1. Introduction
Copper is a widely used and versatile metal in catalysis due to its favorable redox properties, low cost, and recyclability [
1,
2,
3]. It plays a key role in various catalytic processes, including hydrogenation, oxidation, and reduction reactions [
4,
5,
6,
7]. Copper-based catalysts are particularly attractive for industrial applications, where cost and availability are crucial factors. The increasing interest in developing Cu-based heterogeneous catalysts is largely driven by their potential as cost-effective alternatives to precious metals such as platinum and palladium [
1,
8]. Therefore, continued research on innovative and more efficient Cu-based catalysts is essential to achieving superior catalytic performance.
Mesoporous silica materials were identified as highly promising support materials for copper catalysts due to a range of unique properties, such as uniform porous structure and extensive surface area, along with relatively simple synthesis methods. Mesoporous silicas were first synthesized in 1992 by Mobil Oil Company researchers, and their synthesis is based on the surfactant-directed method [
9,
10]. This process involves surfactants self-organizing in solution to form micellar structures, around which the silica source condenses. The final step requires the removal of organic surfactant from the mesoporous silica’s pore system, typically through calcination or, less frequently, solvent extraction [
11,
12]. Mesoporous silica materials, unlike zeolites, possess significantly larger pores, which are crucial for facilitating the effective internal diffusion of larger reactants in catalytic processes [
13]. This advantage allows them to accommodate bulkier species that would otherwise be excluded from the narrower micropores of zeolites, enhancing catalytic efficiency and broadening the scope of potential applications. Nonetheless, a major drawback of these materials is the challenge of uniformly depositing catalytically active components on the surface of mesoporous silicas. In comparison to zeolites, pure mesoporous silica materials lack ion-exchange properties, making it impossible to deposit metal cations (e.g., copper) using traditional ion-exchange methods [
14,
15]. Ion-exchange properties can be achieved by incorporating heteroatoms with a 3+ oxidation state (e.g., aluminum) into the amorphous silicas. However, unlike zeolites, the non-uniform distribution of aluminum cations within the silica matrix results in acid sites with varying strengths and locations. Additionally, the surface density of these acid sites and ion-exchange positions is lower compared to those found in zeolites [
16]. An alternative modification approach is impregnation, which is relatively simple and cost-effective. However, it is very challenging, if not impossible, to control the form and aggregation of the deposited copper species, which is significant for achieving high catalytic selectivity [
17]. Another important method for functionalizing pure silica materials is the molecular designed dispersion (MDD) method, which allows for the deposition of monomeric metal forms into the support [
18,
19]. This method involves grafting organometallic compounds on the silica surface through a selective reaction between the active phase precursor and the support’s silanol groups, followed by drying and calcination. During calcination, the organic functional groups decompose, resulting in a highly dispersed form of metals. However, the grafting method is complex and expensive, requiring a dry, inert atmosphere. Specialized equipment is necessary to maintain these reaction conditions, which greatly limits the feasibility of this method for industrial applications [
17,
20]. A promising and quite simple method used for the preparation of finely dispersed copper species on silica’s surface is the ammonia-driven deposition precipitation (ADP) method [
21,
22]. This technique involves the use of ammonia to form complexes with copper ions, which facilitates the deposition of these ions on various supports, such as mesoporous silicas. The ADP method fundamentally relies on ammonia increasing the solution’s pH above the point of zero charge, leading to the formation of [Cu(NH
3)
4(H
2O)
2]
2⁺ complexes. These complexes then interact electrostatically with the negatively charged surfaces of the support, promoting uniform deposition and preventing metal aggregation during thermal treatment (e.g., calcination) [
22,
23,
24]. In consequence of this, this method allows for high metal dispersion on the support material, resulting in a larger active surface area and improved catalytic activity. Moreover, the ADP process is relatively simple, water-based, and can be performed under mild conditions, making it cost-effective and scalable for industrial applications.
Among the significant applications of copper-modified mesoporous silicas is their role in the low-temperature selective catalytic reduction in NO
x using ammonia (NH
3-SCR) [
25]. This process plays a crucial role in lowering NO
x emissions from industrial and transport sources, contributing to the reduction in air pollution. The traditional catalytic system (V
2O
5–TiO
2) is known to perform well between 300 and 400 °C [
26]. However, modern industrial processes require NO
x reduction at lower temperatures. Since electrostatic precipitators (EPS), which remove particulates from exhaust gases, function at 250 °C or lower, there is a growing need for low-temperature catalysts to improve NH
3-SCR efficiency under these conditions [
25,
26]. A promising solution to this challenge is the use of copper-modified mesoporous silicas as catalysts. These materials demonstrated high activity and selectivity in NO
x reduction at lower temperatures, making them a strong candidate for enhancing NH
3-SCR performance in industrial applications.
The primary focus of the presented studies is the development of catalysts for the low-temperature NH3-SCR process, specifically by testing porous materials as supports for copper catalysts obtained through modification by the ADP method. The regular porous structure of MCM-48 silica as well as its very high surface area provide favorable conditions for copper dispersion, which can contribute to achieving high NO conversion values. The constant development of these catalysts holds promise for more sustainable and cost-effective solutions in industrial catalysis and environmental protection.
2. Results and Discussion
The porous structure of parent MCM-41 and MCM-48, as well as their modifications with copper, was analyzed using the XRD method and low-temperature nitrogen adsorption–desorption. The diffractograms recorded for mesoporous silicas, shown in
Figure 1, confirm the successful synthesis of MCM-41 and MCM-48 materials. In the case of MCM-41 (
Figure 1A), characteristic reflections of (100), (110), and (200) indicate the hexagonal porous structure typical of MCM-41 materials [
27]. The deposition of copper by the ADP method resulted in a shift in the (100) reflection to higher 2-theta positions, as well as disappearance of the (110) and (200) reflections (
Figure 1A). These effects are possibly related to the partial destruction of the ordered porous structure of MCM-41 under the basic conditions of the ADP procedure. In the case of the diffractogram recorded for MCM-48 (
Figure 1B), characteristic reflections of (211) and (220), typical of the regular porous structure of this type of mesoporous material, are observed [
28]. The deposition of copper caused a shift in the location of these reflections into higher 2-theta values and a slight decrease in their intensity, especially in the case of the (220) reflection. These effects, similarly to the MCM-41 series, are possibly attributed to the partial destruction of the MCM-48 porous structure by basic ammonia used in the ADP procedure. The diffractograms presented in the insets of
Figure 1 were recorded in the diffraction range characteristic of copper oxide reflections. No reflections typical of CuO phase were found in this range, indicating the deposition of copper in the form of highly dispersed species onto MCM-41 and MCM-48 silicas.
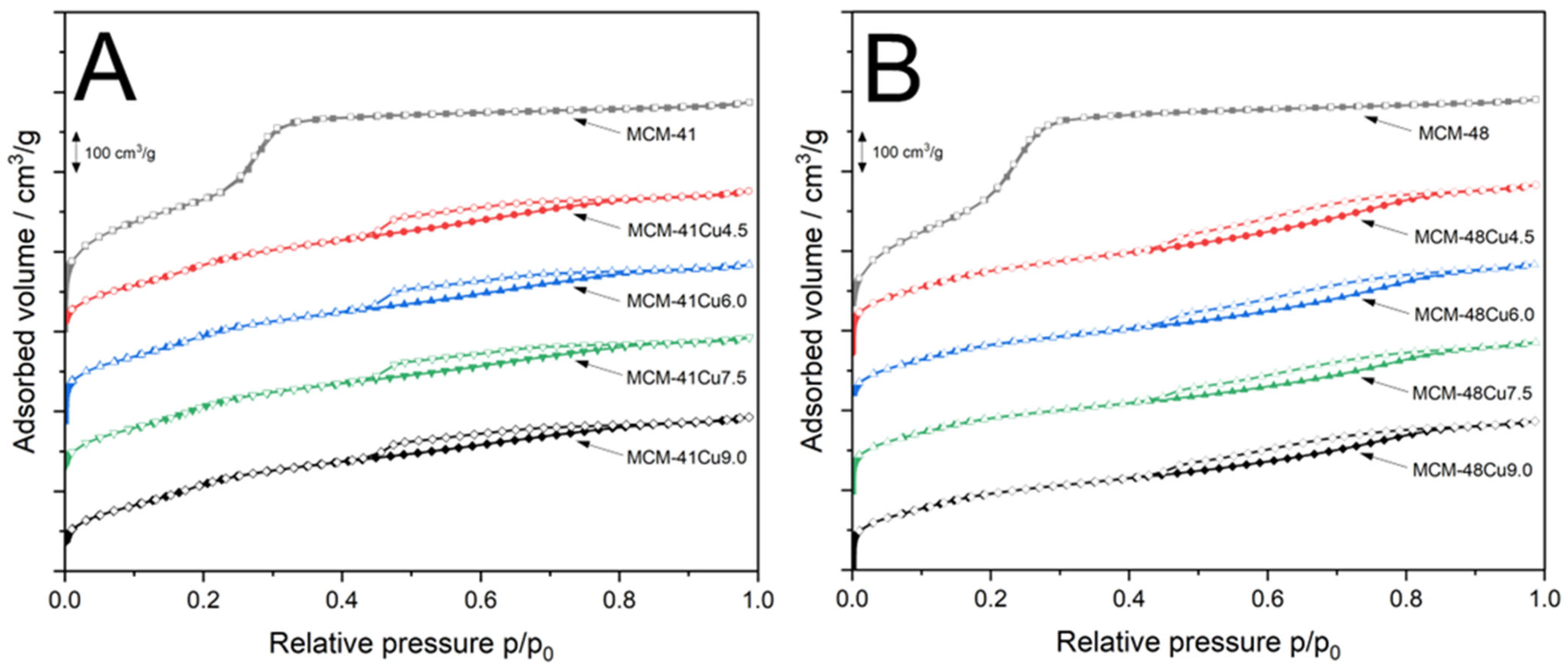
Nitrogen adsorption–desorption isotherms recorded for mesoporous silicas and their modifications with copper are presented in
Figure 2. The isotherms recorded for MCM-41 (
Figure 2A) and MCM-48 (
Figure 2B) belong to type IVb according to the IUPAC classification, characterized by a sharp increase in nitrogen adsorption at relative pressures of about 0.25–0.35, associated with nitrogen capillary condensation inside mesopores [
29]. This type of isotherm is characteristic of materials containing smaller mesopores, formed by conical and cylindrical mesopores that are closed at one end [
29]. The absence of hysteresis loops indicates that the process of nitrogen adsorption and desorption is fully reversible. Moreover, according to Thommes et al. [
29], hysteresis loops start to appear only for pores wider than 4 nm. The deposition of copper into MCM-41 and MCM-48 by the ADP method significantly modified the shape of the isotherms. When it comes to the modified MCM-41 silicas (
Figure 2A), the increase in the nitrogen adsorbed volume, related to the filling of mesopores by adsorbate, is shifted to lower relative pressure and is less intense compared to parent MCM-41. This relates to a decrease in the mesopore volume and average diameter under the conditions of the ADP procedure. Moreover, a gradual increase in the adsorbed volume above a relative pressure of 0.4 with the inflection point at about 0.7–0.8 indicates the formation of larger pores, possibly due to the reaction of ammonia with the silica surface, such as the hydrolysis of siloxane bridges (≡Si-O-Si≡) to silanol groups (≡Si-OH). The hysteresis loops in adsorption–desorption isotherms recorded for MCM-41 modified with copper are classified as category H3 according to the IUPAC standard, typically associated with larger pores that are only partially filled with the condensate [
29]. The isotherms recorded for MCM-48 modified with copper by the ADP method, shown in
Figure 2B, differ significantly from the isotherm of parent MCM-48. The increase in adsorbed volume, related to mesopore filling, was significantly reduced and shifted to lower relative pressure. In this case, the inflection point is hardly visible. This type of isotherm is sometimes called pseudo type I and was reported by Schmidt et al. [
30] for MCM-41 with small pores (2.7 nm). The continuous increase in adsorbed nitrogen volume, especially above p/p
0 = 0.4, with an inflection point at a relative pressure of 0.8–0.9, indicates the formation of larger pores under the conditions of the ADP procedure. The hysteresis loops are classified as category H3 according to the IUPAC standard, characteristic of porous materials with larger pores only partially filled with nitrogen [
29].
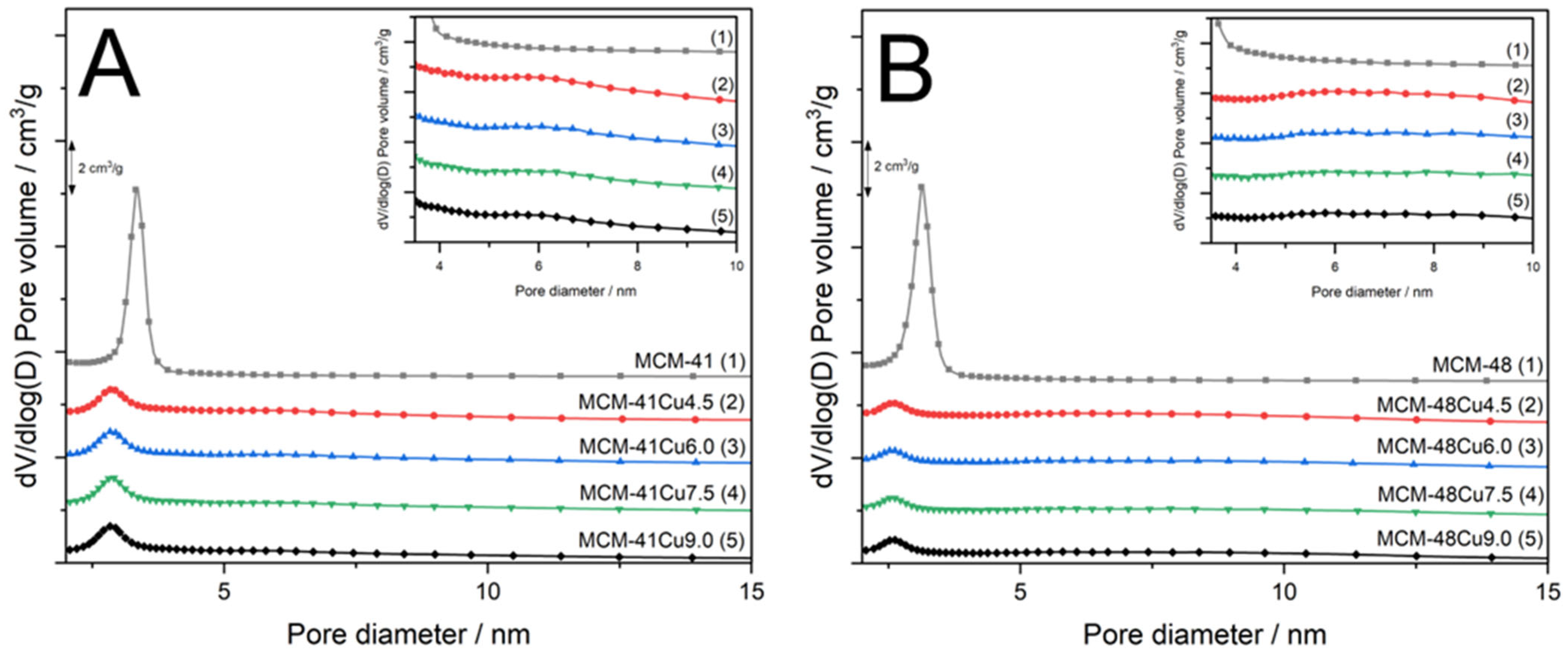
Profiles of pore size distributions (PSD) obtained for parent MCM-41 (
Figure 3A) and MCM-48 (
Figure 3B) prove their uniform mesoporous structures with the PSD maxima at 3.4 and 3.1 nm, respectively. The deposition of copper by the ADP method significantly changed their PSD profiles. In the case of MCM-41 series, the PSD maximum was significantly reduced and shifted to about 2.8–2.9 nm. Moreover, a broad peak with the maximum at about 6.1 nm appeared (
Figure 3A, inset). Similarly, in the case of MCM-48 series, the maximum of the PSD significantly decreased and was shifted to about 2.1 nm (
Figure 3B). In this case as well, an additional broad peak with a maximum at about 6.0 nm can be identified (
Figure 3B, inset). Thus, modifications of mesoporous silicas by the ADP method significantly influenced their porous structure. The effect of the PSD shift could be attributed to the deposition of copper inside pores, reducing their size and volume. However, the analysis of PSD profiles shows that the changes in position and intensity of the PSD distribution are independent of copper loadings. Therefore, the main role in the observed changes in the PSD profile is related to the treatment of the silica samples with ammonia solutions, which, due to basic character, may result in the hydrolysis of siloxane bridges to silanol species or even the extraction of silica species into solution. It should be noted that broad PSD maxima found for the samples modified with copper are close to the double diameter of pores in parent mesoporous silicas. Hence, it is possible that part of the silica walls separating pores was dissolved under the conditions of the ADP procedure. On the other hand, the products of such dissolution, extracted into solution, could then be deposited on the channel walls, resulting in their partial blocking. This hypothesis is very speculative and needs to be evidenced by scientific results.
The textural parameters and copper content in the samples are compared in
Table 1. The specific BET surface area and pore volume decreased following copper deposition, but the copper loading in the samples had only a slight impact on these parameters. The copper content (determined by ICP-OES method) in the samples is very close to the intended values, which can be seen in the samples’ codes. These values confirm the successful deposition of copper on silica surface and demonstrate that the ADP procedure allows for precise control of the copper loading.
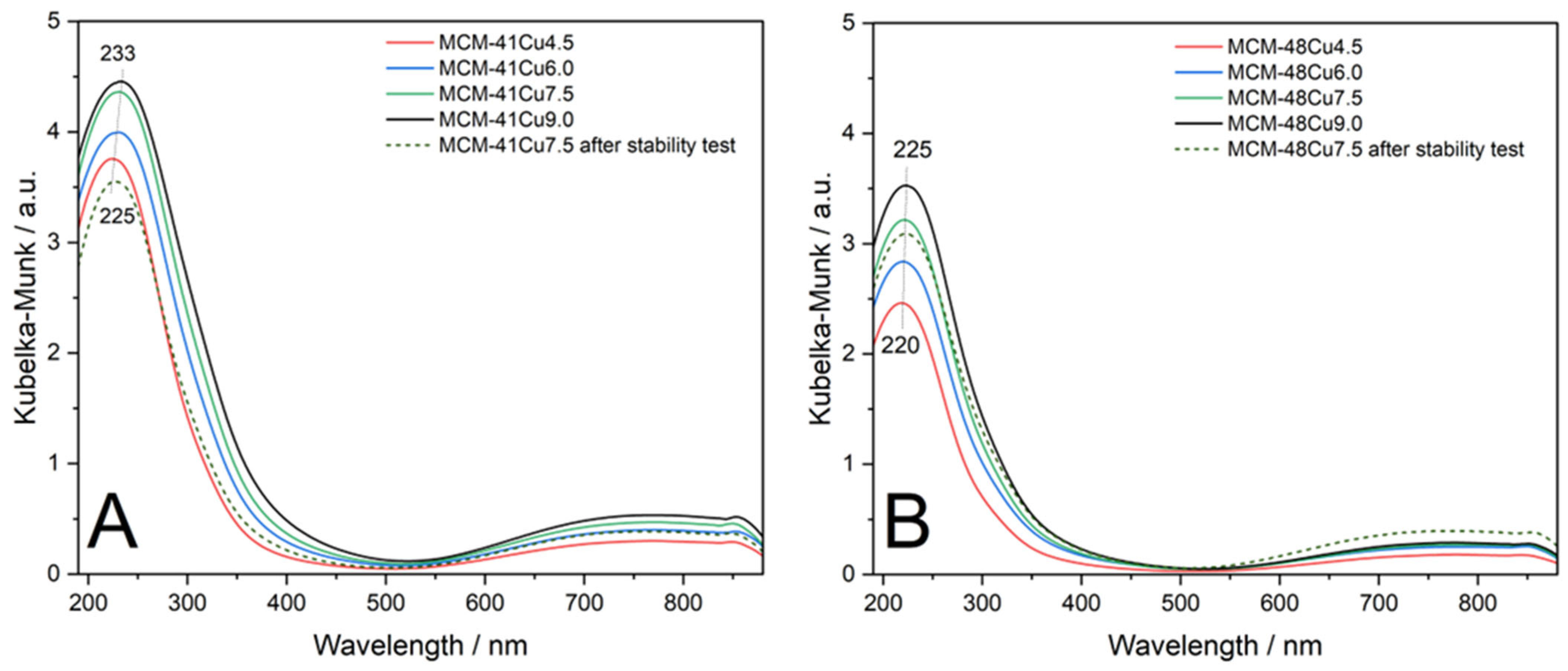
The form and aggregation of the deposited copper species were analyzed by UV-vis-DRS and H
2-TPR measurements. The spectra recorded for copper-modified silicas are shown in
Figure 4. It should be noted that pure silica does not exhibit radiation absorbance in the UV and visible range. Thus, the bands in the presented spectra are attributed only to the presence of copper species. In all the spectra recorded, copper-modified MCM-41 and MCM-48 dominate an intensive and symmetric band at about 220–230 nm, related to the presence of monomeric Cu
2+ ions interacting with oxygens of silica (O
2− → Cu
2+) [
31,
32]. It is noteworthy that the maximum of this band is slightly shifting to higher values of wavelength with increasing copper loading, indicating the formation of a small amount of more aggregated copper species. The presence of monomeric copper cations in the samples is further evidenced by the wide band above 550 nm, associated with the d-d transition of Cu
2+ ions in a pseudo-octahedral arrangement, such as Cu(H
2O)
62+ [
32,
33]. Thus, the results of UV-vis-DRS studies confirm that the ADP method resulted in the predominant deposition of copper in the form of monomeric cations.
The reducibility of copper species deposited on mesoporous silicas was analyzed by the H
2-TPR method (
Figure 5). The reduction in copper species occurs in very specific ways. In the case of CuO aggregates, the reduction in Cu
2+ cations takes place in one step, directly to Cu
0 (Cu
2+ → Cu
0) typically at temperatures below 300–350 °C [
34], while in the case of monomeric copper cations in two separate steps, first, Cu
2+ cations are reduced to Cu
+ (Cu
2+ → Cu
+) at temperatures below 300–350 °C, and then Cu
+ ions are further reduced to Cu
0 (Cu
+ → Cu
0) at temperatures above 400 °C in the second step [
34]. These specific mechanisms of copper species reduction can be used to determine the distribution of copper in the form of monomeric cations and aggregated copper oxide species. The H
2-TPR profiles of the samples contain both a low-temperature reduction peak, associated with the reduction in monomeric Cu
2+ cations to Cu
+ as well as reduction in Cu
2+ cations to Cu
0 in CuO aggregates, and a high-temperature reduction peak, related to the reduction in monomeric Cu
+ cations to Cu
0 (
Figure 5). The calculated distribution of monomeric copper cations (Cu
mon) and copper cations in aggregated copper oxide species (Cu
agr) is presented in
Table 1. As can be seen, in the case of MCM-41-based samples, about 87–88 mol% of copper is present in the form of monomeric copper cation up to 7.5 wt.% of this metal loading. An increase to 9 wt.% of copper loading resulted in a decrease in monomeric Cu
2+ cations’ contribution to 83 mol%. In the series of MCM-48 samples, the highest contribution of monomeric copper cations, on the level of 88 mol%, was determined for the sample with the lowest copper loading, while the lowest contribution of such dispersed cations (76 mol%) for silica had the highest copper content. It should be noted that in the calculation, it was assumed that all copper cations in the samples are in the form of Cu
2+ cations. Of course, the presence of Cu
+ species in the samples (after outgassing but prior to the H
2-TPR runs) cannot be excluded. Thus, the contributions of different forms of copper in the samples, presented in
Table 1, are rather estimates than exact values.
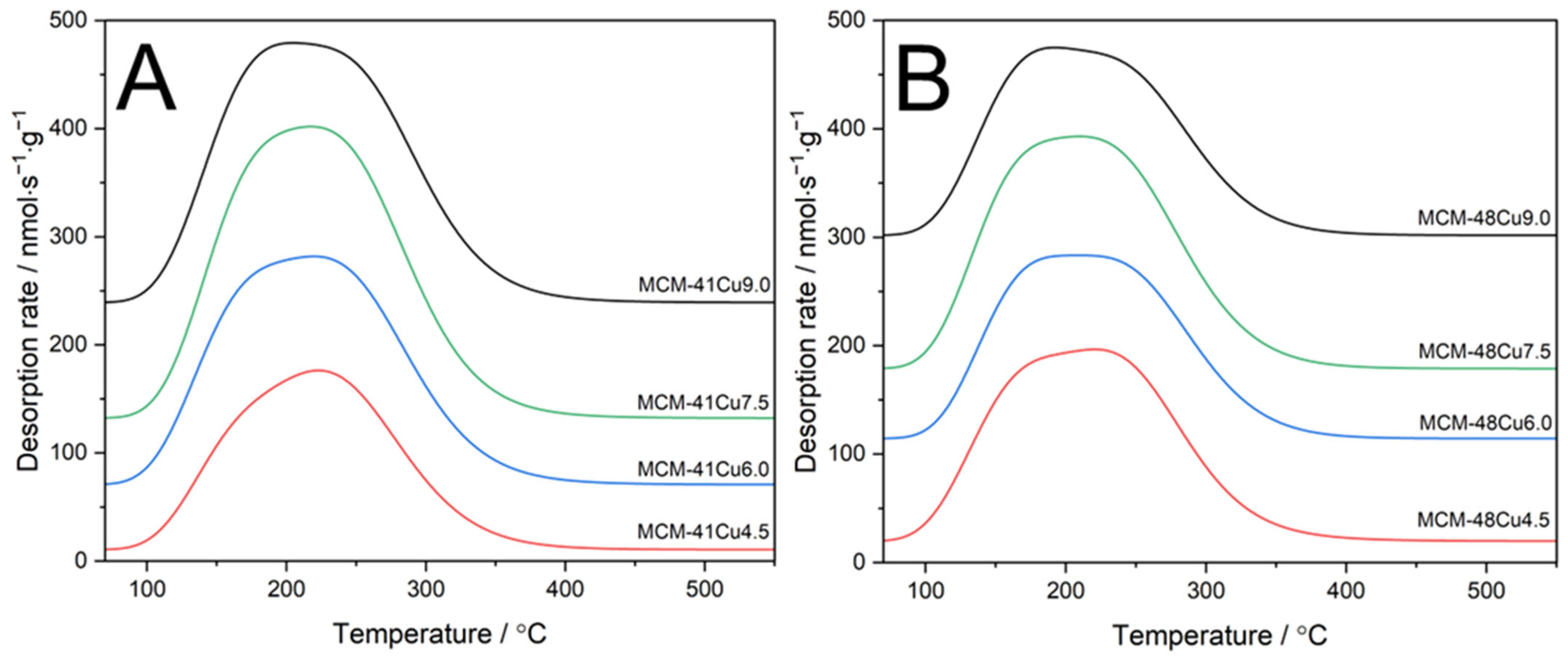
The surface acidity of the copper-modified silicas was analyzed by the NH
3-TPD method. Pure silica MCM-41 and MCM-48 do not exhibit surface acid sites. Such sites are associated with copper species deposited on silicas’ surface, which act as Lewis acid sites [
25]. The copper cations interact with ammonia molecules through the formation of donor–acceptor bonding, a transfer of the free electron pair of ammonia into the vacant d-orbitals of copper (NH
3 → Cu). Therefore, the NH
3-TPD method can be used to determine the surface-available copper cations that can participate in catalytic surface reactions. The majority of the proposed NH
3-SCR mechanisms include chemisorption and activation of ammonia molecules on the active sites [
35,
36]. Thus, the results of NH
3-TPD studies not only provide information about surface acidity but also about the number of surface copper cations involved in the catalytic process. Ammonia desorption profiles, presented in
Figure 6, consist of one peak centered at about 190–230 °C, indicating the presence of the relatively weak acid sites. The intensity of this peak depends on the copper loading, but also their aggregation and surface availability. The surface concentrations of acid sites (C
A) and their density per 1 m
2 (D
A) are compared in
Table 2. It was assumed that one ammonia molecule is bound to one acid site; thus, the number of desorbing ammonia molecules is equal to the number of acid sites, as well as the number of active sites able to chemisorb and activate ammonia molecules to the NH
3-SCR reaction. The surface concentration of acid sites was determined by integrating the areas of the desorption peaks in NH
3-TPD profiles and normalizing them to the catalyst mass. Their density per unit surface area (1 m
2) was calculated by dividing the total concentration of acid sites by the BET surface area of each catalyst.
In both the MCM-41 and MCM-48 series, the surface concentration and surface density of acid sites increased up to copper loading 7.5 wt.% and decreased for the samples with copper content above this level. This can be explained by the increasing contribution of aggregated copper oxide species in the samples with high copper loading, as shown by H
2-TPR studies (
Table 1). The ratio of acid sites’ concentration to copper loading (C
A/Cu), representing the contribution of copper cations available for catalytic process, is relatively low (
Table 2) compared to the contribution of monomeric copper cations in the samples determined by H
2-TPR (
Table 1). The surface availability of copper cations depends on the contribution of aggregated and highly dispersed species deposited on the silica surface, as well as the location of copper species, either on the surface or included in the SiO
2 matrix. It seems possible that under the conditions of the APD procedure, ammonia dissolves some silica, which could then be redeposited on the surface, covering some copper species. This hypothesis should be examined in future studies.
The NH3-SCR process is associated with other reactions, which can either improve or limit its efficiency. One of the important associated reactions is the oxidation of NO to NO2 (2 NO + O2 → 2 NO2), which is necessary for the fast-SCR reaction (2 NH3 + NO2 + NO → 2 N2 + 3 H2O). The conversion of nitrogen oxides according to the fast-SCR reaction is one of the most important low-temperature reaction pathways. Thus, the activity of the catalysts in the oxidation of NO to NO2 could improve their efficiency in the low-temperature NH3-SCR process. On the other hand, the efficiency of the NH3-SCR process at higher temperatures can be significantly limited by the side process of direct ammonia oxidation by residual oxygen present in flue gases. Thus, the NH3-SCR catalysts operating at higher temperatures should be non-active or present only low activity in direct ammonia oxidation. Taking into account the association of the NH3-SCR reaction with other mentioned reactions, the presented catalytic studies include not only NH3-SCR tests but also reactions of NO to NO2 oxidation as well as direct ammonia oxidation.
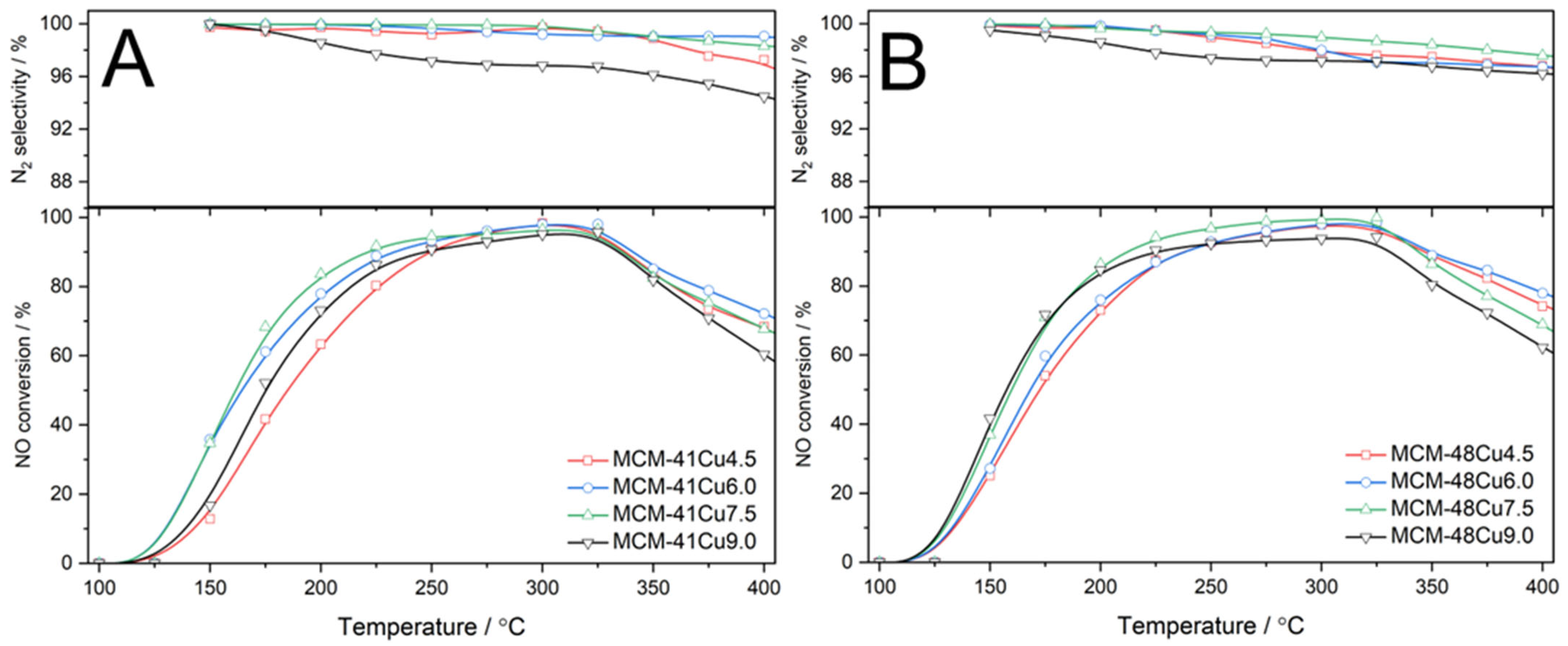
The results of the NH
3-SCR catalytic tests are shown in
Figure 7. In the case of MCM-41 based catalysts, the NO conversion started at about 100–125 °C, gradually increased to 300–325 °C, and then decreased due to the side reaction of direct ammonia oxidation by oxygen present in the reaction mixture (
Figure 7A). At temperatures below 250 °C, the efficiency of the NO conversion increased with increasing copper loadings in the following order: MCM-41Cu4.5 < MCM-41Cu6.0 < MCM-41Cu7.5. An increase in copper content to about 9 wt.% (MCM-41Cu9.0) resulted in a decrease in the NO conversion below the level presented by the MCM-41Cu6.0 catalyst. At higher temperatures, the sample with the highest copper loading was found to be less active than other catalysts of this series and presented the most significant decrease in the NO conversion due to the side reaction of direct ammonia oxidation. This effect could be explained by higher contribution of aggregated copper oxide species compared to other catalysts in this series (
Table 2). On the other hand, the number of copper cations exposed on the catalysts surface and able to chemisorb ammonia molecules (C
A,
Table 2) is higher than for the MCM-41Cu6.0 catalyst. Thus, it could be postulated that monomeric copper cations are more catalytically active than Cu
2+ cations in copper oxide aggregates. A similar hypothesis has been postulated by many authors [
37,
38]. The very high selectivity to nitrogen, which is the desired reaction product, should be noted (
Figure 7A). The outcomes of the NH
3-SCR catalytic testing for a series of MCM-48-based catalysts are shown in
Figure 7B. Similarly to the MCM-41 series, the NO conversion started at about 100–125 °C, gradually increased to 300–325 °C, and then decreased due to the side reaction of direct ammonia oxidation. At temperatures below 175 °C, the efficiency of the NO conversion increases with increasing copper loadings in the catalysts in the following order: MCM-48Cu4.5 < MCM-48Cu6.0 < MCM-48Cu7.5 < MCM-48Cu9.0. The activity order was changed at higher temperatures, and the catalysts with the highest copper loading (MCM-48Cu9.0) presented lower activity in NH
3-SCR than other catalysts of this series. Moreover, direct ammonia oxidation proceeded more intensively compared to other catalysts based on MCM-48. Similarly to the MCM-41 series, this effect was also attributed to the increased contribution of aggregated copper oxide species compared to the other catalysts of this series. It should be noted that all catalysts of this series presented very high selectivity to nitrogen in the studied temperature range (
Figure 7B).
Turn-over-frequency (TOF) values, determined for the reaction conducted at 175 °C, are compared in
Table 2. It was assumed that copper cations that are able to adsorb ammonia molecules (determined by NH
3-TPD) play a role of active sites in the NH
3-SCR process. The TOF was calculated by normalizing the reaction rate (mol of NO
x converted per second) to the number of surface acid sites, expressed as mol of NO
x converted per mol of acid sites per second. The TOF values are in the range of 2.31 × 10
−3 s
−1 to 3.52 × 10
−3 s
−1, which is close to the TOF values reported in the literature for this type of catalysts [
39]. There is no significant trend in TOF changes with increasing copper loading, probably due to the dominant contribution of monomeric copper cations.
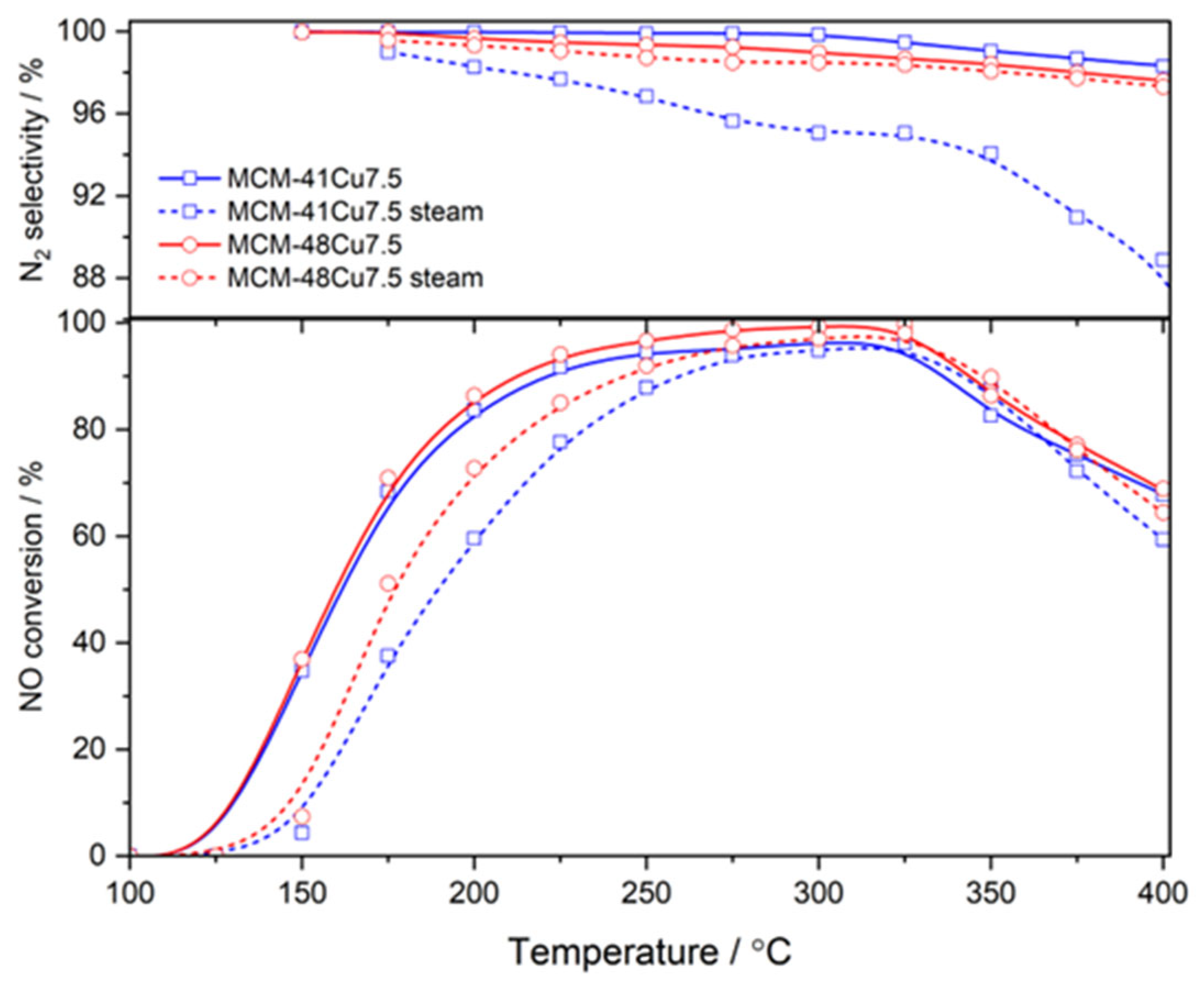
The comparison of NO conversion in wet (with the addition of 5.0 vol.% water vapor) and dry (as in previously presented catalytic tests) reaction mixtures for the selected catalysts of MCM-41 and MCM-48 series is shown in
Figure 8. The introduction of water vapor into the reaction mixture mainly influenced the low-temperature NO conversion and was manifested by a shift in conversion profiles to higher temperatures. In the case of MCM-48Cu7.5, this shift was less significant compared to MCM-41Cu7.5, with shifts of 17 and 26 °C for 50% of the NO conversion, respectively. The possible explanation for this effect is the competitive adsorption of ammonia and water molecules on copper cations, especially at lower temperatures. However, for both tested materials in the presence of moisture, the NO
x conversion at 250 °C remains at a high level, reaching 88% and 92% for MCM-41Cu7.5 and MCM-48Cu7.5, respectively. Moreover, within the temperature window relevant to ESP operation (225–250 °C), the NO
x conversion for MCM-48Cu7.5 stays above 85%. To evaluate the potential for irreversible deactivation under humid conditions, additional catalytic activity tests were performed (
Figure 9). Specifically, catalytic tests were first performed with a wet reaction mixture, followed by cooling of the catalyst samples and subsequent testing in a dry gas atmosphere. Under these dry conditions after exposure to water vapor, an increase in catalytic activity compared to the wet test was observed, indicating that no permanent deactivation of the catalyst occurred and that the catalytic efficiency in the second cycle (dry atmosphere) is similar to that in the first cycle. This enhancement in activity demonstrates that the decrease in NO
x conversion observed under wet conditions results from the competitive adsorption of water and ammonia molecules on the active sites, rather than irreversible changes in the catalyst structure or active species.
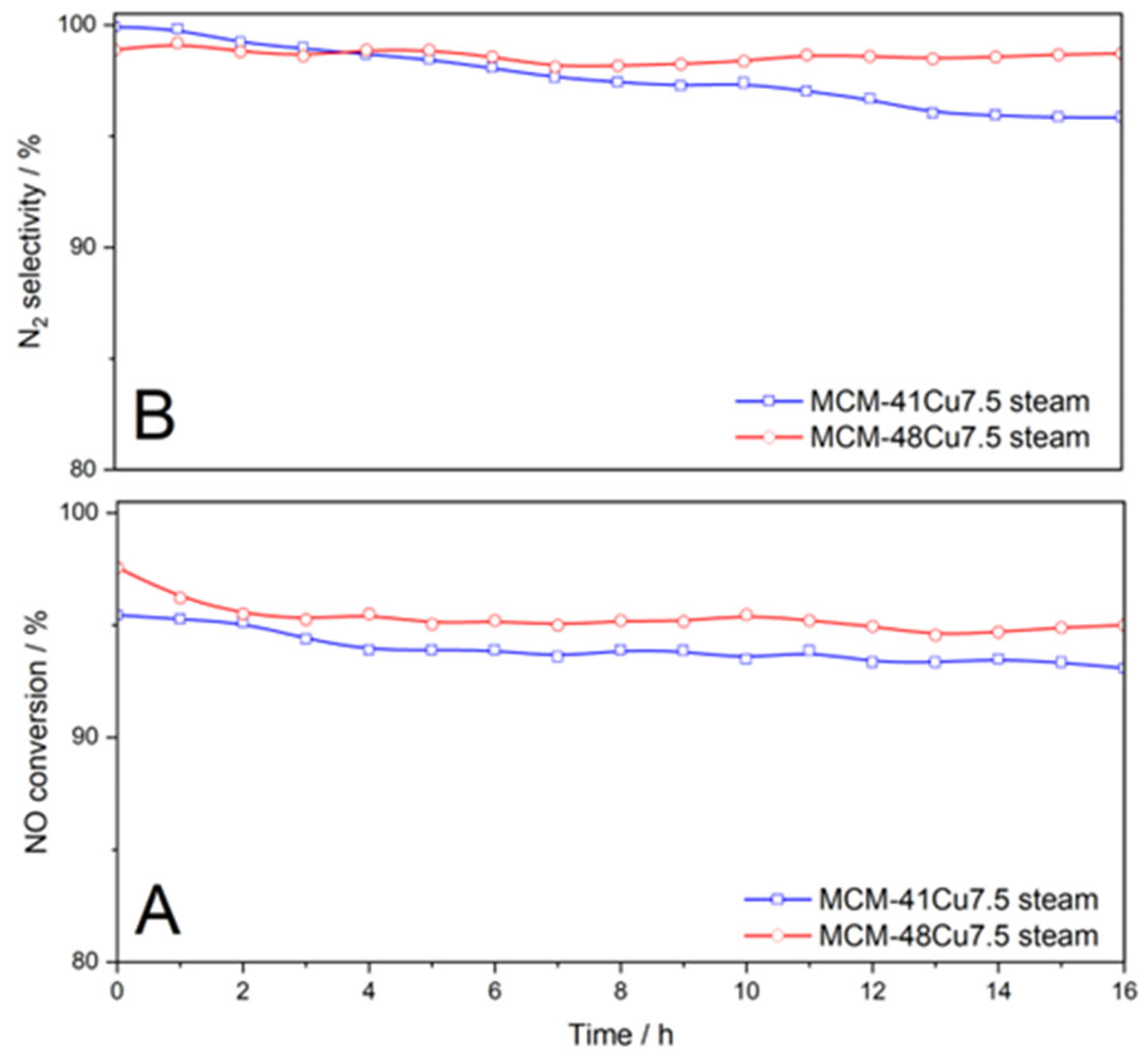
The results of the isothermal (275 °C) catalytic tests conducted in the flow of the wet reaction mixture for MCM-41Cu7.5 and MCM-48Cu7.5 catalysts show only small changes in the NO conversion and N
2 selectivity (
Figure 10). The most significant changes were observed at the beginning of the catalytic tests—the decrease in the NO conversion by about 2% during the first 2 h in the case of MCM-48Cu7.5 and by about 1.5% during the first 4 h in the case of MCM-41Cu7.5. Afterward, NO conversion remained very stable for both catalysts. The selectivity towards N
2 was stable for 16 h of the catalytic test for MCM-48Cu7.5, while in the case of MCM-41Cu7.5, it decreased by about 2%. In conclusion, mesoporous silicas modified with copper by the ADP method presented high stability in the NH
3-SCR process. The catalysts after the stability tests were analyzed with respect to copper species’ aggregation that could occur under conditions of the NH
3-SCR reaction. The diffractograms recorded for the MCM-41Cu7.5 and MCM-48Cu7.5 catalysts after the stability test were compared with the diffractograms of fresh catalysts (
Figure 1, insets). No reflections characteristic of CuO were found in diffractograms recorded for the catalysts after stability tests. Additionally, the UV-vis-DR spectra recorded for the samples after stability tests exhibited nearly the same shape as those obtained for the fresh catalysts and indicate the presence of copper in the form of monomeric cations. Thus, no significant changes in the form or aggregation of copper species were observed under the conditions of the stability catalytic test.
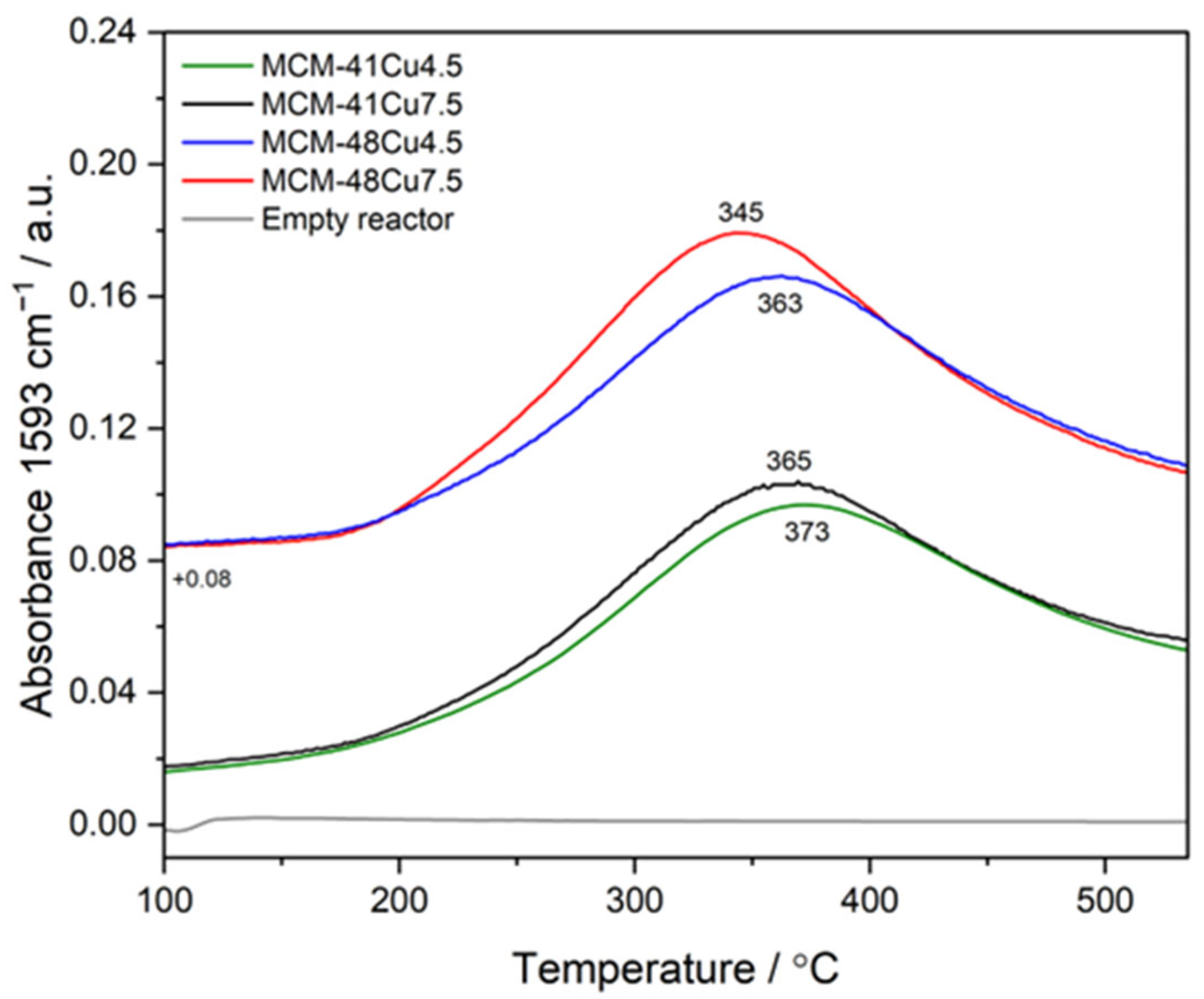
As already mentioned, the overall efficiency of the NH
3-SCR process depends on the associated reactions, such as NO to NO
2 oxidation and direct ammonia oxidation. The results of the NO oxidation for the selected catalysts, presented in
Figure 11, show their activity in this reaction. The maximum of the NO
2 formation occurs at about 245–275 °C and is slightly increased for the catalysts with higher copper loadings. At higher temperatures, the efficiency of NO to NO
2 oxidation decreases due to thermodynamic limitations [
40]. Thus, the conversion of nitrogen oxides according to the fast-SCR pathway (2 NH
3 + NO
2 + NO → 2 N
2 + 3 H
2O) could contribute to the low-temperature NH
3-SCR process.
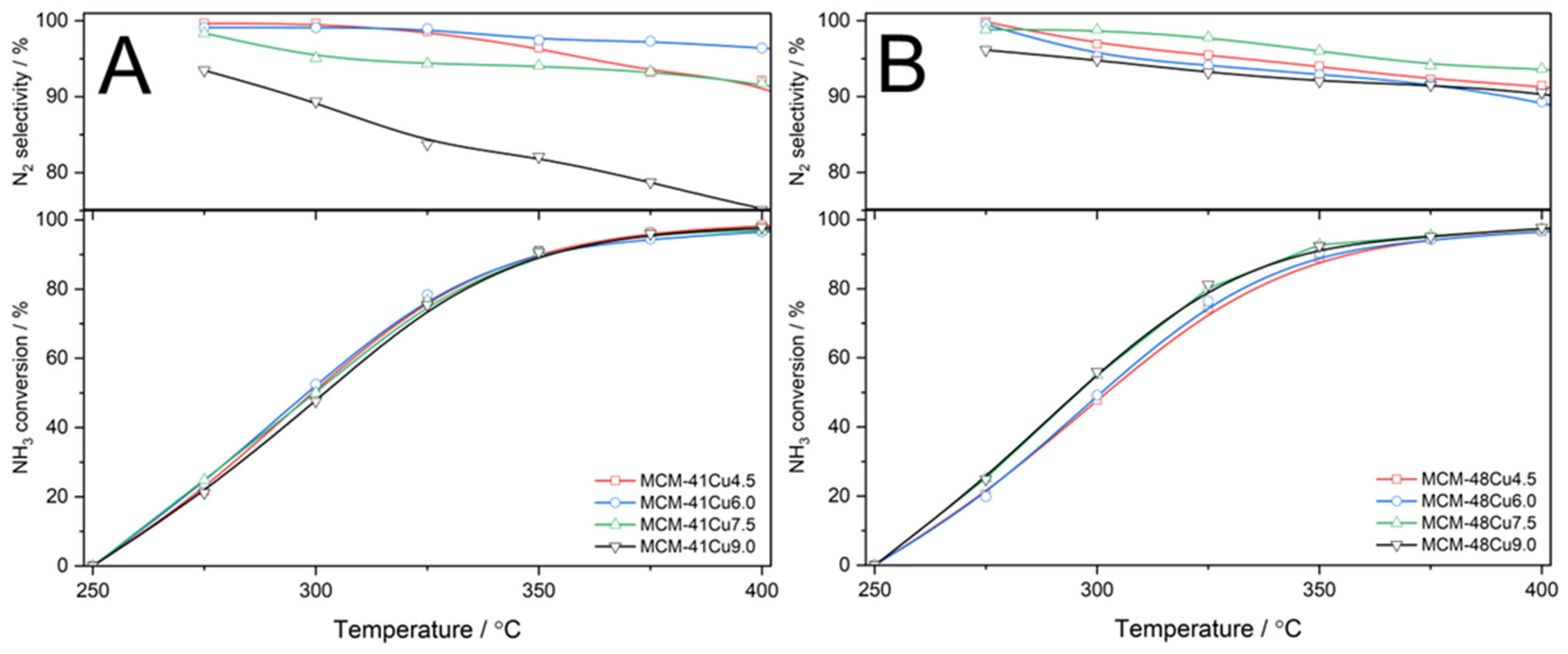
The results of direct ammonia oxidation tests, presented in
Figure 12, show very similar catalytic activity in both series of the catalysts, only slightly dependent on copper loading and form. On the other hand, significant differences in selectivity to nitrogen were identified. In the case of MCM-41 modified with copper, the selectivity to nitrogen was stable at a level above 90% (
Figure 12A. An exception is the catalyst with the highest copper loading, MCM-41Cu9.0, which presented a continuous decrease in selectivity to nitrogen due to the increasing formation of NO as the temperature increased. In the case of MCM-48 modified with copper, the selectivity to nitrogen was stable and above 90% to about 325–350 °C and decreased at higher temperatures due to the increased contribution of NO in the ammonia oxidation products. It should be noted that ammonia oxidation started at about 250 °C, and at 325 °C, the ammonia conversion reached 70–80% (
Figure 12B), while the NO conversion in the NH
3-SCR is above 90% at 325 °C (
Figure 7). The decrease in the efficiency of this process, due to the side process of ammonia oxidation, is observed at higher temperatures. Thus, it can be concluded that the NO reduction with ammonia is the preferential reaction over direct ammonia oxidation in the presence of silicas modified with copper by the ADP method.
3. Materials and Methods
3.1. Catalysts’ Preparation
As catalytic supports, mesoporous silicas of MCM-41 and MCM-48 types were used. MCM-41 material was prepared in accordance with the description presented in our previous paper [
41]. A total of 525 cm
3 of distilled water, 44.0 cm
3 of ammonia (25% solution of NH
3 in water, Chempur, Karlsruhe, Germany), and 45.3 cm
3 hexadecyltrimethylammonium chloride (25% solution in water, Sigma-Aldrich, St. Louis, MO, USA) were blended and thoroughly stirred at room temperature for 30 min. Afterwards, 48.6 cm
3 of tetraethyl orthosilicate (98%, Sigma-Aldrich, St. Louis, MO, USA) was added dropwise (2 drops per s). The procured mixture was then aged for 1 h at room temperature. Finally, the solid product was isolated using filtration, washed with distilled water, and dried at 60 °C for 24 h.
MCM-48 synthesis was performed based on the method described by Machowski et al. [
42]. In brief, 210 cm
3 of methanol (Sigma-Aldrich, St. Louis, MO, USA), 134 cm
3 of distilled water, 32.0 cm
3 of hexadecyltrimethylammonium chloride (25% solution in water, Sigma-Aldrich, St. Louis, MO, USA), and 60.0 cm
3 of ammonia (25% solution of NH
3 in water, Chempur, Karlsruhe, Germany) were mixed at ambient temperature for 30 min. Then, 18.0 cm
3 of tetraethyl orthosilicate (98%, Sigma-Aldrich, St. Louis, MO, USA) was added dropwise at a rate of 3 drops per s. The received slurry was subsequently aged for 2 h at ambient temperature. The solid product was next filtrated, rinsed with distilled water, and dried at 60 °C for 24 h. As-synthesized silicas were then calcined at 550 °C for 8 h in air flow (temperature increase of 1 °C per min).
Mesoporous silicas were modified with copper by the ADP method, as described by Xin et al. [
22]. A specified amount of silica carrier was stirred with an active phase precursor solution, 0.03 M Cu(NO
3)
2·3H
2O, to achieve the desired copper content in weight percent. Subsequently, ammonia (28–30% solution of NH
3 in water, Acros, Belgium) was added to achieve a molar ratio of 1:6 for Cu to NH
3. The resulting slurry was vigorously stirred at ambient temperature for 48 h (with the vessel sealed to prevent ammonia from evaporating). Afterward, the mixture was filtered, washed with distilled water, and air-dried for 24 h. The procured samples were then calcined at 550 °C for 8 h (air flow, with a temperature increase of 1 °C·min
−1).
Table 3 presents the sample codes along with the specific concentrations of reagents used in the modification process. Sample codes follow the general format
MCM-xxCuY, where
xx denotes the mesoporous silica type and
Y represents the copper content in weight percent. For example, MCM-41Cu4.5 refers to the MCM-41 sample modified with 4.5 wt% Cu.
3.2. Characterization
X-ray diffraction patterns were collected using the Bruker D2 Phaser (Bruker, Billerica, MA, USA) diffractometer (Cu-Kα radiation, λ = 1.54056 Å). The measurements were carried out in the 2-theta angle ranges of 1–8 (count time 5 s per step) and 28–42 (count time 1 s per step) with a step of 0.02°.
UV-vis-DR spectroscopy was used to investigate the aggregation and form of copper species deposited on silica carries. The spectra were recorded in the range of 190–900 nm with a resolution of 2 nm using the Lambda 650 S (PerkinElmer, Waltham, MA, USA) spectrophotometer.
To determine the textural parameters of the MCM-41 and MCM-48 samples, low-temperature nitrogen adsorption–desorption was employed. The measurements were conducted using a 3Flex v.1.00 (Micromeritics, Norcross, GA, USA) automated gas adsorption system at −196 °C. Prior to the measurements, the samples were degassed under vacuum at 350 °C for 24 h. The specific surface area was calculated using the BET model, while the pore size was determined using the BJH (Barrett–Joyner–Halenda) model. The pore volume was estimated for the total quantity of adsorbed N2 at p/p0 pressure of 0.98.
The elemental composition of the samples was examined using inductively coupled plasma optical emission spectroscopy (ICP-OES). The measurements were performed with an iCAP 7000 instrument (Thermo Scientific, Waltham, MA, USA). The samples were dissolved in a mixture containing 2 cm3 of HCl (30%, Honeywell, Charlotte, NC, USA), 6 cm3 of HNO3 (67–69%, Honeywell, Charlotte, NC, USA), and 2 cm3 of HF (47–51%, Honeywell, Charlotte, NC, USA) at 190 °C for 1 h utilizing a microwave digestion system (Ethos Easy, Milestone, Sorisole, Italy).
The reducibility of copper species was examined by the temperature-programmed reduction method with hydrogen as a reductant (H2-TPR). The analysis was performed in a flow microreactor with a fixed bed equipped with a thermal conductivity detector (TCD, Valco, Houston, TX, USA). Mass flow controllers (Brooks Instrument, Hatfield, PA, USA) were used to regulate the gas flow to the microreactor. Before the reduction process, the sample (25 mg) was degassed in a pure argon stream at 550 °C for 30 min. Afterward, the temperature was gradually reduced to 50 °C, and H2-TPR runs were carried out with a constant temperature increase of 10 °C per minute from 50 °C to 950 °C in a gas mixture containing 5.0 vol.% H2 diluted in argon.
The surface acidity of the modified silicas was evaluated through the temperature-programmed desorption of ammonia (NH3-TPD). These experiments were performed using a flow quartz microreactor system directly linked to a quadrupole mass spectrometer PREVAC-200 (PREVAC, Rogów, Poland). Prior to desorption, the catalysts (100 mg) were degassed in a helium flow at 550 °C for 30 min. Afterwards, the sample was cooled to 70 °C and subjected to a gas mixture of 1.0 vol.% NH3 diluted in helium for 2.5 h to ensure complete saturation with ammonia. The catalyst was then purged with helium to eliminate any physiosorbed ammonia. NH3-TPD experiments were conducted in a pure helium flow (20 cm3·min−1) with a heating rate of 10 °C·min−1.
3.3. Catalytic Tests
The catalytic activity of copper-doped silicas was assessed for the selective catalytic reduction in NO with ammonia (NH3-SCR, DeNOx). The catalytic experiments were performed in a fixed bed quartz microreactor, operating at temperatures between 100 and 400 °C under atmospheric pressure. The composition of the gas mixture downstream of the microreactor was continuously monitored using a quadrupole mass spectrometer (PREVAC, Rogów, Poland) directly connected to the reactor outlet. The catalyst samples (100 mg), with particle sizes ranging from 0.250 to 0.315 mm, were degassed in a helium flow at 550 °C for 30 min. In the NH3-SCR tests, a reaction mixture containing 0.25 vol.% NO, 0.25 vol.% NH3, and 2.5 vol.% O2 diluted in helium was used. Additionally, the silica-based materials were evaluated in the NH3-SCR process under humid conditions, using a gas mixture comprising 0.25 vol.% NO, 0.25 vol.% NH3, 2.5 vol.% O2, 5.0 vol.% H2O, and 92 vol.% He.
The catalytic performance of modified samples was also evaluated in the selective catalytic oxidation of ammonia (NH3-SCO). These tests were conducted in a fixed-bed quartz microreactor at temperatures ranging from 225 to 450 °C under atmospheric pressure. Similarly to previous catalytic experiments, the concentrations of reactants in the gases were continuously monitored by directly connecting a quadrupole mass spectrometer (PREVAC, Rogów, Poland) to the reactor outlet. The catalyst samples (100 mg), with particle sizes between 0.250 and 0.315 mm, were degassed in a pure helium flow at 550 °C for 30 min. For the NH3-SCO experiments, a gas mixture containing 0.5 vol.% NH3 and 2.5 vol.% O2 diluted in pure helium was utilized.
3.4. Stability Test
For the most active samples, stability tests in the NH3-SCR process were also conducted. The same setup as in the catalytic tests was used for this purpose. Prior to the measurements, the samples were degassed in a helium flow at 550 °C for 30 min. The tests were performed at 275 °C, with a sample exposure time of 16 h. The experiments were conducted in an atmosphere containing water vapor, with a gas mixture comprising 0.25 vol.% NO, 0.25 vol.% NH3, 2.5 vol.% O2, 5.0 vol.% H2O, and 92 vol.% He (flow rate of 40 cm3·min−1). The catalyst mass and particle sizes matched those used in the NH3-SCR and NH3-SCO tests.
3.5. Oxidation of NO to NO2
The oxidation reaction of NO to NO2 was also examined using the same experimental setup as employed for the NH3-SCR tests. The key difference was the substitution of the quadrupole mass spectrometer with an FTIR spectrometer (Nicolet iS5, Thermo Scientific, Waltham, MA, USA), which featured a 10 cm gas cell. Spectra were captured in the wavenumber range of 625–4000 cm−1, with a resolution of 4 cm−1, averaging seven scans per spectrum, recorded every 8 s. NO2 and NO detection relied on the bands at 1593 cm−1 and 1912 cm−1, respectively. The gas mixture used contained 0.5 vol.% NO and 2.5 vol.% O2 and 97 vol.% He. The experiments spanned temperatures from 100 °C to 550 °C, with a temperature increase of 10 °C·min−1


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}