
Confined Catalysis Involving a Palladium Complex and a Self-Assembled Capsule for the Dimerization of Vinyl Arenes and the Formation of Indane and Tribenzo–Pentaphene Derivatives


Abstract

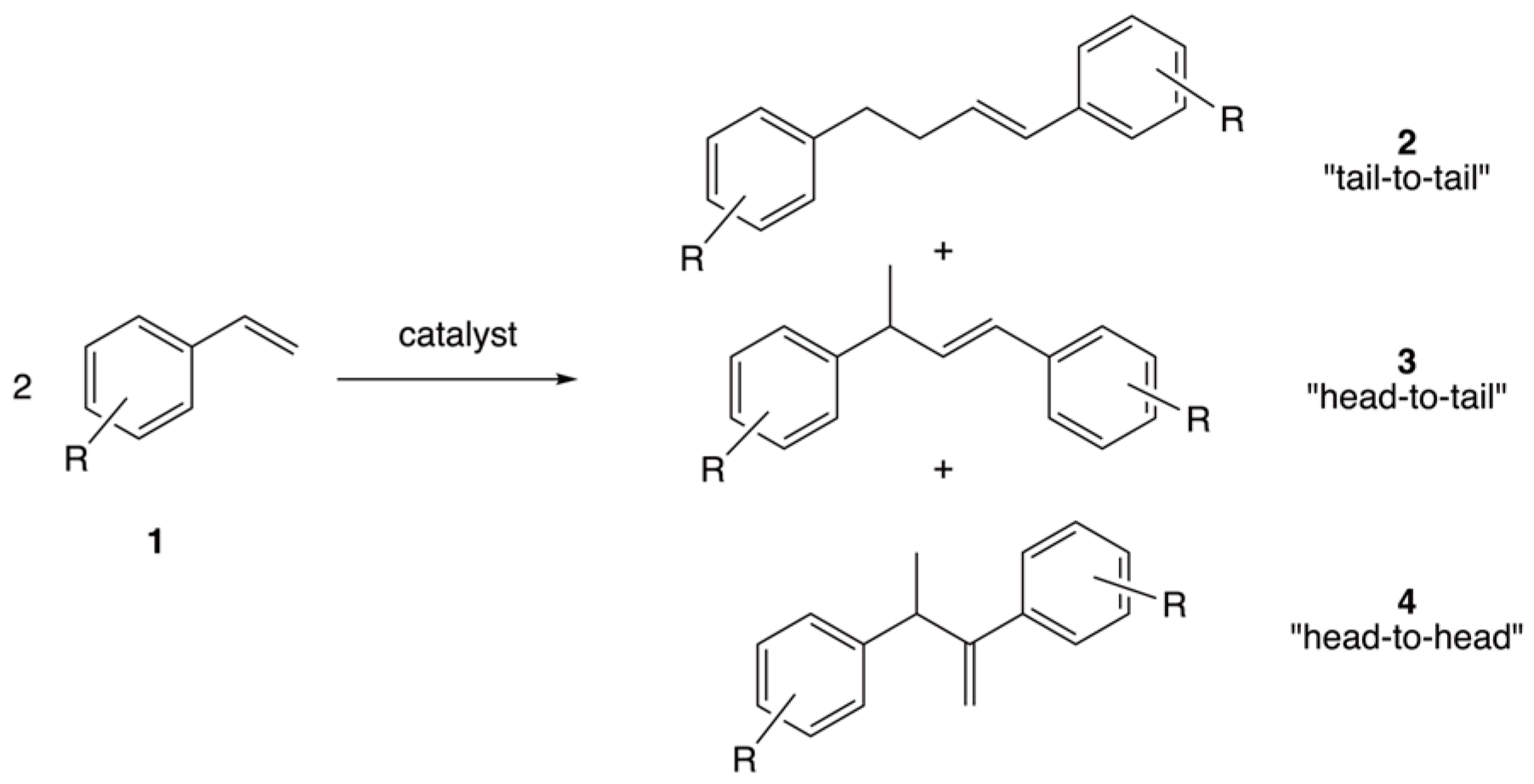
1. Introduction
2. Results and Discussion
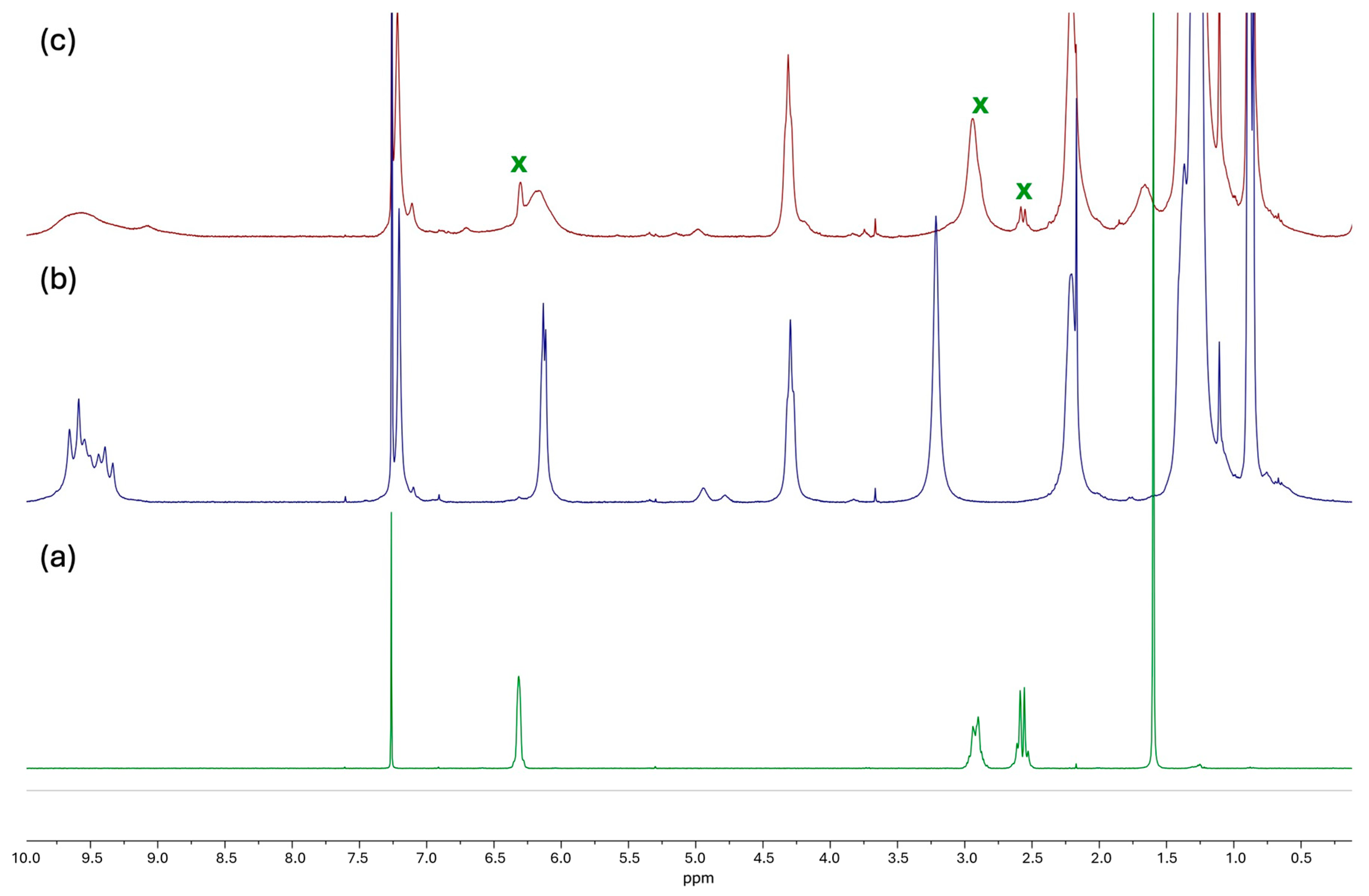
2.1. Formation of Inclusion Complex
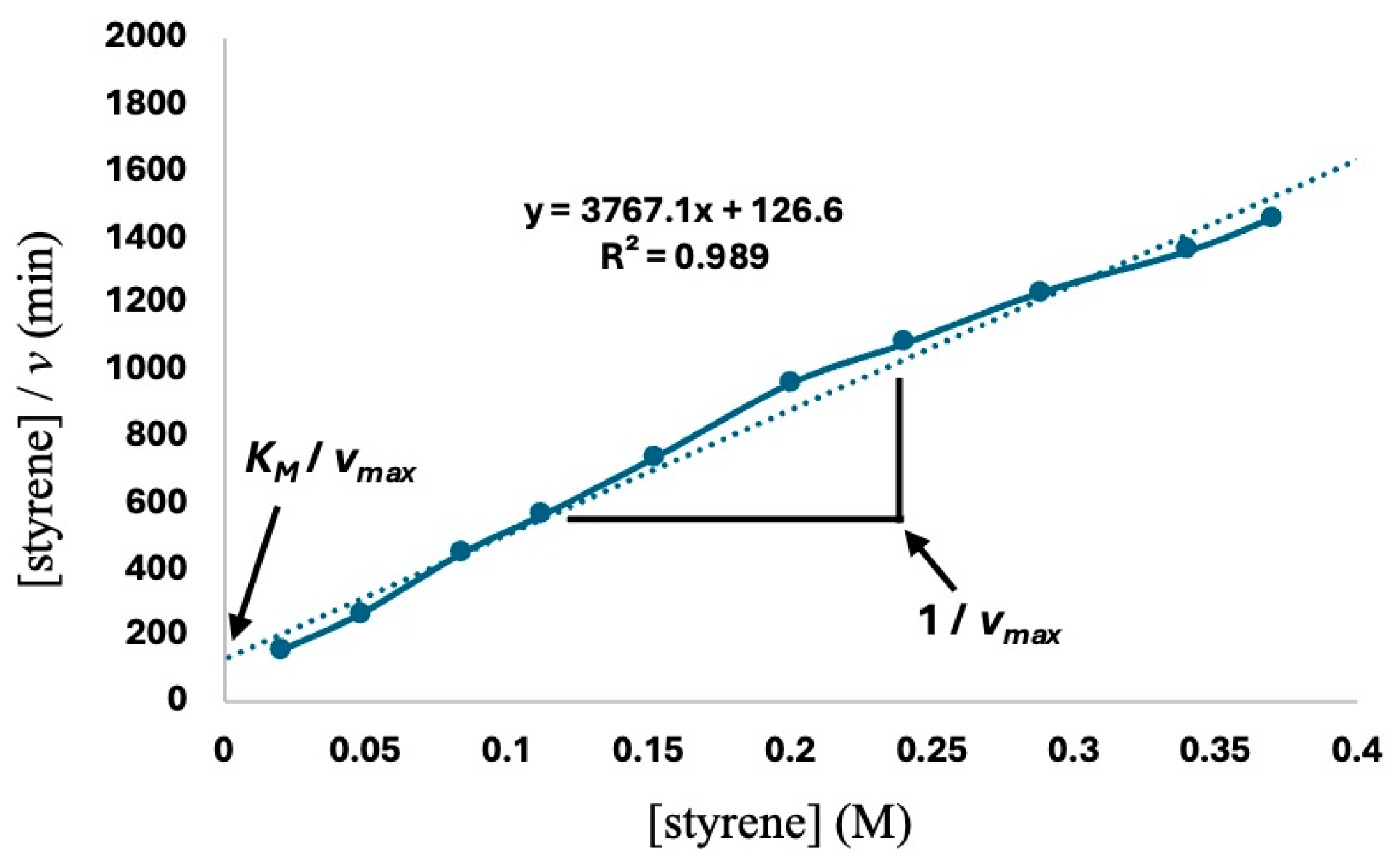
2.2. Catalytic Study
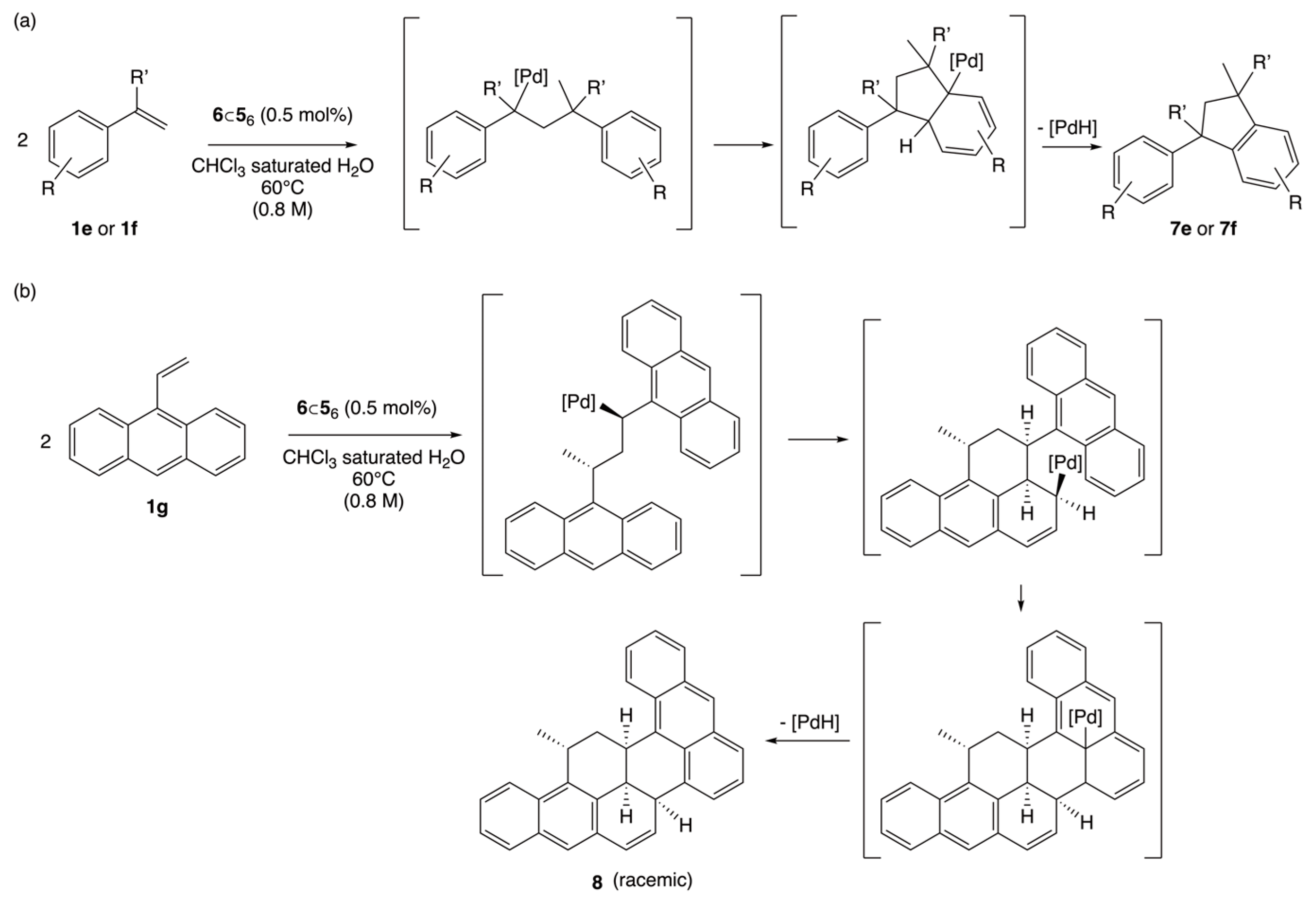
2.3. Mecanistic Considerations
3. Materials and Methods
3.1. General Remarks
3.2. Catalytic Reactions
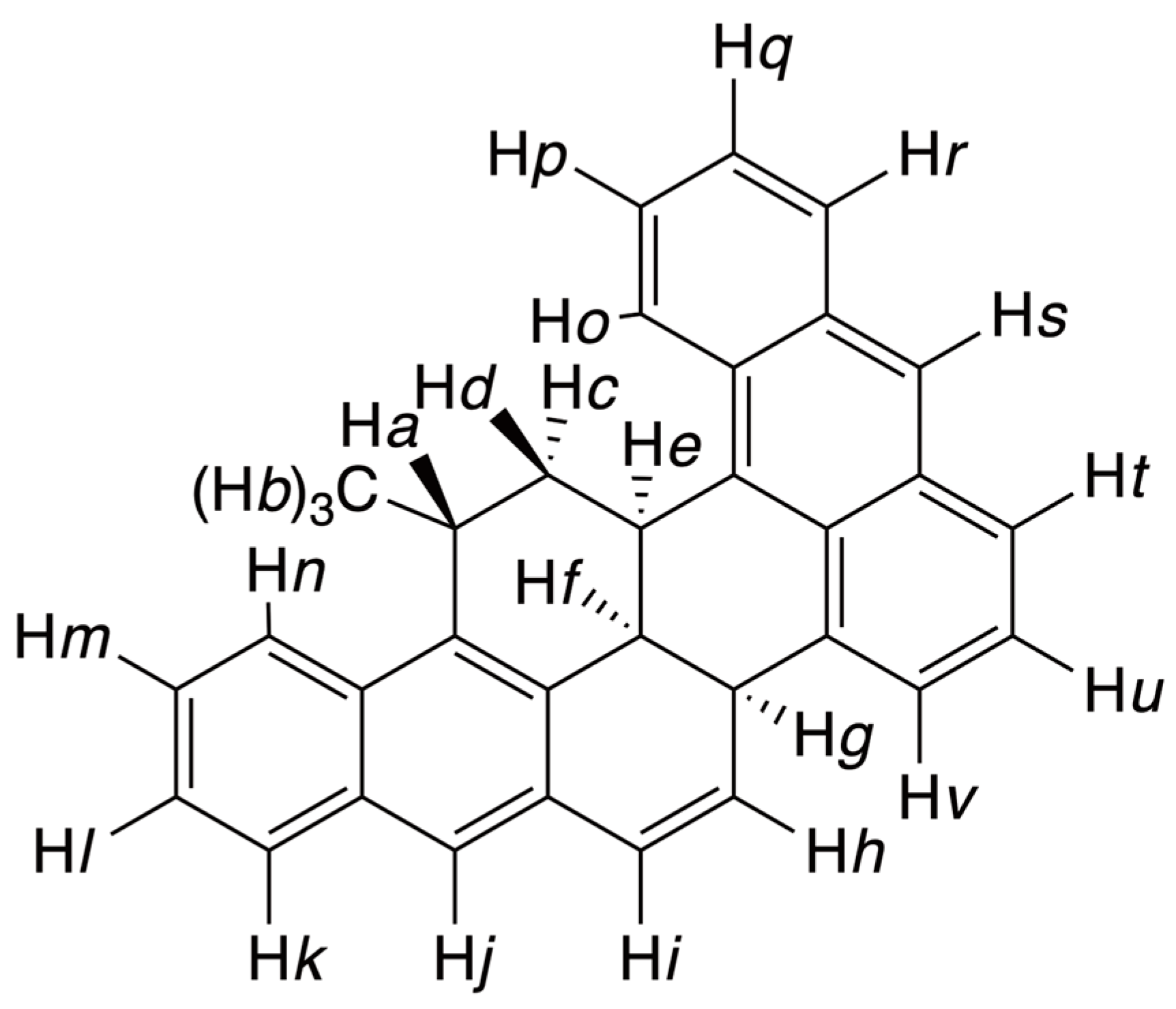
3.2.1. Dimeric Products
3.2.2. Indane Derivatives
3.2.3. Tribenzo–Pentaphene Derivative
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benito, A.; Corma, A.; García, H.; Primo, J. Dimerization of styrene catalyzed by acid 12-membered ring zeolites. Appl. Catal. Gen. 1994, 116, 127–135. [Google Scholar] [CrossRef]
- Grigoréva, N.G.; Talipova, R.R.; Korzhova, L.F.; Vosmerikov, A.V.; Kutepov, B.I.; Dzhemilev, U.M. Dimerization and oligomerization of styrene in the presence of pentasils. Russ. Chem. Bull. 2009, 58, 59–63. [Google Scholar] [CrossRef]
- Yue, H.-L.; Zhao, X.-H.; Jiang, L.; Shao, Y.; Mei, L.-J. Highly efficient TfOH-catalyzed head-to-tail dimerization of vinylarenes. Synth. Commun. 2016, 46, 179–186. [Google Scholar] [CrossRef]
- Cabrero-Antonino, J.R.; Leyva-Pérez, A.; Corma, A. Iron-catalysed regio-and stereoselective head-to-tail dimerisation of styrenes. Adv. Synth. Catal. 2010, 352, 1571–1576. [Google Scholar] [CrossRef]
- Chang, A.S.; Kascoutas, M.A.; Valentine, Q.P.; How, K.I.; Thomas, R.M.; Cook, A.K. Alkene isomerization using a heterogeneous nickel-hydride catalyst. J. Am. Chem. Soc. 2024, 146, 15596–15608. [Google Scholar] [CrossRef]
- Kondo, T.; Takagi, D.; Tsujita, H.; Ura, Y.; Wada, K.; Mitsudo, T. Highly selective dimerization of styrenes and linear co-dimerization of styrenes with ethylene catalyzed by a ruthenium complex. Angew. Chem. Int. Ed. 2007, 46, 5958–5961. [Google Scholar] [CrossRef] [PubMed]
- Shimura, S.; Miura, H.; Tsukada, S.; Wada, K.; Hosokawa, S.; Inoue, M. Highly selective linear dimerization of styrenes by ceria-supported ruthenium catalysts. ChemCatChem 2012, 4, 2062–2067. [Google Scholar] [CrossRef]
- Kretschmer, W.P.; Troyanov, S.I.; Meetsma, A.; Hessen, B.; Teuben, J.H. Regioselective homo-and codimerization of α-olefins catalyzed by bis(2,4,7-trimethylindenyl)yttrium hydride. Organometallics 1998, 17, 284–286. [Google Scholar] [CrossRef]
- Myagmarsuren, G.; Tkach, V.S.; Shmidt, F.K.; Mohamad, M.; Suslov, D.S. Selective dimerization of styrene to 1,3-diphenyl-1-butene with bis(β-diketonato)palladium/boron trifluoride etherate catalyst system. J. Mol. Catal. Chem. 2005, 235, 154–160. [Google Scholar] [CrossRef]
- Tsuchimoto, T.; Kamiyama, S.; Negoro, R.; Shirakawa, E.; Kawakami, Y. Palladium-catalysed dimerization of vinylarenes using indium triflate as an effective co-catalyst. Chem. Commun. 2003, 7, 852–853. [Google Scholar] [CrossRef]
- Bedford, R.B.; Betham, M.; Blake, M.E.; Garcés, A.; Millar, S.L.; Prashar, S. Asymmetric styrene dimerisation using mixed palladium-indium catalysts. Tetrahedron 2005, 61, 9799–9807. [Google Scholar] [CrossRef]
- Peng, J.; Li, J.; Qiu, H.; Jiang, J.; Jiang, K.; Mao, J.; Lai, G. Dimerization of styrene to 1,3-diphenyl-1-butene catalyzed by palladium-Lewis acid in ionic liquid. J. Mol. Catal. A Chem. 2006, 255, 16–18. [Google Scholar] [CrossRef]
- Aloia, A.; Casiello, M.; Nacci, A.; Monopoli, A. Selective palladium(II)-catalyzed dimerization of styrenes and acrylates in molten tetrabutylammonium acetate as an ionic liquid. Eur.J. Org. Chem. 2024, 27, e202400111. [Google Scholar] [CrossRef]
- Ma, H.; Sun, Q.; Li, W.; Wang, J.; Zhang, Z.; Yang, Y.; Lei, Z. Highly efficient Pd(acac)2/TFA catalyzed head-to-tail dimerization of vinylarenes at room temperature. Tetrahedron Lett. 2011, 52, 1569–1573. [Google Scholar] [CrossRef]
- Fanfoni, L.; Meduri, A.; Zangrando, E.; Castillon, S.; Felluga, F.; Milani, B. New chiral PN ligands for the regio-and stereoselective Pd-catalyzed dimerization of styrene. Molecules 2011, 16, 1804–1824. [Google Scholar] [CrossRef]
- Sen, A.; Lai, T.W. Oligomerization and isomerization of olefins by η3-allyl complexes of palladium. The role of the allyl group. Organometallics 1983, 2, 1059–1060. [Google Scholar] [CrossRef]
- Jiang, Z.; Sen, A. Tailored cationic palladium(II) compounds as catalysts for highly selective linear dimerization of styrene and linear polymerization of p-divinylbenzene. J. Am. Chem. Soc. 1990, 112, 9655–9657. [Google Scholar] [CrossRef]
- Choi, J.H.; Kwon, J.K.; RajanBabu, T.V.; Lim, H.J. Highly efficient catalytic dimerization of styrenes via cationic palladium(II) complexes. Adv. Synth. Catal. 2013, 355, 3633–3638. [Google Scholar] [CrossRef]
- Murata, K.; Saito, K.; Kikuchi, S.; Akita, M.; Inagaki, A. Visible-light-controlled homo-and copolymerization of styrenes by a bichromophoric Ir-Pd catalyst. Chem. Commun. 2015, 51, 5717–5720. [Google Scholar] [CrossRef]
- Hkiri, S.; Steinmetz, M.; Schurhammer, R.; Sémeril, D. Encapsulated neutral ruthenium catalyst for substrate-selective oxidation of alcohols. Chem. Eur. J. 2022, 28, e202201887. [Google Scholar] [CrossRef]
- MacGillivray, L.R.; Atwood, J.L. A chiral spherical molecular assembly held together by 60 hydrogen bonds. Nature 1997, 389, 469–472. [Google Scholar] [CrossRef]
- La Manna, P.; Talotta, C.; Floresta, G.; De Rosa, M.; Soriente, A.; Rescifina, A.; Gaeta, C.; Neri, P. Mild Friedel-Crafts reactions inside a hexameric resorcinarene capsule: C-Cl bond activation through hydrogen bonding to bridging water molecules. Angew. Chem. Int. Ed. 2018, 57, 5423–5428. [Google Scholar] [CrossRef] [PubMed]
- Chwastek, M.; Cmoch, P.; Szumna, A. Anion-based self-assembly of resorcin[4]arenes and pyrogallol[4]arenes. J. Am. Chem. Soc. 2022, 144, 5350–5358. [Google Scholar] [CrossRef]
- De Rosa, M.; Gambaro, S.; Soriente, A.; Della Sala, P.; Iuliano, V.; Talotta, C.; Gaeta, C.; Rescifina, A.; Neri, P. Carbocation catalysis in confined space: Activation of trityl chloride inside the hexameric resorcinarene capsule. Chem. Sci. 2022, 13, 8618–8625. [Google Scholar] [CrossRef]
- Iuliano, V.; Talotta, C.; De Rosa, M.; Soriente, A.; Neri, P.; Rescifina, A.; Floresta, G.; Gaeta, C. Insights into the Friedel-Crafts benzoylation of N-methylpyrrole inside the confined space of the self-assembled resorcinarene capsule. Org. Lett. 2023, 25, 6464–6468. [Google Scholar] [CrossRef]
- Merget, S.; Catti, L.; Piccini, G.; Tiefenbacher, K. Requirements for terpene cyclizations inside the supramolecular resorcinarene capsule: Bound water and its protonation determine the catalytic activity. J. Am. Chem. Soc. 2020, 142, 4400–4410. [Google Scholar] [CrossRef]
- Zhang, T.; Le Corre, L.; Reinaud, O.; Colasson, B. A Promising approach for controlling the second coordination sphere of biomimetic metal complexes: Encapsulation in a dynamic hydrogen-bonded capsule. Chem. Eur. J. 2021, 27, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Maverick, A.W.; Fronczek, F.R.; Doyle, J.R.; Baenziger, N.C.; Howells, M.A.M. The Pbca polymorph of dichloro (η4-1,5-cyclooctadiene) palladium (II). Acta Crystallogr. C 1993, 49, 1766–1767. [Google Scholar] [CrossRef]
- Mecozzi, S.; Rebek, J., Jr. The 55% solution: A formula for molecular recognition in the liquid state. Chem. Eur. J. 1998, 4, 1016–1022. [Google Scholar] [CrossRef]
- Avram, L.; Cohen, Y. Spontaneous formation of hexameric resorcinarene capsule in chloroform solution as detected by diffusion NMR. J. Am. Chem. Soc. 2002, 124, 15148–15149. [Google Scholar] [CrossRef]
- Avram, L.; Cohen, Y. The role of water molecules in a resorcinarene capsule as probed by NMR diffusion measurements. Org. Lett. 2002, 4, 4365–4368. [Google Scholar] [CrossRef] [PubMed]
- Shivanyuk, A.; Rebek, J., Jr. Assembly of resorcinarene capsules in wet solvents. J. Am. Chem. Soc. 2003, 125, 3432–3433. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Goody, R.S. The original Michaelis constant: Translation of the 1913 Michaelis-Menten paper. Biochemistry 2011, 50, 8264–8269. [Google Scholar] [CrossRef]
- Chen, W.W.; Niepel, M.; Sorger, P.K. Classic and contemporary approaches to modeling biochemical reactions. Genes Dev. 2010, 24, 1861–1875. [Google Scholar] [CrossRef]
- Rodriguez, J.-M.G.; Hux, N.P.; Philips, S.J.; Towns, M.H. Michaelis-Menten graphs, lineweaver-burk plots, and reaction schemes: Investigating introductory biochemistry students’ conceptions of representations in enzyme kinetics. J. Chem. Educ. 2019, 96, 1833–1845. [Google Scholar] [CrossRef]
- Bisswanger, H. Enzyme Kinetics: Principles and Methods; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Cornish-Bowden, A. Fundamentals of Enzyme Kinetics, 4th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2012. [Google Scholar]
- Hanes, C.S. Studies on plant amylases: The effect of starch concentration upon the velocity of hydrolysis by the amylase of germinated barley. Biochem. J. 1932, 26, 1406–1421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tiefenbacher, K. Hexameric resorcinarene capsule is a Brønsted acid: Investigation and application to synthesis and catalysis. J. Am. Chem. Soc. 2013, 135, 16213–16219. [Google Scholar] [CrossRef]
- Wang, H.; Cui, P.; Zou, G.; Yang, F.; Tang, J. Temperature-controlled highly selective dimerization of α-methylstyrene catalyzed by Brönsted acidic ionic liquid under solvent-free conditions. Tetrahedron 2006, 62, 3985–3988. [Google Scholar] [CrossRef]
- Beesley, R.M.; Ingold, C.K.; Thorpe, J.F. CXIX.-The formation and stability of spiro-compounds. Part I. spiro-Compounds from cyclohexane. J. Chem. Soc. Trans. 1915, 107, 1080–1106. [Google Scholar] [CrossRef]
- Shaw, B.L. Formation of large rings, internal metalation reactions, and internal entropy effects. J. Am. Chem. Soc. 1975, 97, 3856–3857. [Google Scholar] [CrossRef]
- King, R.P.; Krska, S.W.; Buchwald, S.L. A neophyl palladacycle as an air-and thermally stable precursor to oxidative addition complexes. Org. Lett. 2021, 23, 7927–7932. [Google Scholar] [CrossRef] [PubMed]
- Golfmann, M.; Glagow, L.; Giakoumidakis, A.; Golz, C.; Walker, J.C.L. Organophotocatalytic [2+2] cycloaddition of electron-deficient styrenes. Chem. Eur. J. 2023, 29, e202202373. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Wei, W.; Li, M.; Yang, Y.; Ji, J. sp3-sp2 C-C Bond formation via Brønsted acid trifluoromethanesulfonic acid-catalyzed direct coupling reaction of alcohols and alkenes. Adv. Synth. Catal. 2011, 353, 3139–3145. [Google Scholar] [CrossRef]
- Mameda, N.; Gajula, K.S.; Peraka, S.; Kodumuri, S.; Chevella, D.; Banothu, R.; Amrutham, V.; Nama, N. One-pot synthesis of 1, 3-diaryl but-1-enes from 1-arylethanols over Snβ zeolite. Catal. Commun. 2017, 90, 95–99. [Google Scholar] [CrossRef]
- Le Nôtre, J.; Brissieux, L.; Sémeril, D.; Bruneau, C.; Dixneuf, P.H. Tandem isomerization/Claisen transformation of allyl homoallyl and diallyl ethers into γ,δ-unsaturated aldehydes with a new three component catalyst Ru3(CO)12/imidazolinium salt/Cs2CO3. Chem. Commun. 2002, 16, 1772–1773. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar]
- Liu, M.; Zhang, J.; Zhou, H.; Yang, H.; Xia, C.; Jiang, G. Efficient hydroarylation and hydroalkenylation of vinylarenes by Brønsted acid catalysis. RSC Adv. 2016, 6, 76780–76784. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Complex 6 | Capsule 56 | Additive | Conversion (%) 2 | Products Distribution (%) 2 | |
|---|---|---|---|---|---|---|
| 3a | Oligomers | |||||
| 1 | yes | yes | no | 72 | 38 | 62 |
| 2 | no | no | no | 0 | / | / |
| 3 | no | yes | no | 0 | / | / |
| 4 | yes | no | no | 0 | / | / |
| 5 | yes | no | AcOH 3 | traces | / | / |
| 6 | yes | yes | DMSO 4 | 0 | / | / |
| 7 | yes | yes | NEt4Cl 5 | 0 | / | / |
| 8 | yes | yes | D2O 6 | 80 | 35 | 65 |
| Entry | Loading of 6⊂56 (Mol%) | Time (h) | Conversion (%) 2 | Products Distribution (%) 2 | |
|---|---|---|---|---|---|
| 3a | Oligomers | ||||
| 1 | 2 3 | 12 | 88 | 20 | 80 |
| 2 | 16 | 93 | 25 | 75 | |
| 3 | 24 | 95 | 20 | 80 | |
| 4 | 1 4 | 24 | 72 | 38 | 62 |
| 5 | 0.5 5 | 24 | 60 | 48 | 52 |
| 6 | 36 | 82 | 48 | 52 | |
| 7 | 40 | 87 | 48 | 52 | |
| 8 | 44 | 90 | 24 | 76 | |
| Entry | Vinyl Arene (1) | Time (h) | Conversion (%) 2 | Products Distribution 2 | ||
|---|---|---|---|---|---|---|
| Formed Products | Degradation | |||||
| 1 |  | (1b) | 40 | 13 | 3b: 62% | 38% |
| 2 | 72 | 41 | 3b: 20% | 80% | ||
| 3 |  | (1c) | 40 | 17 | 3c: 59% | 41% |
| 4 | 72 | 25 | 3c: 8% | 92% | ||
| 5 |  | (1d) | 24 | 44 | 3d: 34% and 7d: traces | 66% |
| 6 | 32 | 75 | 3d: 28% and 7d: traces | 72% | ||
| 7 |  | (1e) | 40 | 35 | 3e: traces and 7e: 91% | 9% |
| 8 | 72 | 57 | 3e: traces and 7e: 68% | 32% | ||
| 9 |  | (1f) | 24 | 98 | 9: 7%, 10: 7% and 7f: 86% | / |
| 10 | 40 | 100 | 7f: 100% | / | ||
| 11 |  | (1g) | 40 | 95 | 5g: traces and 8: 50% | 50% |
| 12 |  | (1h) | 40 | 0 | / | / |
| 13 |  | (1i) | 40 | 0 | / | / |
| 14 | 1-hexene | 72 | 90 | Isomers | / | |
| Entry | Vinyl Arene (1) | Water | Conversion (%) 2 | Deuterium Incorporation | Products Distribution 2 | ||
|---|---|---|---|---|---|---|---|
| Formed Products | Degradation | ||||||
| 1 |  | (1e) | H2O | 35 | / | 7e: 91% (cis/trans: 60/40) | 9% |
| 2 | D2O | 27 | No | 7e: 89% (cis/trans: 60/40) | 11% | ||
| 3 |  | (1f) | H2O | 100 | / | 7f: 100% | / |
| 4 | D2O | 98 | No | 7f: 100% | / | ||
| 5 |  | (1g) | H2O | 95 | / | 5g: traces and 8: 50% | 50% |
| 6 | D2O | 98 | No | 5g: traces and 8: 54% | 46% | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinmetz, M.; Sémeril, D. Confined Catalysis Involving a Palladium Complex and a Self-Assembled Capsule for the Dimerization of Vinyl Arenes and the Formation of Indane and Tribenzo–Pentaphene Derivatives. Catalysts 2025, 15, 585. https://doi.org/10.3390/catal15060585
Steinmetz M, Sémeril D. Confined Catalysis Involving a Palladium Complex and a Self-Assembled Capsule for the Dimerization of Vinyl Arenes and the Formation of Indane and Tribenzo–Pentaphene Derivatives. Catalysts. 2025; 15(6):585. https://doi.org/10.3390/catal15060585
Chicago/Turabian StyleSteinmetz, Maxime, and David Sémeril. 2025. "Confined Catalysis Involving a Palladium Complex and a Self-Assembled Capsule for the Dimerization of Vinyl Arenes and the Formation of Indane and Tribenzo–Pentaphene Derivatives" Catalysts 15, no. 6: 585. https://doi.org/10.3390/catal15060585
APA StyleSteinmetz, M., & Sémeril, D. (2025). Confined Catalysis Involving a Palladium Complex and a Self-Assembled Capsule for the Dimerization of Vinyl Arenes and the Formation of Indane and Tribenzo–Pentaphene Derivatives. Catalysts, 15(6), 585. https://doi.org/10.3390/catal15060585