
Multi-Enzyme Synergy and Allosteric Regulation in the Shikimate Pathway: Biocatalytic Platforms for Industrial Applications



Abstract

1. Introduction
The Shikimate Pathway: A Central Hub for Aromatic Metabolism
2. Complex Regulation of DAHPS, Which Catalyzes the First Committed Step in Aromatic Amino Acid Biosynthesis
2.1. Evolutionary Distribution, Structural Classification, and Functional Significance of DAHPS
2.2. General Catalytic Mechanism and Active Site Architecture of DAHPS
2.3. Regulatory Mechanisms and Allosteric Control in DAHPS Enzymes
2.4. Enzyme Engineering of DAHPS
2.5. Shikimate Pathway Engineering Involving DAHPS
2.6. Targeting DAHPS in Antimicrobial Drug Discovery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Structural Classification | Allosteric Regulation | Engineering Efforts | Applications | Ref. |
|---|---|---|---|---|---|
| E. coli | Type I; Isoforms: AroF AroG AroH | AroF: Tyr (−) AroG: Phe (−) AroH: Trp (−) | shikimate production (yield 101 g/L) Precursor balancing (E4P/PEP) | [3,17,18,19] | |
| B. subtilis | Type II + CM fusion (AroH/AroQ) | CM substrate/product (−) | aroGD146N: Metabolic Engineering | Menaquinone-7 production (yield 281.4 mg/L) | [20,37] |
| M. tuberculosis | Type II + CM Large subunit, Tetrameric | Inter-enzyme Allostery Phe/Tyr (−) Trp (+) | d-amino acid resistance studies Allosteric hotspot mapping | antimicrobial target | [14,15,28] |
| C. glutamicum | Type II variant | DAHPS activates CM through inter-enzyme allostery | Bypass gene knockouts (qsuB, pyk1) Coordinated pathway engineering | Shikimate production (yield 37 g/L) with bioprocess optimization (yield 141 g/L) | [35,38,39] |
| T. maritima | Type Iβ thermostable | N-terminal ferredoxin-like (FL) regulatory domain. | Thermostability engineering; enhanced thermal adaptation | Proposed thermophilic bioprocessing | [24,25] |
| Geobacillus sp. | Type Iβ + CM fusion | Intramolecular control DAHPS Phe (−) Coordinated with CM activity (+) | Domain interface engineering Sterically controlled regulation | [31] | |
| A. thaliana | Ferredoxin/thioredoxin-mediated control Thioredoxin reduction (+) | DAHPSD154N Reduced inhibitor binding affinity | Plant metabolic engineering; enhanced secondary metabolite production | [21,22,23] | |
| Synechocystis sp. PCC 6803 | Cyanobacterial Type I | Engineered light-dependent control | >30% CO2 redirection to shikimate pathway | Trans-cinnamic acid production (yield 138 mg/L) Carbon-neutral biosynthesis | [40] |
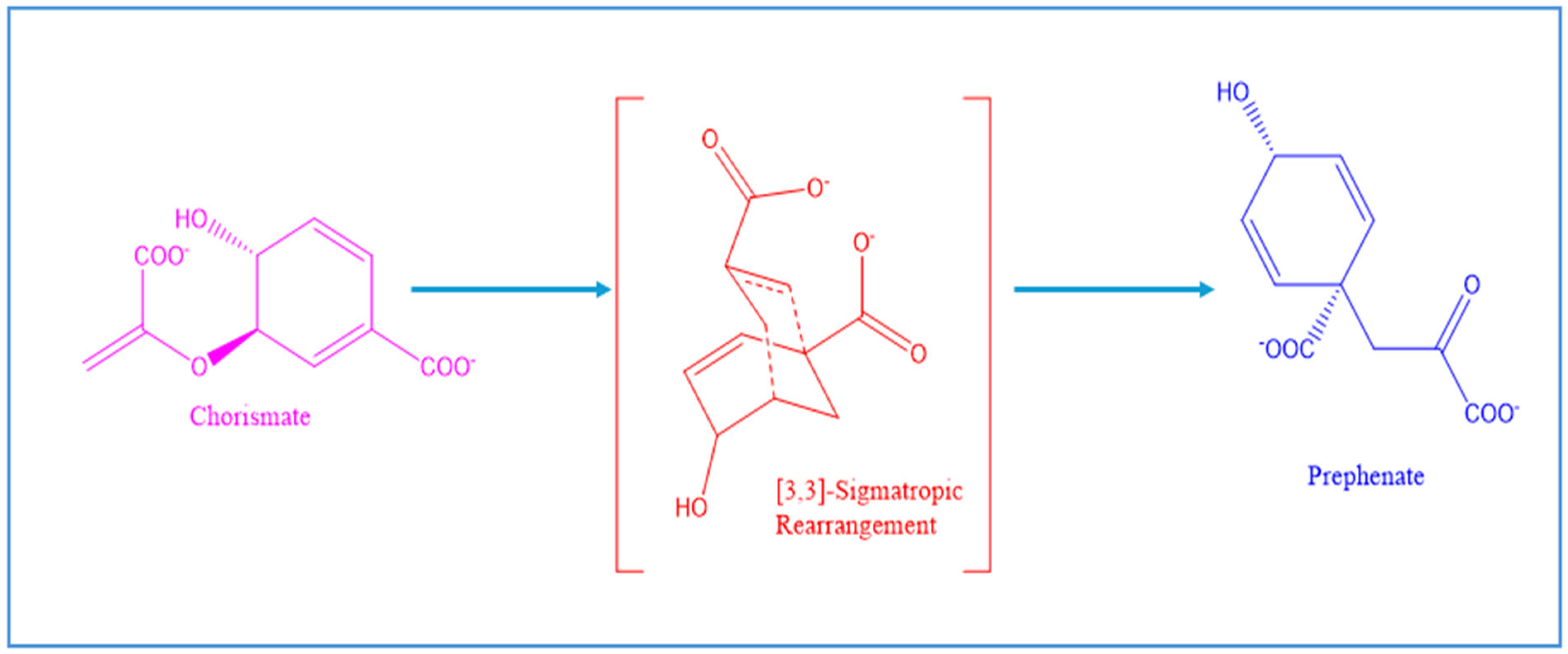
3. Structural Diversity of the Branch-Point Enzyme Chorismate Mutase Provides Insights into Allosteric Regulation and Enzyme Engineering
3.1. Structural Classification and Phylogenetic Distribution of CM Enzymes
3.2. Conserved Catalytic Mechanisms of CM Enzymes Across Structural Families
3.3. Allosteric Regulation in CM as a Metabolic Control Mechanism
3.4. Directed Evolution of CM Enzymes
3.5. Engineering CM: From Protein Design to Industrial Applications
3.6. Allostery in Antimicrobial Therapeutics
| Organism | Structural Classification | Allosteric Regulation | Engineering Efforts | Applications | Ref. |
|---|---|---|---|---|---|
| E. coli | AroQα | Multi-enzyme optimization-TyrC + CM domain (PheACM) | l-Tyrosine production (3 g/L, 66 mg/g glucose, 46–48% improvement) | [60] | |
| M. tuberculosis | AroQδ homodimer with DAHPS complex | Phe (−) | Directed evolution-T52P/V55D loop stabilization, 12-fold catalytic enhancement | Hyperactive variants; validated as a drug target | [54,56,65] |
| S. cerevisiae | AroQ dimer | Trp (+), Tyr (−) | Allosteric reengineering-Interface modifications | Controllable dimerization, programmable regulation | [7,8,46,53] |
| S. cerevisiae (pathway engineered) | Multi-modular systems-ARO7 overexpression + pathway integration | Production of salidroside (yield 26.5 g/L), tyrosol (yield 9.9 g/L) nutraceuticals | [59] | ||
| Arabidopsis thaliana | AtCM1-3 Complex multi-domain architecture | Trp (+), Phe/Tyr (−) (AtCM1) | Multi-isoform analysis-AtCM1, AtCM2, AtCM3 characterization | Controllable biocatalytic systems | [55] |
| C. glutamicum | AroQα Homodimer | Trp bound DAHPS (+) | feedback-resistant CS + chorismate lyase (ubiC) | Amino acid production; 4-Hydroxybenzoic acid production (36.6 g/L, 47.8% C-mol yield) | [30,58] |
4. Engineering of the Channeling Enzyme Tryptophan Synthase Provides Catalytic Utility
4.1. Catalytic Mechanism and Indole Channeling in Tryptophan Synthase
4.2. Evolutionary Distribution and Regulation of TrpS Enzymes
4.3. Allosteric Regulation and Communication Networks in TrpS
4.4. Directed Evolution of TrpS: From Allosteric Regulation to Industrial Biocatalysis
4.5. Engineering the α-Subunit: Decoupling Allosteric Networks
4.6. Recapitulating Allosteric Activation in Stand-Alone β-Subunit
4.7. Engineering of the TrpS Complex
4.8. Pathway Engineering for Biosynthesis of Tryptophan and Derivatives
4.9. Targeting TrpS in Pathogenic Bacteria with Novel Antimicrobials
| Source Organism(s) | Variant | Application | Ref. |
|---|---|---|---|
| E. coli | TrpSV231A/K382G | l-5-hydroxytryptophan synthesis | [92] |
| E. coli and M. aminisulfidivorans | TrpS and MaFMOD197E | Indigo production | [106] |
| P. furiosus | TrpSβmutants | ncAAs production; β-methyltryptophan synthesis | [84] |
| T. maritima | TmTrpS | Synthesis of blue, fluorescent amino acid 4-cyanotryptophan (78% yield) | [103] |
| P. furiosus | PfTrpSβ7E6 | Synthesis of β-branched amino acids; 5-substituted tryptophan analogues | [86] |
| P. furiosus | PfTrpSβquat | Synthesis of alkylation of 3-substituted oxindoles, ncAAs construction with quaternary stereocenters | [91] |
5. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrmann, K.M.; Weaver, L.M. The Shikimate Pathway. Annu. Rev. Plant Biol. 1999, 50, 473–503. [Google Scholar] [CrossRef]
- Wu, S.; Chen, W.; Lu, S.; Zhang, H.; Yin, L. Metabolic Engineering of Shikimic Acid Biosynthesis Pathway for the Production of Shikimic Acid and Its Branched Products in Microorganisms: Advances and Prospects. Molecules 2022, 27, 4779. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-N.; Seo, S.-Y.; Kim, H.-J.; Park, J.-H.; Park, E.; Choi, S.-S.; Lee, S.J.; Kim, E.-S. Artificial cell factory design for shikimate production in Escherichia coli. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab043. [Google Scholar] [CrossRef]
- Mao, J.; Zhang, H.; Chen, Y.; Wei, L.; Liu, J.; Nielsen, J.; Chen, Y.; Xu, N. Relieving metabolic burden to improve robustness and bioproduction by industrial microorganisms. Biotechnol. Adv. 2024, 74, 108401. [Google Scholar] [CrossRef]
- de Oliveira, M.D.; Araujo, J.d.O.; Galúcio, J.M.; Santana, K.; Lima, A.H. Targeting shikimate pathway: In silico analysis of phosphoenolpyruvate derivatives as inhibitors of EPSP synthase and DAHP synthase. J. Mol. Graph. Model. 2020, 101, 107735. [Google Scholar] [CrossRef]
- Shende, V.V.; Bauman, K.D.; Moore, B.S. The shikimate pathway: Gateway to metabolic diversity. Nat. Prod. Rep. 2024, 41, 604–648. [Google Scholar] [CrossRef]
- Winston, D.S.; Gorman, S.D.; Boehr, D.D. Conformational transitions in yeast chorismate mutase important for allosteric regulation as identified by nuclear magnetic resonance spectroscopy. J. Mol. Biol. 2022, 434, 167531. [Google Scholar] [CrossRef]
- Gorman, S.D.; Boehr, D.D. Energy and enzyme activity landscapes of yeast chorismate mutase at cellular concentrations of allosteric effectors. Biochemistry 2019, 58, 4058–4069. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.N.; Boehr, D.D. Allostery, engineering and inhibition of tryptophan synthase. Curr. Opin. Struct. Biol. 2023, 82, 102657. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, N.J.; Nazmi, A.R.; Hutton, R.D.; Webby, M.N.; Baker, E.N.; Jameson, G.B.; Parker, E.J. Complex Formation between Two Biosynthetic Enzymes Modifies the Allosteric Regulatory Properties of Both: An Example of Molecular Symbiosis. J. Biol. Chem. 2015, 290, 18187–18198. [Google Scholar] [CrossRef]
- Watkins-Dulaney, E.; Straathof, S.; Arnold, F. Tryptophan synthase: Biocatalyst extraordinaire. ChemBioChem 2021, 22, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.A. Tyrosine and Phenylalanine Biosynthesis: Relationship between Alternative Pathways, Regulation and Subcellular Location. In The Shikimic Acid Pathway; Conn, E.E., Ed.; Springer: Boston, MA, USA, 1986; pp. 57–81. [Google Scholar]
- Bentley, R. The shikimate pathway--a metabolic tree with many branches. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 307–384. [Google Scholar] [CrossRef]
- Webby, C.J.; Baker, H.M.; Lott, J.S.; Baker, E.N.; Parker, E.J. The Structure of 3-Deoxy-d-arabino-heptulosonate 7-phosphate Synthase from Mycobacterium tuberculosis Reveals a Common Catalytic Scaffold and Ancestry for Type I and Type II Enzymes. J. Mol. Biol. 2005, 354, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Fagbohunka, B.S.; Gauld, J.W. Influence of Cysteine 440 on the Active Site Properties of 3-Deoxy-d-Arabino-Heptulosonate 7-Phosphate Synthase in Mycobacterium tuberculosis (MtDAHPS). ACS Omega 2023, 8, 14401–14409. [Google Scholar] [CrossRef] [PubMed]
- Light, S.H.; Anderson, W.F. The diversity of allosteric controls at the gateway to aromatic amino acid biosynthesis. Protein Sci. 2013, 22, 395–404. [Google Scholar] [CrossRef]
- Ray, J.M.; Yanofsky, C.; Bauerle, R. Mutational analysis of the catalytic and feedback sites of the tryptophan-sensitive 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase of Escherichia coli. J. Bacteriol. 1988, 170, 5500–5506. [Google Scholar] [CrossRef]
- McCandliss, R.J.; Poling, M.D.; Herrmann, K.M. 3-Deoxy-D-arabino-heptulosonate 7-phosphate synthase. Purification and molecular characterization of the phenylalanine-sensitive isoenzyme from Escherichia coli. J. Biol. Chem. 1978, 253, 4259–4265. [Google Scholar] [CrossRef]
- Cui, D.; Deng, A.; Bai, H.; Yang, Z.; Liang, Y.; Liu, Z.; Qiu, Q.; Wang, L.; Liu, S.; Zhang, Y.; et al. Molecular basis for feedback inhibition of tyrosine-regulated 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase from Escherichia coli. J. Struct. Biol. 2019, 206, 322–334. [Google Scholar] [CrossRef]
- Pratap, S.; Dev, A.; Kumar, V.; Yadav, R.; Narwal, M.; Tomar, S.; Kumar, P. Structure of Chorismate Mutase-like Domain of DAHPS from Bacillus subtilis Complexed with Novel Inhibitor Reveals Conformational Plasticity of Active Site. Sci. Rep. 2017, 7, 6364. [Google Scholar] [CrossRef]
- Keith, B.; Dong, X.N.; Ausubel, F.M.; Fink, G.R. Differential induction of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase genes in Arabidopsis thaliana by wounding and pathogenic attack. Proc. Natl. Acad. Sci. USA 1991, 88, 8821–8825. [Google Scholar] [CrossRef]
- Kanaris, M.; Poulin, J.; Shahinas, D.; Johnson, D.; Crowley, V.M.; Fucile, G.; Provart, N.; Christendat, D. Elevated tyrosine results in the cytosolic retention of 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 2022, 109, 789–803. [Google Scholar] [CrossRef]
- Entus, R.; Poling, M.; Herrmann, K.M. Redox regulation of Arabidopsis 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase. Plant Physiol. 2002, 129, 1866–1871. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M. Evolution of DAHP Synthase: From Archaea to Eubacteria; University of Michigan: Ann Arbor, MI, USA, 2007. [Google Scholar]
- Shumilin, I.A.; Bauerle, R.; Wu, J.; Woodard, R.W.; Kretsinger, R.H. Crystal structure of the reaction complex of 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase from Thermotoga maritima refines the catalytic mechanism and indicates a new mechanism of allosteric regulation. J. Mol. Biol. 2004, 341, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.E.; Dunbar, B.; Hunter, I.S.; Nimmo, H.G.; Coggins, J.R. Evidence for a novel class of microbial 3-deoxy-D-arabino-heptulosonate-7-phosphate synthase in Streptomyces coelicolor A3(2), Streptomyces rimosus and Neurospora crassa. Microbiology 1996, 142 Pt 8, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Heyes, L.C.; Reichau, S.; Cross, P.J.; Jameson, G.B.; Parker, E.J. Structural analysis of substrate-mimicking inhibitors in complex with Neisseria meningitidis 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase—The importance of accommodating the active site water. Bioorganic Chem. 2014, 57, 242–250. [Google Scholar] [CrossRef]
- Reichau, S.; Blackmore, N.J.; Jiao, W.; Parker, E.J. Probing the Sophisticated Synergistic Allosteric Regulation of Aromatic Amino Acid Biosynthesis in Mycobacterium tuberculosis Using ᴅ-Amino Acids. PLoS ONE 2016, 11, e0152723. [Google Scholar] [CrossRef]
- Munack, S.; Roderer, K.; Ökvist, M.; Kamarauskaite, J.; Sasso, S.; van Eerde, A.; Kast, P.; Krengel, U. Remote control by inter-enzyme allostery: A novel paradigm for regulation of the shikimate pathway. J. Mol. Biol. 2016, 428, 1237–1255. [Google Scholar] [CrossRef]
- Burschowsky, D.; Thorbjørnsrud, H.V.; Heim, J.B.; Fahrig-Kamarauskaite, J.; Würth-Roderer, K.; Kast, P.; Krengel, U. Inter-enzyme allosteric regulation of chorismate mutase in Corynebacterium glutamicum: Structural basis of feedback activation by Trp. Biochemistry 2018, 57, 557–573. [Google Scholar] [CrossRef]
- Nazmi, A.R.; Lang, E.J.; Bai, Y.; Allison, T.M.; Othman, M.H.; Panjikar, S.; Arcus, V.L.; Parker, E.J. Interdomain Conformational Changes Provide Allosteric Regulation en Route to Chorismate. J. Biol. Chem. 2016, 291, 21836–21847. [Google Scholar] [CrossRef]
- Wang, S.; Liu, D.; Bilal, M.; Wang, W.; Zhang, X. Uncovering the Role of PhzC as DAHP Synthase in Shikimate Pathway of Pseudomonas chlororaphis HT66. Biology 2022, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Johnson, J.L.; Jensen, R.A. The recent evolutionary origin of the phenylalanine-sensitive isozyme of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase in the enteric lineage of bacteria. J. Mol. Evol. 1987, 25, 159–167. [Google Scholar] [CrossRef]
- Yokoyama, R.; Kleven, B.; Gupta, A.; Wang, Y.; Maeda, H.A. 3-Deoxy-D-arabino-heptulosonate 7-phosphate synthase as the gatekeeper of plant aromatic natural product biosynthesis. Curr. Opin. Plant Biol. 2022, 67, 102219. [Google Scholar] [CrossRef]
- Jayaraman, K.; Trachtmann, N.; Sprenger, G.A.; Gohlke, H. Protein engineering for feedback resistance in 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase. Appl. Microbiol. Biotechnol. 2022, 106, 6505–6517. [Google Scholar] [CrossRef]
- Liu, H.; Xiao, Q.; Wu, X.; Ma, H.; Li, J.; Guo, X.; Liu, Z.; Zhang, Y.; Luo, Y. Mechanistic investigation of a D to N mutation in DAHP synthase that dictates carbon flux into the shikimate pathway in yeast. Commun. Chem. 2023, 6, 152. [Google Scholar] [CrossRef]
- Yang, S.; Wang, Y.; Cai, Z.; Zhang, G.; Song, H. Metabolic engineering of Bacillus subtilis for high-titer production of menaquinone-7. AIChE J. 2020, 66, e16754. [Google Scholar] [CrossRef]
- Park, E.; Kim, H.-J.; Seo, S.-Y.; Lee, H.-N.; Choi, S.-S.; Lee, S.J.; Kim, E.-S. Shikimate metabolic pathway engineering in Corynebacterium glutamicum. J. Microbiol. Biotechnol. 2021, 31, 1305. [Google Scholar] [CrossRef]
- Kogure, T.; Kubota, T.; Suda, M.; Hiraga, K.; Inui, M. Metabolic engineering of Corynebacterium glutamicum for shikimate overproduction by growth-arrested cell reaction. Metab. Eng. 2016, 38, 204–216. [Google Scholar] [CrossRef]
- Kukil, K.; Lindberg, P. Metabolic engineering of Synechocystis sp. PCC 6803 for the improved production of phenylpropanoids. Microb. Cell Factories 2024, 23, 57. [Google Scholar] [CrossRef] [PubMed]
- Goldfinger, V.; Spohn, M.; Rodler, J.-P.; Sigle, M.; Kulik, A.; Cryle, M.J.; Rapp, J.; Link, H.; Wohlleben, W.; Stegmann, E. Metabolic engineering of the shikimate pathway in Amycolatopsis strains for optimized glycopeptide antibiotic production. Metab. Eng. 2023, 78, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Frlan, R. An Evolutionary Conservation and Druggability Analysis of Enzymes Belonging to the Bacterial Shikimate Pathway. Antibiotics 2022, 11, 675. [Google Scholar] [CrossRef]
- Brilisauer, K.; Rapp, J.; Rath, P.; Schöllhorn, A.; Bleul, L.; Weiß, E.; Stahl, M.; Grond, S.; Forchhammer, K. Cyanobacterial antimetabolite 7-deoxy-sedoheptulose blocks the shikimate pathway to inhibit the growth of prototrophic organisms. Nat. Commun. 2019, 10, 545. [Google Scholar] [CrossRef]
- Tapas, S.; Kumar Patel, G.; Dhindwal, S.; Tomar, S. In Silico sequence analysis and molecular modeling of the three-dimensional structure of DAHP synthase from Pseudomonas fragi. J. Mol. Model. 2011, 17, 621–631. [Google Scholar] [CrossRef]
- Romero, R.; Roberts, M.; Phillipson, J. Chorismate mutase in microorganisms and plants. Phytochemistry 1995, 40, 1015–1025. [Google Scholar] [CrossRef]
- Russ, W.P.; Figliuzzi, M.; Stocker, C.; Barrat-Charlaix, P.; Socolich, M.; Kast, P.; Hilvert, D.; Monasson, R.; Cocco, S.; Weigt, M. An evolution-based model for designing chorismate mutase enzymes. Science 2020, 369, 440–445. [Google Scholar] [CrossRef]
- Kroll, K.; Holland, C.K.; Starks, C.M.; Jez, J.M. Evolution of allosteric regulation in chorismate mutases from early plants. Biochem. J. 2017, 474, 3705–3717. [Google Scholar] [CrossRef]
- Helmstaedt, K.; Krappmann, S.; Braus, G.H. Allosteric regulation of catalytic activity: Escherichia coli aspartate transcarbamoylase versus yeast chorismate mutase. Microbiol. Mol. Biol. Rev. 2001, 65, 404–421. [Google Scholar] [CrossRef]
- Chook, Y.M.; Ke, H.; Lipscomb, W.N. Crystal structures of the monofunctional chorismate mutase from Bacillus subtilis and its complex with a transition state analog. Proc. Natl. Acad. Sci. USA 1993, 90, 8600–8603. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, D.H.; Bonner, C.A.; Gu, W.; Xie, G.; Jensen, R.A. The emerging periplasm-localized subclass of AroQ chorismate mutases, exemplified by those from Salmonella typhimurium and Pseudomonas aeruginosa. Genome Biol. 2001, 2, research0030.1. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Lipscomb, W.N.; Graf, R.; Schnappauf, G.; Braus, G. The crystal structure of allosteric chorismate mutase at 2.2-A resolution. Proc. Natl. Acad. Sci. USA 1994, 91, 10814–10818. [Google Scholar] [CrossRef] [PubMed]
- MacBeath, G.; Kast, P.; Hilvert, D. A Small, Thermostable, and Monofunctional Chorismate Mutase from the Archeon Methanococcus jannaschii. Biochemistry 1998, 37, 10062–10073. [Google Scholar] [CrossRef]
- Sträter, N.; Schnappauf, G.; Braus, G.; Lipscomb, W.N. Mechanisms of catalysis and allosteric regulation of yeast chorismate mutase from crystal structures. Structure 1997, 5, 1437–1452. [Google Scholar] [CrossRef]
- Fahrig-Kamarauskait, J.; Würth-Roderer, K.; Thorbjørnsrud, H.V.; Mailand, S.; Krengel, U.; Kast, P. Evolving the naturally compromised chorismate mutase from Mycobacterium tuberculosis to top performance. J. Biol. Chem. 2020, 295, 17514–17534. [Google Scholar] [CrossRef] [PubMed]
- Westfall, C.S.; Xu, A.; Jez, J.M. Structural evolution of differential amino acid effector regulation in plant chorismate mutases. J. Biol. Chem. 2014, 289, 28619–28628. [Google Scholar] [CrossRef]
- Thorbjørnsrud, H.V.; Bressan, L.; Khatanbaatar, T.; Carrer, M.; Würth-Roderer, K.; Cordara, G.; Kast, P.; Cascella, M.; Krengel, U. What Drives Chorismate Mutase to Top Performance? Insights from a Combined In Silico and In Vitro Study. Biochemistry 2023, 62, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.; Nasertorabi, F.; Han, G.W.; Stevens, R.C.; Schultz, P.G. Generation of an orthogonal protein–protein interface with a noncanonical amino acid. J. Am. Chem. Soc. 2017, 139, 5728–5731. [Google Scholar] [CrossRef]
- Kitade, Y.; Hashimoto, R.; Suda, M.; Hiraga, K.; Inui, M. Production of 4-hydroxybenzoic acid by an aerobic growth-arrested bioprocess using metabolically engineered Corynebacterium glutamicum. Appl. Environ. Microbiol. 2018, 84, e02587-17. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tian, Y.; Zhou, Y.; Kan, Y.; Wu, T.; Xiao, W.; Luo, Y. Multi-modular engineering of Saccharomyces cerevisiae for high-titre production of tyrosol and salidroside. Microb. Biotechnol. 2021, 14, 2605–2616. [Google Scholar] [CrossRef]
- Chávez-Béjar María, I.; Lara Alvaro, R.; López, H.; Hernández-Chávez, G.; Martinez, A.; Ramírez Octavio, T.; Bolívar, F.; Gosset, G. Metabolic Engineering of Escherichia coli for l-Tyrosine Production by Expression of Genes Coding for the Chorismate Mutase Domain of the Native Chorismate Mutase-Prephenate Dehydratase and a Cyclohexadienyl Dehydrogenase from Zymomonas mobilis. Appl. Environ. Microbiol. 2008, 74, 3284–3290. [Google Scholar] [CrossRef]
- Vitayakritsirikul, V.; Jaemsaeng, R.; Lohmaneeratana, K.; Thanapipatsiri, A.; Daduang, R.; Chuawong, P.; Thamchaipenet, A. Improvement of chloramphenicol production in Streptomyces venezuelae ATCC 10712 by overexpression of the aroB and aroK genes catalysing steps in the shikimate pathway. Antonie van Leeuwenhoek 2016, 109, 379–388. [Google Scholar] [CrossRef]
- Reddy, G.S.; Snehalatha, A.V.; Edwin, R.K.; Hossain, K.A.; Giliyaru, V.B.; Hariharapura, R.C.; Shenoy, G.G.; Misra, P.; Pal, M. Synthesis of 3-indolylmethyl substituted (pyrazolo/benzo) triazinone derivatives under Pd/Cu-catalysis: Identification of potent inhibitors of chorismate mutase (CM). Bioorganic Chem. 2019, 91, 103155. [Google Scholar] [CrossRef]
- Reddy, G.S.; Shukla, S.; Bhuktar, H.; Hossain, K.A.; Edwin, R.K.; Giliyaru, V.B.; Misra, P.; Pal, M. Pd/Cu-catalyzed access to novel 3-(benzofuran-2-ylmethyl) substituted (pyrazolo/benzo) triazinone derivatives: Their in silico/in vitro evaluation as inhibitors of chorismate mutase (CM). RSC Adv. 2022, 12, 26686–26695. [Google Scholar] [CrossRef]
- Khedr, M.A.; Massarotti, A.; Mohamed, M.E. Rational Discovery of (+) (S) Abscisic Acid as a Potential Antifungal Agent: A Repurposing Approach. Sci. Rep. 2018, 8, 8565. [Google Scholar] [CrossRef]
- Shukla, S.; Nishanth Rao, R.; Bhuktar, H.; Edwin, R.K.; Jamma, T.; Medishetti, R.; Banerjee, S.; Giliyaru, V.B.; Shenoy, G.G.; Oruganti, S.; et al. Wang resin catalysed sonochemical synthesis of pyrazolo[4,3-d]pyrimidinones and 2,3-dihydroquinazolin-4(1H)-ones: Identification of chorismate mutase inhibitors having effects on Mycobacterium tuberculosis cell viability. Bioorganic Chem. 2023, 134, 106452. [Google Scholar] [CrossRef]
- Kumar, B.S.; Rao, L.V.; Dhananjaya, G.; Kapavarapu, R.; Pal, M. Wang resin mediated unexpected greener access to 2-substituted quinazoline-4(3H)-ones and their evaluation against chorismate mutase. J. Mol. Struct. 2025, 1321, 139931. [Google Scholar] [CrossRef]
- Ghosh, R.; Hilario, E.; Chang, C.-E.; Mueller, L.; Dunn, M. Allosteric regulation of substrate channeling: Salmonella typhimurium tryptophan synthase. Front. Mol. Biosci. 2022, 9, 923042. [Google Scholar] [CrossRef] [PubMed]
- Hyde, C.C.; Ahmed, S.A.; Padlan, E.A.; Miles, E.W.; Davies, D.R. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J. Biol. Chem. 1988, 263, 17857–17871. [Google Scholar] [CrossRef]
- Bahar, I.; Jernigan, R.L. Cooperative fluctuations and subunit communication in tryptophan synthase. Biochemistry 1999, 38, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Yanofsky, C.; Steven, S.L.L.; Horn, V.; Rowe, J. Structure and Properties of a Hybrid Tryptophan Synthetase; Chain Produced by Genetic Exchange between Escherichia coli and Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 1977, 74, 286–290. [Google Scholar] [CrossRef]
- Ngo, H.; Kimmich, N.; Harris, R.; Niks, D.; Blumenstein, L.; Kulik, V.; Barends, T.R.; Schlichting, I.; Dunn, M.F. Allosteric regulation of substrate channeling in tryptophan synthase: Modulation of the L-serine reaction in stage I of the β-reaction by α-site ligands. Biochemistry 2007, 46, 7740–7753. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; D’Amico, R.; O’Rourke, K.; Bosken, Y.; Sahu, D.; Chang, C.-e. Allosteric networks regulate function in multi-enzyme complexes. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Lane, A.N.; Kirschner, K. The Catalytic Mechanism of Tryptophan Synthase from Escherichia coli: Kinetics of the Reaction of Indole with the Enzyme -l-Serine Complexes. Eur. J. Biochem. 1983, 129, 571–582. [Google Scholar] [CrossRef]
- Ngo, H.; Harris, R.; Kimmich, N.; Casino, P.; Niks, D.; Blumenstein, L.; Barends, T.R.; Kulik, V.; Weyand, M.; Schlichting, I.; et al. Synthesis and characterization of allosteric probes of substrate channeling in the tryptophan synthase bienzyme complex. Biochemistry 2007, 46, 7713–7727. [Google Scholar] [CrossRef]
- Anderson, K.S.; Miles, E.; Johnson, K.A. Serine modulates substrate channeling in tryptophan synthase. A novel intersubunit triggering mechanism. J. Biol. Chem. 1991, 266, 8020–8033. [Google Scholar] [CrossRef]
- Crawford, I.P. Evolution of a biosynthetic pathway: The tryptophan paradigm. Annu. Rev. Microbiol. 1989, 43, 567–600. [Google Scholar] [CrossRef] [PubMed]
- Raboni, S.; Bettati, S.; Mozzarelli, A. Tryptophan synthase: A mine for enzymologists. Cell. Mol. Life Sci. CMLS 2009, 66, 2391–2403. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-X.; McPhie, P.; Miles, E.W. Regulation of tryptophan synthase by temperature, monovalent cations, and an allosteric ligand. Evidence from Arrhenius plots, absorption spectra, and primary kinetic isotope effects. Biochemistry 2000, 39, 4692–4703. [Google Scholar] [CrossRef] [PubMed]
- Weber-Ban, E.; Hur, O.; Bagwell, C.; Banik, U.; Yang, L.-H.; Miles, E.W.; Dunn, M.F. Investigation of allosteric linkages in the regulation of tryptophan synthase: The roles of salt bridges and monovalent cations probed by site-directed mutation, optical spectroscopy, and kinetics. Biochemistry 2001, 40, 3497–3511. [Google Scholar] [CrossRef]
- Dunn, M.F. Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch. Biochem. Biophys. 2012, 519, 154–166. [Google Scholar] [CrossRef]
- Axe, J.M.; Boehr, D.D. Long-range interactions in the alpha subunit of tryptophan synthase help to coordinate ligand binding, catalysis, and substrate channeling. J. Mol. Biol. 2013, 425, 1527–1545. [Google Scholar] [CrossRef]
- D’Amico, R.N.; Bosken, Y.K.; O’Rourke, K.F.; Murray, A.M.; Admasu, W.; Chang, C.-e.A.; Boehr, D.D. Substitution of a surface-exposed residue involved in an allosteric network enhances tryptophan synthase function in cells. Front. Mol. Biosci. 2021, 8, 679915. [Google Scholar] [CrossRef]
- Buller, A.R.; Brinkmann-Chen, S.; Romney, D.K.; Herger, M.; Murciano-Calles, J.; Arnold, F.H. Directed evolution of the tryptophan synthase β-subunit for stand-alone function recapitulates allosteric activation. Proc. Natl. Acad. Sci. USA 2015, 112, 14599–14604. [Google Scholar] [CrossRef] [PubMed]
- Herger, M.; van Roye, P.; Romney, D.K.; Brinkmann-Chen, S.; Buller, A.R.; Arnold, F.H. Synthesis of β-branched tryptophan analogues using an engineered subunit of tryptophan synthase. J. Am. Chem. Soc. 2016, 138, 8388–8391. [Google Scholar] [CrossRef]
- Romney, D.K.; Murciano-Calles, J.; Wehrmüller, J.E.; Arnold, F.H. Unlocking reactivity of TrpB: A general biocatalytic platform for synthesis of tryptophan analogues. J. Am. Chem. Soc. 2017, 139, 10769–10776. [Google Scholar] [CrossRef] [PubMed]
- Boville, C.E.; Scheele, R.A.; Koch, P.; Brinkmann-Chen, S.; Buller, A.R.; Arnold, F.H. Engineered Biosynthesis of β-Alkyl Tryptophan Analogues. Angew. Chem. Int. Ed. Engl. 2018, 57, 14764–14768. [Google Scholar] [CrossRef] [PubMed]
- Almhjell, P.J.; Johnston, K.E.; Porter, N.J.; Kennemur, J.L.; Bhethanabotla, V.C.; Ducharme, J.; Arnold, F.H. The β-subunit of tryptophan synthase is a latent tyrosine synthase. Nat. Chem. Biol. 2024, 20, 1086–1093. [Google Scholar] [CrossRef]
- Francis, D.; Winn, M.; Latham, J.; Greaney, M.F.; Micklefield, J. An engineered tryptophan synthase opens new enzymatic pathways to β-methyltryptophan and derivatives. ChemBioChem 2017, 18, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Murciano-Calles, J.; Romney, D.K.; Brinkmann-Chen, S.; Buller, A.R.; Arnold, F.H. A panel of TrpB biocatalysts derived from tryptophan synthase through the transfer of mutations that mimic allosteric activation. Angew. Chem. Int. Ed. 2016, 55, 11577–11581. [Google Scholar] [CrossRef]
- Watkins-Dulaney, E.J.; Dunham, N.P.; Straathof, S.; Turi, S.; Arnold, F.H.; Buller, A.R. Asymmetric alkylation of ketones catalyzed by engineered TrpB. Angew. Chem. 2021, 133, 21582–21587. [Google Scholar] [CrossRef]
- Dick, M.; Sarai, N.S.; Martynowycz, M.W.; Gonen, T.; Arnold, F.H. Tailoring tryptophan synthase TrpB for selective quaternary carbon bond formation. J. Am. Chem. Soc. 2019, 141, 19817–19822. [Google Scholar] [CrossRef]
- Xu, L.; Li, T.; Huo, Z.; Chen, Q.; Xia, Q.; Jiang, B. Directed evolution improves the enzymatic synthesis of L-5-hydroxytryptophan by an engineered tryptophan synthase. Appl. Biochem. Biotechnol. 2021, 193, 3407–3417. [Google Scholar] [CrossRef]
- Scheele, R.A.; Weber, Y.; Nintzel, F.E.; Herger, M.; Kaminski, T.S.; Hollfelder, F. Ultrahigh throughput evolution of tryptophan synthase in droplets via an aptamer sensor. ACS Catal. 2024, 14, 6259–6271. [Google Scholar] [CrossRef] [PubMed]
- Rix, G.; Watkins-Dulaney, E.J.; Almhjell, P.J.; Boville, C.E.; Arnold, F.H.; Liu, C.C. Scalable continuous evolution for the generation of diverse enzyme variants encompassing promiscuous activities. Nat. Commun. 2020, 11, 5644. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.A.; Ronnebaum, T.A.; Roose, B.W.; Christianson, D.W. Structural basis of substrate promiscuity and catalysis by the reverse prenyltransferase N-dimethylallyl-l-tryptophan synthase from Fusarium fujikuroi. Biochemistry 2022, 61, 2025–2035. [Google Scholar] [CrossRef]
- Fan, A.; Zocher, G.; Stec, E.; Stehle, T.; Li, S.-M. Site-directed mutagenesis switching a dimethylallyl tryptophan synthase to a specific tyrosine C3-prenylating enzyme. J. Biol. Chem. 2015, 290, 1364–1373. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods 2019, 16, 687–694. [Google Scholar] [CrossRef]
- Maria-Solano, M.A.; Iglesias-Fernández, J.; Osuna, S. Deciphering the Allosterically Driven Conformational Ensemble in Tryptophan Synthase Evolution. J. Am. Chem. Soc. 2019, 141, 13049–13056. [Google Scholar] [CrossRef]
- Romney, D.K.; Sarai, N.S.; Arnold, F.H. Nitroalkanes as versatile nucleophiles for enzymatic synthesis of noncanonical amino acids. ACS Catal. 2019, 9, 8726–8730. [Google Scholar] [CrossRef]
- Kneuttinger, A.C.; Zwisele, S.; Straub, K.; Bruckmann, A.; Busch, F.; Kinateder, T.; Gaim, B.; Wysocki, V.H.; Merkl, R.; Sterner, R. Light-regulation of tryptophan synthase by combining protein design and enzymology. Int. J. Mol. Sci. 2019, 20, 5106. [Google Scholar] [CrossRef]
- Dong, Y.; Chen, Z. Systems metabolic engineering of Corynebacterium glutamicum for efficient l-tryptophan production. Synth. Syst. Biotechnol. 2025, 10, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhang, J.; Chen, J.; Li, M.; Chen, H.; Wang, C.; Gong, C. Tryptophan production by catalysis of a putative tryptophan synthase protein. Arch. Microbiol. 2024, 206, 390. [Google Scholar] [CrossRef]
- Boville, C.E.; Romney, D.K.; Almhjell, P.J.; Sieben, M.; Arnold, F.H. Improved synthesis of 4-cyanotryptophan and other tryptophan analogues in aqueous solvent using variants of TrpB from Thermotoga maritima. J. Org. Chem. 2018, 83, 7447–7452. [Google Scholar] [CrossRef]
- Seibold, P.S.; Dörner, S.; Fricke, J.; Schäfer, T.; Beemelmanns, C.; Hoffmeister, D. Genetic regulation of l-tryptophan metabolism in Psilocybe mexicana supports psilocybin biosynthesis. Fungal Biol. Biotechnol. 2024, 11, 4. [Google Scholar] [CrossRef]
- Murciano-Calles, J.; Buller, A.R.; Arnold, F.H. Directed evolution of an allosteric tryptophan synthase to create a platform for synthesis of noncanonical amino acids. In Directed Enzyme Evolution: Advances and Applications; Springer: Berlin/Heidelberg, Germany, 2017; pp. 1–16. [Google Scholar]
- Luo, S.; Wei, W.; Wu, J.; Song, W.; Hu, G.; Liu, L. A dual-enzyme cascade for production of indigo from L-tryptophan. Sheng Wu Gong Cheng Xue Bao Chin. J. Biotechnol. 2024, 40, 2444–2456. [Google Scholar]
- Parmeggiani, F.; Rué Casamajo, A.; Walton, C.J.; Galman, J.L.; Turner, N.J.; Chica, R.A. One-pot biocatalytic synthesis of substituted d-tryptophans from indoles enabled by an engineered aminotransferase. ACS Catal. 2019, 9, 3482–3486. [Google Scholar] [CrossRef]
- Xu, L.; Wu, F.; Li, T.; Zhang, X.; Chen, Q.; Jiang, B.; Xia, Q. Ultrasound-assisted l-cysteine whole-cell bioconversion by recombinant Escherichia coli with tryptophan synthase. Green Process. Synth. 2021, 10, 842–850. [Google Scholar] [CrossRef]
- Michalska, K.; Chang, C.; Maltseva, N.I.; Jedrzejczak, R.; Robertson, G.T.; Gusovsky, F.; McCarren, P.; Schreiber, S.L.; Nag, P.P.; Joachimiak, A. Allosteric inhibitors of Mycobacterium tuberculosis tryptophan synthase. Protein Sci. 2020, 29, 779–788. [Google Scholar] [CrossRef]
- Naz, S.; Farooq, U.; Ali, S.; Sarwar, R.; Khan, S.; Abagyan, R. Identification of new benzamide inhibitor against α-subunit of tryptophan synthase from Mycobacterium tuberculosis through structure-based virtual screening, anti-tuberculosis activity and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 37, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Farooq, U.; Khan, S.; Sarwar, R.; Mabkhot, Y.N.; Saeed, M.; Alsayari, A.; Muhsinah, A.B.; Ul-Haq, Z. Pharmacophore model-based virtual screening, docking, biological evaluation and molecular dynamics simulations for inhibitors discovery against α-tryptophan synthase from Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2021, 39, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Cox, J.A.G.; Fütterer, K.; Rullas, J.; Ortega-Muro, F.; Loman, N.J.; Moynihan, P.J.; Pérez-Herrán, E.; Jiménez, E.; Esquivias, J.; et al. Inhibiting mycobacterial tryptophan synthase by targeting the inter-subunit interface. Sci. Rep. 2017, 7, 9430. [Google Scholar] [CrossRef]
- Wellington, S.; Nag, P.P.; Michalska, K.; Johnston, S.E.; Jedrzejczak, R.P.; Kaushik, V.K.; Clatworthy, A.E.; Siddiqi, N.; McCarren, P.; Bajrami, B. A small-molecule allosteric inhibitor of Mycobacterium tuberculosis tryptophan synthase. Nat. Chem. Biol. 2017, 13, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Bosken, Y.K.; Ai, R.; Hilario, E.; Ghosh, R.K.; Dunn, M.F.; Kan, S.H.; Niks, D.; Zhou, H.; Ma, W.; Mueller, L.J. Discovery of antimicrobial agent targeting tryptophan synthase. Protein Sci. 2022, 31, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Simeth, N.A.; Kinateder, T.; Rajendran, C.; Nazet, J.; Merkl, R.; Sterner, R.; König, B.; Kneuttinger, A.C. Towards photochromic azobenzene-based inhibitors for tryptophan synthase. Chem. A Eur. J. 2021, 27, 2439–2451. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.; Boehr, D.D. Multi-Enzyme Synergy and Allosteric Regulation in the Shikimate Pathway: Biocatalytic Platforms for Industrial Applications. Catalysts 2025, 15, 718. https://doi.org/10.3390/catal15080718
Khan S, Boehr DD. Multi-Enzyme Synergy and Allosteric Regulation in the Shikimate Pathway: Biocatalytic Platforms for Industrial Applications. Catalysts. 2025; 15(8):718. https://doi.org/10.3390/catal15080718
Chicago/Turabian StyleKhan, Sara, and David D. Boehr. 2025. "Multi-Enzyme Synergy and Allosteric Regulation in the Shikimate Pathway: Biocatalytic Platforms for Industrial Applications" Catalysts 15, no. 8: 718. https://doi.org/10.3390/catal15080718
APA StyleKhan, S., & Boehr, D. D. (2025). Multi-Enzyme Synergy and Allosteric Regulation in the Shikimate Pathway: Biocatalytic Platforms for Industrial Applications. Catalysts, 15(8), 718. https://doi.org/10.3390/catal15080718