1. Introduction
The rapid expansion of the global economy coupled with the continual growth of the human population has led to a steady increase in energy demand. Traditional fossil fuels are the predominant source of energy. These fossil fuels face challenges related to not only resource depletion, but also their combustion, which releases substantial amounts of carbon dioxide (CO
2), thereby exacerbating the greenhouse effect. Moreover, the oxidation and combustion of nitrogen, sulfur, and other constituents within the fuel generate hazardous gases such as nitrogen dioxide (NO
2), nitric oxide (NO), and sulfur dioxide (SO
2). When these gases interact with water vapor in the atmosphere, they can cause acid rain, which has significant adverse effects on both the ecological environment and human activities [
1,
2]. Consequently, the demand for clean energy has become increasingly urgent [
3,
4].
Water splitting is a crucial technique for generating clean energy because it facilitates the conversion of water into hydrogen and oxygen without producing harmful byproducts. This process involves two primary reactions: hydrogen evolution reaction (HER) [
5], which is responsible for hydrogen production, and oxygen evolution reaction (OER), which produces oxygen. Furthermore, the reverse reaction of the OER is known as the oxygen reduction reaction (ORR) [
6]. Therefore, the development of efficient, cost-effective, and stable electrocatalysts for water splitting is of paramount importance. However, conventional electrocatalysts such as platinum and palladium face limitations owing to their high cost, scarcity, and vulnerability to poisoning and deactivation. These challenges hinder their widespread adoption and practical applications in energy systems [
7,
8,
9].
In response to these issues, two-dimensional biphenyl-based materials such as graphenylene (GPN) have emerged as a novel class of catalytic materials with significant research potential [
10,
11].
GPN is synthesized via the polycondensation of 1,3,5-trihydroxybenzene, yielding a two-dimensional carbon-based framework characterized by an alternating arrangement of quadrilateral and hexagonal rings in equimolar stoichiometry, termed 4–6 carbophene. Structural analysis reveals that all carbon centers within this lattice predominantly exhibit sp
2 hybridization with slight geometric distortions, collectively generating an extended conjugated π-electron system across the planar architecture. The σ-bond angles at these hybridized carbon sites are distributed across three distinct configurations: approximately 120°, 90°, and 150°, consistent with theoretical predictions for strained sp
2 systems. Topological connectivity studies demonstrate that each hexagonal unit shares three non-consecutive edges with adjacent tetragonal rings, whereas every quadrilateral structure bonds covalently to two diametrically opposed hexagons. This periodic arrangement produces uniformly distributed dodecagonal pores with an average diameter of 5.8 Å, which are aligned in long-range ordered arrays throughout the crystalline monolayer [
12,
13].
Inspired by advances in graphite [
14] and carbon nanotubes (CNTs) [
15], our TM
3@GPN design leverages an innovation. Synergistic metal–substrate interactions: while carbon materials exhibit inherent catalytic activity, the integration of transition metal (TM) clusters with GPN introduces localized d-orbital states near the Fermi level, enabling simultaneous optimization of H* and O* intermediate adsorption—a critical challenge for bifunctional water splitting.
Compared to traditional electrocatalysts, two-dimensional biphenyl-based materials have several notable advantages. First, their large surface area enhances reaction activity, promoting more efficient catalytic processes. Secondly, these materials exhibit a highly tunable electronic structure, enabling the optimization of their catalytic performance through adjustments to both their composition and structural configuration [
16,
17,
18,
19]. Specifically, by loading a small number of atoms onto a material, the catalytic properties can be significantly enhanced. This approach offers a highly efficient and cost-effective solution, because even a few atoms can significantly improve the performance of a material. Such atom-loading strategies provide a way to optimize the catalytic activity without requiring large quantities of expensive materials; this characteristic renders it an attractive strategy for engineering next-generation catalytic systems [
20,
21,
22,
23,
24]. Therefore, exploring the use of two-dimensional biphenyl-based materials to support multi-atom electrocatalysts for overall water splitting reactions is of significant scientific importance. This approach has the potential to improve the catalytic efficiency, stability, and cost-effectiveness, making it highly promising for practical applications in clean energy production. The integration of multi-atom electrocatalysts with these materials can lead to more efficient and sustainable methods for hydrogen and oxygen production, further advancing the development of water splitting technologies.
In-depth studies on the properties and structure of GPN can provide valuable insights into their role in electrocatalytic reactions. The present investigation establishes a conceptual framework for developing electrocatalysts with enhanced performance and durability, which facilitates significant progress in sustainable energy transformation systems. Furthermore, the development of low-cost renewable catalysts that can replace traditional precious metals will significantly reduce the cost of water splitting technologies, making them more accessible for large-scale applications. This shift is essential to promote the adoption of sustainable energy solutions. Moreover, this research has the potential to address pressing energy supply challenges. Hydrogen produced by water splitting can be used as a clean and renewable energy source, particularly for fuel cells, hydrogen energy storage, and other energy applications. Ultimately, this will advance the development of clean energy technologies, help reduce dependence on conventional, non-renewable energy sources, and support a more sustainable energy future [
25,
26].
The application prospects of GPN-supported multi-atom electrocatalysts for water splitting are promising. Within contemporary power systems, this innovation serves as a critical enabler for optimized hydrogen generation—a zero-emission energy carrier. The electrolytic dissociation-derived hydrogen demonstrates versatile applicability in electrochemical conversion devices, delivering stable and environmentally benign power solutions across industrial domains ranging from vehicular propulsion to grid-scale storage architectures. Such technological integration substantially diminishes dependence on carbon-intensive resources while accelerating the worldwide shift toward decarbonized energy matrices, thereby fostering synergistic advancements in renewable energy paradigms and low-carbon power infrastructure evolution [
27,
28]. For environmental protection, water splitting offers significant advantages because it does not produce greenhouse gases or pollutants during the process. These characteristics make it an environmentally friendly method for hydrogen production, helping to reduce air pollution and combat global climate change. Water splitting can play a pivotal role in decreasing the carbon footprint of traditional energy production systems by providing a clean energy solution. Furthermore, this technology can be widely applied in synthetic chemistry, catalytic conversion, and various industrial fields. This enables the efficient synthesis of important chemicals essential for the production of sustainable materials and chemicals used in everyday life. Additionally, it enhances resource utilization by improving the efficiency of chemical reactions and reducing waste, thereby contributing to more sustainable and circular industrial processes. These applications have the potential to reshape industries, making them more energy efficient and environmentally conscious.
In conclusion, research on GPN-supported multi-atom catalysts for overall water splitting is of significant importance for addressing clean energy demands and key technological challenges [
29,
30,
31]. By thoroughly investigating the properties and structures of GPN-supported multi-atom electrocatalysts, valuable theoretical insights can be gained to guide the design and synthesis of high-performance, cost-effective electrocatalysts. This approach is crucial for advancing the efficiency and affordability of water splitting technologies and enabling their broader application in various sectors. The success of this research will contribute significantly to the growth of the clean energy industry. By improving the efficiency of hydrogen production and making it more cost-effective, water splitting technologies can become key players in the transition to renewable energy sources. This can foster sustainable development, benefiting both the national economy and society by reducing the dependence on fossil fuels, enhancing energy security, and contributing to environmental preservation. Widespread adoption of such technologies could play a transformative role in achieving long-term sustainability and advancing global efforts to combat climate change.
2. Results and Discussion
2.1. Selective Adsorption of TM3 on GPN
The geometric configuration of the intrinsic GPN monolayer was systematically optimized using DFT calculations. Subsequently, comprehensive structural characterization was performed, focusing on the crystallographic symmetry, lattice parameters, and topological distribution of the carbon rings within the unit cell.
To investigate the substrate-mediated stabilization effects, 28 distinct tri-atomic clusters comprising 3d, 4d, and 5d transition metals (excluding lanthanides and radioactive technetium) were anchored onto the GPN substrate. The selection of metallic species followed a periodic-table-based screening strategy, omitting Tc owing to its radiological instability and lanthanides owing to their distinct electronic configurations.
Guided by coordination chemistry principles and prior studies on trimetallic single-cluster catalysts (SCCs), two representative adsorption configurations were identified for the three-atom metal clusters on GPN. For illustrative purposes, the Fe
3@GPN system was selected as a prototypical model, and its optimized adsorption architecture is depicted in
Figure 1. The Fe clusters were chosen owing to their balanced electronic structures and catalytic relevance to analogous systems.
The adsorption energetics of 28 tri-atomic clusters were systematically evaluated across two distinct coordination sites on the GPN substrate using spin-polarized DFT calculations (
Figure 2).
To establish periodic trends, the transition metal elements were categorized according to their d-block groups (3d: Sc-Zn; 4d: Y-Cd; 5d: Hf-Hg), followed by a comparative analysis of their respective ground-state configurations (
Figure 3).
The calculated adsorption energy landscapes demonstrated a periodic-dependent configuration preference.
Intraperiod trend: Early transition metals (lower atomic numbers) exhibit thermodynamic stabilization in configuration 1, whereas late transition metals (higher atomic numbers) preferentially adopt configuration 2.
Interperiod consistency: This configuration-selective behavior remained robust across the 3d, 4d, and 5d series.
2.2. Stability Screening of Catalysts
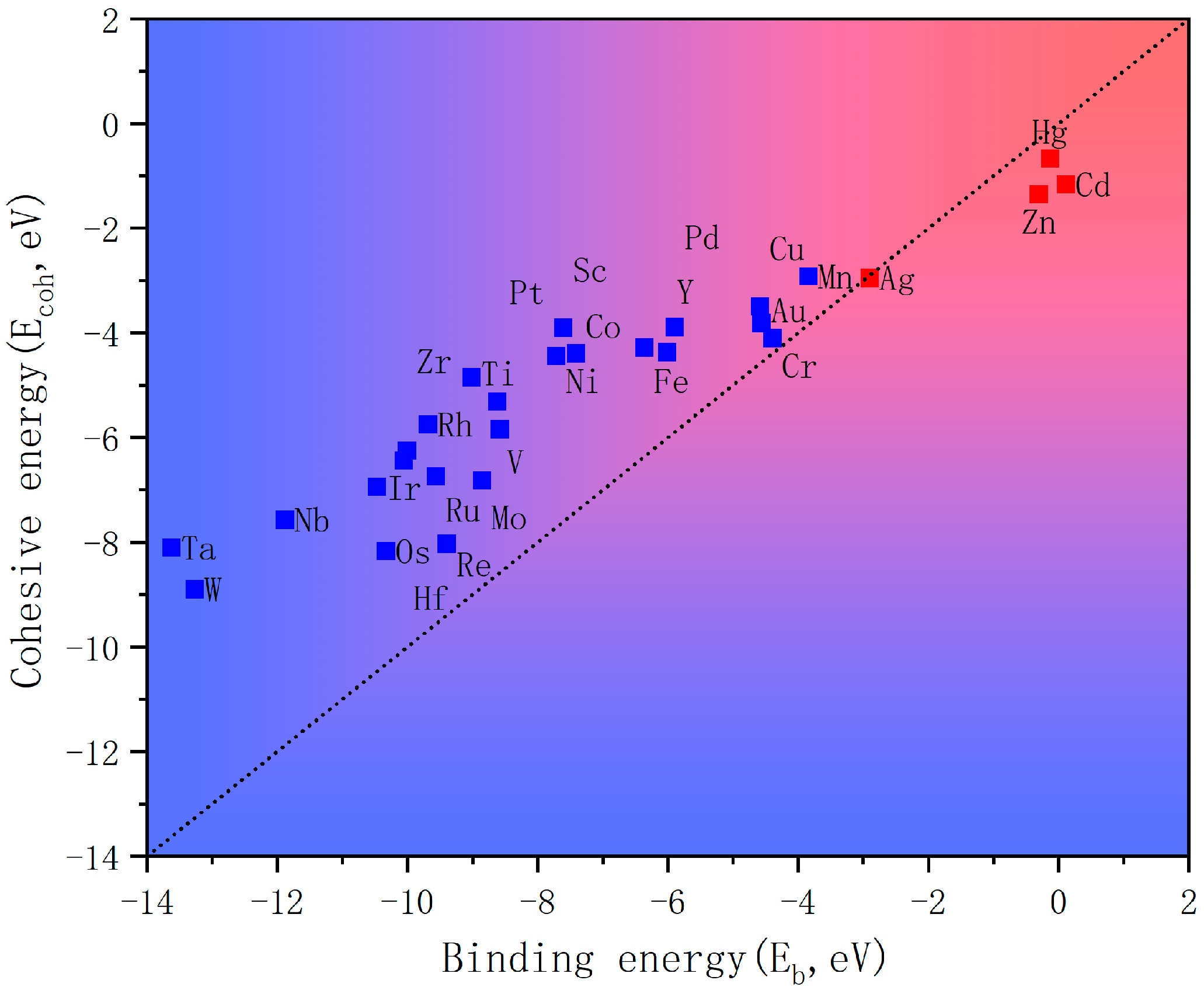
The structural stability of a catalyst is a critical determinant of sustainable catalytic reduction applications. To screen viable candidates among the 28 TM
3@GPN systems, we implemented a thermodynamic stability criterion based on the comparative relationship between metal–substrate binding energy (E
b) and intrinsic metal cluster cohesion energy (E
coh) (
Figure 4).
Systems satisfying Eb < Ecoh were identified as thermodynamically stable, where cluster–substrate interactions sufficiently counteracted metal aggregation tendencies. This screening protocol effectively excluded Ag-, Hg-, Cd-, and Zn-based clusters owing to their pronounced instability, attributed to their weak d-orbital hybridization capabilities and low metal–carbon affinity.
The retained transition metal clusters demonstrated robust adhesion to the GPN substrate, suggesting feasible operational durability under the reduction reaction conditions.
To evaluate the operational thermal stability, molecular dynamics simulations were performed at 500 K for 20 ps (10,000 steps). As shown in
Figure 5, the representative TM
3@GPN systems (TM = Au, Pt, Pd) exhibited minimal energy fluctuations and negligible structural degradation throughout the simulation.
Key observations:
No metal-cluster detachment or significant distortion was observed. The stable adsorption configurations were maintained for over 90% of the simulation time. The energy variance was within 1% of the initial value.
These results confirm the robust thermal stability of the screened catalysts under the simulated reaction conditions, which is consistent with prior thermodynamic stability analysis.
2.3. HER, OER, and ORR Catalytic Performance of TM3@GPN
To investigate the overall water splitting performance of the different TM
3@GPN catalysts, their HER performances were evaluated separately. As shown in
Figure 5a, among the 3d elements, the ΔG(H) values for Mn, Cr, and Cu were 0.09, 0.10, and 0.22 eV, respectively. As shown in
Figure 5b, among the 4d elements, the ΔG(H) values for Pd, Rh, and Ru were −0.11, −0.27, and −0.30 eV, respectively. As shown in
Figure 5c, among the 5d elements, the ΔG(H) values for Au, Pt, and Os were 0.03, −0.16, and 0.21 eV, respectively.
Based on the computational results for the HER (
Figure 6), OER, and ORR (
Figure 7), we can obtain three-dimensional plots of the different elements of TM
3@GPN. If only the overall water splitting performance is considered, the result will be a two-dimensional plot.
In electrocatalysis for overall water splitting, the overpotentials for the OER, ORR, and HER are critical for evaluating the catalytic performance. Lower overpotentials for the OER and ORR are desirable because they indicate reduced energy losses. For the HER, a minimal overpotential close to 0 was sought, reflecting a high hydrogen production efficiency.
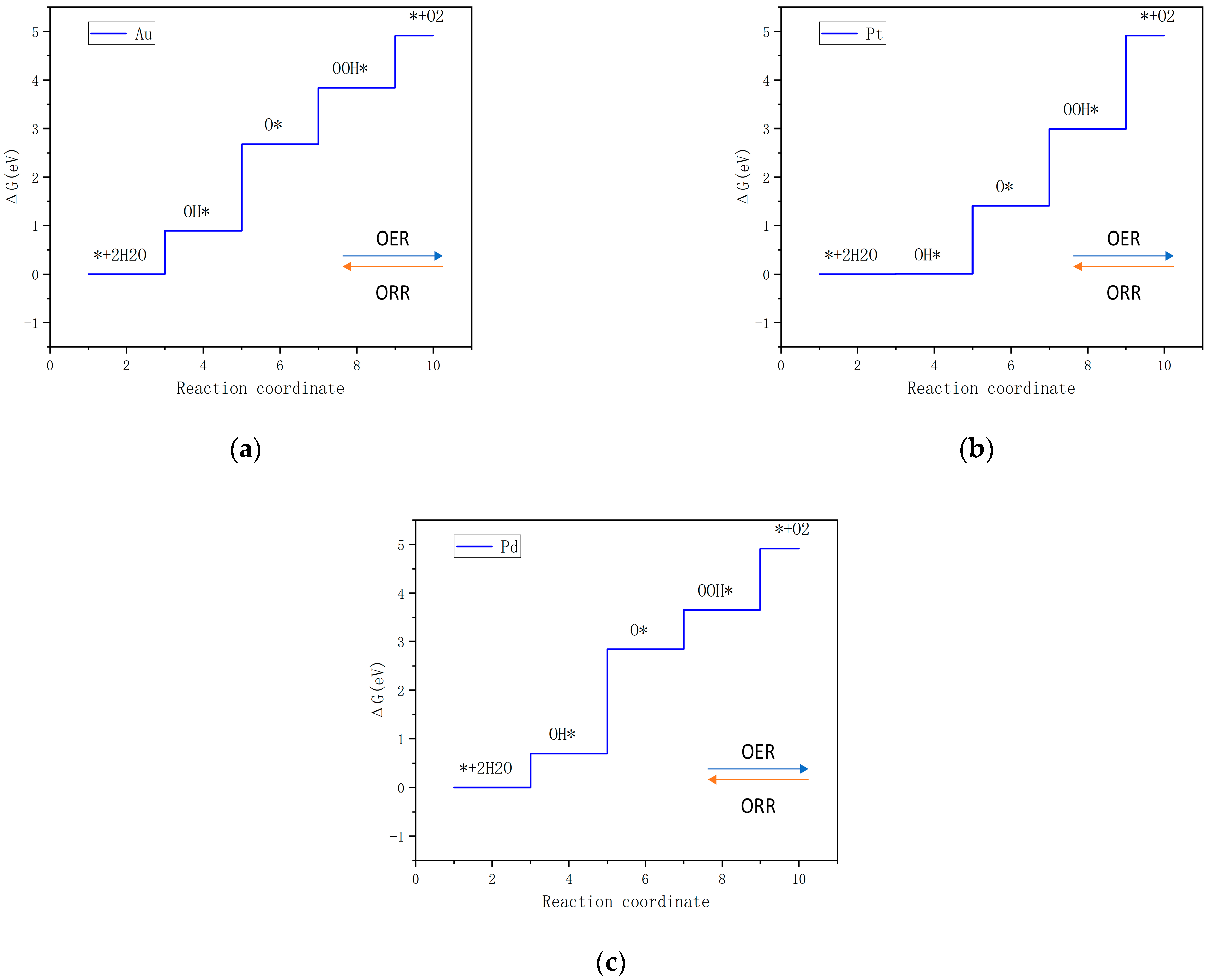
In
Figure 8a, for TM
3@GPN, the plane where the red dots are located is the projection of HER and OER, the plane where the green dots are located is the projection of OER and ORR, and the plane where the blue dots are the projection of HER and ORR. Depicting the performance of various electrocatalysts, elements that are closer to the center are generally considered to exhibit better catalytic performance across all three reactions (OER, ORR, and HER) because they effectively balance the overpotentials. Au
3@GPN exhibits balanced performance with near-ideal HER activity (η HER = −0.03 V), superior ORR efficiency (η ORR = 0.34 V), and competitive OER kinetics (η OER = 0.56 V), outperforming Pt
3@GPN in OER (η OER = 0.70 V). While Pd
3@GPN shows strong HER activity (η HER = −0.11 V), its higher OER overpotential (η OER = 0.91 V) limits bifunctionality. Notably, Ni
3@GPN (η OER = 0.60 V) and Rh
3@GPN (η ORR = 1.06 V) demonstrate reaction-specific limitations, highlighting Au
3@GPN as the most versatile candidate across all three electrocatalytic processes.
In
Figure 8b, focused on overall water splitting, elements positioned closer to the top-left corner are preferred because they show better bifunctional catalytic performance for both the OER and HER, which are the two key reactions for overall water splitting.
3. Computational Details
All computational investigations were conducted employing the Vienna ab initio simulation package (VASP) [
32,
33], implementing the projector-augmented wave (PAW) formalism [
34] within a spin-polarized DFT framework.
The exchange-correlation interactions were modeled by implementing the Perdew–Burke–Ernzerhof (PBE) functional [
35], with plane-wave basis sets truncated at 300 eV. Convergence thresholds were rigorously defined as 0.02 eV Å
−1 for atomic forces and 10
−4 eV for total energy minimization. A 2 × 2 × 1 GPN supercell was chosen, and a vacuum gap of 20 Å was set to preclude interplay between two adjacent surfaces. For structural relaxation procedures, the Brillouin zone integration was configured using a 3 × 3 × 1 k-point mesh. First-principles molecular dynamics (AIMD) [
36] calculations were conducted at 500 K under canonical ensemble conditions to assess the catalyst’s structural integrity under thermal excitation.
We compared the average binding energies (E
b) of the precious atoms on GPN [
37]. The descriptor is defined as follows:
where E
TM3@GPN, E
GPN, and E
TM are the energies of TM
3@GPN, basic GPN, and the isolated metal atom, respectively.
We compared the cohesive energies (E
coh) of TM
3@GPN. The descriptor is defined as follows:
where E
bulk denotes the energy of the metallic bulk crystalline phase, E
TM represents the isolated metallic species energy, and n corresponds to the atomic coordination number within the pristine metallic lattice. Thermodynamic analysis reveals that the TM
3@GPN configuration exhibits enhanced stability when the calculated E
b values approach or remain below the material’s E
coh, indicating favorable interfacial energetics for structural preservation. The binding energetics of oxygen-containing intermediates (O*, OH*, OOH*, H*) chemisorbed on active catalytic surfaces were systematically evaluated through first-principles calculations. Specifically, the adsorption thermodynamic parameters were quantified using the following computational protocol:
The thermodynamic descriptors (E*, EO*, EOH*, EOOH*, and EH*) correspond to the total electronic energies of the catalytic system with and without specific adsorbates. The energies were calculated from the energies of H2O and H2 molecules in the gas phases.
The thermodynamic analysis employed the computational hydrogen electrode (CHE) framework pioneered by Nørskov et al. [
38], where adsorbate Gibbs free energies (ΔG) were evaluated through a thermodynamic formalism. Free energy variations were determined via the established proton-coupled electron transfer (PCET) equation:
The thermodynamic formalism incorporates three principal components: ΔE (reaction enthalpy), ΔZPE (zero-point vibrational energy correction), and TΔS (entropic contribution at 298.15 K). Gas-phase molecular entropies and vibrational spectra were curated from the NIST Chemistry for precise thermodynamic profiling, where standard acidic conditions (pH = 0) were explicitly defined to emulate proton-exchange membrane fuel cell operational environments. ΔG
U were calculated using the following equation:
The limiting potential (U), a descriptor of the catalyst activity, was calculated using the following equation:
Under acidic conditions (pH = 0), the hydrogen evolution reaction (HER) proceeds through proton reduction at the electrocatalytic interface, governed by the fundamental electrochemical equation:
This reaction pathway adheres to the Volmer–Tafel mechanism under standard thermodynamic conditions (298.15 K, 1 atm), where hydrated protons (H3O+) from the electrolyte undergo sequential adsorption and recombination on active catalytic sites.
The Gibbs free energy variation (ΔG) for elementary reaction steps is thermodynamically defined as:
The enthalpy component (ΔE) is computed via spin-polarized DFT employing the PBE exchange-correlation functional, with dispersion interactions explicitly accounted for at adsorbate–substrate interfaces. Zero-point vibrational energy corrections (ΔZPE) are integrated through harmonic frequency analysis at 298.15 K, while entropic contributions (TΔS) utilize NIST-derived thermodynamic tables. The proton–electron pair (H+ + e−) energy reference state under standard conditions (pH = 0, 0 V vs. RHE) corresponds to half the H2 molecule’s energy. Oxygen’s free energy is determined via the equilibrium O2 + 2H2 → 2H2O(l), yielding ΔG = 4.92 eV at standard temperature (298.15 K), under atmospheric pressure, and in acidic conditions.
The oxygen evolution (OER) and reduction (ORR) mechanisms on graphene-based porous nanostructures (GPN) were analyzed through the first-principles electrochemical model pioneered by Nørskov et al. [
38]. Under proton-rich conditions (pH = 0), the OER proceeds via four proton-coupled electron transfer (PCET) steps governed by the following thermodynamic sequence:
Here, * represents an active site on the base site, and (l) and (g) refer to the liquid and gas phases, respectively.
ORR consists of four basic steps in reverse.
The free energy of each step of OER is defined in order as ΔG9, ΔG10, ΔG11, and ΔG12.
The free energy of each step of ORR is defined in order as ΔG13, ΔG14, ΔG15, and ΔG16.
The theoretical overpotentials of OER (η
OER), ORR (η
ORR) and HER (η
HER) are obtained as follows:


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}