
Combination of aza-Friedel Crafts MCR with Other MCRs Under Heterogeneous Conditions



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
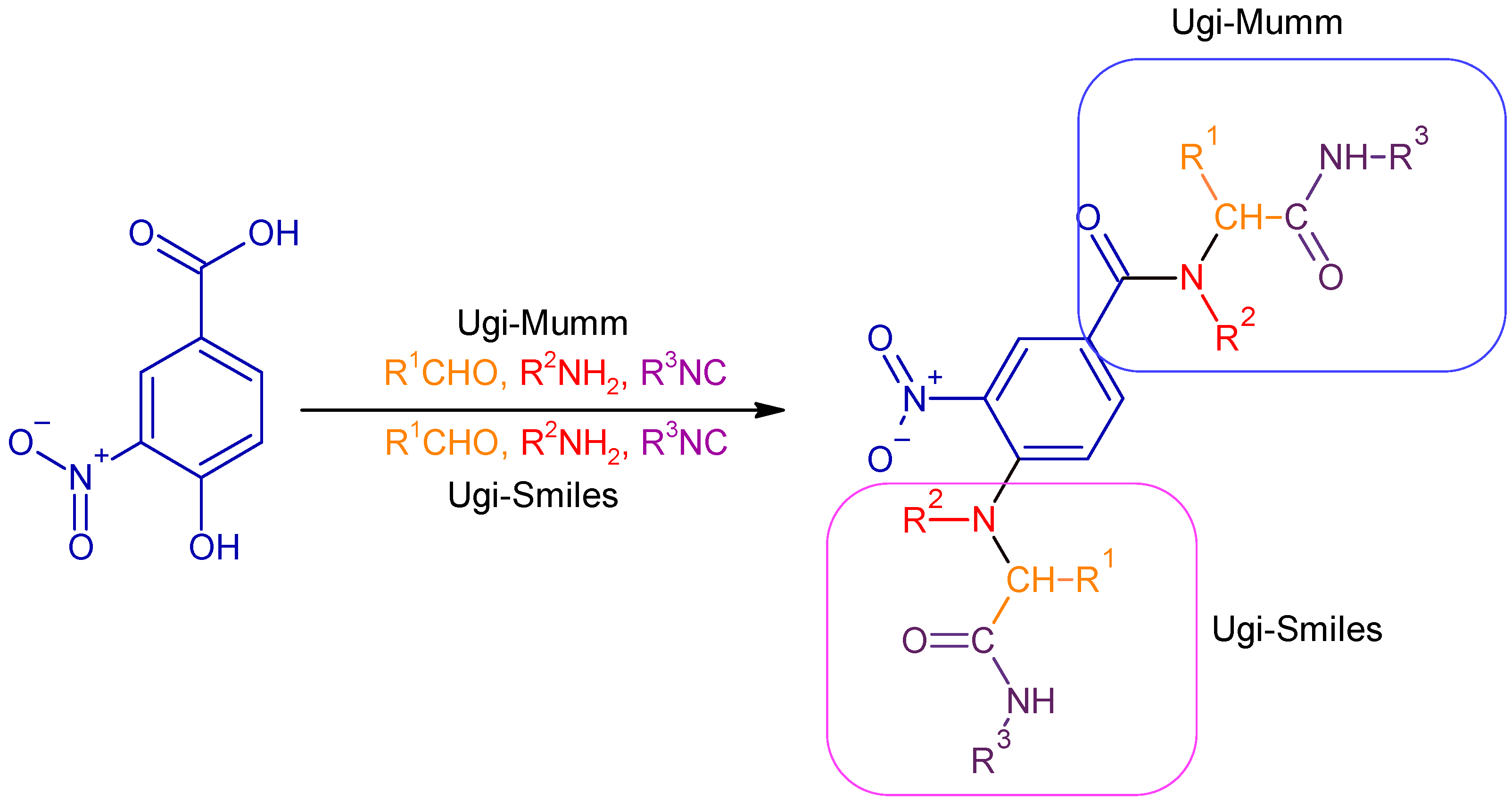
1. Introduction
2. Results and Discussion
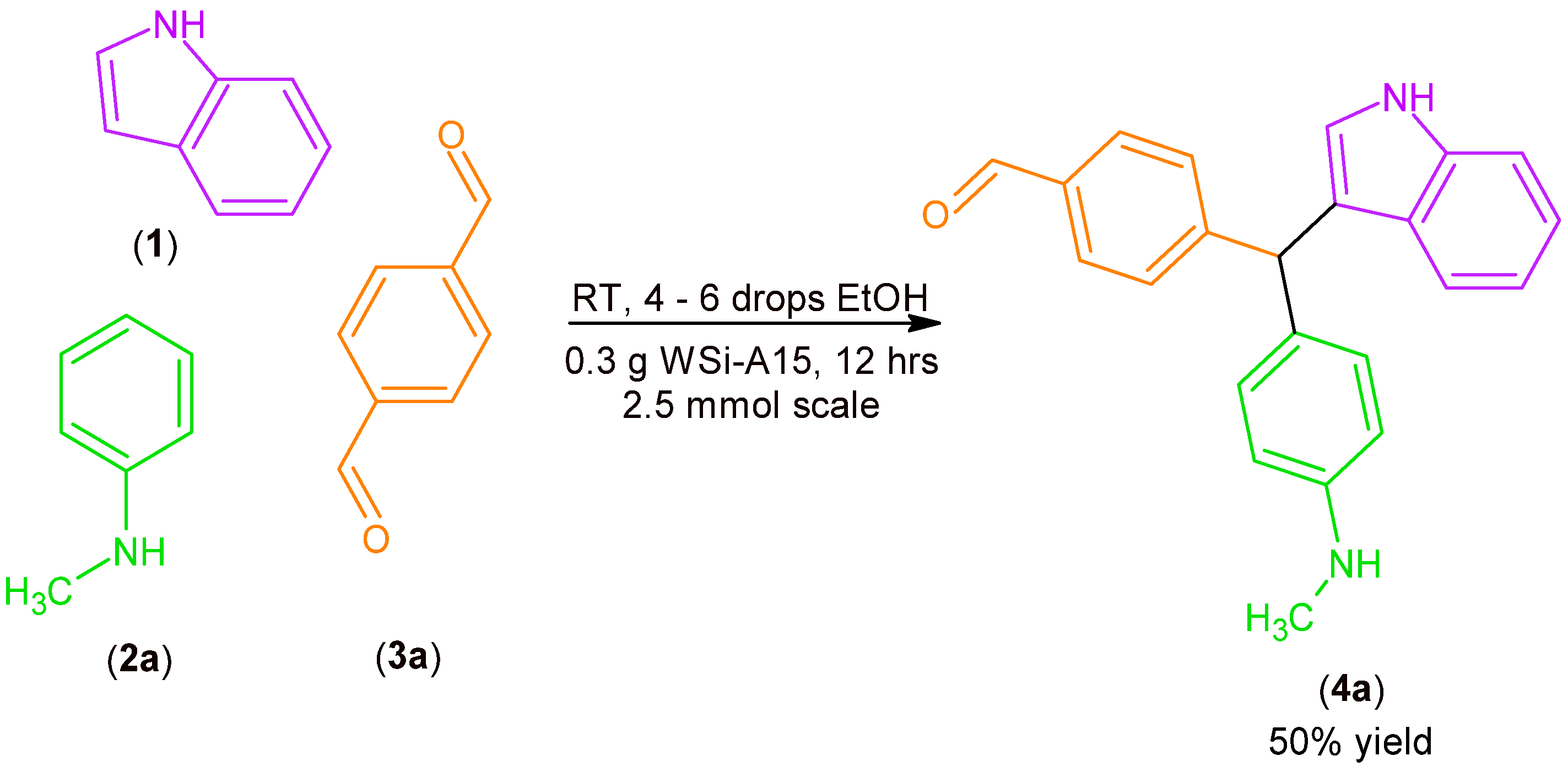
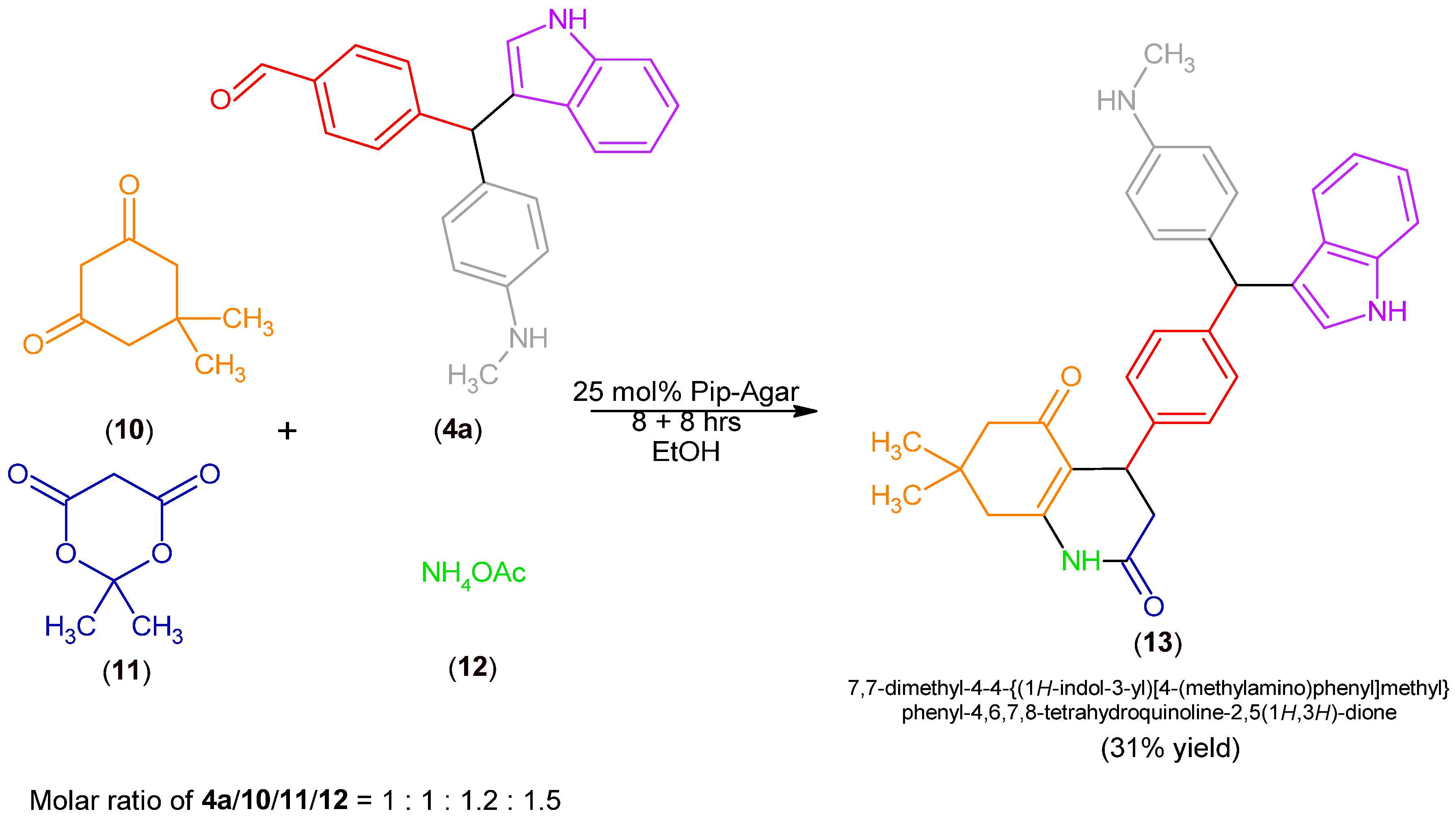
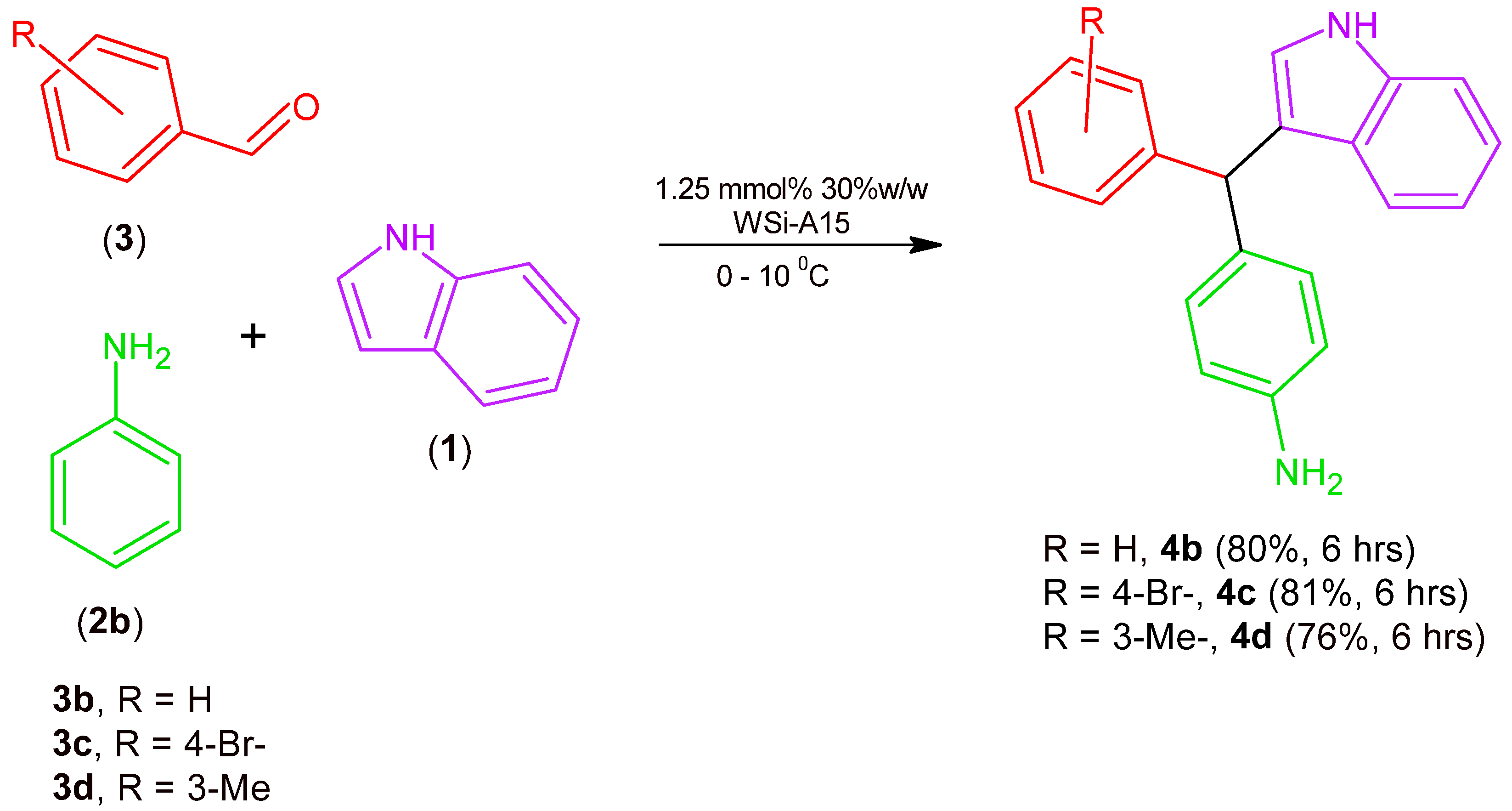
2.1. Synthesis of MCR Products Using an aza-Friedel Crafts Product with a Free Aldehyde Group
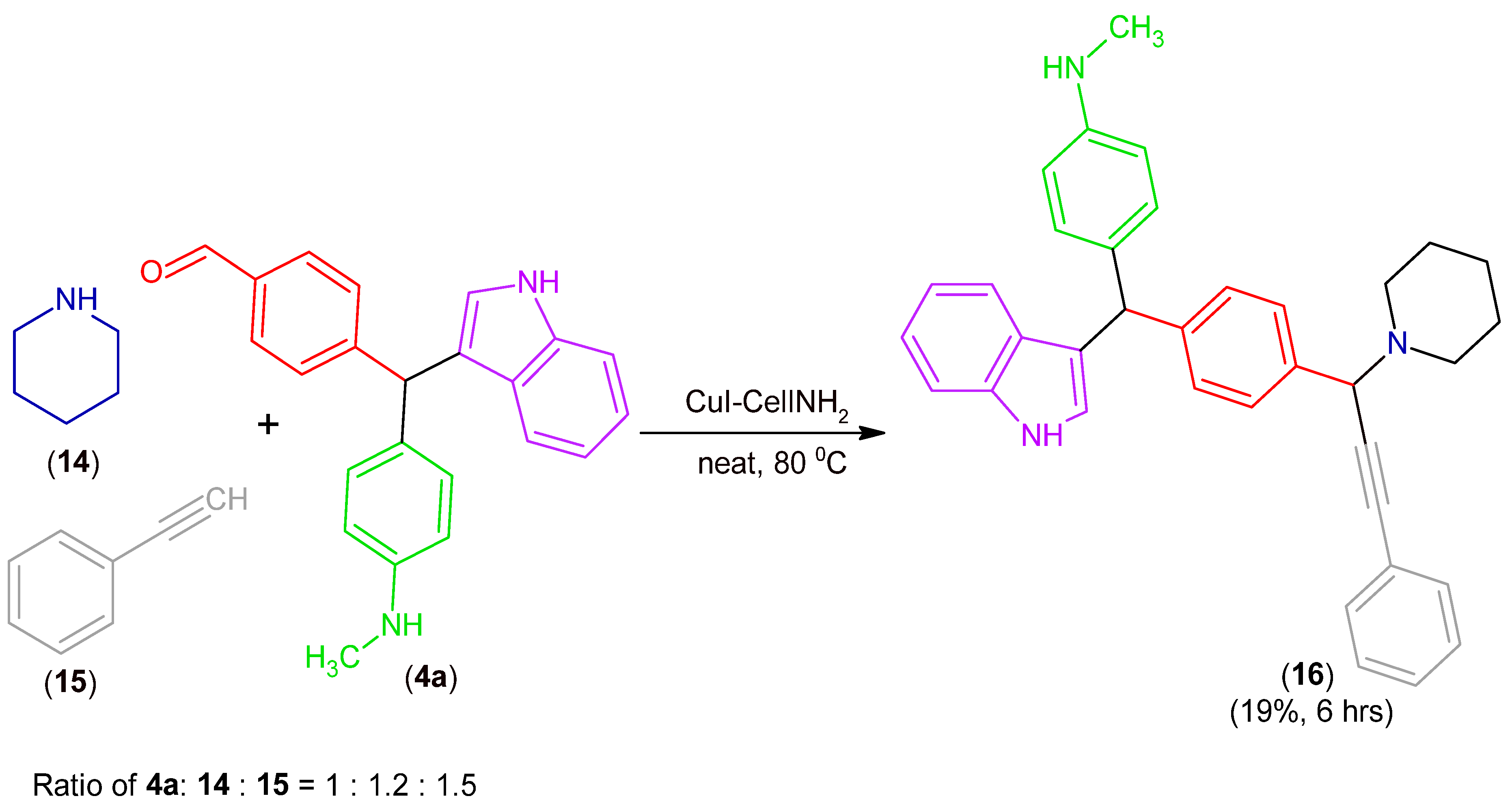
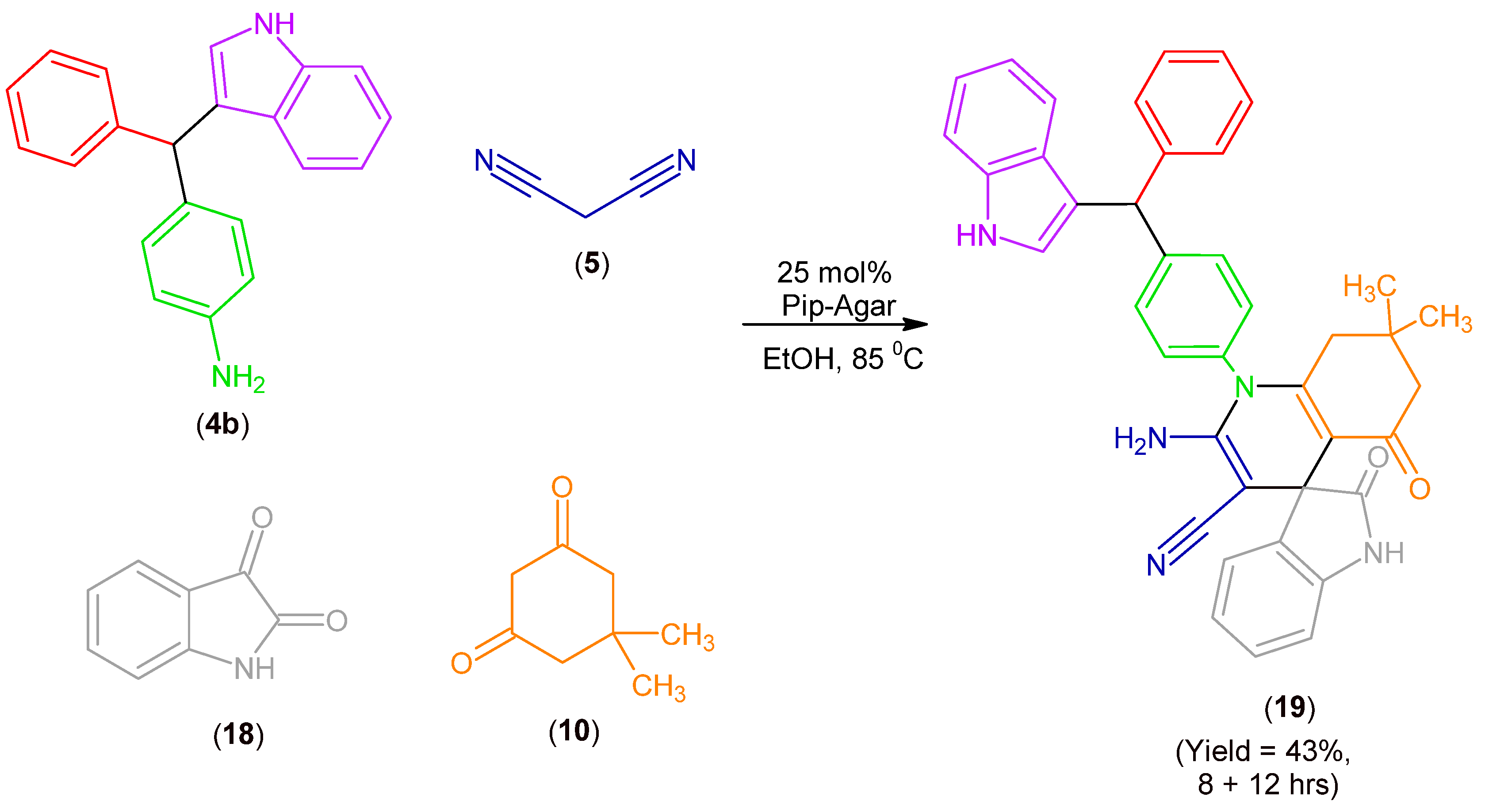
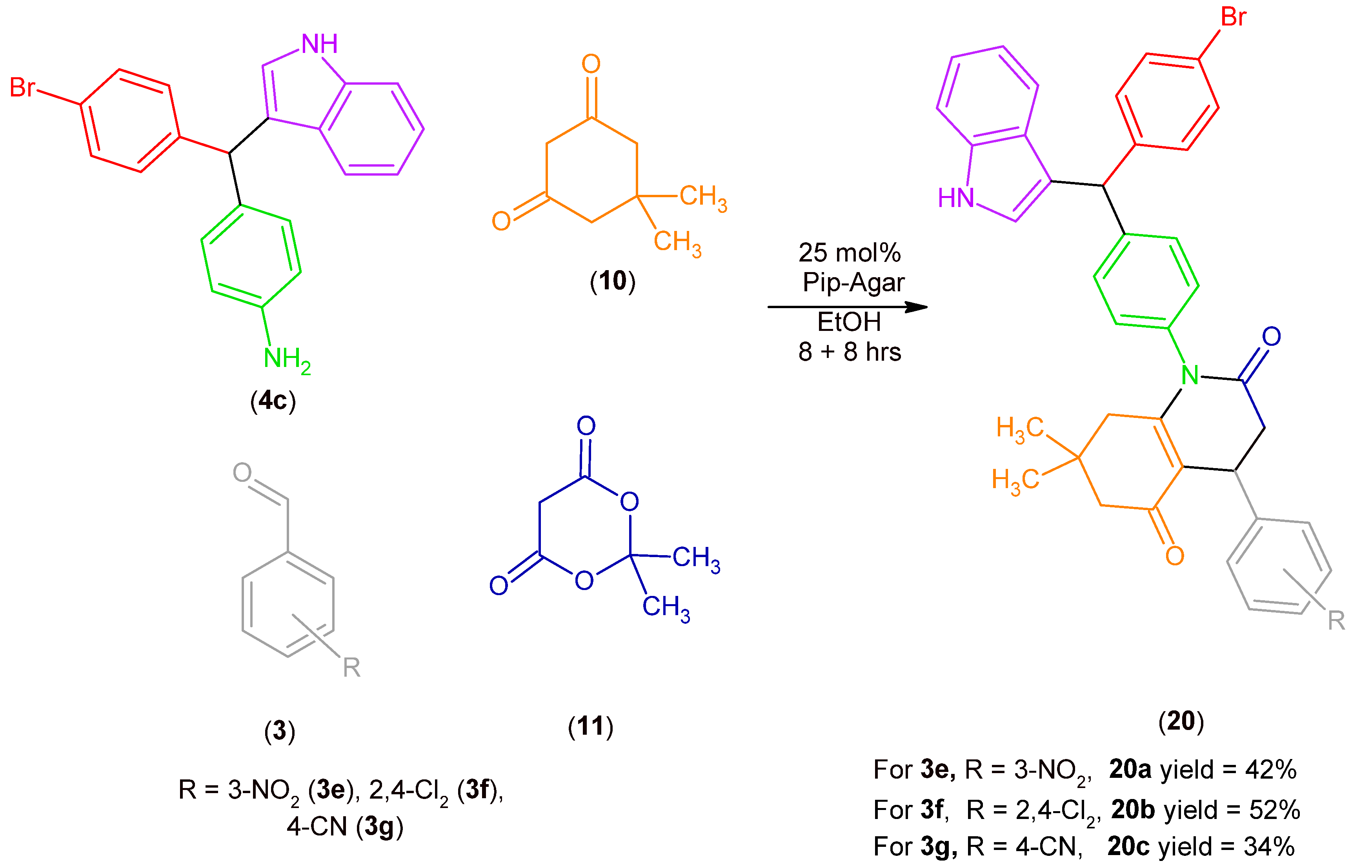
2.2. Synthesis of MCR Products Using an aza-Friedel Crafts Product with a Terminal Amino Group
2.3. Advanced Product Characterization
3. Methodology
3.1. Instrumentation
3.2. Reaction Methods
3.2.1. Synthesis of aza-Friedel Crafts Products (4a–d)
3.2.2. Synthesis of Product (8)
3.2.3. Synthesis of Product (9)
3.2.4. Synthesis of Product (13)
3.2.5. Synthesis of Product (16)
3.2.6. Synthesis of Products (17), (19), (20)
3.2.7. Catalyst Synthesis
3.3. Selected Product Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, M.S.; Chowdhury, S. Recent developments in solvent-free multicomponent reactions: A perfect synergy for eco-compatible organic synthesis. RSC Adv. 2012, 2, 4547–4592. [Google Scholar] [CrossRef]
- Bosica, G.; Abdilla, R. Recent advances in multicomponent reactions under operationally heterogeneous conditions. Catalysts 2022, 12, 725. [Google Scholar] [CrossRef]
- Damera, T.; Pagadala, R.; Rana, S.; Jonnalagadda, S.B. A concise review of multicomponent reactions using novel heterogeneous catalysts under microwave irradiation. Catalysts 2023, 13, 1034. [Google Scholar] [CrossRef]
- Graziano, G.; Stefanachi, A.; Contino, M.; Prieto-Díaz, R.; Ligresti, A.; Kumar, P.; Scilimati, A.; Sotelo, E.; Leonetti, F. Multicomponent reaction-assisted drug Discovery: A time- and cost-effective green approach speeding up identification and optimization of anticancer drugs. Int. J. Mol. Sci. 2023, 24, 6581. [Google Scholar] [CrossRef]
- John, S.E.; Gulati, S.; Shankaraiah, N. Recent advances in multi-component reactions and their mechanistic insights: A triennium review. Org. Chem. Front. 2021, 8, 4237–4287. [Google Scholar] [CrossRef]
- Neto, B.A.D.; Rocha, R.O.; Rodrigues, M.O. Catalytic approaches to multicomponent reactions: A critical review and perspectives on the roles of catalysis. Molecules 2021, 27, 132. [Google Scholar] [CrossRef]
- Brauch, S.; Van Berkel, S.S.; Westermann, B. Higher-order multicomponent reactions: Beyond four reactants. Chem. Soc. Rev. 2013, 42, 4948–4962. [Google Scholar] [CrossRef]
- Akritopoulou-Zanze, I.; Djuric, S.W. Recent advances in the development and applications of post-Ugi transformations. Heterocycles 2007, 73, 125–147. [Google Scholar] [CrossRef]
- Dömling, A.; Ugi, I. The seven-component reaction. Angew. Chem. Int. Ed. Engl. 1993, 32, 563–564. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Chandgude, A.L.; Dömling, A. Multicomponent reactions, union of MCRs and beyond. Chem. Rec. 2015, 15, 981–996. [Google Scholar] [CrossRef]
- Elders, N.; Van der Born, D.; Hendrickx, L.J.D.; Timmer, B.J.J.; Krause, A.; Janssen, E.; De Kanter, F.J.J.; Ruijter, E.; Orru, R.V.A. The efficient one-pot reaction of up to eight components by the union of multicomponent reactions. Angew. Chem. Int. Ed. 2009, 48, 5856–5859. [Google Scholar] [CrossRef]
- Al-Tel, T.J.; Al-Qawasmeh, R.; Voelter, W. Rapid assembly of polyfunctional structures using a one-pot five- and six-component sequential Groebke–Blackburn/Ugi/Passerini process. Eur. J. Org. Chem. 2010, 29, 5586–5593. [Google Scholar] [CrossRef]
- Brauch, S.; Gabriel, L.; Westermann, B. Seven-component reactions by sequential chemoselective Ugi-Mumm/Ugi-Smiles reactions. Chem. Commun. 2010, 46, 3387–3389. [Google Scholar] [CrossRef]
- Innocenti, R.; Lenci, E.; Menchi, G.; Trabocchi, A. Combination of multicomponent KA2 and Pauson-Khand reactions: Short synthesis of spirocyclic pyrrolocyclopentenones. Beilstein, J. Org. Chem. 2020, 16, 200–211. [Google Scholar] [CrossRef]
- He, Y.; Li, Z.; Robeyns, K.; Van Meervelt, L.; Van der Eycken, E.V. A gold-catalyzed domino cyclization enabling rapid construction of diverse polyheterocyclic frameworks. Angew. Chem. Int. Ed. 2018, 57, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Halimehjani, A.Z.; Sharifi, M. Synthesis of a novel category of Ugi adducts using succinic acid, succinic anhydride and maleic anhydride and their application in post-Ugi reactions for synthesis of functionalized piperazine 2,5-diones. Tetrahedron 2017, 73, 5778–5783. [Google Scholar] [CrossRef]
- Bosica, G.; Abdilla, R. DABCO-Amberlyst®15: A versatile heterogeneous catalyst in the multicomponent synthesis of tetrahydronaphthalenes and tetrahydroquinolines. Tetrahedron Green Chem 2024, 3, 100033. [Google Scholar] [CrossRef]
- Bosica, G.; Abdilla, R. Novel biopolymer-based catalyst for the multicomponent synthesis of N-aryl-4-aryl-substituted dihydropyridines derived from simple and complex anilines. Molecules 2024, 29, 1884. [Google Scholar] [CrossRef]
- Bosica, G.; Abdilla, R. Regioselective one-pot aza-Friedel-Crafts reaction for primary, secondary and tertiary anilines using heterogeneous catalyst. Green Chem. 2017, 19, 5683–5690. [Google Scholar] [CrossRef]
- Bosica, G.; Gabarretta, J. Unprecedented one-pot multicomponent synthesis of propargylamines using Amberlyst A-21 supported CuI under solvent-free conditions. RSC Adv. 2015, 57, 46074–46087. [Google Scholar] [CrossRef]
- Sindhu, K.S.; Anilkumar, G. Recent advances and applications of Glaser coupling employing greener protocols. RSC Adv. 2014, 4, 27867–27887. [Google Scholar] [CrossRef]
- Sabaqian, S.; Nemati, F.; Heravi, M.M.; Nahzomi, H.T. Copper(I) iodide supported on modified cellulose-based nano-magnetite composite as a biodegradable catalyst for the synthesis of 1,2,3-triazoles. Appl. Organomet. Chem. 2016, 31, e3660. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosica, G.; Abdilla, R. Combination of aza-Friedel Crafts MCR with Other MCRs Under Heterogeneous Conditions. Catalysts 2025, 15, 657. https://doi.org/10.3390/catal15070657
Bosica G, Abdilla R. Combination of aza-Friedel Crafts MCR with Other MCRs Under Heterogeneous Conditions. Catalysts. 2025; 15(7):657. https://doi.org/10.3390/catal15070657
Chicago/Turabian StyleBosica, Giovanna, and Roderick Abdilla. 2025. "Combination of aza-Friedel Crafts MCR with Other MCRs Under Heterogeneous Conditions" Catalysts 15, no. 7: 657. https://doi.org/10.3390/catal15070657
APA StyleBosica, G., & Abdilla, R. (2025). Combination of aza-Friedel Crafts MCR with Other MCRs Under Heterogeneous Conditions. Catalysts, 15(7), 657. https://doi.org/10.3390/catal15070657