
Mesostructured Silica–Zirconia–Tungstophosphoric Acid Composites as Catalyst in Calcium Channel Blocker Nifedipine Synthesis

 , ,
, ,  and
and
Abstract

1. Introduction
2. Results and Discussion
2.1. Characterization of Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vibration Modes | Band (cm−1) | |
|---|---|---|
| Bending vibration Oa-P-Oa | 524 | [34,35,36] |
| Stretching vibrations W-OC-W | 793 | |
| Stretching vibrations W-Ob-W | 896 | |
| Stretching vibrations W=Od | 982 | |
| Stretching vibrations P-Oa | 1081 | |
| Bending vibration O-Si-O | 480 | |
| Stretching Si-O-Si group | 800 | |
| Stretching Si-OH group | 965 | |
| Angular vibration of H2O | 1650 | |
| Asymmetric stretching siloxane group | 1220–1100 | |
| Stretching OH group | 3700–3200 | |
| Stretching O-Zr-O | 460 | |
| Asymmetric stretching Si-O-Zr | 980 | |
| TEA C-H vibration | 1400 |
2.2. Catalytic Test
3. Materials and Methods
3.1. Reagents
3.2. Synthesis of Mesostructured SiO2-ZrO2-TPA Composites
3.3. Catalyst Characterization
3.4. Catalytic Test and Product Identification
3.5. General Procedure for the Synthesis of Nifedipine (Thermal Procedure)
3.6. General Procedure for the Synthesis of Nifedipine (Microwave Procedure)
3.7. Catalyst Recycling Under Microwave Irradiation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TPA | Tungstophosphoric acid |
| TEA | Triethanolamine |
| TEOS | Tetraethyl orthosilicate |
| HPA | Heteropolyacid |
| MCR | Reaction multicomponent |
| DHP | Dihydropyridines |
| DTG | Derivative thermogravimetry |
| SBET | Surface Area Brunauer–Emmett–Teller |
| Dp | Pore diameter |
| FT-IR | Infrared spectroscopy |
| (NMR-MAS) | 31P solid-state nuclear magnetic resonance |
| XRD | X-ray diffraction |
| TGA | Thermogravimetric analysis |
| XPS | X-ray photoelectron spectra |
References
- Kajal, K.; Shakya, R.; Rashid, M.; Nigam, V.; Das Kurmi, B.; Das Gupta, G.; Patel, P. Recent Green Chemistry Approaches for Pyrimidine Derivatives as a Potential Anti-Cancer Agent: An Overview (2013–2023). Sustain. Chem. Pharm. 2024, 37, 101374. [Google Scholar] [CrossRef]
- Osman, A.I.; Zhang, Y.; Farghali, M.; Rashwan, A.K.; Eltaweil, A.S.; El-Monaem, E.M.A.; Mohamed, I.M.A.; Badr, M.M.; Ihara, I.; Rooney, D.W.; et al. Synthesis of Green Nanoparticles for Energy, Biomedical, Environmental, Agricultural, and Food Applications: A Review; Springer International Publishing: Cham, Switzerland, 2024; Volume 22, ISBN 0123456789. [Google Scholar]
- Nardi, M.; Cano, N.C.H.; Simeonov, S.; Bence, R.; Kurutos, A.; Scarpelli, R.; Wunderlin, D.; Procopio, A. A Review on the Green Synthesis of Benzimidazole Derivatives and Their Pharmacological Activities. Catalysts 2023, 13, 392. [Google Scholar] [CrossRef]
- Castiello, C.; Junghanns, P.; Mergel, A.; Jacob, C.; Ducho, C.; Valente, S.; Rotili, D.; Fioravanti, R.; Zwergel, C.; Mai, A. GreenMedChem: The Challenge in the next Decade toward Eco-Friendly Compounds and Processes in Drug Design. Green Chem. 2023, 25, 2109–2169. [Google Scholar] [CrossRef]
- Zimmerman, J.B.; Anastas, P.T.; Erythropel, H.C.; Leitner, W. Designing for a Green Chemistry Future. Science 2020, 367, 397–400. [Google Scholar] [CrossRef]
- Zaera, F. Designing Sites in Heterogeneous Catalysis: Are We Reaching Selectivities Competitive with Those of Homogeneous Catalysts? Chem. Rev. 2022, 122, 8594–8757. [Google Scholar] [CrossRef]
- Alvim, H.G.O.; Júnior, E.N.d.S.; Neto, B.A.D. What Do We Know about Multicomponent Reactions? Mechanisms and Trends for the Biginelli, Hantzsch, Mannich, Passerini and Ugi MCRs. RSC Adv. 2014, 4, 54282–54299. [Google Scholar] [CrossRef]
- Younus, H.A.; Al-Rashida, M.; Hameed, A.; Uroos, M.; Salar, U.; Rana, S.; Khan, K.M. Multicomponent Reactions (MCR) in Medicinal Chemistry: A Patent Review (2010–2020). Expert Opin. Ther. Pat. 2021, 31, 267–289. [Google Scholar] [CrossRef]
- Mohlala, R.L.; Rashamuse, T.J.; Coyanis, E.M. Highlighting Multicomponent Reactions as an Efficient and Facile Alternative Route in the Chemical Synthesis of Organic-Based Molecules: A Tremendous Growth in the Past 5 Years. Front. Chem. 2024, 12, 1469677. [Google Scholar] [CrossRef]
- Hantzsch, A. Condensationsprodukte aus Aldehydammoniak und Ketonartigen Verbindungen. Berichte Dtsch. Chem. Ges. 1881, 14, 1637–1638. [Google Scholar] [CrossRef]
- Palermo, V.; Sathicq, Á.G.; Constantieux, T.; Rodríguez, J.; Vázquez, P.G.; Romanelli, G.P. First Report About the Use of Micellar Keggin Heteropolyacids as Catalysts in the Green Multicomponent Synthesis of Nifedipine Derivatives. Catal. Lett. 2016, 146, 1634–1647. [Google Scholar] [CrossRef]
- Kiowski, W.; Erne, P.; Bühler, F.R. Use of Nifedipine in Hypertension and Raynaud’s Phenomenon. Cardiovasc. Drugs Ther. 1990, 4, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Block, J.A.; Sequeira, W. Raynaud’s Phenomenon. Lancet 2001, 357, 2042–2048. [Google Scholar] [CrossRef]
- Khrustalev, D.; Yedrissov, A.; Khrustaleva, A.; Mustafin, M.; Bekisheva, K. An Efficient Method for the Synthesis of 1,4-Dihydropyridine Derivatives in a Flow Microwave Reactor. In Materials Today: Proceedings; Elsevier Ltd.: Amsterdam, The Netherlands, 2023; Volume 81, pp. 1204–1208. [Google Scholar]
- D’Alessandro, O.; Sathicq, Á.G.; Sambeth, J.E.; Thomas, H.J.; Romanelli, G.P. A Study of the Temperature Effect on Hantzsch Reaction Selectivity Using Mn and Ce Oxides under Solvent-Free Conditions. Catal. Commun. 2015, 60, 65–69. [Google Scholar] [CrossRef]
- Vargas, A.Y.; Rojas, H.A.; Romanelli, G.P.; Martínez, J.J. Synthesis of 1,4-Dihydropyrimidines with Immobilized Urease: Effect of Method Immobilization on Magnetic Supports. Green Process. Synth. 2017, 6, 377–384. [Google Scholar] [CrossRef]
- Sanchez, L.M.; Sathicq, Á.G.; Romanelli, G.P.; González, L.M.; Villa, A.L. Activity of Immobilized Metallic Phthalocyanines in the Multicomponent Synthesis of Dihydropyridine Derivatives and Their Subsequent Aromatization. Mol. Catal. 2017, 435, 1–12. [Google Scholar] [CrossRef]
- Cheng, M.; Shi, T.; Guan, H.; Wang, S.; Wang, X.; Jiang, Z. Clean Production of Glucose from Polysaccharides Using a Micellar Heteropolyacid as a Heterogeneous Catalyst. Appl. Catal. B Environ. 2011, 107, 104–109. [Google Scholar] [CrossRef]
- Toufaily, J.; Soulard, M.; Guth, J.L.; Patarin, J.; Delmote, L.; Hamieh, T.; Kodeih, M.; Naoufal, D.; Hamad, H. Synthesis and Characterization of New Catalysts Formed by Direct Incorporation of Heteropolyacids into Organized Mesoporous Silica. Colloids Surf. A Physicochem. Eng. Asp. 2008, 316, 285–291. [Google Scholar] [CrossRef]
- Gorsd, M.; Sathicq, G.; Romanelli, G.; Pizzio, L.; Blanco, M. Tungstophosphoric acid supported on core-shell polystyrene-silica microspheres or hollow silica spheres catalyzed trisubstituted imidazole synthesis by multicomponent reaction. J. Mol. Catal. A Chem. 2016, 420, 294–302. [Google Scholar] [CrossRef]
- Pizzio, L.R.; Cáceres, V.C.; Blanco, M.N. Adsorption of Tungstophosphoric or Tungstosilicic Acids from Ethanol–Water Solutions on Carbon. J. Colloid Interface Sci. 1997, 190, 318–326. [Google Scholar] [CrossRef]
- Rivera, T.S.; Sosa, A.; Romanelli, G.P.; Blanco, M.N.; Pizzio, L.R. Tungstophosphoric Acid/Zirconia Composites Prepared by the Sol-Gel Method: An Efficient and Recyclable Green Catalyst for the One-Pot Synthesis of 14-Aryl-14H-Dibenzo[a,j]Xanthenes. Appl. Catal. A Gen. 2012, 443–444, 207–213. [Google Scholar] [CrossRef]
- Méndez, F.J.; Llanos, A.; Echeverría, M.; Jáuregui, R.; Villasana, Y.; Díaz, Y.; Liendo-Polanco, G.; Ramos-García, M.A.; Zoltan, T.; Brito, J.L. Mesoporous Catalysts Based on Keggin-Type Heteropolyacids Supported on MCM-41 and Their Application in Thiophene Hydrodesulfurization. Fuel 2013, 110, 249–258. [Google Scholar] [CrossRef]
- Morey, M.S.; Stucky, G.D.; Schwarz, S.; Fröba, M. Isomorphic Substitution and Postsynthesis Incorporation of Zirconium into MCM-48 Mesoporous Silica. J. Phys. Chem. B 1999, 103, 2037–2041. [Google Scholar] [CrossRef]
- Lai, F.; Yan, F.; Wang, Y.; Li, C.; Cai, J.; Zhang, Z. Tungstophosphoric Acid Supported on Metal/Si-Pillared Montmorillonite for Conversion of Biomass-Derived Carbohydrates into Methyl Levulinate. J. Clean. Prod. 2021, 314, 128072. [Google Scholar] [CrossRef]
- Ramanathan, A.; Villalobos, M.C.C.; Kwakernaak, C.; Telalovic, S.; Hanefeld, U. Zr-TUD-1: A Lewis Acidic, Three-Dimensional, Mesoporous, Zirconium-Containing Catalyst. Chem. A Eur. J. 2008, 14, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Z.; Zeng, H.C. The Self-Catalytic Role of Zirconium w-Propoxide in Sol-Gel Syntheses of ZrO2-SiO2 Mixed Oxides. J. Mater. Chem. 1999, 9, 2647–2652. [Google Scholar] [CrossRef]
- Sawant, D.P.; Vinu, A.; Jacob, N.E.; Lefebvre, F.; Halligudi, S.B. Formation of Nanosized Zirconia-Supported 12-Tungstophosphoric Acid in Mesoporous Silica SBA-15: A Stable and Versatile Solid Acid Catalyst for Benzylation of Phenol. J. Catal. 2005, 235, 341–352. [Google Scholar] [CrossRef]
- Colmenares-Zerpa, J.; Gajardo, J.; Peixoto, A.F.; Silva, D.S.A.; Silva, J.A.; Gispert-Guirado, F.; Llorca, J.; Urquieta-Gonzalez, E.A.; Santos, J.B.O.; Chimentão, R.J. High Zirconium Loads in Zr-SBA-15 Mesoporous Materials Prepared by Direct-Synthesis and PH-Adjusting Approaches. J. Solid State Chem. 2022, 312, 123296. [Google Scholar] [CrossRef]
- Gorsd, M.N.; Sosa, A.A.; Frenzel, R.A.; Pizzio, L.R. Synthesis and Characterization of Tungstophosphoric Acid-Modified Mesoporous Sponge-like TUD-1 Materials. J. Sol-Gel Sci. Technol. 2018, 87, 204–215. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, Q.; Xu, M.; Wang, S.; You, J. In Situ Spectroscopic Studies of Decomposition of ZrSiO4 during Alkali Fusion Process Using Various Hydroxides. RSC Adv. 2015, 5, 11658–11666. [Google Scholar] [CrossRef]
- Escobar, A.M.; Blanco, M.N.; Martínez, J.J.; Cubillos, J.A.; Romanelli, G.P.; Pizzio, L.R. Biomass Derivative Valorization Using Nano Core-Shell Magnetic Materials Based on Keggin-Heteropolyacids: Levulinic Acid Esterification Kinetic Study with n-Butanol. J. Nanomater. 2019, 2019, 5710708. [Google Scholar] [CrossRef]
- Sosa, A.A.; Gorsd, M.N.; Blanco, M.N.; Pizzio, L.R. Synthesis and Characterization of Tungstophosphoric Acid-Modified Mesoporous Silica Nanoparticles with Tuneable Diameter and Pore Size Distribution. J. Sol-Gel Sci. Technol. 2017, 83, 355–364. [Google Scholar] [CrossRef]
- Morales, M.D.; Infantes-Molina, A.; Lázaro-Martínez, J.M.; Romanelli, G.P.; Pizzio, L.R.; Rodríguez-Castellón, E. Heterogeneous Acid Catalysts Prepared by Immobilization of H3PW12O40 on Silica through Impregnation and Inclusion, Applied to the Synthesis of 3H-1,5-Benzodiazepines. Mol. Catal. 2020, 485, 110842. [Google Scholar] [CrossRef]
- Méndez, L.; Torviso, R.; Pizzio, L.; Blanco, M. 2-Methoxynaphthalene acylation using aluminum or copper salts of tungstophosphoric and tungstosilicic acids as catalysts. Catal. Today 2011, 173, 32–37. [Google Scholar] [CrossRef]
- Khder, A.E.R.S.; Hassan, H.M.A.; El-Shall, M.S. Acid Catalyzed Organic Transformations by Heteropoly Tungstophosphoric Acid Supported on MCM-41. Appl. Catal. A Gen. 2012, 411–412, 77–86. [Google Scholar] [CrossRef]
- Salas, P.; Wang, J.A.; Armendariz, H.; Angeles-Chavez, C.; Chen, L.F. Effect of the Si/Zr Molar Ratio on the Synthesis of Zr-Based Mesoporous Molecular Sieves. Mater. Chem. Phys. 2009, 114, 139–144. [Google Scholar] [CrossRef]
- Li, H.; Ren, J.; Qin, X.; Qin, Z.; Lin, J.; Li, Z. Ni/SBA-15 Catalysts for CO Methanation: Effects of V, Ce, and Zr Promoters. RSC Adv. 2015, 5, 96504–96517. [Google Scholar] [CrossRef]
- Wang, L.; Yang, J. Zirconia-Doped Methylated Silica Membranes via Sol-Gel Process: Microstructure and Hydrogen Permselectivity. Nanomaterials 2022, 12, 2159. [Google Scholar] [CrossRef]
- Ardizzone, S.; Bianchi, C.L. XPS Characterization of Sulphated Zirconia Catalysts: The Role of Iron. In Surface and Interface Analysis; Wiley Analytical Science: Weinheim, Germany, 2000; Volume 30. [Google Scholar]
- Lackner, P.; Zou, Z.; Mayr, S.; Diebold, U.; Schmid, M. Using Photoelectron Spectroscopy to Observe Oxygen Spillover to Zirconia. Phys. Chem. Chem. Phys. 2019, 21, 17613–17620. [Google Scholar] [CrossRef]
- Yang, G.; Wang, L.; Jiang, H. Zr-Incorporating SBA-15 for Conversion of the Ethanol-Acetaldehyde Mixture to Butadiene. React. Chem. Eng. 2020, 5, 1833–1844. [Google Scholar] [CrossRef]
- Li, W.; Nie, X.; Jiang, X.; Zhang, A.; Ding, F.; Liu, M.; Liu, Z.; Guo, X.; Song, C. ZrO2 Support Imparts Superior Activity and Stability of Co Catalysts for CO2 Methanation. Appl. Catal. B Environ. 2018, 220, 397–408. [Google Scholar] [CrossRef]
- Gu, J.; Xin, Z.; Tao, M.; Lv, Y.; Gao, W.; Si, Q. Effect of Si-Modified Zirconia on the Properties of MoO3/Si-ZrO2 Catalysts for Sulfur-Resistant CO Methanation. Appl. Catal. A Gen. 2019, 575, 230–237. [Google Scholar] [CrossRef]
- Bosman, H.J.M.; Pijpers, A.P.; Jaspers, A.W.M.A. An X-Ray Photoelectron Spectroscopy Study of the Acidity of SiO2-ZrO2 Mixed Oxides. J. Catal. 1996, 161, 551–559. [Google Scholar] [CrossRef]
- Alam, A.U.; Howlader, M.M.R.; Deen, M.J. Oxygen Plasma and Humidity Dependent Surface Analysis of Silicon, Silicon Dioxide and Glass for Direct Wafer Bonding. ECS J. Solid State Sci. Technol. 2013, 2, P515–P523. [Google Scholar] [CrossRef]
- You, X.; Yu, L.-L.; Xiao, F.-F.; Wu, S.-C.; Yang, C.; Cheng, J.-H. Synthesis of Phosphotungstic Acid-Supported Bimodal Mesoporous Silica-Based Catalyst for Defluorination of Aqueous Perfluorooctanoic Acid under Vacuum UV Irradiation. Chem. Eng. J. 2018, 335, 812–821. [Google Scholar] [CrossRef]
- Rengifo-Herrera, J.A.; Blanco, M.; Wist, J.; Florian, P.; Pizzio, L.R. TiO2 Modified with Polyoxotungstates Should Induce Visible-Light Absorption and High Photocatalytic Activity through the Formation of Surface Complexes. Appl. Catal. B Environ. 2016, 189, 99–109. [Google Scholar] [CrossRef]
- Legagneux, N.; Basset, J.M.; Thomas, A.; Lefebvre, F.; Goguet, A.; Sá, J.; Hardacre, C. Characterization of Silica-Supported Dodecatungstic Heteropolyacids as a Function of Their Dehydroxylation Temperature. Dalton Trans. 2009, 2235–2240. [Google Scholar] [CrossRef]
- Fuchs, V.; Méndez, L.; Blanco, M.; Pizzio, L. Mesoporous Titania Directly Modified with Tungstophosphoric Acid: Synthesis, Characterization and Catalytic Evaluation. Appl. Catal. A Gen. 2009, 358, 73–78. [Google Scholar] [CrossRef]
- Massart, R.; Contant, R.; Fruchart, J.M.; Ciabrini, J.P.; Fournier, M. 31P NMR Studies on Molybdic and Tungstic Heteropolyanions. Correlation between Structure and Chemical Shift. Inorg. Chem. 1977, 16, 2916–2921. [Google Scholar] [CrossRef]
- Sosa, A.A.; Palermo, V.; Langer, P.; Luque, R.; Romanelli, G.P.; Pizzio, L.R. Tungstophosphoric Acid/Mesoporous Silicas as Suitable Catalysts in Quinoxaline Synthesis. Mol. Catal. 2022, 517, 112046. [Google Scholar] [CrossRef]
- Rengifo-Herrera, J.A.; Blanco, M.; Pizzio, L.R. Photocatalytic bleaching of aqueous malachite green solutions by UV-A and blue-light-illuminated TiO2 spherical nanoparticles modified with tungstophosphoric acid. Appl. Catal. B Environ. 2011, 110, 126–132. [Google Scholar] [CrossRef]
- Sosa, A.A.; Rivera, T.S.; Blanco, M.N.; Pizzio, L.R.; Romanelli, G.P. Tungstophosphoric Acid Supported on Zirconia: A Recyclable Catalyst for the Green Synthesis on Quinoxaline Derivatives under Solvent-Free Conditions. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 1071–1079. [Google Scholar] [CrossRef]
- Pizzio, L.R.; Cáceres, C.V.; Blanco, M.N. Equilibrium Adsorption of 11-Tungstophosphate Anion on Different Supports. Appl. Surf. Sci. 1999, 151, 91–101. [Google Scholar] [CrossRef]
- Escobar, A.; Sathicq, Á.; Pizzio, L.; Blanco, M.; Romanelli, G. Biomass Valorization Derivatives: Clean Esterification of 2-Furoic Acid Using Tungstophosphoric Acid/Zirconia Composites as Recyclable Catalyst. Process Saf. Environ. Prot. 2015, 98, 176–186. [Google Scholar] [CrossRef]
- Mokhtar, M.; Saleh, T.S.; Narasimharao, K.; Al-Mutairi, E. New Green Perspective to Dihydropyridines Synthesis Utilizing Modified Heteropoly Acid Catalysts. Catal. Today 2022, 397–399, 484–496. [Google Scholar] [CrossRef]
- Abdella, A.M.; Abdelmoniem, A.M.; Abdelhamid, I.A.; Elwahy, A.H.M. Synthesis of Heterocyclic Compounds via Michael and Hantzsch Reactions. J. Heterocycl. Chem. 2020, 57, 1476–1523. [Google Scholar] [CrossRef]
- Shen, L.; Cao, S.; Wu, J.; Zhang, J.; Li, H.; Liu, N.; Qian, X. A Revisit to the Hantzsch Reaction: Unexpected Products beyond 1,4-Dihydropyridines Supporting Information: Spectra of the Pyridine Derivatives: Diethyl 2-Phenyl-4, 6-Dimethylpyridine-3, 5-Dicarboxylate (3a). Green Chem. 2009, 11, 1414–1420. [Google Scholar] [CrossRef]
- Bosica, G.; Demanuele, K.; Padrón, J.M.; Puerta, A. One-Pot Multicomponent Green Hantzsch Synthesis of 1,2-Dihydropyridine Derivatives with Antiproliferative Activity. Beilstein J. Org. Chem. 2020, 16, 2862–2869. [Google Scholar] [CrossRef]
- Wei, J.; Xing, Y.; Ye, X.; Nguyen, B.; Wojtas, L.; Hong, X.; Shi, X. Gold-Catalyzed Amine Cascade Addition to Diyne-Ene: Enantioselective Synthesis of 1,2-Dihydropyridines. Angew. Chem. Int. Ed. 2023, 62, e202305409. [Google Scholar] [CrossRef]
- To, T.H.; Tran, D.B.; Pham, T.C.; Tran, P.D. A New Approach to Pyrimidine-Type Heterocycles Based on Petrenko–Kritschenko Synthesis. Chem. Heterocycl. Compd. 2022, 58, 608–614. [Google Scholar] [CrossRef]
- Kukreja, S. Copper-Catalyzed Ambient Synthesis of Functionalized 1,4-Dihydropyridines. J. Integr. Sci. Technol. 2023, 11, 4–8. [Google Scholar]
- Gallego-Villada, L.A.; Alarcón, E.A.; Cerrutti, C.; Blustein, G.; Sathicq, Á.G.; Romanelli, G.P. Levulinic Acid Esterification with N-Butanol over a Preyssler Catalyst in a Microwave-Assisted Batch Reactor: A Kinetic Study. Ind. Eng. Chem. Res. 2023, 62, 10915–10929. [Google Scholar] [CrossRef]
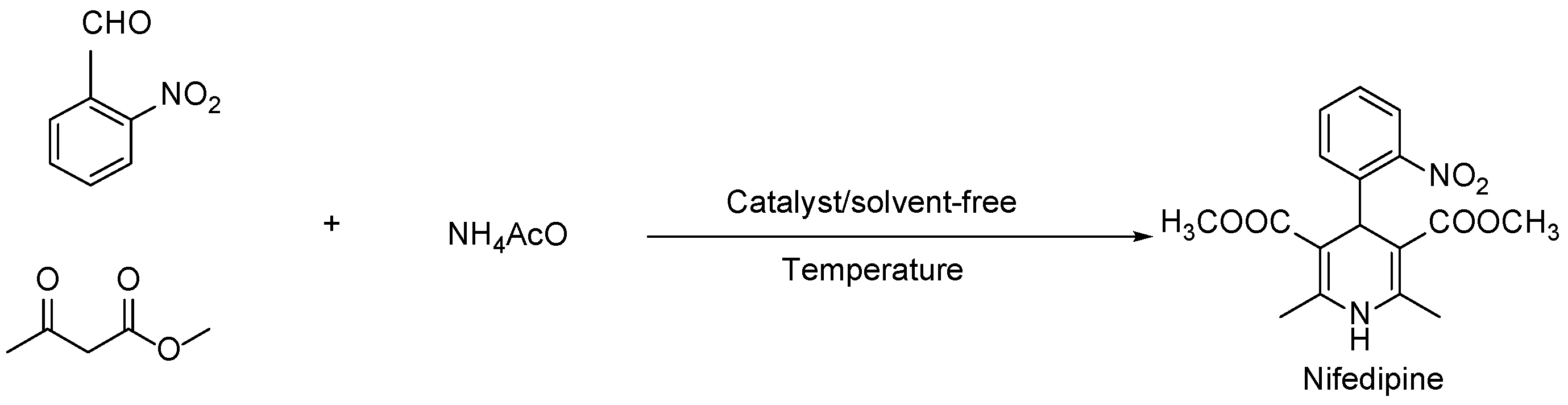

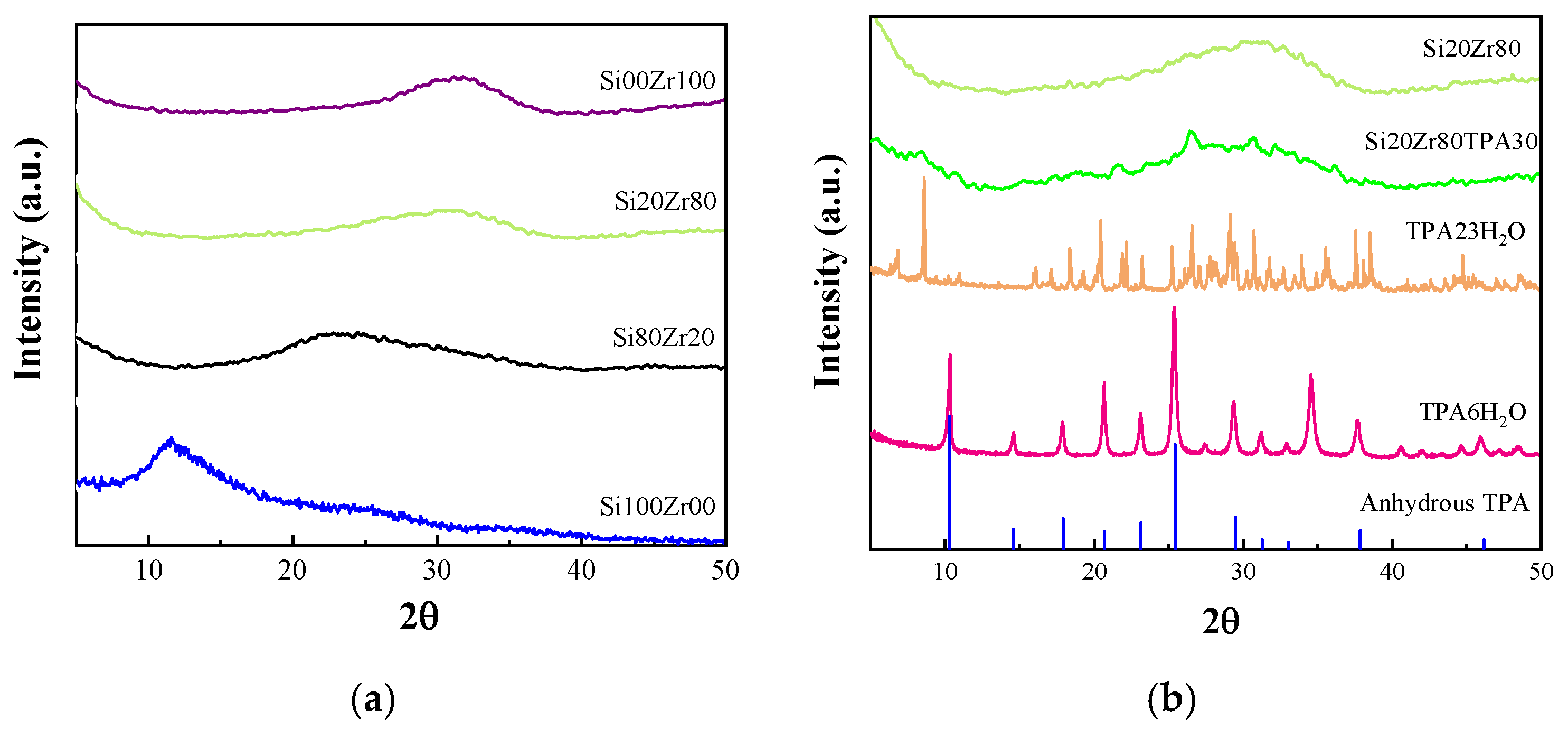
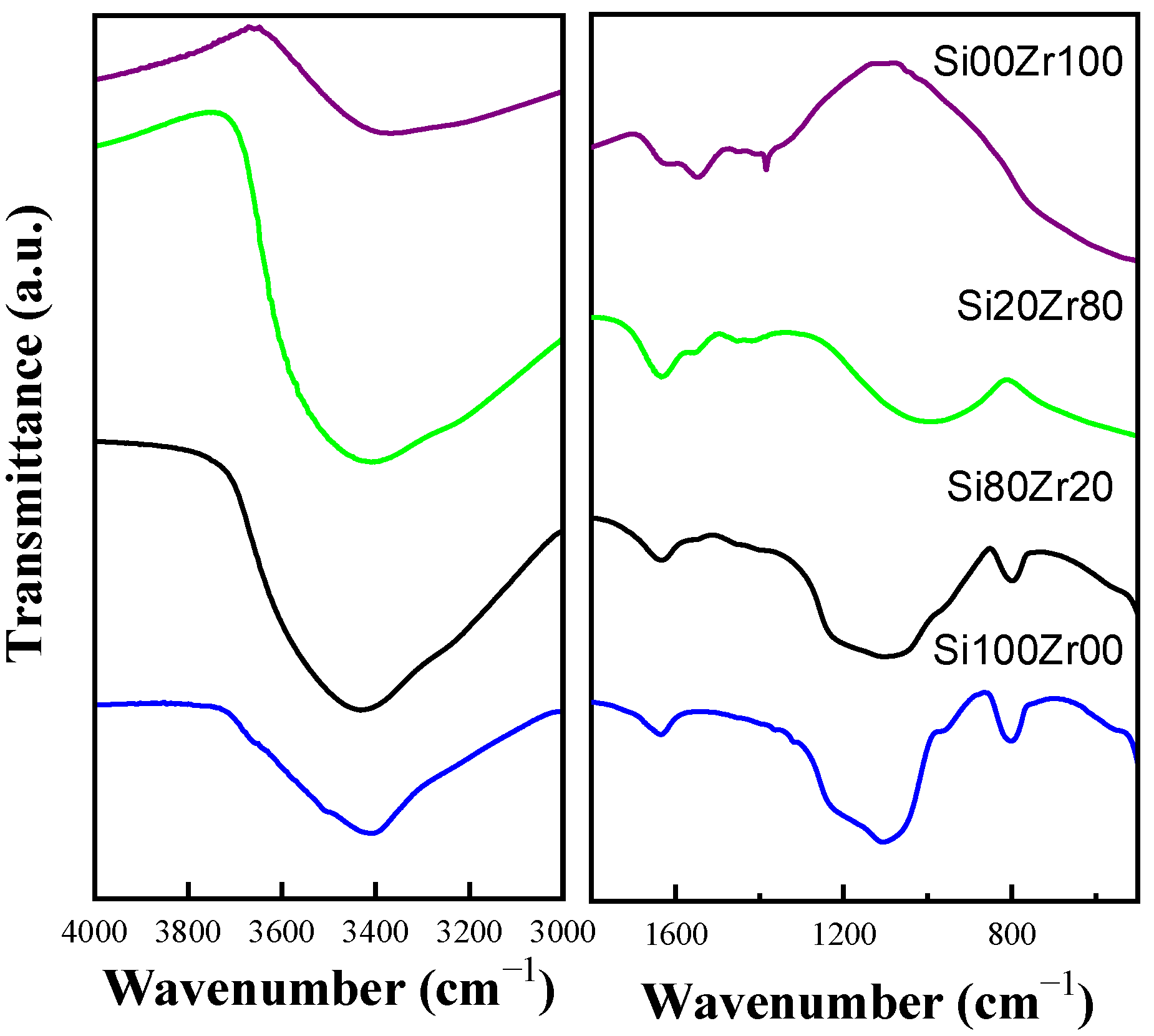
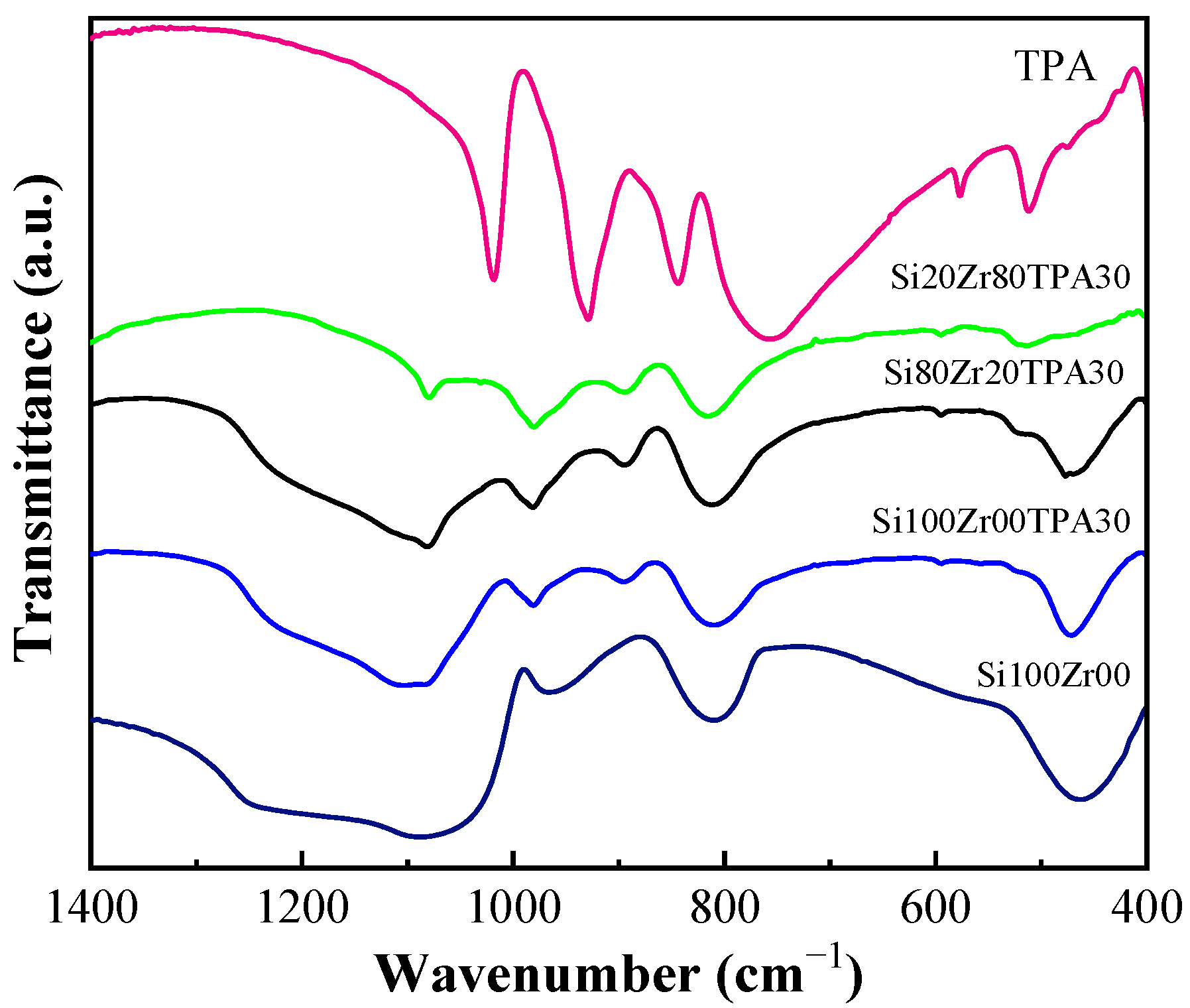



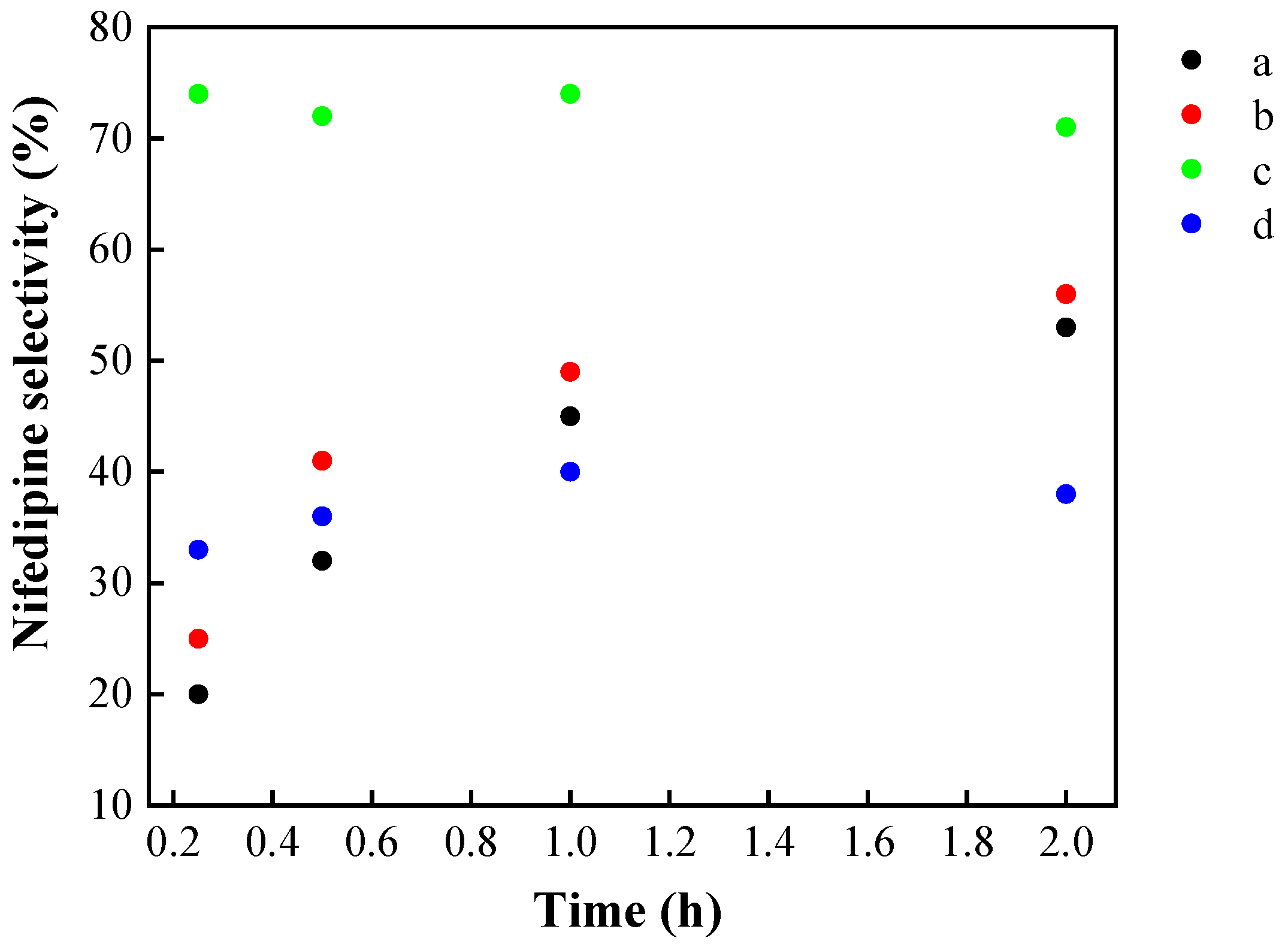
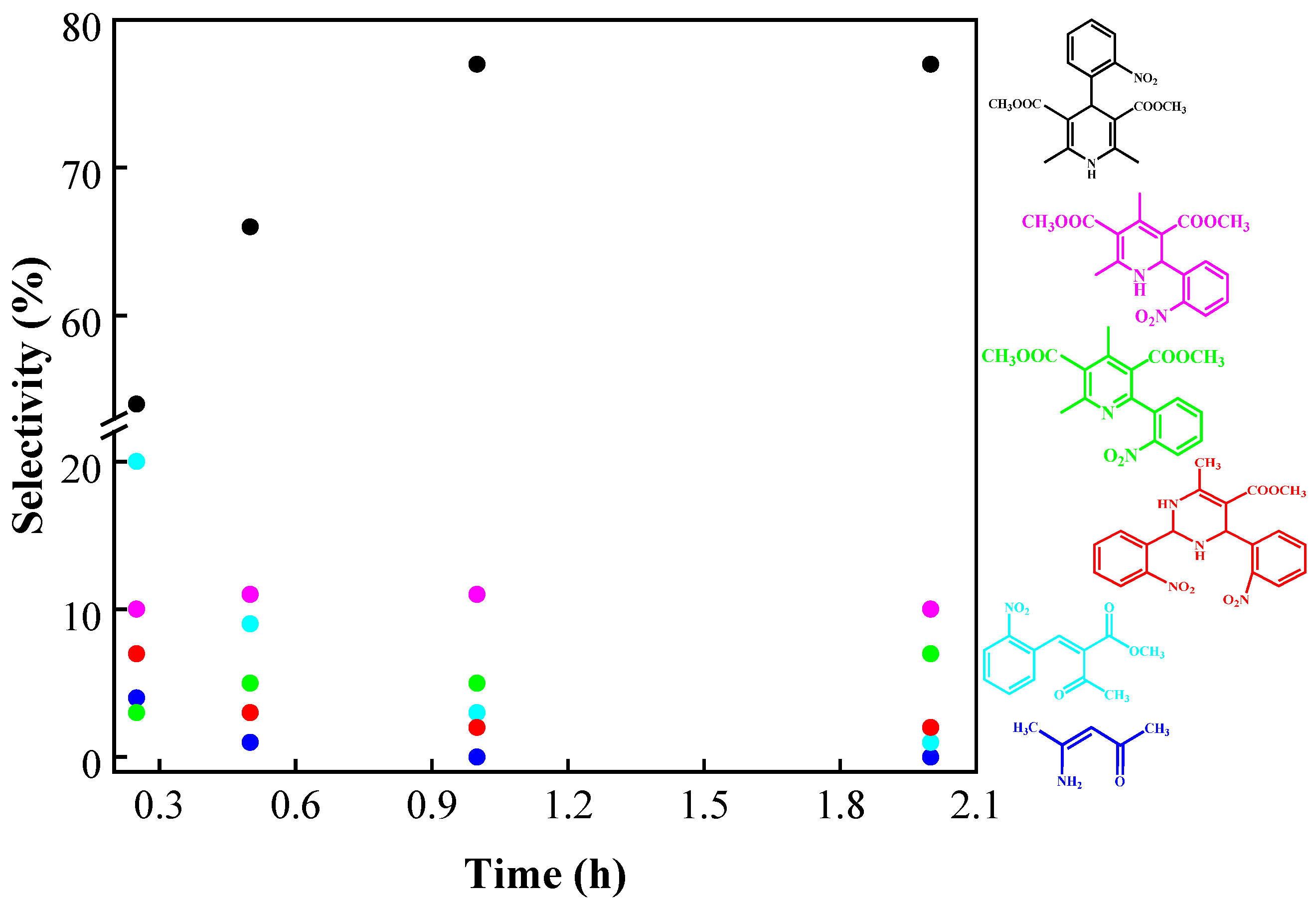
| Catalyst | % Ratio (SiO2:ZrO2) | SBET (m2 g−1) | Pore volume (cm3 g−1) | Dp (nm) |
|---|---|---|---|---|
| Si100Zr00TPA30 | 100:0 | 75 | 0.32 | 17.10 |
| Si80Zr20TPA30 | 80:20 | 149 | 0.37 | 14.42 |
| Si20Zr80TPA30 | 20:80 | 181 | 0.39 | 3.28 |
| Si00Zr100TPA30 | 0:100 | 116 | 0.32 | 2.96 |
| Sample | Binding Energy (eV) | |||||||
|---|---|---|---|---|---|---|---|---|
| Zr 3d | O 1s | Si 2p | W 4f | |||||
| d5/2 | d3/2 | W7/2 | W5/2 | |||||
| Si00Zr100TPA30 | 182.4 | 184.7 | 532.5 | 530.6 | - | 35.5 | 37.6 | |
| Si20Zr80TPA30 | 182.8 | 185.2 | 532.9 | 530.8 | 103.4 | 35.6 | 37.8 | |
| Si80Zr20TPA30 | 182.7 | 185.1 | 530.9 | 532.6 | 102.4 | 35.8 | 37.9 | |
| Si100Zr00TPA30 | - | - | 534.7 | 533.0 | 530.9 | 103.6 | 35.8 | 37.9 |
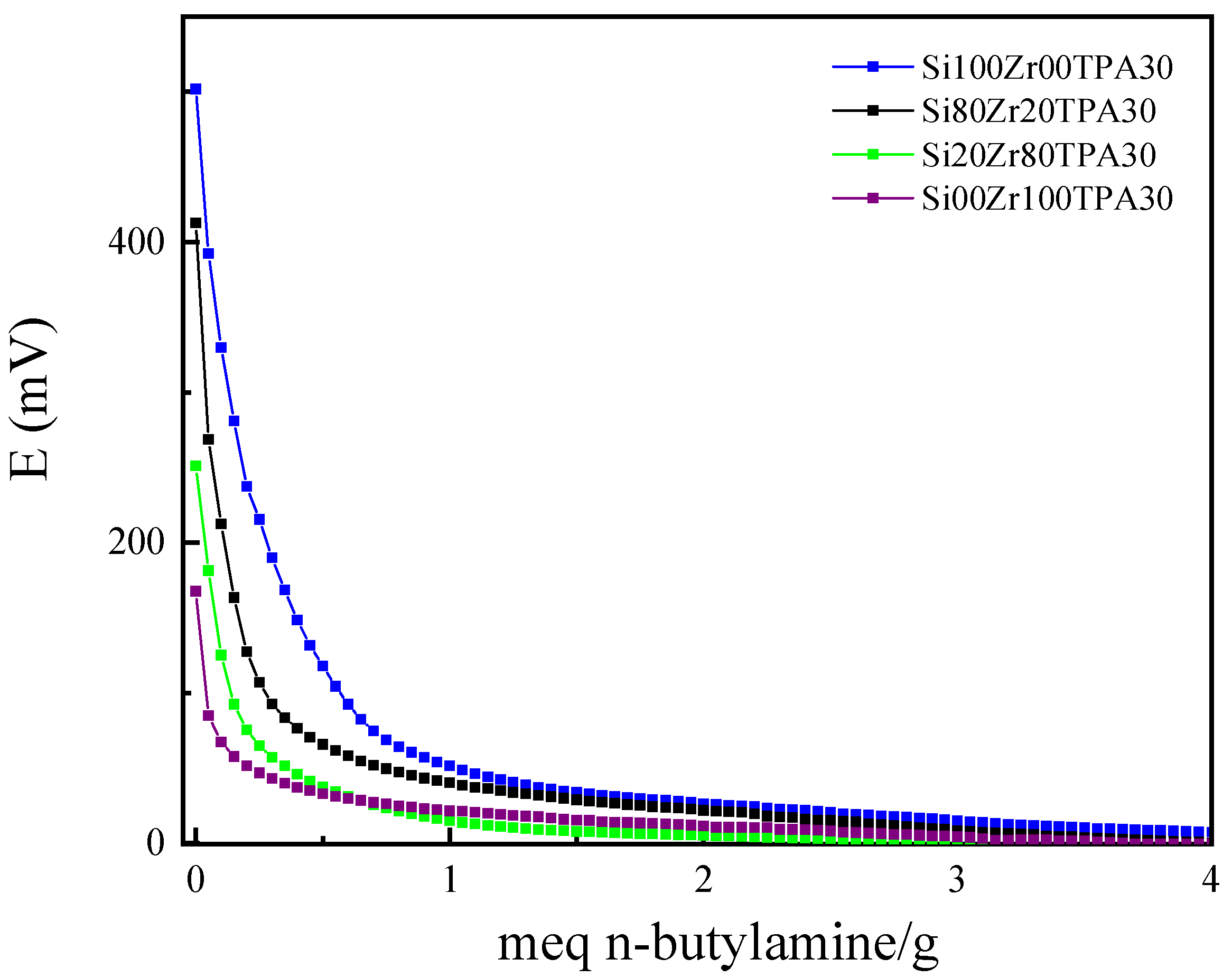
| Sample | Ei (mV) | Number of Acid Sites (Meq n-Butylamine/g) |
|---|---|---|
| Si100Zr00TPA30 | 502 | 232 |
| Si80Zr20TPA30 | 413 | 146 |
| Si20Zr80TPA30 | 251 | 68 |
| Si00Zr100TPA30 | 168 | 66 |
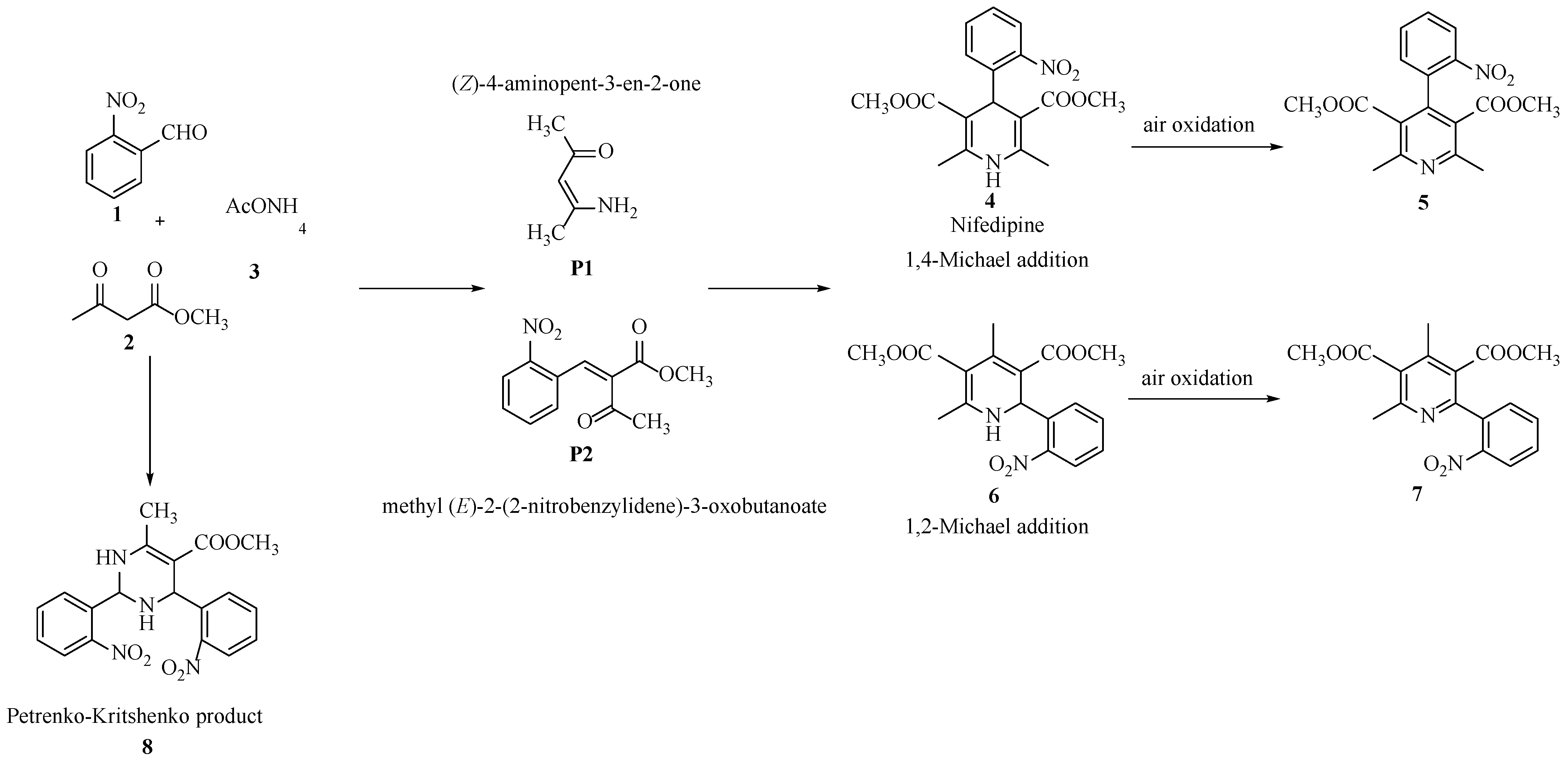
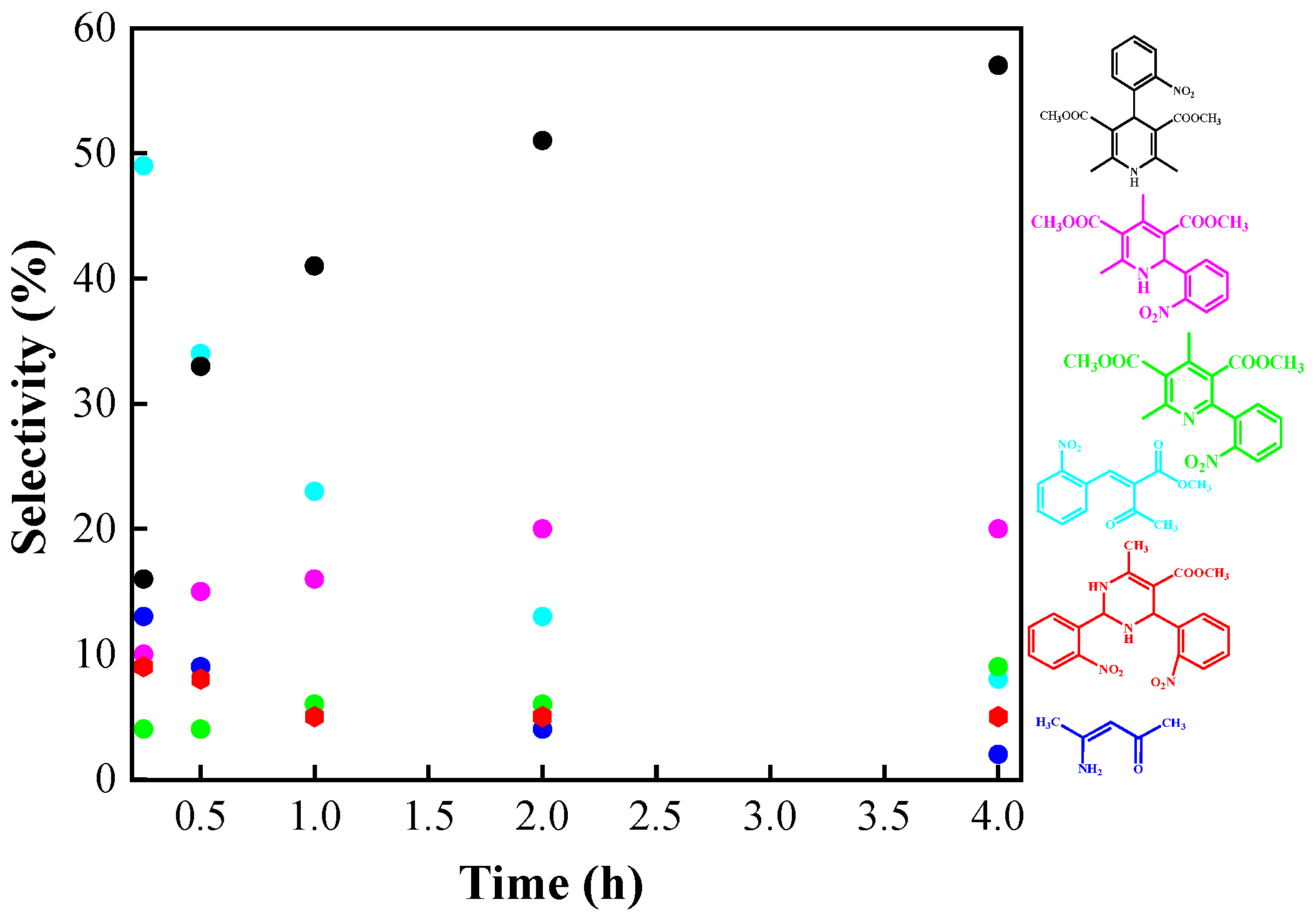
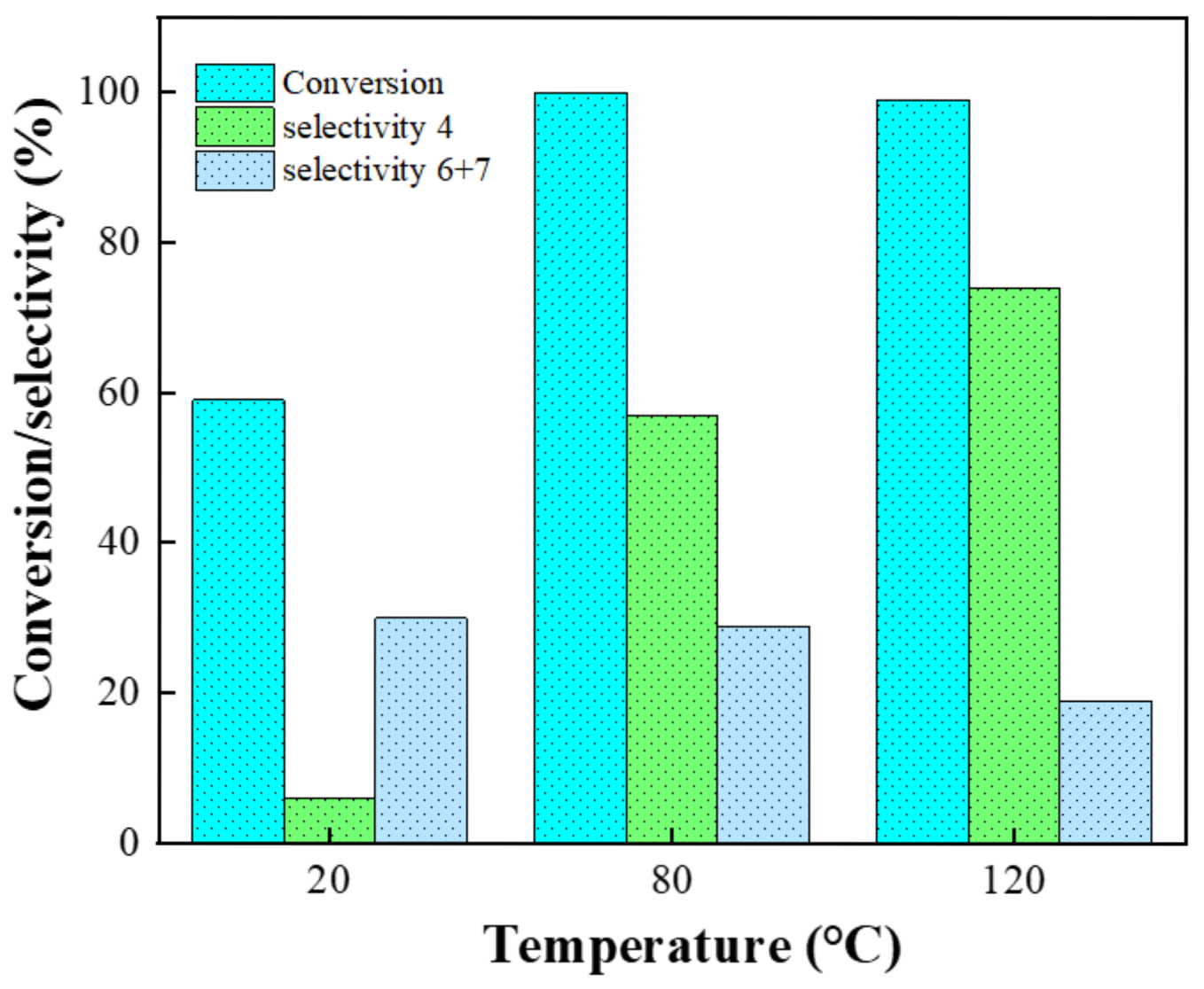
| Entry | Catalysts | NBC a (%) | NS b (%) | R c = 4 + 5/6 + 7 |
|---|---|---|---|---|
| 1 | None | 99 | 35 | 1.0 |
| 2 | Si100Zr00 | 99 | 42 | 1.0 |
| 3 | Si00Zr100 | 100 | 50 | 1.1 |
| 4 | Si100Zr0TPA30 | 100 | 48 | 1.5 |
| 5 | Si80Zr20TPA30 | 100 | 52 | 1.7 |
| 6 | Si20Zr80TPA30 | 100 | 57 | 1.9 |
| 7 | Si00Zr100TPA30 | 100 | 53 | 1.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilera, E.X.; Sathicq, Á.G.; Sosa, A.; Murguía, M.C.; Martínez, J.J.; Pizzio, L.R.; Romanelli, G.P. Mesostructured Silica–Zirconia–Tungstophosphoric Acid Composites as Catalyst in Calcium Channel Blocker Nifedipine Synthesis. Catalysts 2025, 15, 537. https://doi.org/10.3390/catal15060537
Aguilera EX, Sathicq ÁG, Sosa A, Murguía MC, Martínez JJ, Pizzio LR, Romanelli GP. Mesostructured Silica–Zirconia–Tungstophosphoric Acid Composites as Catalyst in Calcium Channel Blocker Nifedipine Synthesis. Catalysts. 2025; 15(6):537. https://doi.org/10.3390/catal15060537
Chicago/Turabian StyleAguilera, Edna X., Ángel G. Sathicq, Alexis Sosa, Marcelo C. Murguía, José J. Martínez, Luis R. Pizzio, and Gustavo P. Romanelli. 2025. "Mesostructured Silica–Zirconia–Tungstophosphoric Acid Composites as Catalyst in Calcium Channel Blocker Nifedipine Synthesis" Catalysts 15, no. 6: 537. https://doi.org/10.3390/catal15060537
APA StyleAguilera, E. X., Sathicq, Á. G., Sosa, A., Murguía, M. C., Martínez, J. J., Pizzio, L. R., & Romanelli, G. P. (2025). Mesostructured Silica–Zirconia–Tungstophosphoric Acid Composites as Catalyst in Calcium Channel Blocker Nifedipine Synthesis. Catalysts, 15(6), 537. https://doi.org/10.3390/catal15060537