
Active Sites in Carbocatalysis: Tuning Their Activity



Abstract
1. Introduction
2. Overview of Carbon-Catalyzed Reactions
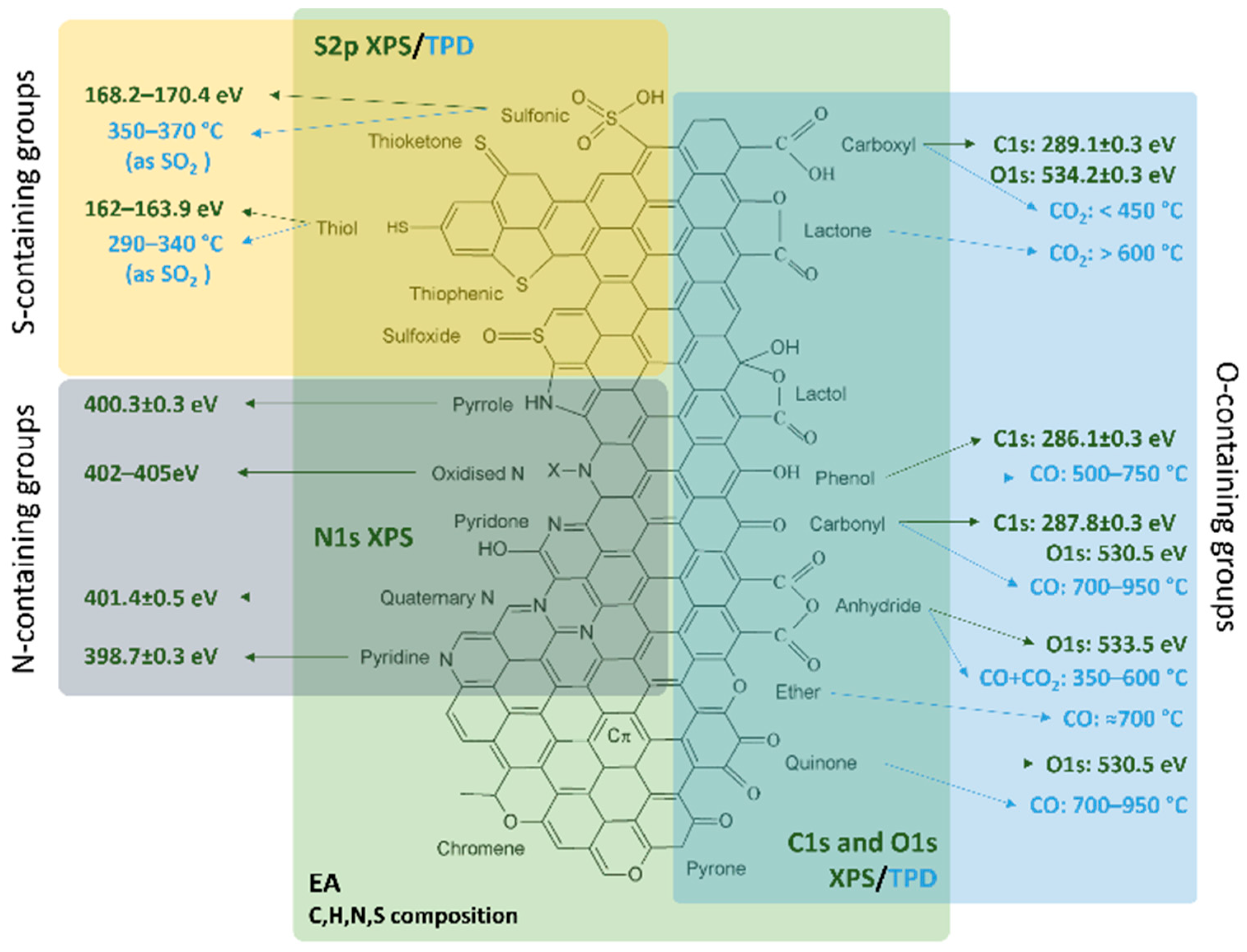
3. Case Studies
3.1. Oxidative Dehydrogenation of Hydrocarbons

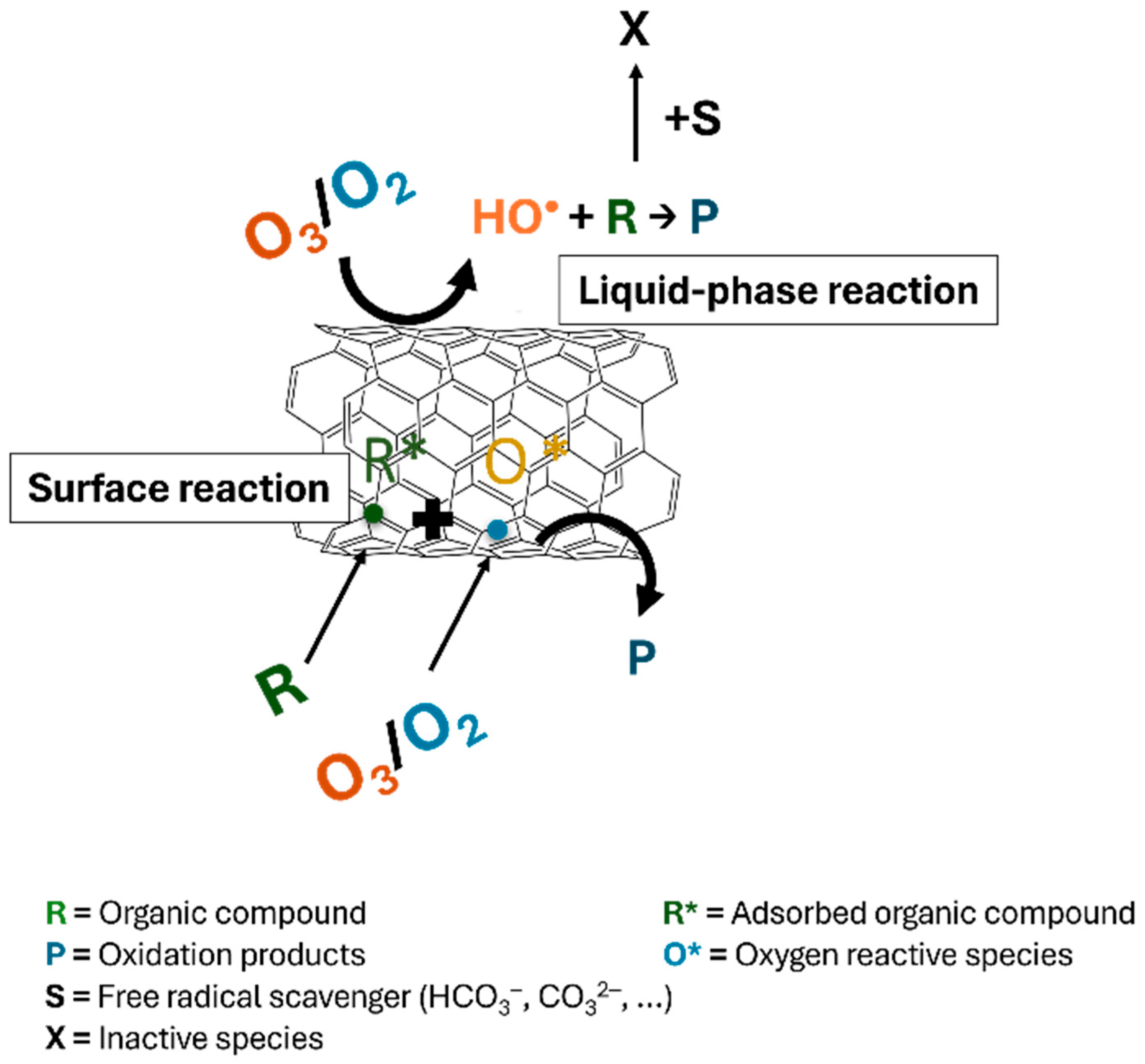
3.2. Advanced Oxidation Processes (AOPs)
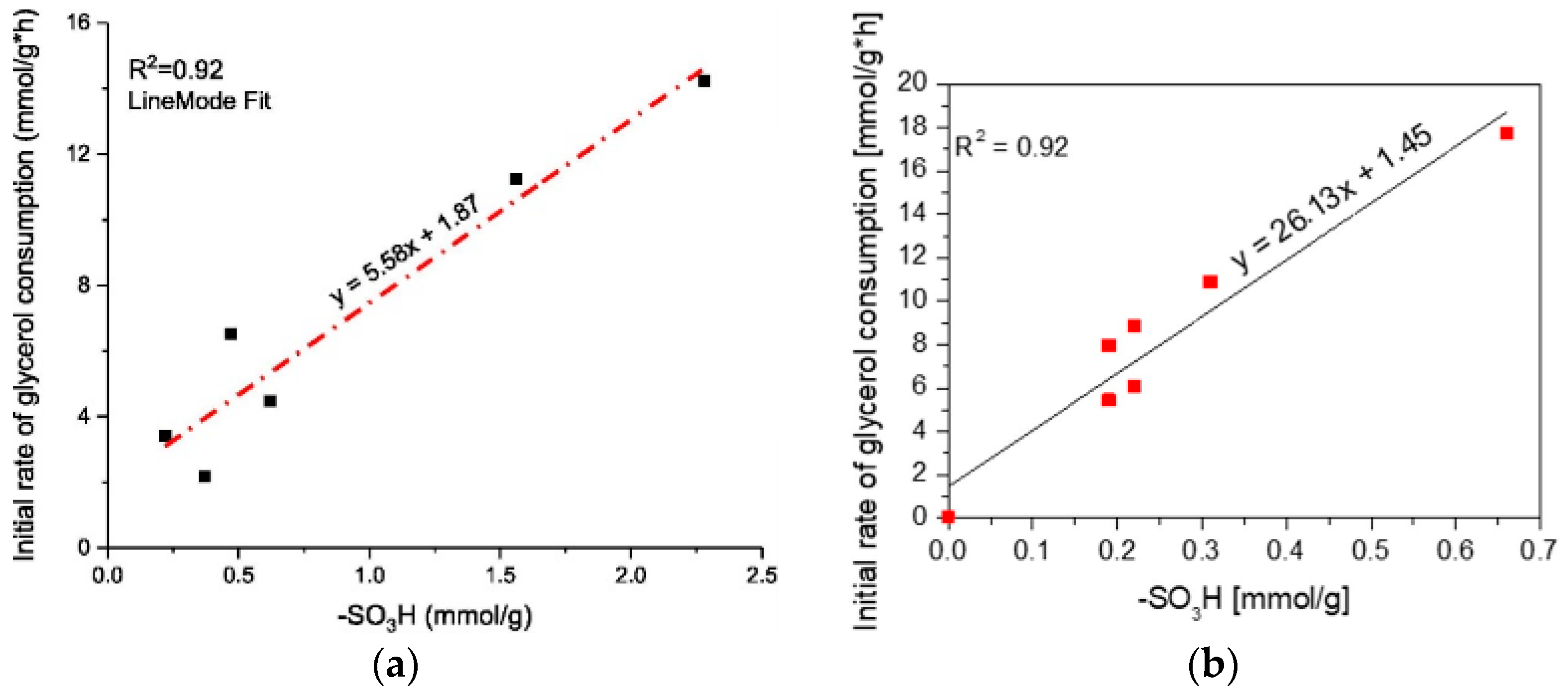
3.3. Acid Catalysis: Esterification and Etherification
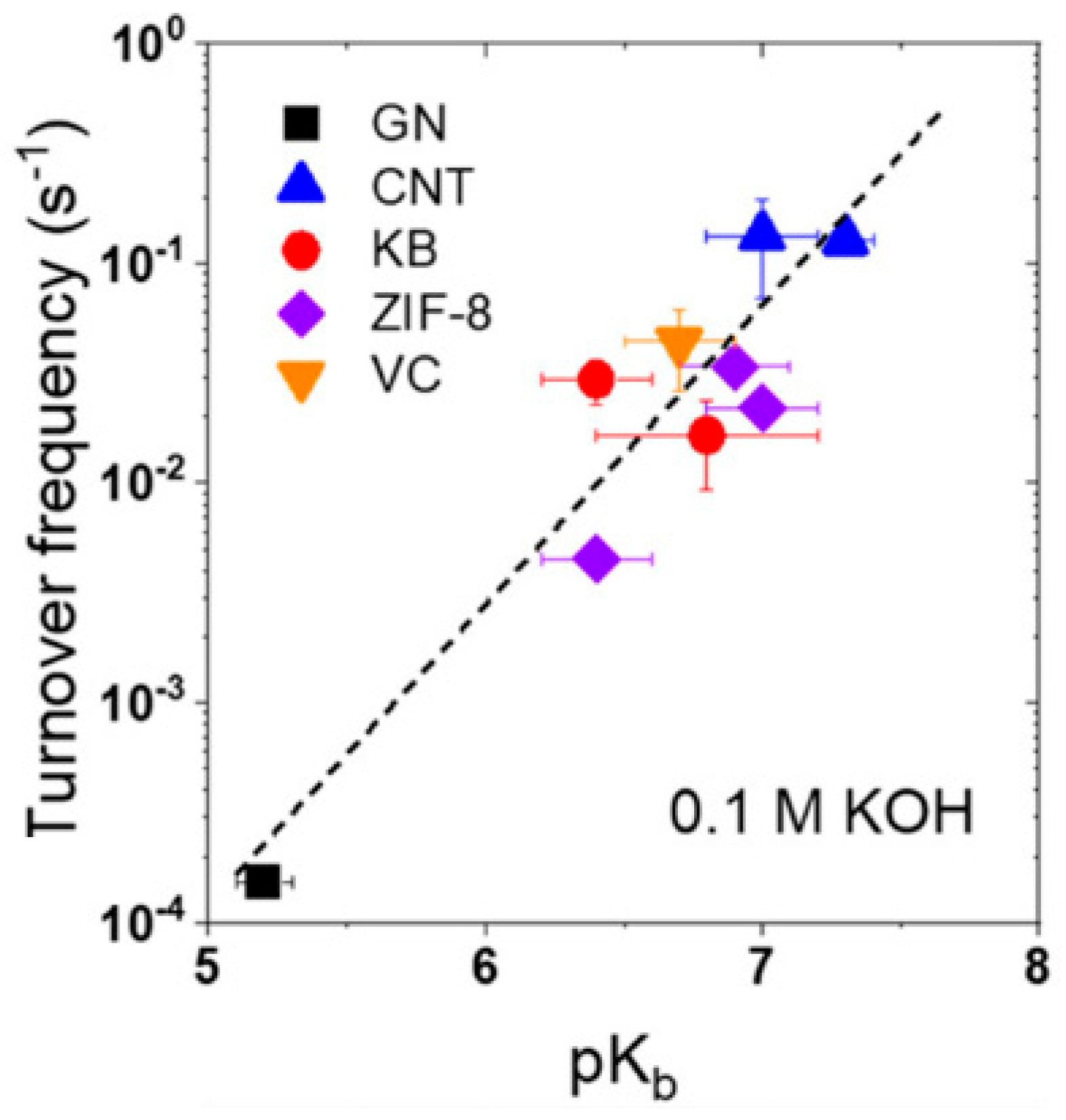
3.4. Electrocatalysis: The Oxygen Reduction Reaction (ORR)
4. Summary and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rodríguez-Reinoso, F. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R. Carbon as Catalyst. In Carbon Materials for Catalysis; Serp, P., Figueiredo, J.L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 177–217. [Google Scholar] [CrossRef]
- Coughlin, R.W. Carbon as Adsorbent and Catalyst. Ind. Eng. Chem. Prod. Res. Dev. 1969, 8, 12–23. [Google Scholar] [CrossRef]
- Su, D.S.; Perathoner, S.; Centi, G. Nanocarbons for the Development of Advanced Catalysts. Chem. Rev. 2013, 113, 5782–5816. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Nagelli, E.; Du, F.; Dai, L. Metal-Free Carbon Nanomaterials Become More Active than Metal Catalysts and Last Longer. J. Phys. Chem. Lett. 2010, 1, 2165–2173. [Google Scholar] [CrossRef]
- Su, D.S.; Wen, G.; Wu, S.; Peng, F.; Schlögl, R. Carbocatalysis in Liquid-Phase Reactions. Angew. Chem. Int. Ed. 2017, 56, 936–964. [Google Scholar] [CrossRef]
- Duan, X.; Sun, H.; Wang, S. Metal-Free Carbocatalysis in Advanced Oxidation Reactions. Acc. Chem. Res. 2018, 51, 678–687. [Google Scholar] [CrossRef]
- Chua, C.K.; Pumera, M. Carbocatalysis: The State of “Metal-Free” Catalysis. Chem.—A Eur. J. 2015, 21, 12550–12562. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R. The role of surface chemistry in catalysis with carbons. Catal. Today 2010, 150, 2–7. [Google Scholar] [CrossRef]
- Figueiredo, J.L. Functionalization of porous carbons for catalytic applications. J. Mater. Chem. A 2013, 1, 9351–9364. [Google Scholar] [CrossRef]
- Pereira, M.F.R.; Órfão, J.J.M.; Figueiredo, J.L. Oxidative dehydrogenation of ethylbenzene on activated carbon catalysts. I. Influence of surface chemical groups. Appl. Catal. A Gen. 1999, 184, 153–160. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Carrasco-Marín, F.; Parejo-Pérez, C.; López Ramón, M.V. Dehydration of methanol to dimethyl ether catalyzed by oxidized activated carbons with varying surface acidic character. Carbon 2001, 39, 869–875. [Google Scholar] [CrossRef]
- Szymański, G.S.; Rychlicki, G. Catalytic conversion of propan-2-ol on carbon catalysts. Carbon 1993, 31, 247–257. [Google Scholar] [CrossRef]
- Szymański, G.S.; Grzybek, T.; Papp, H. Influence of nitrogen surface functionalities on the catalytic activity of activated carbon in low temperature SCR of NOx with NH3. Catal. Today 2004, 90, 51–59. [Google Scholar] [CrossRef]
- Mochida, I.; Kawabuchi, Y.; Kawano, S.; Matsumura, Y.; Yoshikawa, M. High catalytic activity of pitch-based activated carbon fibres of moderate surface area for oxidation of NO to NO2 at room temperature. Fuel 1997, 76, 543–548. [Google Scholar] [CrossRef]
- Raymundo-Piñero, E.; Cazorla-Amorós, D.; Linares-Solano, A. The role of different nitrogen functional groups on the removal of SO2 from flue gases by N-doped activated carbon powders and fibres. Carbon 2003, 41, 1925–1932. [Google Scholar] [CrossRef]
- Adib, F.; Bagreev, A.; Bandosz, T.J. Adsorption/Oxidation of Hydrogen Sulfide on Nitrogen-Containing Activated Carbons. Langmuir 2000, 16, 1980–1986. [Google Scholar] [CrossRef]
- Sotowa, C.; Watanabe, Y.; Yatsunami, S.; Korai, Y.; Mochida, I. Catalytic dehydrochlorination of 1,2-dichloroethane into vinyl chloride over polyacrylonitrile-based active carbon fiber. Appl. Catal. A Gen. 1999, 180, 317–323. [Google Scholar] [CrossRef]
- Zhou, K.; Li, B.; Zhang, Q.; Huang, J.-Q.; Tian, G.-L.; Jia, J.-C.; Zhao, M.-Q.; Luo, G.-H.; Su, D.S.; Wei, F. The Catalytic Pathways of Hydrohalogenation over Metal-Free Nitrogen-Doped Carbon Nanotubes. ChemSusChem 2014, 7, 723–728. [Google Scholar] [CrossRef]
- Rocha, R.P.; Sousa, J.P.S.; Silva, A.M.T.; Pereira, M.F.R.; Figueiredo, J.L. Catalytic activity and stability of multiwalled carbon nanotubes in catalytic wet air oxidation of oxalic acid: The role of the basic nature induced by the surface chemistry. Appl. Catal. B Environ. 2011, 104, 330–336. [Google Scholar] [CrossRef]
- Gomes, H.T.; Miranda, S.M.; Sampaio, M.J.; Silva, A.M.T.; Faria, J.L. Activated carbons treated with sulphuric acid: Catalysts for catalytic wet peroxide oxidation. Catal. Today 2010, 151, 153–158. [Google Scholar] [CrossRef]
- Gonçalves, A.G.; Figueiredo, J.L.; Órfão, J.J.M.; Pereira, M.F.R. Influence of the surface chemistry of multi-walled carbon nanotubes on their activity as ozonation catalysts. Carbon 2010, 48, 4369–4381. [Google Scholar] [CrossRef]
- Budarin, V.L.; Clark, J.H.; Luque, R.; Macquarrie, D.J. Versatile mesoporous carbonaceous materials for acid catalysis. Chem. Commun. 2007, 634–636. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, H.; Zhao, Y.; Shen, J. Preparation of a novel sulfonated carbon catalyst for the etherification of isopentene with methanol to produce tert-amyl methyl ether. Catal. Commun. 2010, 11, 824–828. [Google Scholar] [CrossRef]
- Liang, X.; Li, C.; Qi, C. Novel carbon-based strong acid catalyst from starch and its catalytic activities for acetalization. J. Mater. Sci. 2011, 46, 5345–5349. [Google Scholar] [CrossRef]
- Onda, A.; Ochi, T.; Yanagisawa, K. Hydrolysis of Cellulose Selectively into Glucose Over Sulfonated Activated-Carbon Catalyst Under Hydrothermal Conditions. Top. Catal. 2009, 52, 801–807. [Google Scholar] [CrossRef]
- Matos, I.; Neves, P.D.; Castanheiro, J.E.; Perez-Mayoral, E.; Martin-Aranda, R.; Duran-Valle, C.; Vital, J.; Botelho do Rego, A.M.; Fonseca, I.M. Mesoporous carbon as an efficient catalyst for alcoholysis and aminolysis of epoxides. Appl. Catal. A Gen. 2012, 439-440, 24–30. [Google Scholar] [CrossRef]
- van Dommele, S.; de Jong, K.P.; Bitter, J.H. Nitrogen-containing carbon nanotubes as solid base catalysts. Chem. Commun. 2006, 46, 4859–4861. [Google Scholar] [CrossRef]
- Guo, D.; Shibuya, R.; Akiba, C.; Saji, S.; Kondo, T.; Nakamura, J. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 2016, 351, 361–365. [Google Scholar] [CrossRef]
- Boehm, H.P. Some aspects of the surface chemistry of carbon blacks and other carbons. Carbon 1994, 32, 759–769. [Google Scholar] [CrossRef]
- Boehm, H.P. Surface oxides on carbon and their analysis: A critical assessment. Carbon 2002, 40, 145–149. [Google Scholar] [CrossRef]
- Menéndez, J.A.; Phillips, J.; Xia, B.; Radovic, L.R. On the Modification and Characterization of Chemical Surface Properties of Activated Carbon: In the Search of Carbons with Stable Basic Properties. Langmuir 1996, 12, 4404–4410. [Google Scholar] [CrossRef]
- Montes-Morán, M.A.; Suárez, D.; Menéndez, J.A.; Fuente, E. On the nature of basic sites on carbon surfaces: An overview. Carbon 2004, 42, 1219–1225. [Google Scholar] [CrossRef]
- Boehm, H.-P. Catalytic Properties of Nitrogen-Containing Carbons. In Carbon Materials for Catalysis; Serp, P., Figueiredo, J.L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 219–265. [Google Scholar] [CrossRef]
- Gupta, P.; Paul, S. Solid acids: Green alternatives for acid catalysis. Catal. Today 2014, 236, 153–170. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfão, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Rocha, R.P.; Pereira, M.F.R.; Figueiredo, J.L. Characterisation of the surface chemistry of carbon materials by temperature-programmed desorption: An assessment. Catal. Today 2023, 418, 114136. [Google Scholar] [CrossRef]
- Kiciński, W.; Szala, M.; Bystrzejewski, M. Sulfur-doped porous carbons: Synthesis and applications. Carbon 2014, 68, 1–32. [Google Scholar] [CrossRef]
- Rocha, R.P.; Soares, O.S.G.P.; Figueiredo, J.L.; Pereira, M.F.R. Tuning CNT Properties for Metal-Free Environmental Catalytic Applications. C 2016, 2, 17. [Google Scholar] [CrossRef]
- Pels, J.R.; Kapteijn, F.; Moulijn, J.A.; Zhu, Q.; Thomas, K.M. Evolution of nitrogen functionalities in carbonaceous materials during pyrolysis. Carbon 1995, 33, 1641–1653. [Google Scholar] [CrossRef]
- Jansen, R.J.J.; van Bekkum, H. XPS of nitrogen-containing functional groups on activated carbon. Carbon 1995, 33, 1021–1027. [Google Scholar] [CrossRef]
- Zhou, J.-H.; Sui, Z.-J.; Zhu, J.; Li, P.; Chen, D.; Dai, Y.-C.; Yuan, W.-K. Characterization of surface oxygen complexes on carbon nanofibers by TPD, XPS and FT-IR. Carbon 2007, 45, 785–796. [Google Scholar] [CrossRef]
- Kundu, S.; Xia, W.; Busser, W.; Becker, M.; Schmidt, D.A.; Havenith, M.; Muhler, M. The formation of nitrogen-containing functional groups on carbon nanotube surfaces: A quantitative XPS and TPD study. Phys. Chem. Chem. Phys. 2010, 12, 4351–4359. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Castilla, C.; Ferro-Garcia, M.A.; Joly, J.P.; Bautista-Toledo, I.; Carrasco-Marin, F.; Rivera-Utrilla, J. Activated Carbon Surface Modifications by Nitric Acid, Hydrogen Peroxide, and Ammonium Peroxydisulfate Treatments. Langmuir 1995, 11, 4386–4392. [Google Scholar] [CrossRef]
- Newcombe, G.; Hayes, R.; Drikas, M. Granular activated carbon: Importance of surface properties in the adsorption of naturally occurring organics. Colloids Surf. A Physicochem. Eng. Asp. 1993, 78, 65–71. [Google Scholar] [CrossRef]
- Iwasawa, Y.; Nobe, H.; Ogasawara, S. Reaction mechanism for styrene synthesis over polynaphthoquinone. J. Catal. 1973, 31, 444–449. [Google Scholar] [CrossRef]
- Emig, G.; Hofmann, H. Action of zirconium phosphate as a catalyst for the oxydehydrogenation of ethylbenzene to styrene. J. Catal. 1983, 84, 15–26. [Google Scholar] [CrossRef]
- Schraut, A.; Emig, G.; Sockel, H.G. Composition and structure of active coke in the oxydehydrogenation of ethylbenzene. Appl. Catal. 1987, 29, 311–326. [Google Scholar] [CrossRef]
- Schraut, A.; Emig, G.; Hofmann, H. Kinetic investigations of the oxydehydrogenation of ethylbenzene. J. Catal. 1988, 112, 221–228. [Google Scholar] [CrossRef]
- de Jesús Díaz Velásquez, J.; Suárez, L.M.C.; Figueiredo, J.L. Oxidative dehydrogenation of isobutane over activated carbon catalysts. Appl. Catal. A Gen. 2006, 311, 51–57. [Google Scholar] [CrossRef]
- Pelech, I.; Soares, O.S.G.P.; Pereira, M.F.R.; Figueiredo, J.L. Oxidative dehydrogenation of isobutane on carbon xerogel catalysts. Catal. Today 2015, 249, 176–183. [Google Scholar] [CrossRef]
- Oturan, M.A.; and Aaron, J.-J. Advanced Oxidation Processes in Water/Wastewater Treatment: Principles and Applications. A Review. Crit. Rev. Environ. Sci. Technol. 2014, 44, 2577–2641. [Google Scholar] [CrossRef]
- Gao, J.; Qin, T.; Wacławek, S.; Duan, X.; Huang, Y.; Liu, H.; Dionysiou, D.D. The application of advanced oxidation processes (AOPs) to treat unconventional water for fit-for-purpose reuse. Curr. Opin. Chem. Eng. 2023, 42, 100974. [Google Scholar] [CrossRef]
- Esplugas, S.; Giménez, J.; Contreras, S.; Pascual, E.; Rodríguez, M. Comparison of different advanced oxidation processes for phenol degradation. Water Res. 2002, 36, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Stüber, F.; Font, J.; Fortuny, A.; Bengoa, C.; Eftaxias, A.; Fabregat, A. Carbon materials and catalytic wet air oxidation of organic pollutants in wastewater. Top. Catal. 2005, 33, 3–50. [Google Scholar] [CrossRef]
- Rocha, R.P.; Silva, A.M.T.; Romero, S.M.M.; Pereira, M.F.R.; Figueiredo, J.L. The role of O- and S-containing surface groups on carbon nanotubes for the elimination of organic pollutants by catalytic wet air oxidation. Appl. Catal. B Environ. 2014, 147, 314–321. [Google Scholar] [CrossRef]
- Chen, H.; Yang, G.; Feng, Y.; Shi, C.; Xu, S.; Cao, W.; Zhang, X. Biodegradability enhancement of coking wastewater by catalytic wet air oxidation using aminated activated carbon as catalyst. Chem. Eng. J. 2012, 198–199, 45–51. [Google Scholar] [CrossRef]
- Yang, G.; Chen, H.; Qin, H.; Feng, Y. Amination of activated carbon for enhancing phenol adsorption: Effect of nitrogen-containing functional groups. Appl. Surf. Sci. 2014, 293, 299–305. [Google Scholar] [CrossRef]
- Restivo, J.; Rocha, R.P.; Silva, A.M.T.; Órfão, J.J.M.; Pereira, M.F.R.; Figueiredo, J.L. Catalytic performance of heteroatom-modified carbon nanotubes in advanced oxidation processes. Cuihua Xuebao/Chin. J. Catal. 2014, 35, 896–905. [Google Scholar] [CrossRef]
- Rocha, R.P.; Restivo, J.; Sousa, J.P.S.; Órfão, J.J.M.; Pereira, M.F.R.; Figueiredo, J.L. Nitrogen-doped carbon xerogels as catalysts for advanced oxidation processes. Catal. Today 2015, 241, 73–79. [Google Scholar] [CrossRef]
- Rocha, R.P.; Gonçalves, A.G.; Pastrana-Martínez, L.M.; Bordoni, B.C.; Soares, O.S.G.P.; Órfão, J.J.M.; Faria, J.L.; Figueiredo, J.L.; Silva, A.M.T.; Pereira, M.F.R. Nitrogen-doped graphene-based materials for advanced oxidation processes. Catal. Today 2015, 249, 192–198. [Google Scholar] [CrossRef]
- Santiago, M.; Stüber, F.; Fortuny, A.; Fabregat, A.; Font, J. Modified activated carbons for catalytic wet air oxidation of phenol. Carbon 2005, 43, 2134–2145. [Google Scholar] [CrossRef]
- Pollak, E.; Salitra, G.; Soffer, A.; Aurbach, D. On the reaction of oxygen with nitrogen-containing and nitrogen-free carbons. Carbon 2006, 44, 3302–3307. [Google Scholar] [CrossRef]
- Santos, D.F.M.; Soares, O.S.G.P.; Silva, A.M.T.; Figueiredo, J.L.; Pereira, M.F.R. Catalytic wet oxidation of organic compounds over N-doped carbon nanotubes in batch and continuous operation. Appl. Catal. B Environ. 2016, 199, 361–371. [Google Scholar] [CrossRef]
- Peng, F.; Zhang, L.; Wang, H.; Lv, P.; Yu, H. Sulfonated carbon nanotubes as a strong protonic acid catalyst. Carbon 2005, 43, 2405–2408. [Google Scholar] [CrossRef]
- Yu, H.; Jin, Y.; Li, Z.; Peng, F.; Wang, H. Synthesis and characterization of sulfonated single-walled carbon nanotubes and their performance as solid acid catalyst. J. Solid State Chem. 2008, 181, 432–438. [Google Scholar] [CrossRef]
- Giménez, J.; Costa, J.; Cervera-March, S. Catalysis by ion-exchange sulfonated resins: Comparative study of gel and macroporous types and influence of divinylbenzene concentration. Appl. Catal. 1987, 31, 221–234. [Google Scholar] [CrossRef]
- Rocha, R.P.; Pereira, M.F.R.; Figueiredo, J.L. Carbon as a catalyst: Esterification of acetic acid with ethanol. Catal. Today 2013, 218-219, 51–56. [Google Scholar] [CrossRef]
- Hara, M.; Yoshida, T.; Takagaki, A.; Takata, T.; Kondo, J.N.; Hayashi, S.; Domen, K. A Carbon Material as a Strong Protonic Acid. Angew. Chem. Int. Ed. 2004, 43, 2955–2958. [Google Scholar] [CrossRef]
- Goscianska, J.; Malaika, A. A facile post-synthetic modification of ordered mesoporous carbon to get efficient catalysts for the formation of acetins. Catal. Today 2020, 357, 84–93. [Google Scholar] [CrossRef]
- Malaika, A.; Ptaszyńska, K.; Morawa Eblagon, K.; Pereira, M.F.R.; Figueiredo, J.L.; Kozłowski, M. Solid acid carbon catalysts for sustainable production of biofuel enhancers via transesterification of glycerol with ethyl acetate. Fuel 2021, 304, 121381. [Google Scholar] [CrossRef]
- Malaika, A.; Ptaszyńska, K.; Kozłowski, M. Conversion of renewable feedstock to bio-carbons dedicated for the production of green fuel additives from glycerol. Fuel 2021, 288, 119609. [Google Scholar] [CrossRef]
- Ptaszyńska, K.; Eblagon, K.M.; Malaika, A.; Figueiredo, J.L.; Kozłowski, M. The role of mechanochemical treatment of carbon nanotubes in promoting glycerol etherification. Catal. Sci. Technol. 2024, 14, 3184–3200. [Google Scholar] [CrossRef]
- Ptaszyńska, K.; Malaika, A.; Morawa Eblagon, K.; Figueiredo, J.L.; Kozłowski, M. Promoting Effect of Ball Milling on the Functionalization and Catalytic Performance of Carbon Nanotubes in Glycerol Etherification. Molecules 2024, 29, 1623. [Google Scholar] [CrossRef] [PubMed]
- Rocha, I.M.; Soares, O.S.G.P.; Figueiredo, J.L.; Freire, C.; Pereira, M.F.R. Bifunctionality of the pyrone functional group in oxidized carbon nanotubes towards oxygen reduction reaction. Catal. Sci. Technol. 2017, 7, 1868–1879. [Google Scholar] [CrossRef]
- Ma, R.; Lin, G.; Zhou, Y.; Liu, Q.; Zhang, T.; Shan, G.; Yang, M.; Wang, J. A review of oxygen reduction mechanisms for metal-free carbon-based electrocatalysts. npj Comput. Mater. 2019, 5, 78. [Google Scholar] [CrossRef]
- Tao, L.; Wang, Q.; Dou, S.; Ma, Z.; Huo, J.; Wang, S.; Dai, L. Edge-rich and dopant-free graphene as a highly efficient metal-free electrocatalyst for the oxygen reduction reaction. Chem. Commun. 2016, 52, 2764–2767. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, L.; Sun, T.; Zhao, J.; Lyu, Z.; Zhuo, O.; Wang, X.; Wu, Q.; Ma, J.; Hu, Z. Significant Contribution of Intrinsic Carbon Defects to Oxygen Reduction Activity. ACS Catal. 2015, 5, 6707–6712. [Google Scholar] [CrossRef]
- Rocha, I.M.; Soares, O.S.G.P.; Fernandes, D.M.; Freire, C.; Figueiredo, J.L.; Pereira, M.F.R. N-doped Carbon Nanotubes for the Oxygen Reduction Reaction in Alkaline Medium: Synergistic Relationship between Pyridinic and Quaternary Nitrogen. ChemistrySelect 2016, 1, 2522–2530. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Origin of the Electrocatalytic Oxygen Reduction Activity of Graphene-Based Catalysts: A Roadmap to Achieve the Best Performance. J. Am. Chem. Soc. 2014, 136, 4394–4403. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, F.; Song, Z.; Zhang, L. Oxygen Reduction Reaction on Pyridinic Nitrogen-Functionalized Carbon: Active Site Quantification and Effects of Lewis Basicity. ACS Catal. 2025, 15, 296–309. [Google Scholar] [CrossRef]
- Chakraborty, A.; Devivaraprasad, R.; Bera, B.; Neergat, M. Electrochemical estimation of the active site density on metal-free nitrogen-doped carbon using catechol as an adsorbate. Phys. Chem. Chem. Phys. 2017, 19, 25414–25422. [Google Scholar] [CrossRef]
- Jaouen, F.; Dodelet, J.-P. Average turn-over frequency of O2 electro-reduction for Fe/N/C and Co/N/C catalysts in PEFCs. Electrochim. Acta 2007, 52, 5975–5984. [Google Scholar] [CrossRef]
- Wang, J.; Kong, H.; Zhang, J.; Hao, Y.; Shao, Z.; Ciucci, F. Carbon-based electrocatalysts for sustainable energy applications. Prog. Mater. Sci. 2021, 116, 100717. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, L.; Xia, Z.; Roy, A.; Chang, D.W.; Baek, J.-B.; Dai, L. BCN Graphene as Efficient Metal-Free Electrocatalyst for the Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 2012, 51, 4209–4212. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Jiao, Y.; Jaroniec, M.; Qiao, S.Z. Sulfur and Nitrogen Dual-Doped Mesoporous Graphene Electrocatalyst for Oxygen Reduction with Synergistically Enhanced Performance. Angew. Chem. Int. Ed. 2012, 51, 11496–11500. [Google Scholar] [CrossRef]
- Zhao, Z.; Xia, Z. Design Principles for Dual-Element-Doped Carbon Nanomaterials as Efficient Bifunctional Catalysts for Oxygen Reduction and Evolution Reactions. ACS Catal. 2016, 6, 1553–1558. [Google Scholar] [CrossRef]
- Goldsmith, B.R.; Esterhuizen, J.; Liu, J.-X.; Bartel, C.J.; Sutton, C. Machine learning for heterogeneous catalyst design and discovery. AIChE J. 2018, 64, 2311–2323. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactions | Surface Chemistry/Active Sites [Reference] |
|---|---|
| Gas phase | |
| Oxidative dehydrogenation | Carbonyl/Quinone groups [11] |
| Dehydration of alcohols | Carboxylic acids [12] |
| Dehydrogenation of alcohols | Lewis acids and basic sites [13] |
| NOx reduction (SCR with NH3) | Acidic surface oxides (carboxylic and lactonic) + basic sites (N6) [14] |
| NO oxidation | Basic sites [15] |
| SO2 oxidation | Basic sites, N6 [16] |
| H2S oxidation | Basic N groups [17] |
| Dehydrohalogenation | Pyridinic nitrogen (N6) [18] |
| Hydrohalogenation | Quaternary nitrogen (NQ) [19] |
| Liquid phase | |
| Advanced Oxidation Processes (CWAO; WPO; Ozonation) | Basic sites [20,21,22] |
| Esterification | Sulfonic acid groups [23] |
| Etherification | Sulfonic acid groups [24] |
| Alkylation | Sulfonic acid groups [23] |
| Acylation | Sulfonic acid groups [23] |
| Acetalization | Sulfonic acid groups [25] |
| Hydrolysis of cellulose | Sulfonic acid groups [26] |
| Alcoholysis of epoxides | Sulfonic acid groups [27] |
| Knoevenagel condensation | Pyridinic nitrogen (N6) [28] |
| Oxygen reduction (ORR) | C atoms next to pyridinic N [29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, R.P.; Figueiredo, J.L. Active Sites in Carbocatalysis: Tuning Their Activity. Catalysts 2025, 15, 443. https://doi.org/10.3390/catal15050443
Rocha RP, Figueiredo JL. Active Sites in Carbocatalysis: Tuning Their Activity. Catalysts. 2025; 15(5):443. https://doi.org/10.3390/catal15050443
Chicago/Turabian StyleRocha, Raquel Pinto, and José Luís Figueiredo. 2025. "Active Sites in Carbocatalysis: Tuning Their Activity" Catalysts 15, no. 5: 443. https://doi.org/10.3390/catal15050443
APA StyleRocha, R. P., & Figueiredo, J. L. (2025). Active Sites in Carbocatalysis: Tuning Their Activity. Catalysts, 15(5), 443. https://doi.org/10.3390/catal15050443