
Improving the Catalytic Selectivity of Reverse Water–Gas Shift Reaction Catalyzed by Ru/CeO2 Through the Addition of Yttrium Oxide

 , ,
, ,
Abstract

1. Introduction
2. Results and Discussion
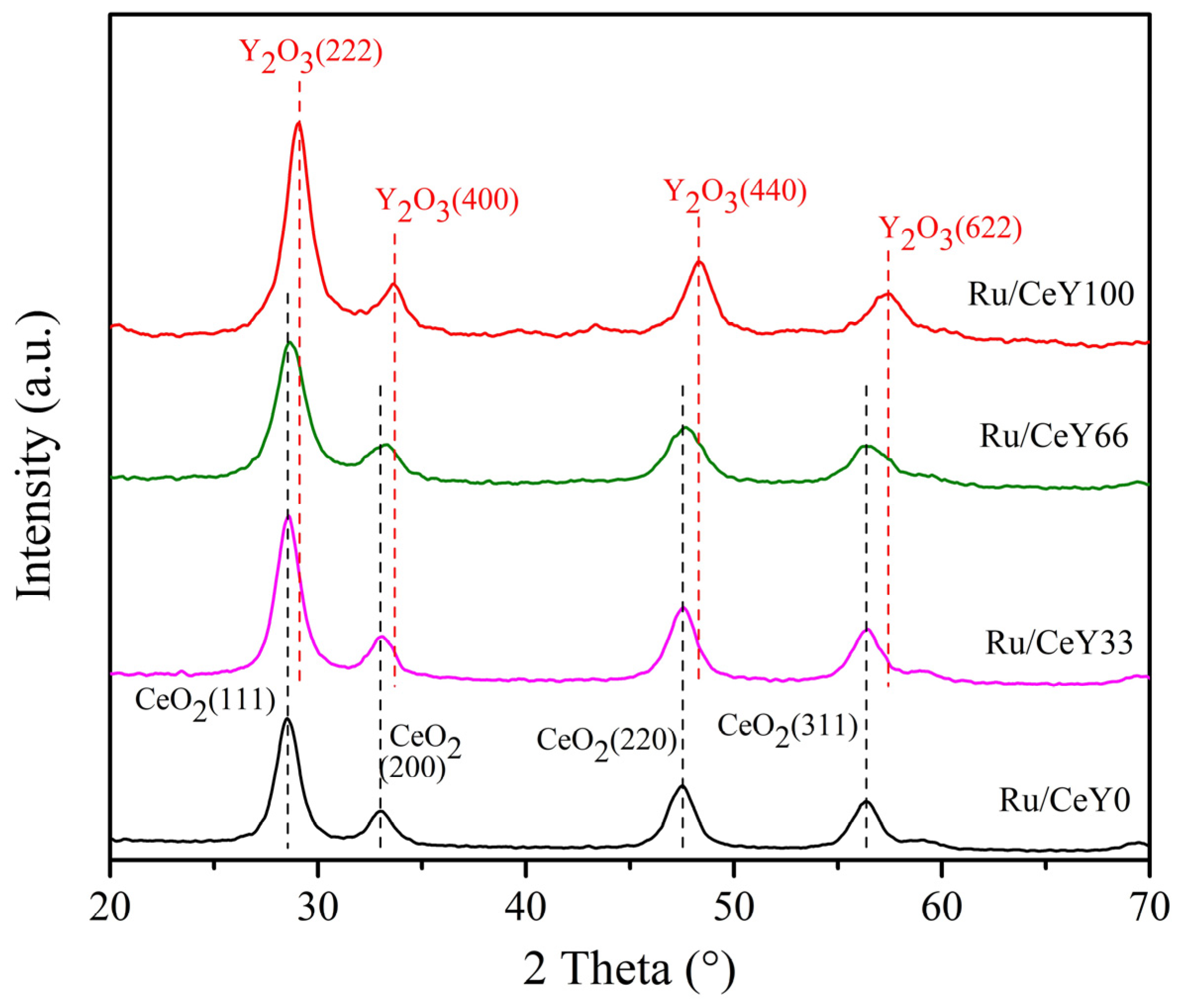
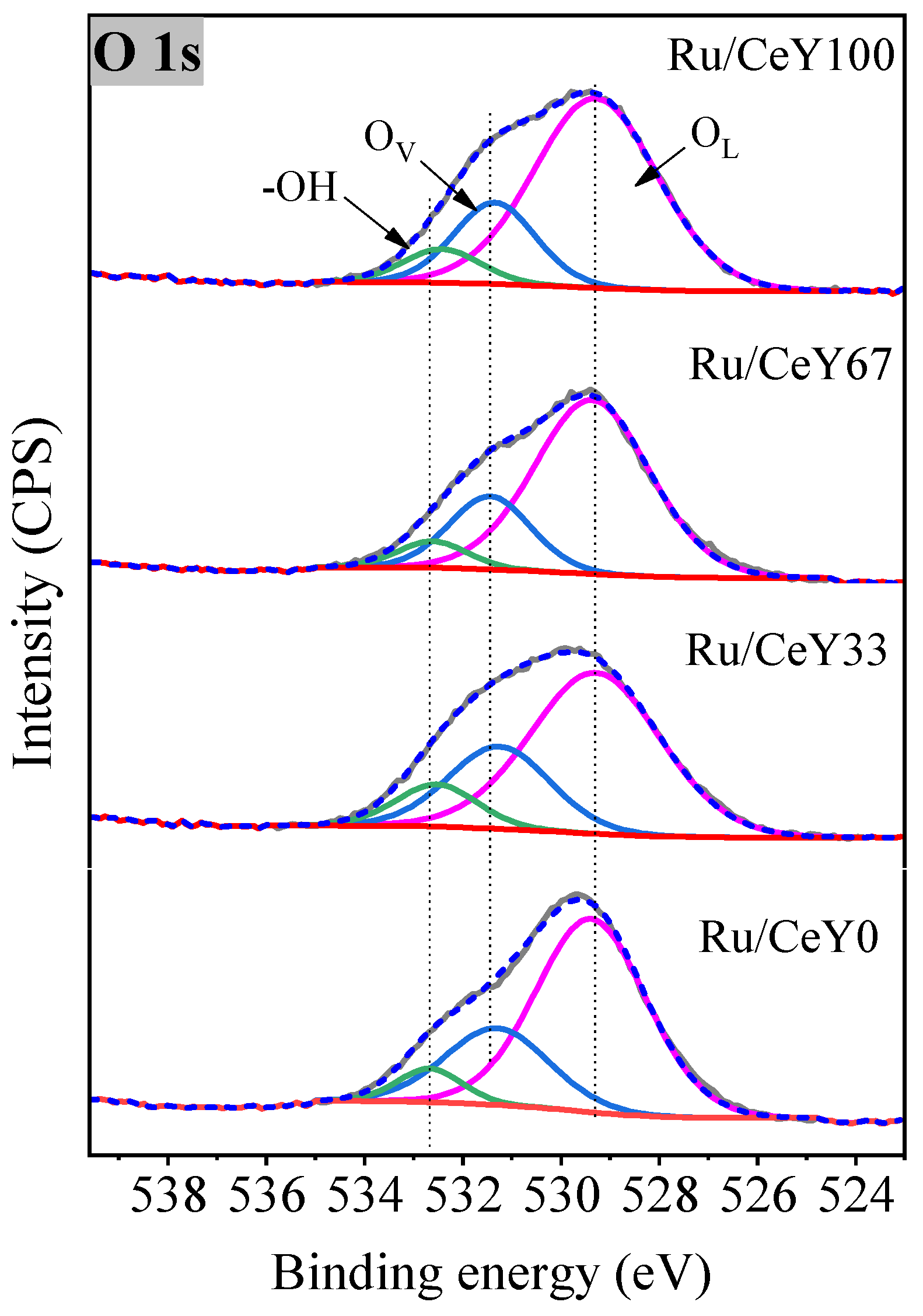
2.1. Catalysts Characterization
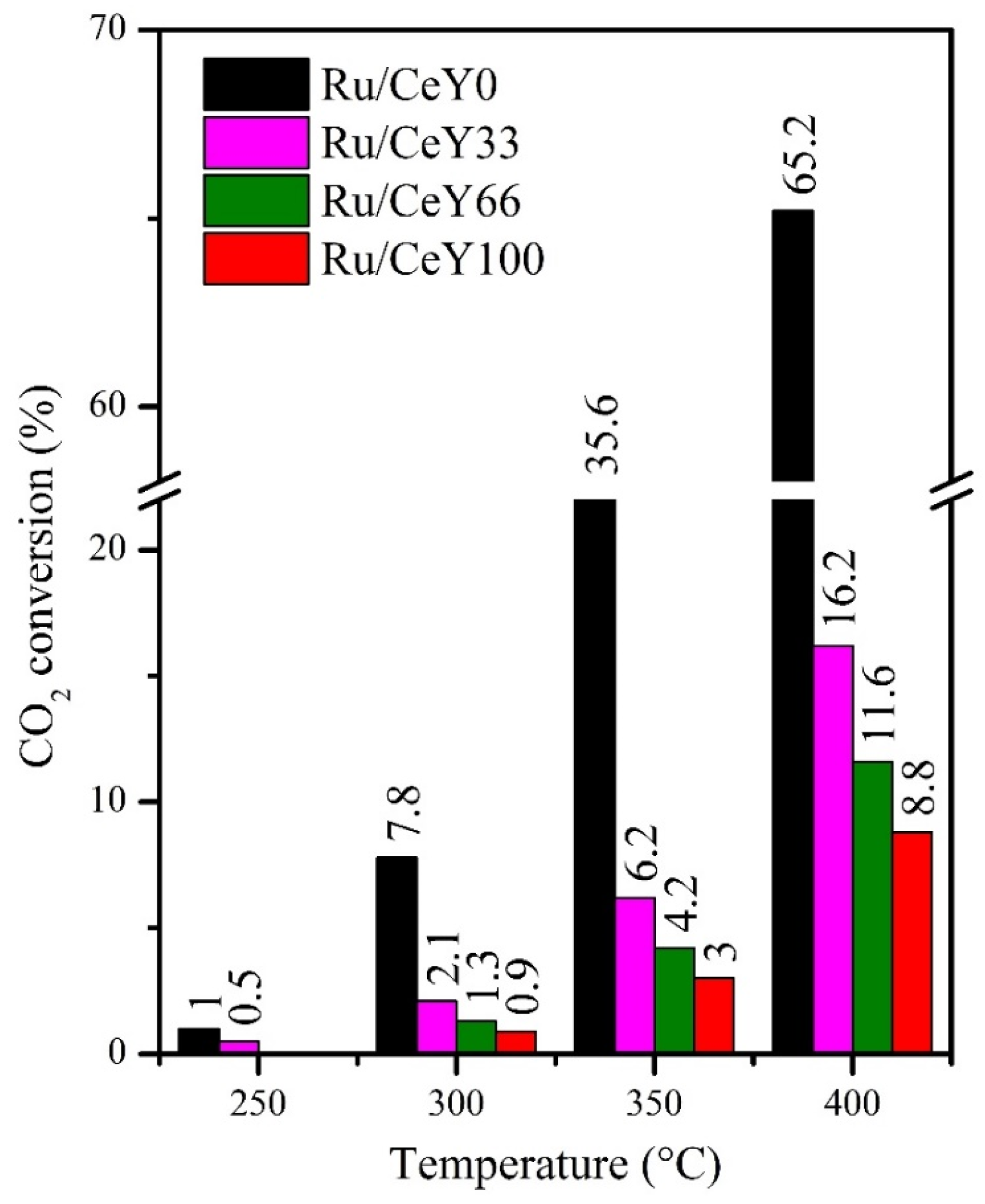
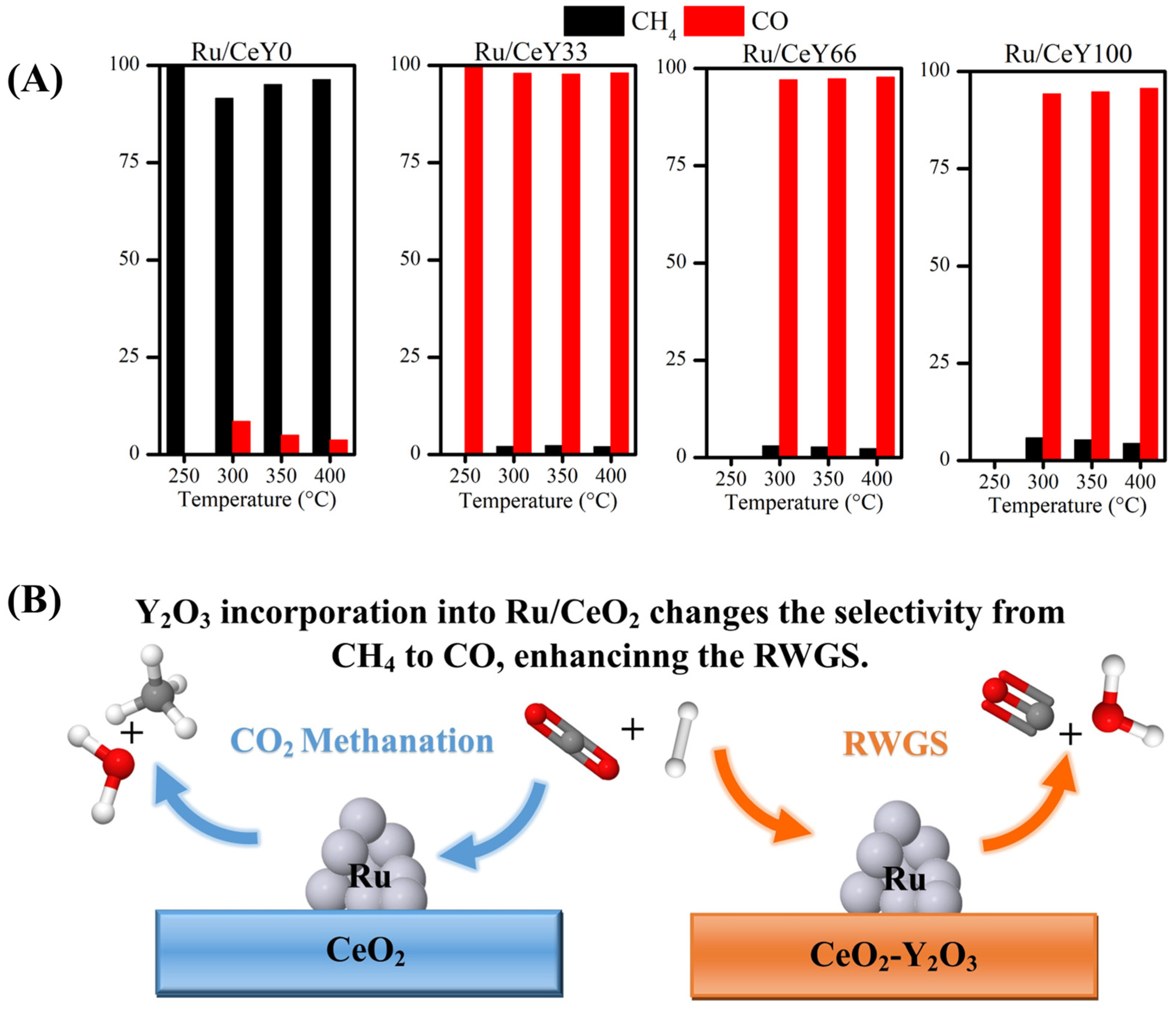
2.2. Catalytic Activity
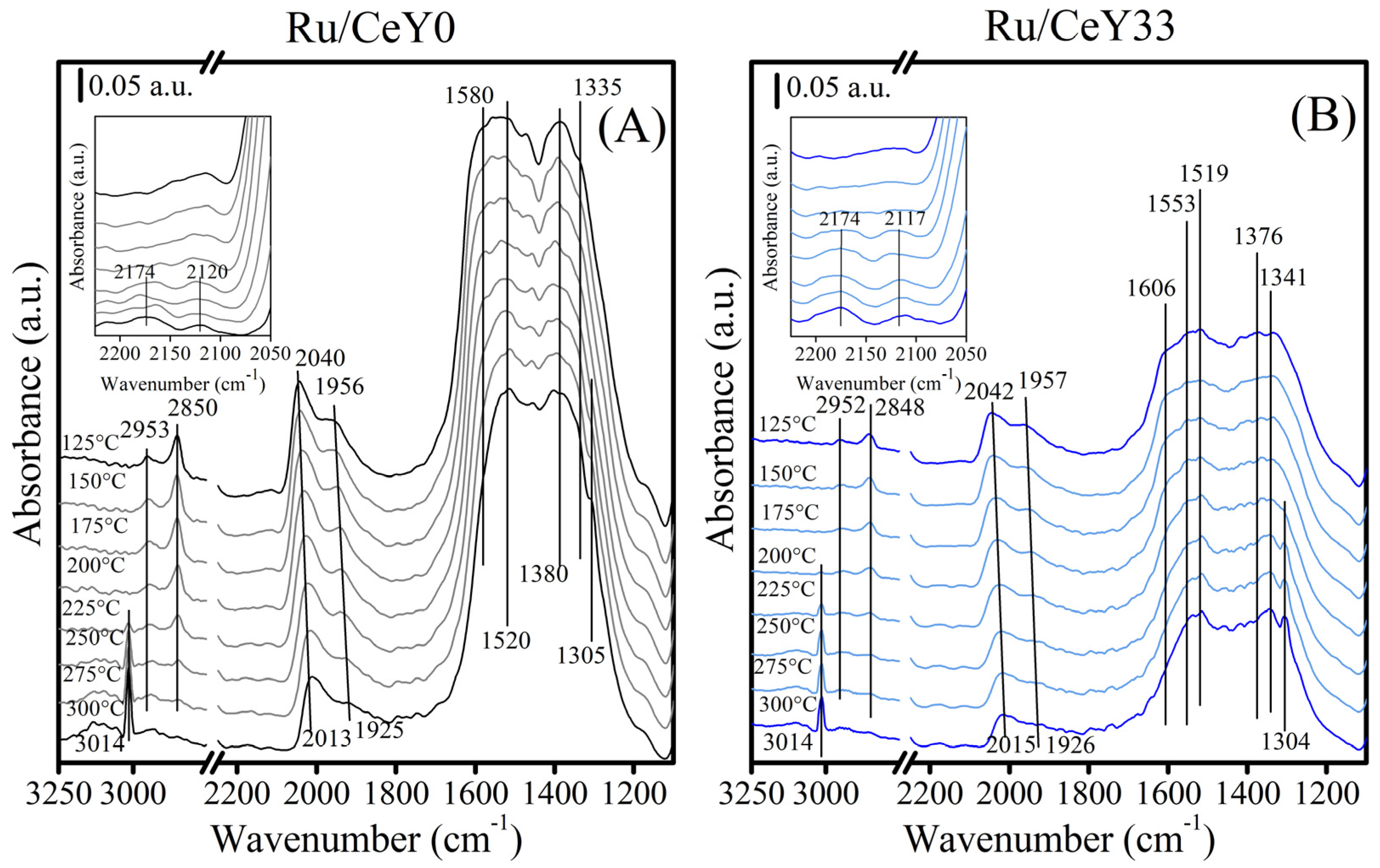
2.3. FTIR Characterization Under Reaction Conditions
2.3.1. Ru/CeY0
2.3.2. Ru/CeY33
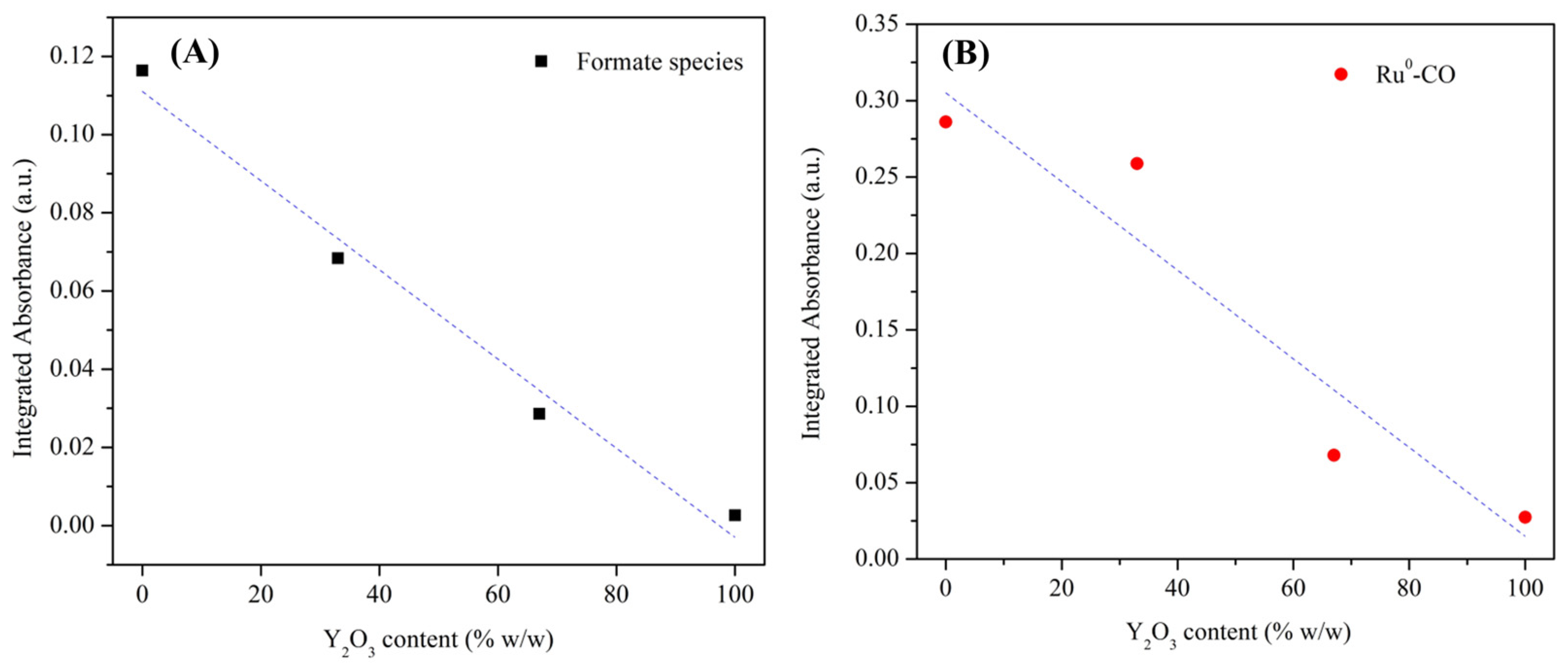
2.3.3. Effect of Y2O3 Addition to Ru/CeO2 over the RWGS Reaction
3. Materials and Methods
3.1. Synthesis of Catalyst Powders
3.2. Catalysts Characterization
3.3. Catalytic Tests
3.4. FTIR Characterization Under Reaction Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Ghussain, L. Global Warming: Review on Driving Forces and Mitigation. Environ. Prog. Sustain. Energy 2019, 38, 13–21. [Google Scholar] [CrossRef]
- Faria, C.; Rocha, C.; Miguel, C.; Rodrigues, A.; Madeira, L.M. Process Intensification Concepts for CO2 Methanation—A Review. Fuel 2025, 386, 134269. [Google Scholar] [CrossRef]
- Müller, K.; Mokrushina, L.; Arlt, W. Thermodynamic Constraints for the Utilization of CO2. Chem. Ing. Tech. 2014, 86, 497–503. [Google Scholar] [CrossRef]
- Ye, R.P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q. CO2 Hydrogenation to High-Value Products via Heterogeneous Catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef]
- Saeidi, S.; Najari, S.; Hessel, V.; Wilson, K.; Keil, F.J.; Concepción, P.; Suib, S.L.; Rodrigues, A.E. Recent Advances in CO2 Hydrogenation to Value-Added Products—Current Challenges and Future Directions. Prog. Energy Combust. Sci. 2021, 85, 100905. [Google Scholar] [CrossRef]
- Zhou, W.; Cheng, K.; Kang, J.; Zhou, C.; Subramanian, V.; Zhang, Q.; Wang, Y. New Horizon in C1 Chemistry: Breaking the Selectivity Limitation in Transformation of Syngas and Hydrogenation of CO2 into Hydrocarbon Chemicals and Fuels. Chem. Soc. Rev. 2019, 48, 3193–3228. [Google Scholar] [CrossRef]
- Bahmanpour, A.M.; Signorile, M.; Kröcher, O. Recent Progress in Syngas Production via Catalytic CO2 Hydrogenation Reaction. Appl. Catal. B 2021, 295, 120319. [Google Scholar] [CrossRef]
- Yamaoka, M.; Tomozawa, K.; Sumiyoshi, K.; Ueda, T.; Ogo, S. Efficient Reverse Water Gas Shift Reaction at Low Temperatures over an Iron Supported Catalyst under an Electric Field. Sci. Rep. 2024, 14, 10216. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sim, G.D.; Jung, U.; Park, Y.; Youn, M.H.; Chun, D.H.; Rhim, G.B.; Kim, K.Y.; Koo, K.Y. Copper Catalysts for CO2 Hydrogenation to CO through Reverse Water–Gas Shift Reaction for e-Fuel Production: Fundamentals, Recent Advances, and Prospects. Chem. Eng. J. 2024, 492, 152283. [Google Scholar] [CrossRef]
- González-Castaño, M.; Dorneanu, B.; Arellano-García, H. The Reverse Water Gas Shift Reaction: A Process Systems Engineering Perspective. React. Chem. Eng. 2021, 6, 954–976. [Google Scholar] [CrossRef]
- Oshima, K.; Shinagawa, T.; Nogami, Y.; Manabe, R.; Ogo, S.; Sekine, Y. Low Temperature Catalytic Reverse Water Gas Shift Reaction Assisted by an Electric Field. Catal. Today 2014, 232, 27–32. [Google Scholar] [CrossRef]
- Zhu, M.; Ge, Q.; Zhu, X. Catalytic Reduction of CO2 to CO via Reverse Water Gas Shift Reaction: Recent Advances in the Design of Active and Selective Supported Metal Catalysts. Trans. Tianjin Univ. 2020, 26, 172–187. [Google Scholar]
- Zamljen, A.; Lavrič, Ž.; Prašnikar, A.; Teržan, J.; Grilc, M.; Meden, A.; Likozar, B. Understanding Platinum-Based H2 Adsorption/Desorption Kinetics during Catalytic Hydrogenation or Hydrogen Storage-Related Reactions. Renew. Energy 2024, 226, 120467. [Google Scholar]
- Tierney, H.L.; Baber, A.E.; Kitchin, J.R.; Sykes, E.C.H. Hydrogen Dissociation and Spillover on Individual Isolated Palladium Atoms. Phys. Rev. Lett. 2009, 103, 246102. [Google Scholar] [CrossRef]
- Liu, X.; Sun, L.; Deng, W.-Q. Theoretical Investigation of CO2 Adsorption and Dissociation on Low Index Surfaces of Transition Metals. J. Phys. Chem. C 2018, 122, 8306–8314. [Google Scholar]
- Halasi, G.; Schubert, G.; Solymosi, F. Comparative Study on the Photocatalytic Decomposition of Methanol on TiO2 Modified by N and Promoted by Metals. J. Catal. 2012, 294, 199–206. [Google Scholar]
- Kattel, S.; Yan, B.; Chen, J.G.; Liu, P. CO2 Hydrogenation on Pt, Pt/SiO2 and Pt/TiO2: Importance of Synergy between Pt and Oxide Support. J. Catal. 2016, 343, 115–126. [Google Scholar] [CrossRef]
- Pachecka, M.; Sturm, J.M.; Lee, C.J.; Bijkerk, F. Adsorption and Dissociation of CO2 on Ru (0001). J. Phys. Chem. C 2017, 121, 6729–6735. [Google Scholar]
- López-Rodríguez, S.; Davo-Quinonero, A.; Bailón-García, E.; Lozano-Castello, D.; Herrera, F.C.; Pellegrin, E.; Escudero, C.; García-Melchor, M.; Bueno-López, A. Elucidating the Role of the Metal Catalyst and Oxide Support in the Ru/CeO2-Catalyzed CO2 Methanation Mechanism. J. Phys. Chem. C 2021, 125, 25533–25544. [Google Scholar] [CrossRef]
- Rose, F.; Tatarkhanov, M.; Fomin, E.; Salmeron, M. Nature of the Dissociation Sites of Hydrogen Molecules on Ru (001). J. Phys. Chem. C 2007, 111, 19052–19057. [Google Scholar]
- Peng, Z.; Wang, H.; Zhou, L.; Wang, Y.; Gao, J.; Liu, G.; Redfern, S.A.T.; Feng, X.; Lu, S.; Li, B. Hollow Carbon Shells Enhanced by Confined Ruthenium as Cost-Efficient and Superior Catalysts for the Alkaline Hydrogen Evolution Reaction. J. Mater. Chem. A Mater. 2019, 7, 6676–6685. [Google Scholar] [CrossRef]
- Ehlers, A.W.; Frenking, G. Structures and Bond Energies of the Transition-Metal Carbonyls M(CO)5 (M = Fe, Ru, Os) and M(CO)4 (M = Ni, Pd, Pt). Organometallics 1995, 14, 423–426. [Google Scholar] [CrossRef]
- Ziegler, T.; Tschinke, V.; Ursenbach, C. Thermal Stability and Kinetic Lability of the Metal Carbonyl Bond. A Theoretical Study on M(CO)6 (M = Chromium, Molybdenum, Tungsten), M(CO)5 (M = Iron, Ruthenium, Osmium), and M(CO)4 (M = Nickel, Palladium, Platinum). J. Am. Chem. Soc. 1987, 109, 4825–4837. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Qian, K.; Huang, W. Metal–Support Interactions in Metal/Oxide Catalysts and Oxide–Metal Interactions in Oxide/Metal Inverse Catalysts. ACS Catal. 2022, 12, 1268–1287. [Google Scholar] [CrossRef]
- Li, M.; Yan, W.; Liu, M.; Liu, C.-J. Improved Activity of Ru/CeO2 Catalyst for CO2 Methanation by Enhanced Electronic Metal–Support Interaction. Energy Fuels 2024, 39, 604–613. [Google Scholar] [CrossRef]
- Dai, L.; Li, B.; Peng, Y.; Hao, X.; Xuan, R.; Huang, H.; Bi, Y.; Xu, L.; Zhang, Y.; Guo, Y. Tuning Interaction Strength between CeO2 and Iridium to Promote CO Oxidation over Ir/TiO2. J. Rare Earths 2024, 42, 651–659. [Google Scholar] [CrossRef]
- Ro, I.; Resasco, J.; Christopher, P. Approaches for Understanding and Controlling Interfacial Effects in Oxide-Supported Metal Catalysts. ACS Catal. 2018, 8, 7368–7387. [Google Scholar] [CrossRef]
- van Deelen, T.W.; Hernández Mejía, C.; de Jong, K.P. Control of Metal-Support Interactions in Heterogeneous Catalysts to Enhance Activity and Selectivity. Nat. Catal. 2019, 2, 955–970. [Google Scholar] [CrossRef]
- Liu, H.-X.; Li, S.-Q.; Wang, W.-W.; Yu, W.-Z.; Zhang, W.-J.; Ma, C.; Jia, C.-J. Partially Sintered Copper—Ceria as Excellent Catalyst for the High-Temperature Reverse Water Gas Shift Reaction. Nat. Commun. 2022, 13, 867. [Google Scholar] [CrossRef]
- Qiao, Y.; He, J.; Zhou, Y.; Wu, S.; Li, X.; Jiang, G.; Jiang, G.; Demir, M.; Ma, P. Flexible All-Solid-State Asymmetric Supercapacitors Based on PPy-Decorated SrFeO3−δ Perovskites on Carbon Cloth. ACS Appl. Mater. Interfaces 2023, 15, 52381–52391. [Google Scholar] [CrossRef]
- Zhang, B.; Huang, S.; Li, Y.; Shen, J.; Tian, X.; Ding, M. Highly Dispersed Cu on Hollow Spherical CeO2: An Efficient and Stable Catalyst for the RWGS Reaction. Appl. Catal. B Environ. Energy 2025, 366, 125003. [Google Scholar] [CrossRef]
- Fan, Y.; Li, R.; Wang, B.; Feng, X.; Du, X.; Liu, C.; Wang, F.; Liu, C.; Dong, C.; Ning, Y. Water-Assisted Oxidative Redispersion of Cu Particles through Formation of Cu Hydroxide at Room Temperature. Nat. Commun. 2024, 15, 3046. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, B.; Chen, D.; Li, Y.; Feng, C.; Du, W. Mechanochemistry Strategy in Metal/Fe3O4 with High Stability for Superior Chemoselective Catalysis. ACS Appl. Mater. Interfaces 2024, 16, 66219–66229. [Google Scholar] [CrossRef]
- Yu, Y.; Xia, W.; Yu, A.; Simakov, D.S.A.; Ricardez-Sandoval, L. Transition-Metal-Doped CeO2 for the Reverse Water-Gas Shift Reaction: An Experimental and Theoretical Study on CO2 Adsorption and Surface Vacancy Effects. ChemSusChem 2025, 18, e202400681. [Google Scholar] [CrossRef]
- Lin, L.; Yao, S.; Liu, Z.; Zhang, F.; Li, N.; Vovchok, D.; Martínez-Arias, A.; Castaneda, R.; Lin, J.; Senanayake, S.D. In Situ Characterization of Cu/CeO2 Nanocatalysts for CO2 Hydrogenation: Morphological Effects of Nanostructured Ceria on the Catalytic Activity. J. Phys. Chem. C 2018, 122, 12934–12943. [Google Scholar] [CrossRef]
- Quindimil, A.; De-La-Torre, U.; Pereda-Ayo, B.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castello, D.; González-Marcos, J.A.; Bueno-Lopez, A.; González-Velasco, J.R. Effect of Metal Loading on the CO2 Methanation: A Comparison between Alumina Supported Ni and Ru Catalysts. Catal. Today 2020, 356, 419–432. [Google Scholar] [CrossRef]
- Wang, F.; He, S.; Chen, H.; Wang, B.; Zheng, L.; Wei, M.; Evans, D.G.; Duan, X. Active Site Dependent Reaction Mechanism over Ru/CeO2 Catalyst toward CO2 Methanation. J. Am. Chem. Soc. 2016, 138, 6298–6305. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, G.J.; Lee, S.H.; Hwang, I.H.; Hong, S.C.; Kim, S.S. Catalytic Performance of Ce0.6Y0.4O2-Supported Platinum Catalyst for Low-Temperature Water–Gas Shift Reaction. ACS Omega 2018, 3, 3156–3163. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Domínguez, M.C.; Solis-García, A.; Venezia, A.M.; Jimenez-Lam, S.; Fuentes-Moyado, S.; Ponce-Pérez, R.; Guerrero-Sanchez, J.; Pacheco-Sosa, J.G.; de León, J.N.D. The Role of Ga and Y on Binary Al2O3-Y2O3 and Al2O3-Ga2O3 Mixed Oxides Nanoparticles towards Potential Ni Water-Gas Shift Catalysts. Nano-Struct. Nano-Objects 2024, 38, 101165. [Google Scholar] [CrossRef]
- Lee, Y.H.; Ahn, J.Y.; Nguyen, D.D.; Chang, S.W.; Kim, S.S.; Lee, S.M. Role of Oxide Support in Ni Based Catalysts for CO2 Methanation. RSC Adv. 2021, 11, 17648–17657. [Google Scholar] [CrossRef]
- Oemar, U.; Hidajat, K.; Kawi, S. High Catalytic Stability of Pd-Ni/Y2O3 Formed by Interfacial Cl for Oxy-CO2 Reforming of CH4. Catal. Today 2017, 281, 276–294. [Google Scholar] [CrossRef]
- Hasan, M.; Asakoshi, T.; Muroyama, H.; Matsui, T.; Eguchi, K. CO2 Methanation Mechanism over Ni/Y2O3: An in Situ Diffuse Reflectance Infrared Fourier Transform Spectroscopic Study. Phys. Chem. Chem. Phys. 2021, 23, 5551–5558. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Shih, P.-C.; Tsao, K.-C.; Pan, Y.-T.; Yin, X.; Sun, C.-J.; Yang, H. High-Performance Pyrochlore-Type Yttrium Ruthenate Electrocatalyst for Oxygen Evolution Reaction in Acidic Media. J. Am. Chem. Soc. 2017, 139, 12076–12083. [Google Scholar] [CrossRef] [PubMed]
- Lao, M.; Zhao, G.; Li, P.; Ma, T.; Jiang, Y.; Pan, H.; Dou, S.X.; Sun, W. Manipulating the Coordination Chemistry of Ru—N(O)—C Moieties for Fast Alkaline Hydrogen Evolution Kinetics. Adv. Funct. Mater. 2021, 31, 2100698. [Google Scholar] [CrossRef]
- Caudillo-Flores, U.; Carrales-Alvarado, D.H.; Rodríguez-Ramos, I.; Fernández-García, M.; Kubacka, A. Synergistic, Dual Energy Source Production of Hydrogen Using Ru-Promoted Nitrogen-Doped Carbon Nanotubes. Carbon 2024, 221, 118903. [Google Scholar] [CrossRef]
- Liu, M.; Liu, W.; Liu, X.; Ouyang, Y.; Hou, H.; Lei, M.; Wei, Z. Yttrium Oxide as a Q-Switcher for the near-Infrared Erbium-Doped Fiber Laser. Nanophotonics 2020, 9, 2887–2894. [Google Scholar] [CrossRef]
- Mongstad, T.; Thøgersen, A.; Subrahmanyam, A.; Karazhanov, S. The Electronic State of Thin Films of Yttrium, Yttrium Hydrides and Yttrium Oxide. Sol. Energy Mater. Sol. Cells 2014, 128, 270–274. [Google Scholar] [CrossRef]
- Song, J.-B.; Kim, J.-T.; Oh, S.-G.; Yun, J.-Y. Contamination Particles and Plasma Etching Behavior of Atmospheric Plasma Sprayed Y2O3 and YF3 Coatings under NF3 Plasma. Coatings 2019, 9, 102. [Google Scholar] [CrossRef]
- Li, N.; Huo, L.; Dong, Q.; Zhu, B.; Huang, L.; Ma, J. RuSe2/CeO2 Heterostructure as a Novel Electrocatalyst for Highly Efficient Alkaline Hydrogen Evolution. Nanotechnology 2023, 35, 115602. [Google Scholar] [CrossRef]
- Izquierdo-Colorado, A.; Torres-Torres, G.; Gamboa-Rodríguez, M.T.; Silahua-Pavón, A.A.; Arévalo-Pérez, J.C.; Cervantes-Uribe, A.; Cordero-García, A.; Beltramini, J.N. Catalytic Wet Air Oxidation (CWAO) of Phenol in a Fixed Bed Reactor Using Supported Ru and Ru-Au Catalysts: Effect of Gold and Ce Loading. ChemistrySelect 2019, 4, 1275–1284. [Google Scholar] [CrossRef]
- Huang, H.; Dai, Q.; Wang, X. Morphology Effect of Ru/CeO2 Catalysts for the Catalytic Combustion of Chlorobenzene. Appl. Catal. B 2014, 158, 96–105. [Google Scholar] [CrossRef]
- Mena-Saucedo, A.; Caudillo-Flores, U.; Kubacka, A.; Fernández-García, M. In2O3-TiO2 Composites for CO2 Photothermocatalytic Valorization. J. Phys. Chem. C 2024, 128, 20923–20932. [Google Scholar] [CrossRef]
- Sun, S.; Sun, H.; Guan, S.; Xu, S.; Wu, C. Integrated CO2 Capture and Methanation on Ru/CeO2-MgO Combined Materials: Morphology Effect from CeO2 Support. Fuel 2022, 317, 123420. [Google Scholar] [CrossRef]
- López-Rodríguez, S.; Davo-Quinonero, A.; Bailón-García, E.; Lozano-Castello, D.; Bueno-Lopez, A. Effect of Ru Loading on Ru/CeO2 Catalysts for CO2 Methanation. Mol. Catal. 2021, 515, 111911. [Google Scholar]
- Sun, C.; Beaunier, P.; La Parola, V.; Liotta, L.F.; Da Costa, P. Ni/CeO2 Nanoparticles Promoted by Yttrium Doping as Catalysts for CO2 Methanation. ACS Appl. Nano Mater. 2020, 3, 12355–12368. [Google Scholar] [CrossRef]
- Ro, I.; Sener, C.; Stadelman, T.M.; Ball, M.R.; Venegas, J.M.; Burt, S.P.; Hermans, I.; Dumesic, J.A.; Huber, G.W. Measurement of Intrinsic Catalytic Activity of Pt Monometallic and Pt-MoOx Interfacial Sites over Visible Light Enhanced PtMoOx/SiO2 Catalyst in Reverse Water Gas Shift Reaction. J. Catal. 2016, 344, 784–794. [Google Scholar] [CrossRef]
- He, Y.; Huang, D. Single-Atom Platinum Catalyst for Efficient CO2 Conversion via Reverse Water Gas Shift Reaction. Molecules 2023, 28, 6630. [Google Scholar] [CrossRef]
- Chen, L.; Kovarik, L.; Szanyi, J. Temperature-Dependent Communication between Pt/Al2O3 Catalysts and Anatase TiO2 Dilutant: The Effects of Metal Migration and Carbon Transfer on the Reverse Water–Gas Shift Reaction. ACS Catal. 2021, 11, 12058–12067. [Google Scholar] [CrossRef]
- Dongapure, P.; Bagchi, S.; Mayadevi, S.; Devi, R.N. Variations in Activity of Ru/TiO2 and Ru/Al2O3 Catalysts for CO2 Hydrogenation: An Investigation by in-Situ Infrared Spectroscopy Studies. Mol. Catal. 2020, 482, 110700. [Google Scholar]
- Hu, J.; Brooks, K.P.; Holladay, J.D.; Howe, D.T.; Simon, T.M. Catalyst Development for Microchannel Reactors for Martian in Situ Propellant Production. Catal. Today 2007, 125, 103–110. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Sato, H.; Kaneeda, M.; Kondo, J.N. Synthesis and Analysis of CO2 Adsorbents Based on Cerium Oxide. J. CO2 Util. 2014, 8, 34–38. [Google Scholar] [CrossRef]
- Cárdenas-Arenas, A.; Quindimil, A.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castello, D.; De-La-Torre, U.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R.; Bueno-López, A. Isotopic and in Situ DRIFTS Study of the CO2 Methanation Mechanism Using Ni/CeO2 and Ni/Al2O3 Catalysts. Appl. Catal. B 2020, 265, 118538. [Google Scholar] [CrossRef]
- Fan, L.; Zhang, J.; Ma, K.; Zhang, Y.; Hu, Y.-M.; Kong, L.; Jia, A.; Zhang, Z.; Huang, W.; Lu, J.-Q. Ceria Morphology-Dependent Pd-CeO2 Interaction and Catalysis in CO2 Hydrogenation into Formate. J. Catal. 2021, 397, 116–127. [Google Scholar] [CrossRef]
- Wang, Y.; Arandiyan, H.; Scott, J.; Aguey-Zinsou, K.-F.; Amal, R. Single Atom and Nanoclustered Pt Catalysts for Selective CO2 Reduction. ACS Appl. Energy Mater. 2018, 1, 6781–6789. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Xu, H.; Han, Y. Reverse Water–Gas Shift Reaction over Ceria Nanocube Synthesized by Hydrothermal Method. Catal. Commun. 2016, 76, 1–6. [Google Scholar] [CrossRef]
- Shido, T.; Iwasawa, Y. Regulation of Reaction Intermediate by Reactant in the Water-Gas Shift Reaction on CeO2, in Relation to Reactant-Promoted Mechanism. J. Catal. 1992, 136, 493–503. [Google Scholar] [CrossRef]
- Lustemberg, P.G.; Bosco, M.V.; Bonivardi, A.; Busnengo, H.F.; Ganduglia-Pirovano, M. V Insights into the Nature of Formate Species in the Decomposition and Reaction of Methanol over Cerium Oxide Surfaces: A Combined Infrared Spectroscopy and Density Functional Theory Study. J. Phys. Chem. C 2015, 119, 21452–21464. [Google Scholar] [CrossRef]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.; Onishi, T. Adsorption of Carbon Monoxide and Carbon Dioxide on Cerium Oxide Studied by Fourier-Transform Infrared Spectroscopy. Part 2.—Formation of Formate Species on Partially Reduced CeO2 at Room Temperature. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1989, 85, 1451–1461. [Google Scholar] [CrossRef]
- González-Rangulan, V.V.; Reyero, I.; Bimbela, F.; Romero-Sarria, F.; Daturi, M.; Gandía, L.M. CO2 Methanation over Nickel Catalysts: Support Effects Investigated through Specific Activity and Operando IR Spectroscopy Measurements. Catalysts 2023, 13, 448. [Google Scholar] [CrossRef]
- Falbo, L.; Visconti, C.G.; Lietti, L.; Szanyi, J. The Effect of CO on CO2 Methanation over Ru/Al2O3 Catalysts: A Combined Steady-State Reactivity and Transient DRIFT Spectroscopy Study. Appl. Catal. B 2019, 256, 117791. [Google Scholar] [CrossRef]
- Chin, S.Y.; Williams, C.T.; Amiridis, M.D. FTIR Studies of CO Adsorption on Al2O3-and SiO2-Supported Ru Catalysts. J. Phys. Chem. B 2006, 110, 871–882. [Google Scholar] [PubMed]
- Hadjiivanov, K.; Lavalley, J.-C.; Lamotte, J.; Maugé, F.; Saint-Just, J.; Che, M. FTIR Study of CO Interaction with Ru/TiO2Catalysts. J. Catal. 1998, 176, 415–425. [Google Scholar]
- Solis-Garcia, A.; Zepeda, T.A.; Fierro-Gonzalez, J.C. Spectroscopic Evidence of Surface Species during CO2 Methanation Catalyzed by Supported Metals: A Review. Catal. Today 2022, 394, 2–12. [Google Scholar] [CrossRef]
- Podrojková, N.; Sans, V.; Oriňak, A.; Oriňaková, R. Recent Developments in the Modelling of Heterogeneous Catalysts for CO2 Conversion to Chemicals. ChemCatChem 2020, 12, 1802–1825. [Google Scholar]
- Zhang, S.-T.; Yan, H.; Wei, M.; Evans, D.G.; Duan, X. Hydrogenation Mechanism of Carbon Dioxide and Carbon Monoxide on Ru (0001) Surface: A Density Functional Theory Study. RSC Adv. 2014, 4, 30241–30249. [Google Scholar]
- Fisher, I.A.; Bell, A.T. A Comparative Study of CO and CO2Hydrogenation over Rh/SiO2. J. Catal. 1996, 162, 54–65. [Google Scholar]
- Panayotov, D.; Mihaylov, M.; Nihtianova, D.; Spassov, T.; Hadjiivanov, K. Spectral Evidence for Hydrogen-Induced Reversible Segregation of CO Adsorbed on Titania-Supported Rhodium. Phys. Chem. Chem. Phys. 2014, 16, 13136–13144. [Google Scholar] [PubMed]
- Azzolina-Jury, F.; Thibault-Starzyk, F. Mechanism of Low Pressure Plasma-Assisted CO2 Hydrogenation over Ni-USY by Microsecond Time-Resolved FTIR Spectroscopy. Top. Catal. 2017, 60, 1709–1721. [Google Scholar] [CrossRef]
- Huang, B.; Kobayashi, H.; Yamamoto, T.; Toriyama, T.; Matsumura, S.; Nishida, Y.; Sato, K.; Nagaoka, K.; Haneda, M.; Xie, W. A CO Adsorption Site Change Induced by Copper Substitution in a Ruthenium Catalyst for Enhanced CO Oxidation Activity. Angew. Chem. Int. Ed. 2019, 58, 2230–2235. [Google Scholar]
- Köck, E.M.; Kogler, M.; Bielz, T.; Klötzer, B.; Penner, S. In Situ FT-IR Spectroscopic Study of CO2 and CO Adsorption on Y2O3, ZrO2, and Yttria-Stabilized ZrO2. J. Phys. Chem. C 2013, 117, 17666–17673. [Google Scholar] [CrossRef]
- Campani, P.; Fang, C.S.; Prengle, H.W. Infrared Absorption Coefficients for Certain Pollutant Gases. Appl. Spectrosc. 1972, 26, 372–378. [Google Scholar] [CrossRef]
- Li, Z.-X.; Xu, K.; Wang, W.-W.; Fu, X.-P.; Jia, C. Stabilized Inverse Y2O3/Cu Interfaces Boost the Performance of the Reverse Water–Gas Shift Reaction. Catal. Sci. Technol. 2024, 14, 3483–3492. [Google Scholar] [CrossRef]
- Dreyer, J.A.H.; Li, P.; Zhang, L.; Beh, G.K.; Zhang, R.; Sit, P.H.-L.; Teoh, W.Y. Influence of the Oxide Support Reducibility on the CO2 Methanation over Ru-Based Catalysts. Appl. Catal. B 2017, 219, 715–726. [Google Scholar] [CrossRef]
- Wang, X.; Hong, Y.; Shi, H.; Szanyi, J. Kinetic Modeling and Transient DRIFTS–MS Studies of CO2 Methanation over Ru/Al2O3 Catalysts. J. Catal. 2016, 343, 185–195. [Google Scholar] [CrossRef]
- Eckle, S.; Anfang, H.-G.; Behm, R.J. Reaction Intermediates and Side Products in the Methanation of CO and CO2 over Supported Ru Catalysts in H2-Rich Reformate Gases. J. Phys. Chem. C 2011, 115, 1361–1367. [Google Scholar] [CrossRef]
- Yamano, R.; Ogo, S.; Nakano, N.; Higo, T.; Sekine, Y. Non-Conventional Low-Temperature Reverse Water–Gas Shift Reaction over Highly Dispersed Ru Catalysts in an Electric Field. EES Catal. 2023, 1, 125–133. [Google Scholar] [CrossRef]
- Chen, L.; Filot, I.A.W.; Hensen, E.J.M. Elucidation of the Reverse Water–Gas Shift Reaction Mechanism over an Isolated Ru Atom on CeO2 (111). J. Phys. Chem. C 2023, 127, 20314–20324. [Google Scholar]
- Aitbekova, A.; Wu, L.; Wrasman, C.J.; Boubnov, A.; Hoffman, A.S.; Goodman, E.D.; Bare, S.R.; Cargnello, M. Low-Temperature Restructuring of CeO2-Supported Ru Nanoparticles Determines Selectivity in CO2 Catalytic Reduction. J. Am. Chem. Soc. 2018, 140, 13736–13745. [Google Scholar] [CrossRef]
- Caudillo-Flores, U.; Barba-Nieto, I.; Muñoz-Batista, M.J.; Meira, D.M.; Fernández-García, M.; Kubacka, A. Thermo-Photo Production of Hydrogen Using Ternary Pt-CeO2-TiO2 Catalysts: A Spectroscopic and Mechanistic Study. Chem. Eng. J. 2021, 425, 130641. [Google Scholar] [CrossRef]
- Atkins, P.W.; De Paula, J.; Keeler, J. Atkins’ Physical Chemistry; Oxford University Press: Oxford, UK, 2023; ISBN 0198847815. [Google Scholar]
- Fogler, H.S. Essentials of Chemical Reaction Engineering: Essenti Chemica Reactio Engi; Pearson Education: London, UK, 2010; ISBN 0132317176. [Google Scholar]
- Satterfield, C.N. Heterogeneous Catalysis in Industrial Practice. 1991. Available online: https://www.osti.gov/biblio/5495428 (accessed on 19 March 2025).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | CeO2-Y2O3 b (%w/w) | BET Area c (m2 g−1) | Pore Volume c (cm3 g−1) | Pore Diameter c (nm) | a0 CeO2 d (Å) |
|---|---|---|---|---|---|
| Ru/CeY0 | 100-0 | 106.3 | 0.247 | 7.5 | 5.41 |
| Ru/CeY33 | 67-33 | 65.9 | 0.106 | 7.4 | 5.40 |
| Ru/CeY66 | 34-66 | 51.8 | 0.111 | 8.7 | 5.40 |
| Ru/CeY100 | 0-100 | 41.7 | 0.107 | 9.4 | 10.64 Y2O3 |
| Sample | Y/Ce | Ce3+/Ce4+ | Ov (%) |
|---|---|---|---|
| Ru/CeY0 | ----- | ----- | 25.1 |
| Ru/CeY33 | 1.1 | 0.10 | 22.7 |
| Ru/CeY66 | 2.3 | 0.11 | 18.3 |
| Ru/CeY100 | ----- | 0.11 | 16.8 |
| Sample | Temp (°C) | GHSV (h−1) | Inlet Gas Composition (%v/v) | XCO2 (%) | SCO (%) | REF |
|---|---|---|---|---|---|---|
| 0.5% Ru/TiO2 | 325 | 14,400 | 10% CO2/40% H2/50% H2 | 60 | 0 | [59] |
| 0.5% Ru/Al2O3 | 325 | 14,400 | 10% CO2/40% H2/50% H2 | 30 | 0 | [59] |
| 3.0% Ru/SiO2 | 350 | 45,000 | 20% CO2/80% H2 | 46 | 0.1 | [60] |
| 3.0% Ru/TiO2 | 350 | 45,000 | 20% CO2/80% H2 | 64.1 | 0.1 | [60] |
| 4.0% Ru/CeO2 | 325 | 9000 | 16% CO2/64% H2/20% N2 | 90 | 0.1 | [19] |
| Ru/CeY0 | 350 | 27,450 | 16% CO2/64% H2/20% N2 | 35.6 | 4.9% | This work |
| Ru/CeY33 | 350 | 27,450 | 16% CO2/64% H2/20% N2 | 6.2 | 97.7% | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solís-García, A.; Portillo-Cortez, K.; Domínguez, D.; Fuentes-Moyado, S.; Díaz de León, J.N.; Zepeda, T.A.; Caudillo-Flores, U. Improving the Catalytic Selectivity of Reverse Water–Gas Shift Reaction Catalyzed by Ru/CeO2 Through the Addition of Yttrium Oxide. Catalysts 2025, 15, 301. https://doi.org/10.3390/catal15040301
Solís-García A, Portillo-Cortez K, Domínguez D, Fuentes-Moyado S, Díaz de León JN, Zepeda TA, Caudillo-Flores U. Improving the Catalytic Selectivity of Reverse Water–Gas Shift Reaction Catalyzed by Ru/CeO2 Through the Addition of Yttrium Oxide. Catalysts. 2025; 15(4):301. https://doi.org/10.3390/catal15040301
Chicago/Turabian StyleSolís-García, Alfredo, Karina Portillo-Cortez, David Domínguez, Sergio Fuentes-Moyado, Jorge N. Díaz de León, Trino A. Zepeda, and Uriel Caudillo-Flores. 2025. "Improving the Catalytic Selectivity of Reverse Water–Gas Shift Reaction Catalyzed by Ru/CeO2 Through the Addition of Yttrium Oxide" Catalysts 15, no. 4: 301. https://doi.org/10.3390/catal15040301
APA StyleSolís-García, A., Portillo-Cortez, K., Domínguez, D., Fuentes-Moyado, S., Díaz de León, J. N., Zepeda, T. A., & Caudillo-Flores, U. (2025). Improving the Catalytic Selectivity of Reverse Water–Gas Shift Reaction Catalyzed by Ru/CeO2 Through the Addition of Yttrium Oxide. Catalysts, 15(4), 301. https://doi.org/10.3390/catal15040301