and the challenge of bacterial cell membranes. Annu. Rev. Biochem. 2017, 86, 799–823. [Google Scholar] [CrossRef] [PubMed]
- Morales, J. Eggshell biliverdin as an antioxidant maternal effect: Biliverdin as an antioxidant resource in oviparous animals. Bioessays 2020, 42, e2000010. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Xu, G.Y.; Liu, Z.Z.; Li, J.Y.; Yang, N. A study on eggshell pigmentation: Biliverdin in blue-shelled chickens. Poult. Sci. 2006, 85, 546–549. [Google Scholar] [CrossRef]
- Ding, Z.K.; Xu, Y.Q. Purification and characterization of biliverdin IXα from Atlantic salmon (Salmo salar) bile. Biochemistry (Moscow) 2002, 67, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Wu, X.; Zheng, S.; Yi, Y.; Wang, X.; Ying, G. Production of bilirubin by biotransformation of biliverdin using recombinant Escherichia coli cells. Bioprocess Biosyst. Eng. 2022, 45, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Brunelle, J.L.; Green, R. Chapter Twelve–One-dimensional SDS-Polyacrylamide Gel Electrophoresis (1D SDS-PAGE). In Methods in Enzymology; Lorsch, J., Ed.; Academic Press: New York, NY, USA, 2014; Volume 541, pp. 151–159. [Google Scholar] [CrossRef]
- Hayes, J.M.; Mantle, T.J. The effect of pH on the initial rate kinetics of the dimeric biliverdin-IXα reductase from the cyanobacterium Synechocystis PCC6803. FEBS J. 2009, 276, 4414–4425. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).