
Production of 5-Hydroxymethylfurfural (HMF) from Sucrose in Aqueous Phase Using S, N-Doped Hydrochars

 ,
,  ,
,  , ,
, ,  and
and
Abstract

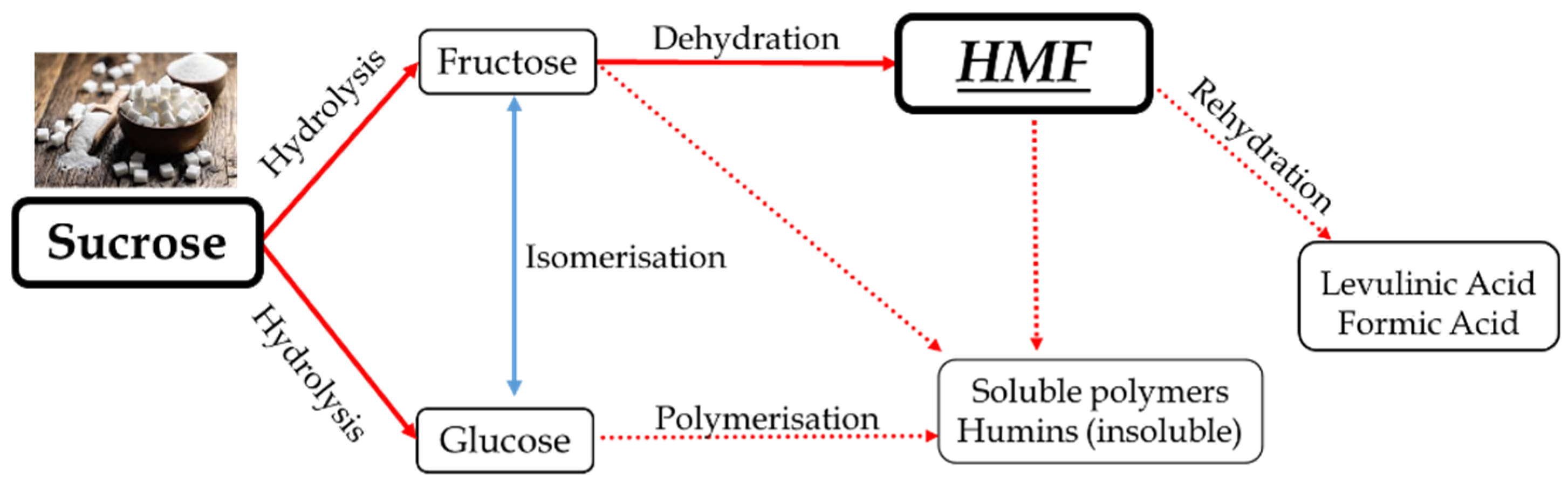
1. Introduction
2. Results
2.1. Elemental Analysis
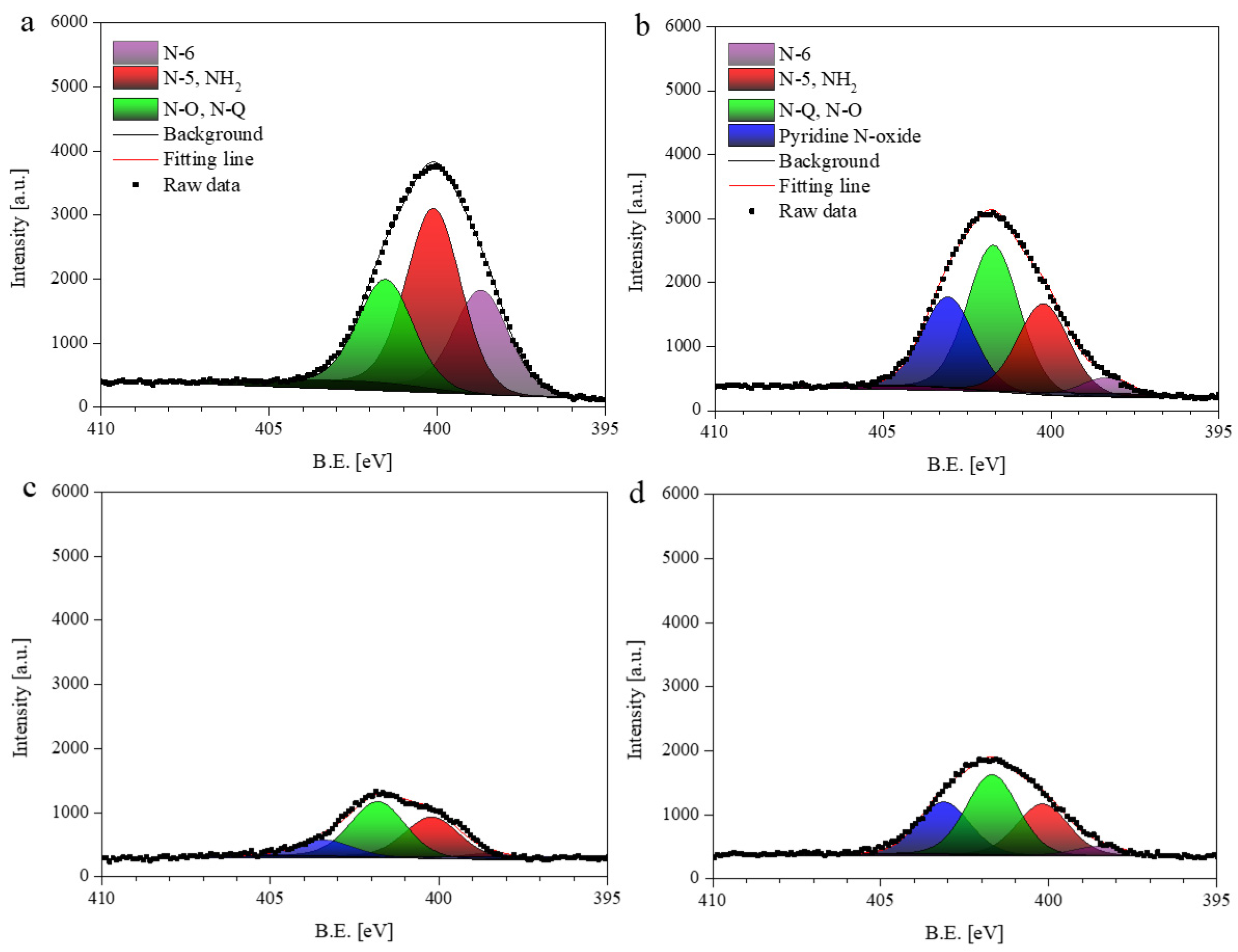
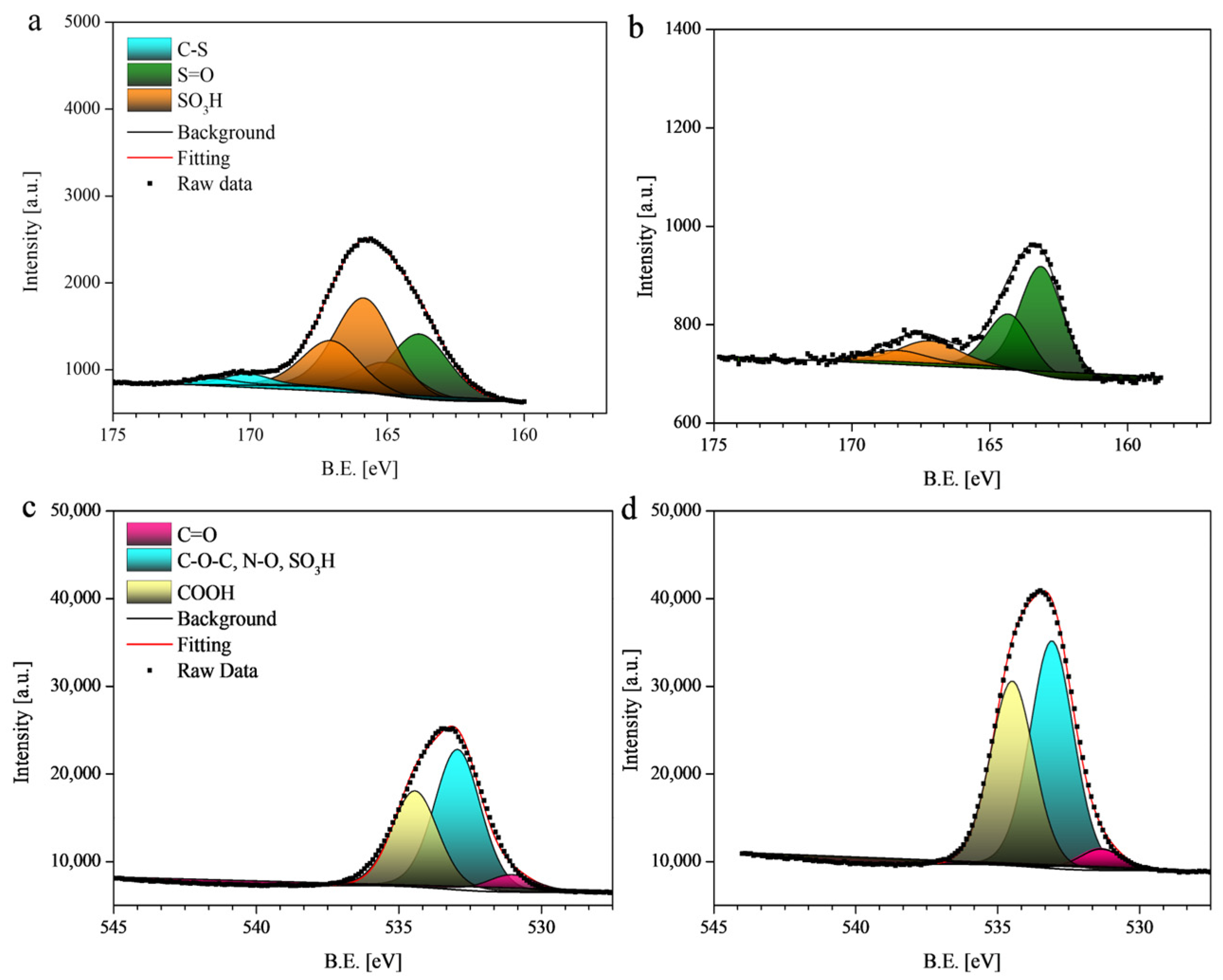
2.2. X-Ray Photoelectron Spectroscopy
2.3. Total Acidity Measurement
2.4. Scanning Electron Microscopy (SEM-EDX)
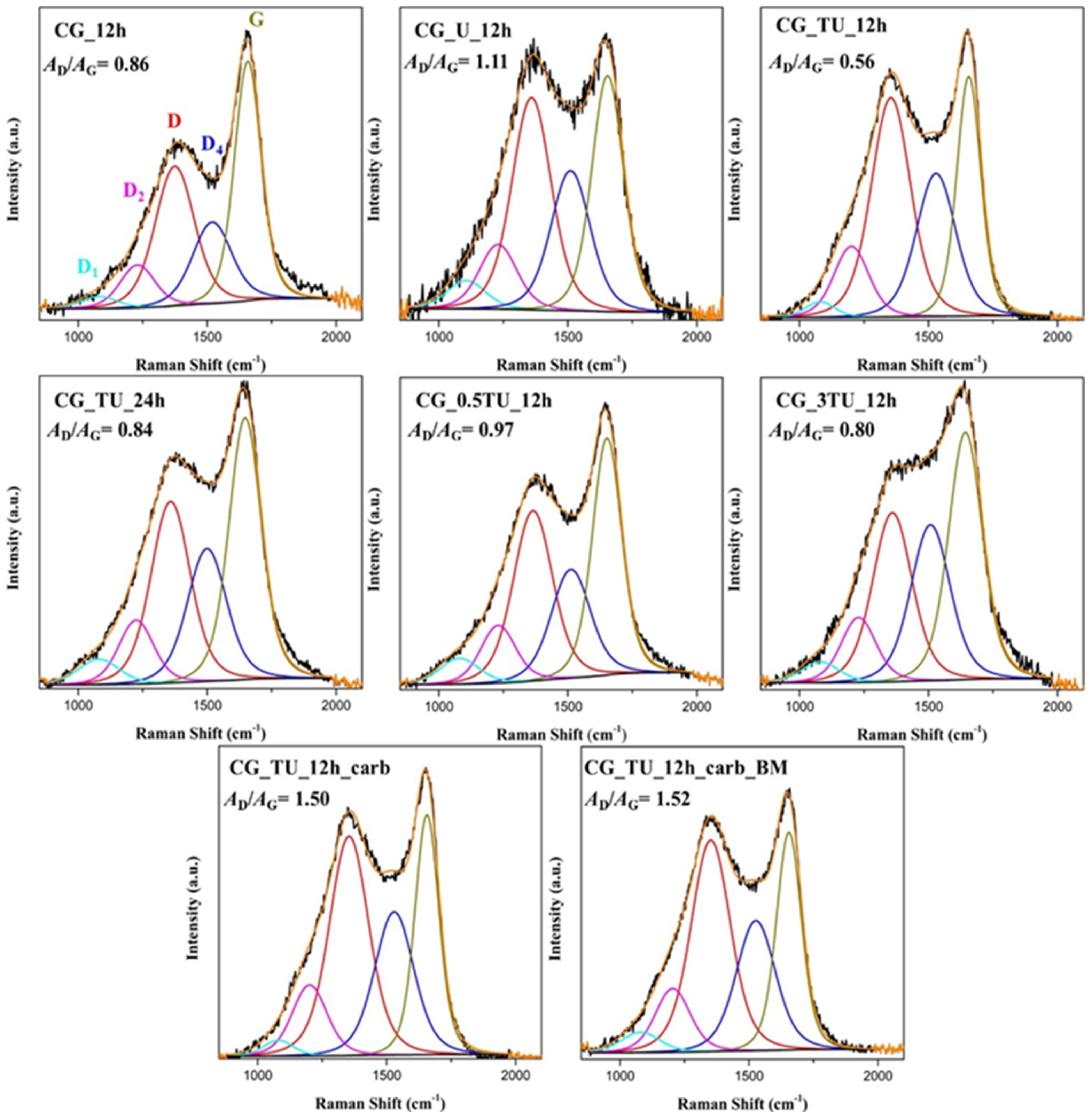
2.5. Raman Spectroscopy
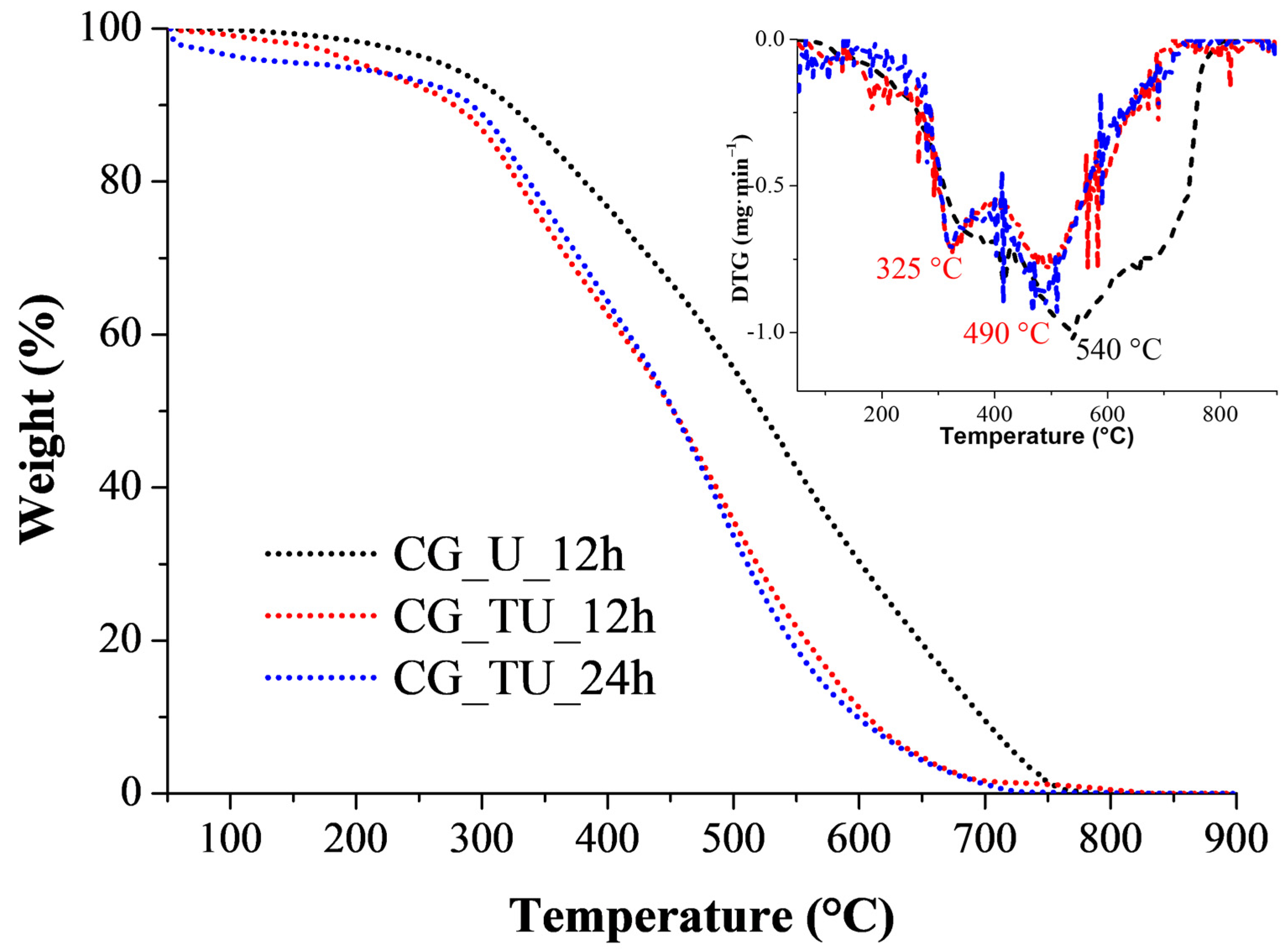
2.6. Thermogravimetric (TG) and Differential Thermogravimetric (DTG) Analysis
2.7. Catalytic Results
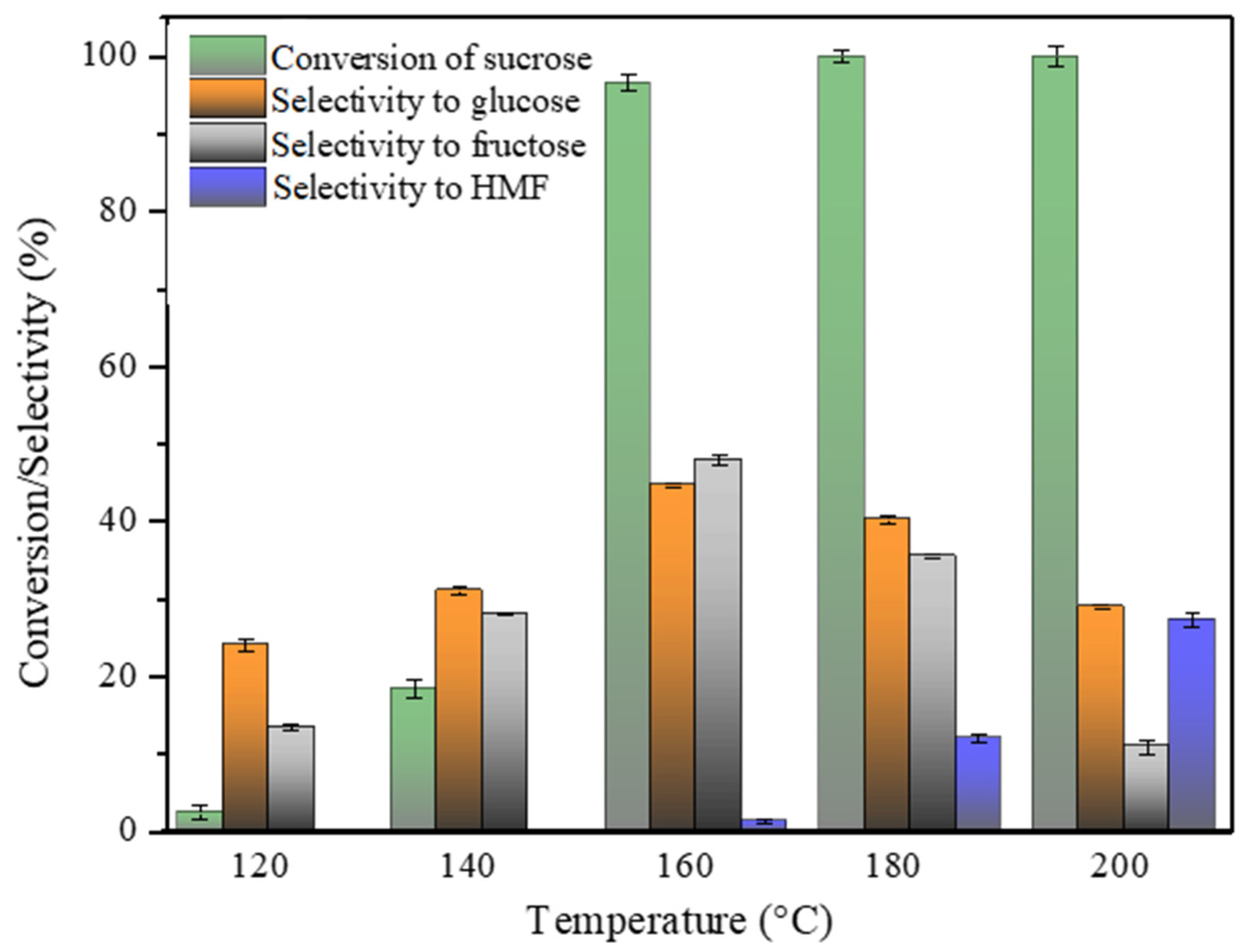
2.7.1. Impact of the Reaction Temperature on the Reaction
2.7.2. Catalysts Screening
2.7.3. The Influence of Different Variables on the Reaction Kinetics
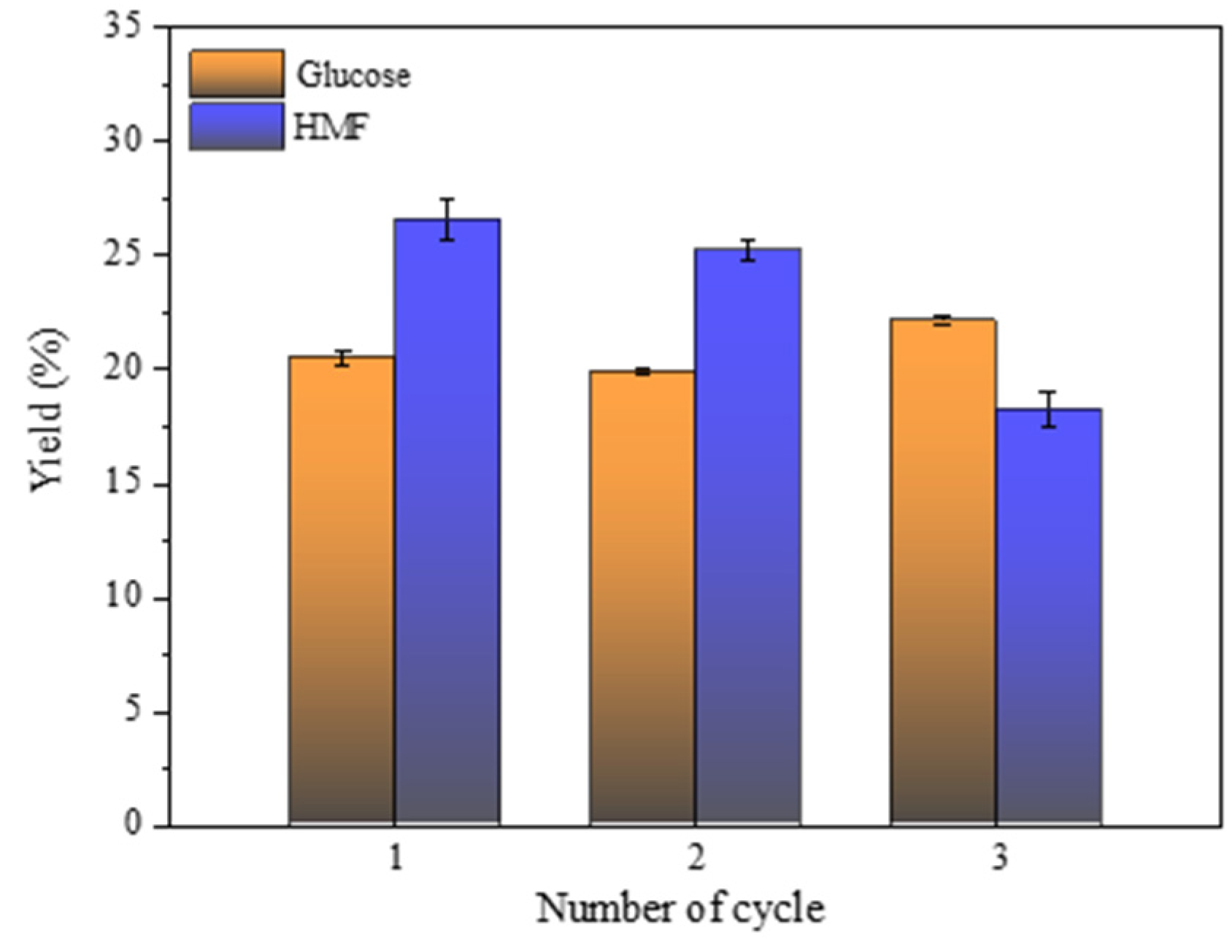
2.7.4. Recyclability of the Catalyst
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterisation
3.3. Catalyst Testing
3.4. Reusability Tests
3.5. Kinetic Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agency, E.E. Greenhouse Gas Emissions from Transport in Europe. 2024. Available online: https://www.eea.europa.eu/en/analysis/indicators/greenhouse-gas-emissions-from-transport (accessed on 5 May 2025).
- Dataintelo. Industrial 5-Hydroxymethylfurfural (5-HMF) Market Research Report 2033. Dataintelo Consulting Pvt. Ltd.: Pune, India, 2024. [Google Scholar]
- AVA Biochem. Available online: https://ava-biochem.com/ (accessed on 1 April 2025).
- Hu, L.; Wu, Z.; Jiang, Y.; Wang, X.; He, A.; Song, J.; Xu, J.; Zhou, S.; Zhao, Y.; Xu, J. Recent advances in catalytic and autocatalytic production of biomass-derived 5-hydroxymethylfurfural. Renew. Sustain. Energy Rev. 2020, 134, 110317. [Google Scholar] [CrossRef]
- Zuo, M.; Lin, L.; Zeng, X. The synthesis of potential biofuel 5-ethoxymethylfurfural: A review. Fuel 2023, 343, 127863. [Google Scholar] [CrossRef]
- Hoang, A.T.; Nižetić, S.; Ölçer, A.I. 2,5-Dimethylfuran (DMF) as a promising biofuel for the spark ignition engine application: A comparative analysis and review. Fuel 2021, 285, 119140. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; Luque, R.; Sepúlveda-Escribano, A. Transformations of biomass-derived platform molecules: From high added-value chemicals to fuels via aqueous-phase processing. Chem. Soc. Rev. 2011, 40, 5266–5281. [Google Scholar] [CrossRef]
- Toftgaard Pedersen, A.; Ringborg, R.; Grotkjær, T.; Pedersen, S.; Woodley, J.M. Synthesis of 5-hydroxymethylfurfural (HMF) by acid catalyzed dehydration of glucose–fructose mixtures. Chem. Eng. J. 2015, 273, 455–464. [Google Scholar] [CrossRef]
- Eblagon, K.M.; Figueiredo, J.L.; Fernando, R.; Pereira, M. Catalytic valorization of industrial grade sugarcane molasses to 5-hydroxymethylfurfural in water. Catal. Today 2024, 441, 114898. [Google Scholar] [CrossRef]
- Souzanchi, S.; Nazari, L.; Rao, K.T.V.; Yuan, Z.; Tan, Z.; Xu, C. 5-HMF production from industrial grade sugar syrups derived from corn and wood using niobium phosphate catalyst in a biphasic continuous-flow tubular reactor. Catal. Today 2023, 407, 274–280. [Google Scholar] [CrossRef]
- Tao, J.; Pan, Y.; Zhou, H.; Tang, Y.; Ren, G.; Yu, Z.; Li, J.; Zhang, R.; Li, X.; Qiao, Y.; et al. Catalytic Systems for 5-Hydroxymethylfurfural Preparation from Different Biomass Feedstocks: A Review. Catalysts 2024, 14, 30. [Google Scholar] [CrossRef]
- Baruah, J.; Nath, B.K.; Sharma, R.; Kumar, S.; Chandra Deka, R.; Chandra Baruah, D.; Kalita, E. Recent Trends in the Pretreatment of Lignocellulosic Biomass for Value-Added Products. Front. Energy Res. 2018, 6, 141. [Google Scholar] [CrossRef]
- Souzanchi, S.; Nazari, L.; Rao, K.T.V.; Yuan, Z.; Tan, Z.; Xu, C. Catalytic isomerization of glucose to fructose using heterogeneous solid Base catalysts in a continuous-flow tubular reactor: Catalyst screening study. Catal. Today 2019, 319, 76–83. [Google Scholar] [CrossRef]
- Mulk, S.; Sajid, M.; Wang, L.; Liu, F.; Pan, G. Catalytic conversion of sucrose to 5-hydroxymethylfurfural in green aqueous and organic medium. J. Environ. Chem. Eng. 2022, 10, 106613. [Google Scholar] [CrossRef]
- Steinbach, D.; Kruse, A.; Sauer, J.; Vetter, P. Sucrose Is a Promising Feedstock for the Synthesis of the Platform Chemical Hydroxymethylfurfural. Energies 2018, 11, 645. [Google Scholar] [CrossRef]
- Tian, X.; Qi, B.; Zhang, S.; Luo, J.; Wan, Y. Catalytic production of 5-hydroxymethylfurfural from sucrose and molasses by aluminum chloride in green aqueous γ-valerolactone system. Biomass Convers. Biorefinery 2021, 11, 1931–1941. [Google Scholar] [CrossRef]
- Kreissl, H.T.; Nakagawa, K.; Peng, Y.-K.; Koito, Y.; Zheng, J.; Tsang, S.C.E. Niobium oxides: Correlation of acidity with structure and catalytic performance in sucrose conversion to 5-hydroxymethylfurfural. J. Catal. 2016, 338, 329–339. [Google Scholar] [CrossRef]
- Tabtimtong, N.; Padungwat, A.; Lertna, N.; Athikaphan, P.; Neramittagapong, A.; Minato, T.; Neramittagapong, S. Sucrose conversion to 5-hydroxymethylfurfural over commercial ion-exchange resin. Biomass Bioenergy 2025, 199, 107940. [Google Scholar] [CrossRef]
- Tong, X.; Wang, Y.; Nie, G.; Yan, Y. Selective dehydration of fructose and sucrose to 5-hydroxymethyl-2-furfural with heterogeneous ge (IV) catalysts. Environ. Prog. Sustain. Energy 2015, 34, 207–210. [Google Scholar] [CrossRef]
- Tongtummachat, T.; Akkarawatkhoosith, N.; Jaree, A. Process intensification for 5-hydroxymethylfurfural production from sucrose in a continuous fixed-bed reactor. Chem. Eng. Res. Des. 2022, 182, 312–323. [Google Scholar] [CrossRef]
- Zhang, M.; Tong, X.; Ma, R.; Li, Y. Catalytic transformation of carbohydrates into 5-hydroxymethyl furfural over tin phosphate in a water-containing system. Catal. Today 2016, 264, 131–135. [Google Scholar] [CrossRef]
- Perez, G.a.P.; Pandey, S.; Dumont, M.-J. Sulfosuccinic acid-based metal-center catalysts for the synthesis of HMF from carbohydrates. Catal. Today 2023, 418, 114127. [Google Scholar] [CrossRef]
- Morais, R.G.; Rey-Raap, N.; Figueiredo, J.L.; Pereira, M.F.R. Glucose-derived carbon materials with tailored properties as electrocatalysts for the oxygen reduction reaction. Beilstein J. Nanotechnol. 2019, 10, 1089–1102. [Google Scholar] [CrossRef]
- Nahavandi, M.; Kasanneni, T.; Yuan, Z.S.; Xu, C.C.; Rohani, S. Efficient Conversion of Glucose into 5-Hydroxymethylfurfural Using a Sulfonated Carbon-Based Solid Acid Catalyst: An Experimental and Numerical Study. ACS Sustain. Chem. Eng. 2019, 7, 11970–11984. [Google Scholar] [CrossRef]
- Eblagon, K.M.; Arenillas, A.; Malaika, A.; Pereira, M.F.R.; Figueiredo, J.L. The influence of the surface chemistry of phosphorylated carbon xerogel catalysts on the production of HMF from fructose in water. Fuel 2023, 334, 126610. [Google Scholar] [CrossRef]
- Johnson, R.L.; Anderson, J.M.; Shanks, B.H.; Schmidt-Rohr, K. Simple One-Step Synthesis of Aromatic-Rich Materials with High Concentrations of Hydrothermally Stable Catalytic Sites, Validated by NMR. Chem. Mater. 2014, 26, 5523–5532. [Google Scholar] [CrossRef]
- Chen, S.S.; Yu, I.K.M.; Cho, D.-W.; Song, H.; Tsang, D.C.W.; Tessonnier, J.-P.; Ok, Y.S.; Poon, C.S. Selective Glucose Isomerization to Fructose via a Nitrogen-doped Solid Base Catalyst Derived from Spent Coffee Grounds. ACS Sustain. Chem. Eng. 2018, 6, 16113–16120. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Zhang, Y.; Song, F.; Xie, Y.; Wang, M.; Cui, H.; Yi, W. N-Doped Carbon Materials as Heterogeneous Catalysts for High Efficiency Isomerization Glucose to Fructose in Aqueous Media. Catal. Lett. 2020, 150, 493–504. [Google Scholar] [CrossRef]
- Xiong, X.; Yu, I.K.M.; Chen, S.S.; Tsang, D.C.W.; Cao, L.; Song, H.; Kwon, E.E.; Ok, Y.S.; Zhang, S.; Poon, C.S. Sulfonated biochar as acid catalyst for sugar hydrolysis and dehydration. Catal. Today 2018, 314, 52–61. [Google Scholar] [CrossRef]
- Palma, V.; Barba, D.; Cortese, M.; Martino, M.; Renda, S.; Meloni, E. Microwaves and Heterogeneous Catalysis: A Review on Selected Catalytic Processes. Catalysts 2020, 10, 246. [Google Scholar] [CrossRef]
- Gomes, G.R.; Pastre, J.C. Microwave-assisted HMF production from water-soluble sugars using betaine-based natural deep eutectic solvents (NADES). Sustain. Energy Fuels 2020, 4, 1891–1898. [Google Scholar] [CrossRef]
- Rocha, S.; Marzialetti, T.; Kopp, M.; Cea, M. Reaction Mechanism of the Microwave-Assisted Synthesis of 5-Hydroxymethylfurfural from Sucrose in Sugar Beet Molasses. Catalysts 2021, 11, 1458. [Google Scholar] [CrossRef]
- Rustamaji, H.; Prakoso, T.; Devianto, H.; Widiatmoko, P.; Febriyanto, P.; Eviani, M. Modification of hydrochar derived from palm waste with thiourea to produce N, S co-doped activated carbon for supercapacitor. Sustain. Chem. Environ. 2024, 7, 100132. [Google Scholar] [CrossRef]
- Morawa Eblagon, K.; Rey-Raap, N.; Figueiredo, J.L.; Pereira, M.F.R. Relationships between texture, surface chemistry and performance of N-doped carbon xerogels in the oxygen reduction reaction. Appl. Surf. Sci. 2021, 548, 149242. [Google Scholar] [CrossRef]
- Kiani, A.; Acocella, M.R.; Granata, V.; Mazzotta, E.; Malitesta, C.; Guerra, G. Green Oxidation of Carbon Black by Dry Ball Milling. ACS Sustain. Chem. Eng. 2022, 10, 16019–16026. [Google Scholar] [CrossRef]
- Sharma, R.; Rana, D.S.; Awasthi, A.; Singh, D.; Ibrahim, A.A.; Umar, A.; Baskoutas, S. Nitrogen functionalized biomass derived mesoporous carbon nanomaterials for electrochemical detection of lead (II) ions. Heliyon 2024, 10, e39090. [Google Scholar] [CrossRef]
- Unglaube, N.; Florent, M.; Otto, T.; Stötzer, M.; Grothe, J.; Kaskel, S.; Bandosz, T.J. Doping of porous carbons with sulfur and nitrogen markedly enhances their surface activity for formaldehyde removal. J. Colloid Interface Sci. 2024, 653, 594–605. [Google Scholar] [CrossRef]
- Gorgulho, H.F.; Gonçalves, F.R.; Pereira, M.F.R.; Figueiredo, J.L. Synthesis and characterization of nitrogen-doped carbon xerogels. Carbon 2009, 47, 2032–2039. [Google Scholar] [CrossRef]
- Pels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, K.M. Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis. Carbon 1995, 33, 1641–1653. [Google Scholar] [CrossRef]
- Falco, C.; Sevilla, M.; White, R.J.; Rothe, R.; Titirici, M.-M. Renewable Nitrogen-Doped Hydrothermal Carbons Derived from Microalgae. ChemSusChem 2012, 5, 1834–1840. [Google Scholar] [CrossRef]
- Eblagon, K.M.; Malaika, A.; Ptaszyńska, K.; Pereira, M.F.R.; Kozłowski, M.; Figueiredo, J.L. Niobium oxide-phosphorylated carbon xerogel composites as solid acid catalysts for cascade conversion of glucose to 5-hydroxymethylfurfural (HMF) in pure water. Catal. Today 2023, 418, 114070. [Google Scholar] [CrossRef]
- Feng, Y.; Jia, W.; Yan, G.; Zeng, X.; Sperry, J.; Xu, B.; Sun, Y.; Tang, X.; Lei, T.; Lin, L. Insights into the active sites and catalytic mechanism of oxidative esterification of 5-hydroxymethylfurfural by metal-organic frameworks-derived N-doped carbon. J. Catal. 2020, 381, 570–578. [Google Scholar] [CrossRef]
- Chhabra, T.; Bahuguna, A.; Dhankhar, S.S.; Nagaraja, C.M.; Krishnan, V. Sulfonated graphitic carbon nitride as a highly selective and efficient heterogeneous catalyst for the conversion of biomass-derived saccharides to 5-hydroxymethylfurfural in green solvents. Green Chem. 2019, 21, 6012–6026. [Google Scholar] [CrossRef]
- Qiu, S.; Fu, Y.; Lei, W.; Chang, J. Preparation of nitrogen and sulfur co-doped tubular porous carbon derived from Ceiba speciosa flowers for supercapacitors. J. Energy Storage 2025, 112, 115536. [Google Scholar] [CrossRef]
- Titirici, M.-M. Chapter 12—Hydrothermal Carbons: Synthesis, Characterization, and Applications. In Novel Carbon Adsorbents; Tascón, J.M.D., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 351–399. [Google Scholar]
- Malaika, A.; Eblagon, K.M.; Matuszek, N.; Pereira, M.F.R.; Kozłowski, M. Towards valorization of glycerol and molasses: Carbon-based catalysts from molasses for the synthesis of acetins. Bioresour. Technol. 2025, 417, 131834. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, M.; Qi, L.; Qi, Y. Mechanism for the formation and growth of carbonaceous spheres from sucrose by hydrothermal carbonization. RSC Adv. 2016, 6, 20814–20823. [Google Scholar] [CrossRef]
- Titirici, M.-M.; Antonietti, M.; Baccile, N. Hydrothermal carbon from biomass: A comparison of the local structure from poly- to monosaccharides and pentoses/hexoses. Green Chem. 2008, 10, 1204–1212. [Google Scholar] [CrossRef]
- Kong, X.; Vinju Vasudevan, S.; Cao, M.; Cai, J.; Mao, H.; Bu, Q. Microwave-Assisted Efficient Fructose–HMF Conversion in Water over Sulfonated Carbon Microsphere Catalyst. ACS Sustain. Chem. Eng. 2021, 9, 15344–15356. [Google Scholar] [CrossRef]
- Hirsch, E.; Pataki, H.; Farkas, A.; Bata, H.; Vass, P.; Fehér, C.; Barta, Z.; Párta, L.; Csontos, I.; Ballagi, A. Raman-Based Feedback Control of the Enzymatic Hydrolysis of Lactose. Org. Process Res. Dev. 2016, 20, 1721–1727. [Google Scholar] [CrossRef]
- Yadav, R.; Joshi, P.; Hara, M.; Yana, T.; Hashimoto, S. Intercorrelation between physical and electrochemical behavior of nitrogen-doping in graphene for symmetric supercapacitor electrode. SN Appl. Sci. 2020, 2, 1630. [Google Scholar] [CrossRef]
- Straten, J.W.; Schleker, P.; Krasowska, M.; Veroutis, E.; Granwehr, J.; Auer, A.A.; Hetaba, W.; Becker, S.; Schlögl, R.; Heumann, S. Nitrogen-Functionalized Hydrothermal Carbon Materials by Using Urotropine as the Nitrogen Precursor. Chem. A Eur. J. 2018, 24, 12298–12317. [Google Scholar] [CrossRef]
- Chen, H.; Sun, F.; Wang, J.; Li, W.; Qiao, W.; Ling, L.; Long, D. Nitrogen Doping Effects on the Physical and Chemical Properties of Mesoporous Carbons. J. Phys. Chem. C 2013, 117, 8318–8328. [Google Scholar] [CrossRef]
- Kastner, J.R.; Miller, J.; Geller, D.P.; Locklin, J.; Keith, L.H.; Johnson, T. Catalytic esterification of fatty acids using solid acid catalysts generated from biochar and activated carbon. Catal. Today 2012, 190, 122–132. [Google Scholar] [CrossRef]
- Florent, M.; Hashmi, R.; Bandosz, T.J. The extent of carbon surface oxygen affinity and its effects on the activity of metal-free carbon catalysts in the oxygen reduction reaction: The interplay of porosity and N-, O- and S-enriched surface chemistry. Mater. Adv. 2022, 3, 8567–8578. [Google Scholar] [CrossRef]
- Eblagon, K.M.; Malaika, A.; Pereira, M.F.R.; Figueiredo, J.L. Cutting the Green Waste. Structure-Performance Relationship in Functionalized Carbon Xerogels for Hydrolysis of Cellobiose. ChemCatChem 2018, 10, 4934–4946. [Google Scholar] [CrossRef]
- Portillo Perez, G.; Mukherjee, A.; Dumont, M.-J. Insights into HMF catalysis. J. Ind. Eng. Chem. 2019, 70, 1–34. [Google Scholar] [CrossRef]
- Reis, G.M.; Machado, L.F.L.; Nunes, R.S.; Mandelli, D.; Carvalho, W.A. Tailored sulfonated carbons: Unraveling enhanced catalytic dynamics for fructose dehydration under conventional and microwave heating. RSC Sustain. 2024, 2, 1456–1471. [Google Scholar] [CrossRef]
- Mason, J.B.; Sun, Y. Microwave-Assisted Production of 5-Hydroxymethylfurfural from Glucose. Chem. Sel. 2021, 6, 10582–10586. [Google Scholar] [CrossRef]
- Morawa Eblagon, K.; Pereira, M.F.R.; Figueiredo, J.L. One-pot oxidation of cellobiose to gluconic acid. Unprecedented high selectivity on bifunctional gold catalysts over mesoporous carbon by integrated texture and surface chemistry optimization. Appl. Catal. B Environ. 2016, 184, 381–396. [Google Scholar] [CrossRef]
- Pinheiro Torres, A.; Oliveira, F.a.R. Application of the acid hydrolysis of sucrose as a temperature indicator in continuous thermal processes. J. Food Eng. 1999, 40, 181–188. [Google Scholar] [CrossRef]
- Tombari, E.; Salvetti, G.; Ferrari, C.; Johari, G.P. Kinetics and Thermodynamics of Sucrose Hydrolysis from Real-Time Enthalpy and Heat Capacity Measurements. J. Phys. Chem. B 2007, 111, 496–501. [Google Scholar] [CrossRef]
- Liu, S.; Zhu, Y.; Liao, Y.; Wang, H.; Liu, Q.; Ma, L.; Wang, C. Advances in understanding the humins: Formation, prevention and application. Appl. Energy Combust. Sci. 2022, 10, 100062. [Google Scholar] [CrossRef]
- Konwar, L.J.; Mäki-Arvela, P.; Mikkola, J.-P. SO3H-Containing Functional Carbon Materials: Synthesis, Structure, and Acid Catalysis. Chem. Rev. 2019, 119, 11576–11630. [Google Scholar] [CrossRef]
- Li, X.; Shao, Z.; Shan, H.; Liu, L. Evolution process of humins derived from cellulose by a humin extraction approach. Green Chem. 2025, 27, 5322–5331. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | C (wt%) | H (wt%) | N (wt%) | S (wt%) | O * (wt%) |
|---|---|---|---|---|---|
| CG_12h | 68.6 | 4.5 | 0.0 | 0.0 | 28.0 |
| CG_U_12h | 63.7 | 5.3 | 8.1 | 0.0 | 22.9 |
| CG_TU_12h | 57.1 | 4.6 | 5.2 | 6.1 | 27.3 |
| CG_TU_24h | 60.1 | 4.3 | 5.7 | 6.0 | 23.9 |
| CG_0.5TU_12h | 66.5 | 4.4 | 3.5 | 3.9 | 22.1 |
| CG_3TU_12h | 61.5 | 4.4 | 10.4 | 12.7 | 11.5 |
| CG_TU_12h_carb | 83.7 | 1.9 | 5.6 | 2.1 | 7.0 |
| CG_TU_12h_carb_BM | 79.6 | 1.9 | 4.8 | 1.5 | 13.0 |
| Sample | C (wt%) | N (wt%) | O (wt%) | S (wt%) | Atot (mmol H+/g) |
|---|---|---|---|---|---|
| CG_12h | 79.7 | 0.0 | 20.3 | 0.0 | 2.32 |
| CG_U_12h | 78.0 | 6.1 | 15.9 | 0.0 | - |
| CG_TU_12h | 71.7 | 4.2 | 16.6 | 7.5 | 1.64 |
| CG_TU_24h | 70.6 | 4.3 | 18.4 | 6.8 | 2.23 |
| CG_0.5TU_12h | 74.5 | 2.3 | 18.6 | 4.7 | 1.62 |
| CG_3TU_12h | 64.1 | 7.8 | 11.0 | 17.1 | 0.29 |
| CG_TU_12h_carb_BM | 77.2 | 1.2 | 21.0 | 0.6 | 0.03 * |
| Catalyst | Selectivity to Glucose (%) | Selectivity to Fructose (%) | Selectivity to HMF (%) |
|---|---|---|---|
| Blank test * | 29.0 | 10.8 | 27.4 |
| CG_12h * | 31.2 | 1.3 | 28.8 |
| CG_U_12h # | 16.5 | 25.2 | 26.1 |
| CG_TU_12h * | 27.0 | 5.2 | 33.8 |
| CG_TU_24h * | 29.5 | 4.6 | 30.3 |
| CG_TU_12h_carb # | 23.5 | 19.2 | 25.8 |
| CG_TU_12h_carb_BM * | 23.5 | 12.5 | 27.0 |
| CG_0.5_TU * | 23.1 | 3.3 | 30.2 |
| CG_3_TU * | 25.2 | 7.3 | 29.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morawa Eblagon, K.; Morais, R.G.; Malaika, A.; Castro Bravo, M.A.; Rey-Raap, N.; Pereira, M.F.R.; Kozłowski, M. Production of 5-Hydroxymethylfurfural (HMF) from Sucrose in Aqueous Phase Using S, N-Doped Hydrochars. Catalysts 2025, 15, 656. https://doi.org/10.3390/catal15070656
Morawa Eblagon K, Morais RG, Malaika A, Castro Bravo MA, Rey-Raap N, Pereira MFR, Kozłowski M. Production of 5-Hydroxymethylfurfural (HMF) from Sucrose in Aqueous Phase Using S, N-Doped Hydrochars. Catalysts. 2025; 15(7):656. https://doi.org/10.3390/catal15070656
Chicago/Turabian StyleMorawa Eblagon, Katarzyna, Rafael G. Morais, Anna Malaika, Manuel Alejandro Castro Bravo, Natalia Rey-Raap, M. Fernando R. Pereira, and Mieczysław Kozłowski. 2025. "Production of 5-Hydroxymethylfurfural (HMF) from Sucrose in Aqueous Phase Using S, N-Doped Hydrochars" Catalysts 15, no. 7: 656. https://doi.org/10.3390/catal15070656
APA StyleMorawa Eblagon, K., Morais, R. G., Malaika, A., Castro Bravo, M. A., Rey-Raap, N., Pereira, M. F. R., & Kozłowski, M. (2025). Production of 5-Hydroxymethylfurfural (HMF) from Sucrose in Aqueous Phase Using S, N-Doped Hydrochars. Catalysts, 15(7), 656. https://doi.org/10.3390/catal15070656