
Design of Multicatalytic Systems Through Self-Assembly


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- cooperative (or synergistic) catalysis, wherein different catalytic groups or moieties (co-catalysts) share their catalytic cycles in a single catalyzed reaction affecting a single bond;
- tandem (or relay catalysis), wherein two or more catalytic systems are involved in non-interfering catalytic cycles to execute a sequence of independent reactions.
2. Design Considerations for Immobilized Multicatalysis and Self-Assembly
3. Weak and Non-Directional Interactions
3.1. Van der Waals Interactions Between Alkyl Chains in Monolayers
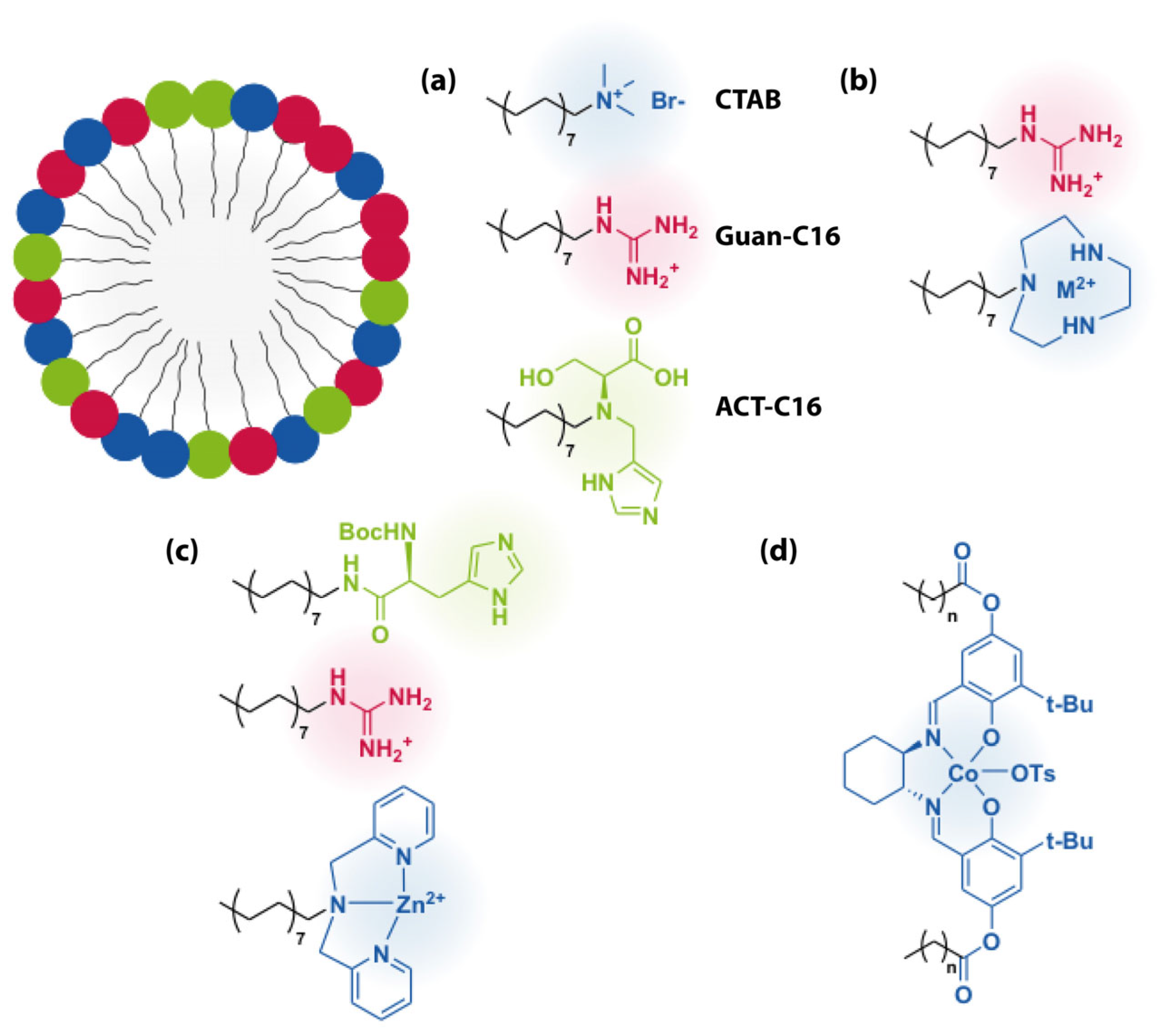
3.2. Hydrophobic Interactions
4. Intermediate Strength and Directional Interactions (Ionic Interactions)
5. Strong and Highly Directional Interactions
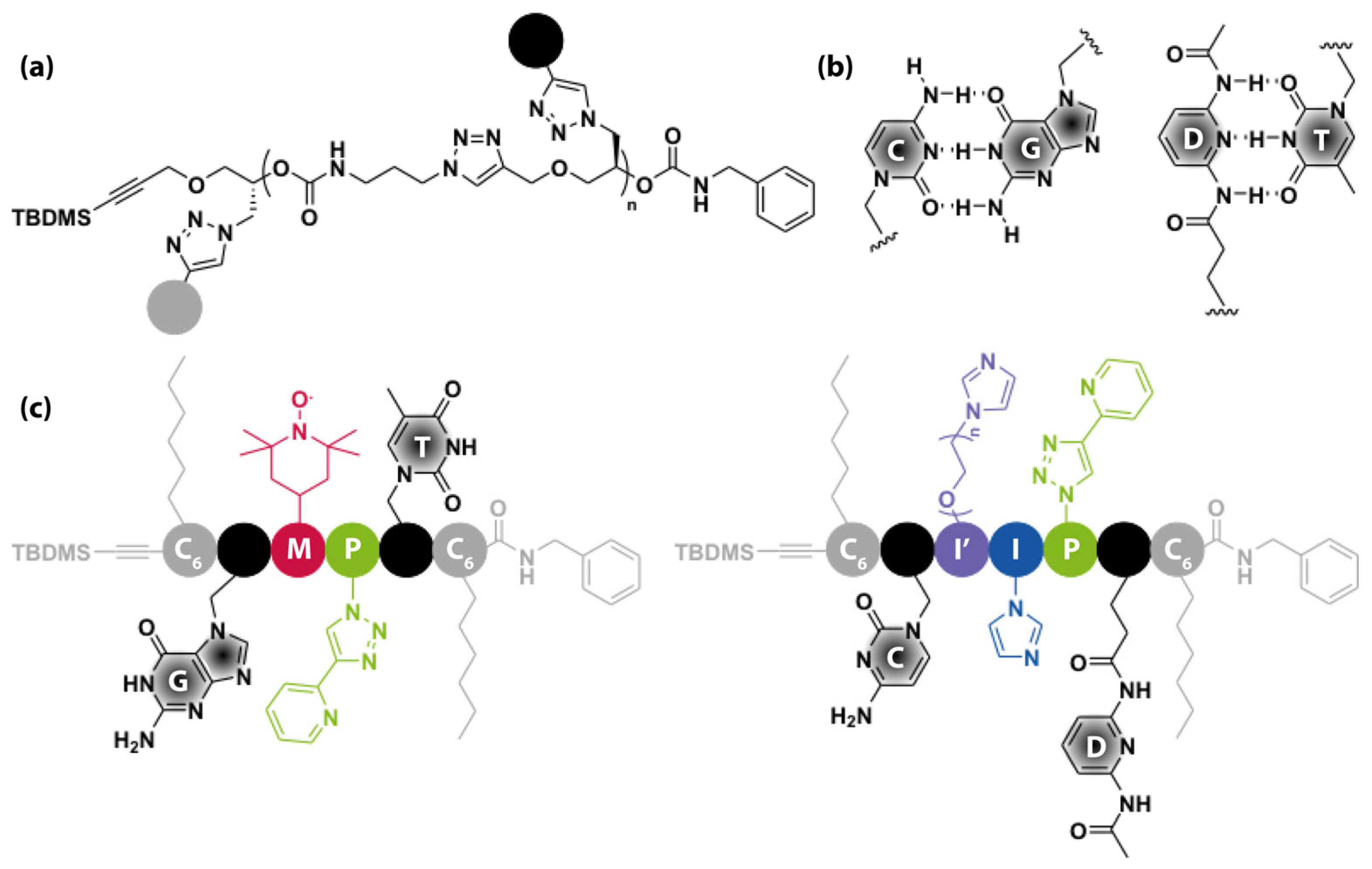
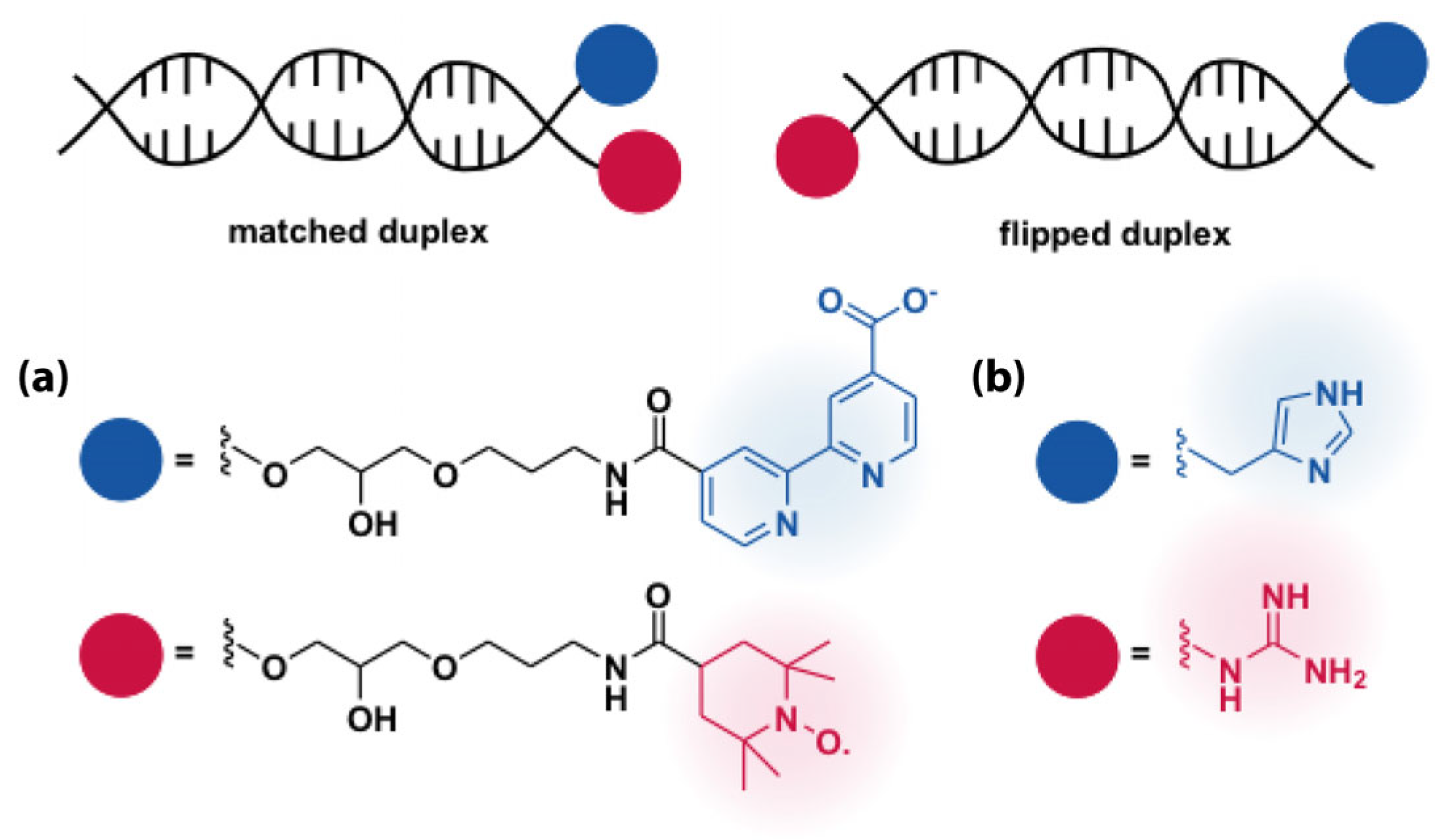
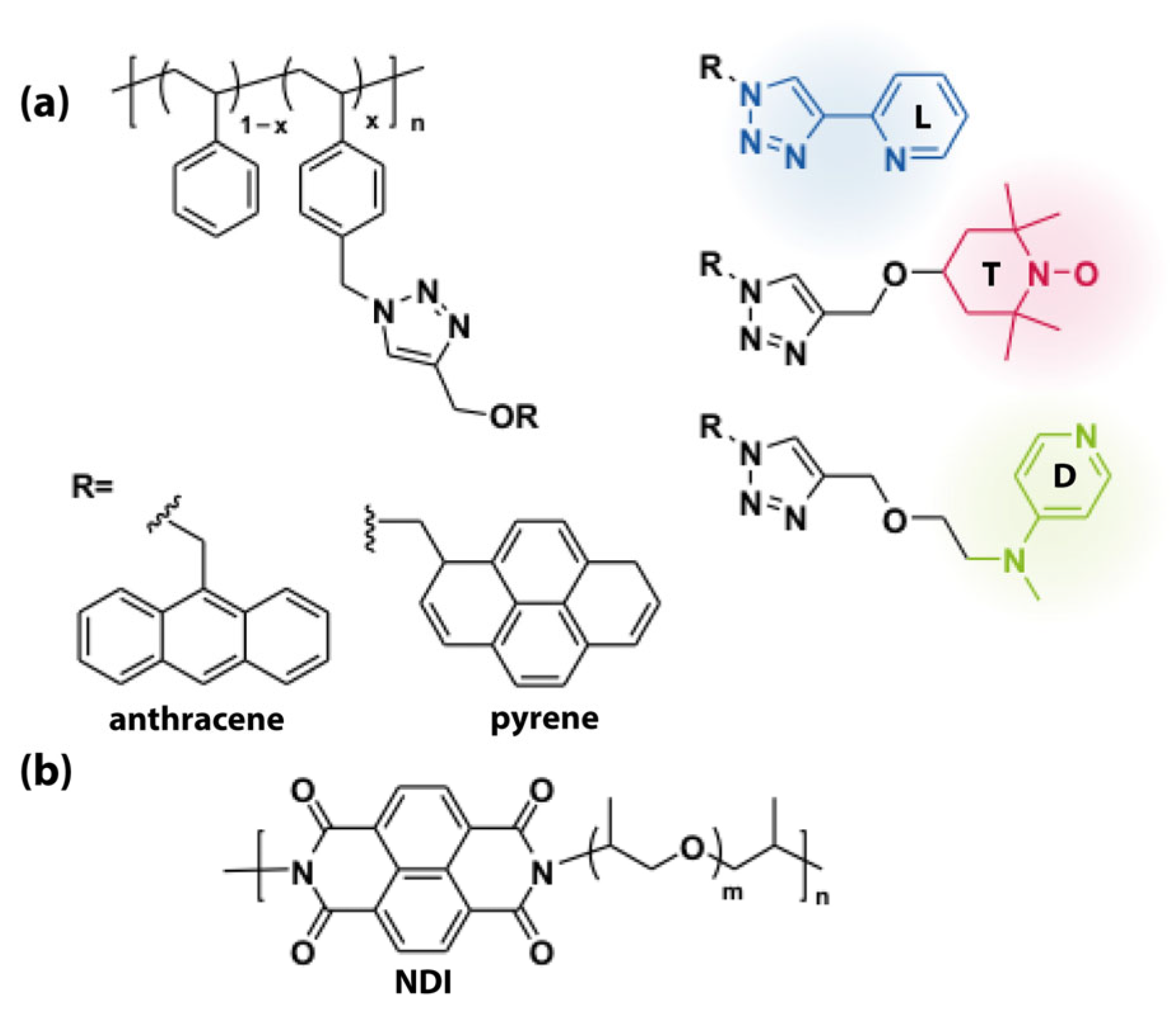
5.1. Hydrogen Bonding
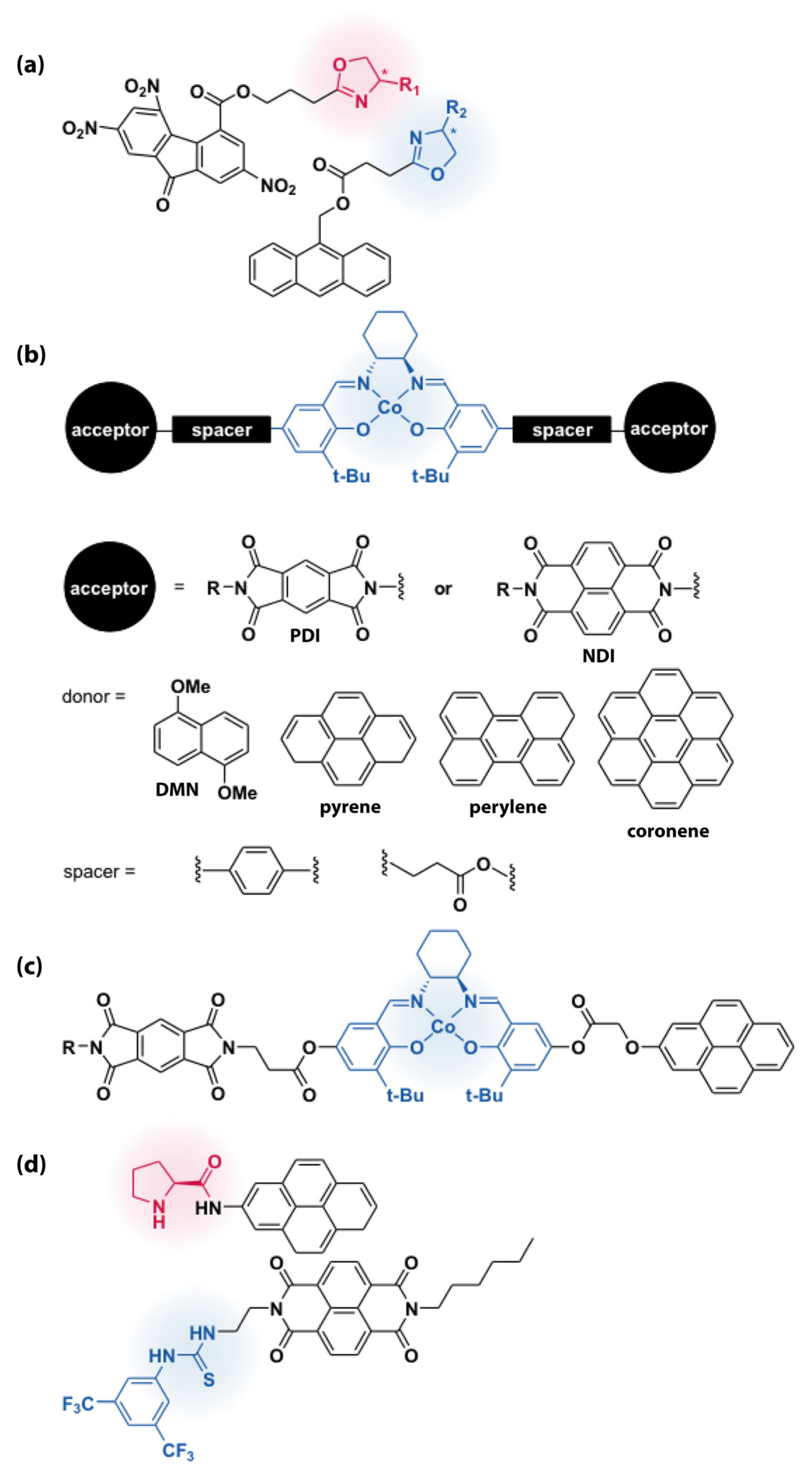
5.2. Aromatic Donor–Acceptor and π-Stacking Interactions
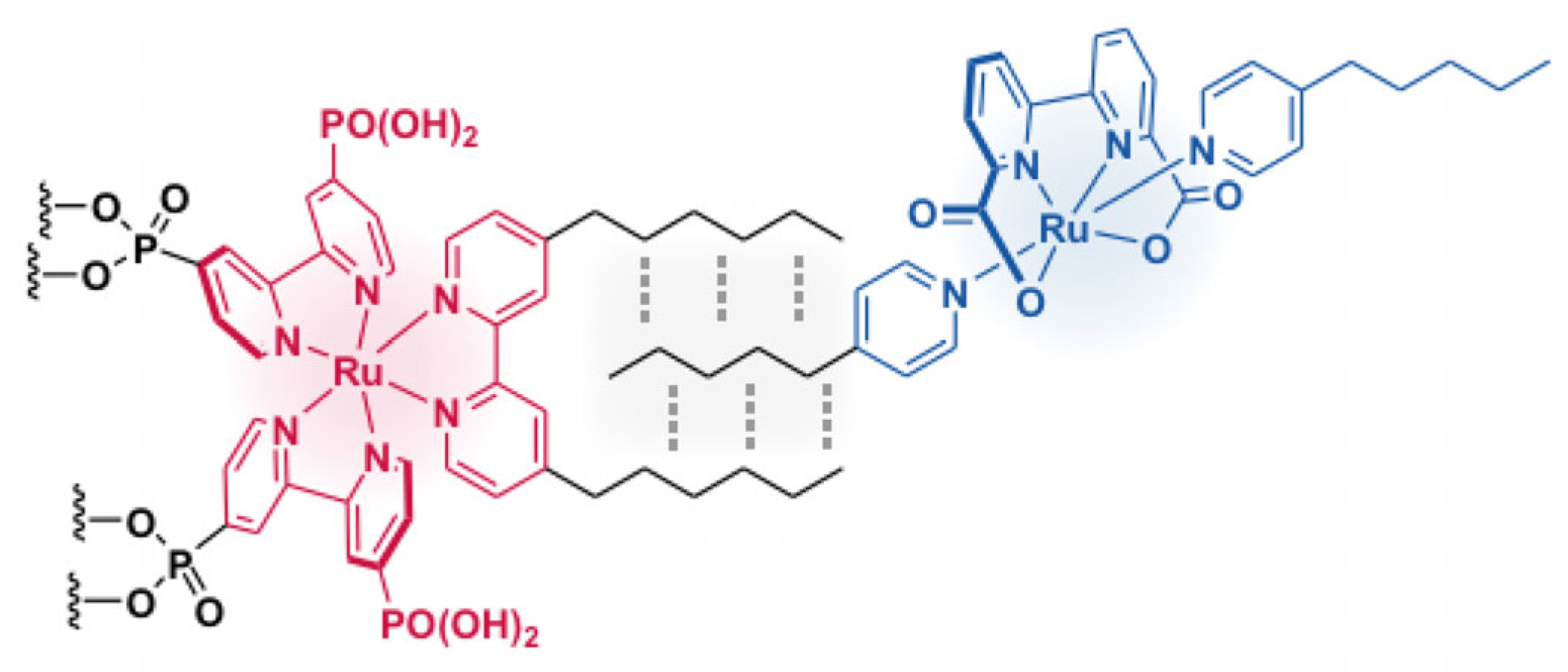
5.3. Metal–Ligand Coordination

6. Conclusions
Funding
Conflicts of Interest
References
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef]
- Chen, K.; Arnold, F.H. Engineering new catalytic activities in enzymes. Nat. Catal. 2020, 3, 203–213. [Google Scholar] [CrossRef]
- Rocha, R.A.; Speight, R.E.; Scott, C. Engineering Enzyme Properties for Improved Biocatalytic Processes in Batch and Continuous Flow. Org. Process Res. Dev. 2022, 26, 1914–1924. [Google Scholar] [CrossRef]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.L.; Texier, H.; Gavalda, S.; et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 2020, 580, 216–219. [Google Scholar] [CrossRef]
- Ingram, A.A.; Oike, K. Artificial Biocatalysis: Quo Vadis? ChemCatChem 2024, 16, e202301759. [Google Scholar] [CrossRef]
- Buller, R.; Lutz, S.; Kazlauskas, R.J.; Snajdrova, R.; Moore, J.C.; Bornscheuer, U.T. From nature to industry: Harnessing enzymes for biocatalysis. Science 2023, 382, eadh8615. [Google Scholar] [CrossRef]
- Lyu, Y.; Scrimin, P. Mimicking Enzymes: The Quest for Powerful Catalysts from Simple Molecules to Nanozymes. ACS Catal. 2021, 11, 11501–11509. [Google Scholar] [CrossRef]
- Kuah, E.; Toh, S.; Yee, J.; Ma, Q.; Gao, Z. Enzyme Mimics: Advances and Applications. Chemistry 2016, 22, 8404–8430. [Google Scholar] [CrossRef]
- Rebilly, J.-N.; Colasson, B.; Bistri, O.; Over, D.; Reinaud, O. Biomimetic cavity-based metal complexes. Chem. Soc. Rev. 2015, 44, 467–489. [Google Scholar] [CrossRef]
- Marchetti, L.; Levine, M. Biomimetic Catalysis. ACS Catal. 2011, 1, 1090–1118. [Google Scholar] [CrossRef]
- Nothling, M.D.; Ganesan, A.; Condic-Jurkic, K.; Pressly, E.; Davalos, A.; Gotrik, M.R.; Xiao, Z.; Khoshdel, E.; Hawker, C.J.; O’Mara, M.L.; et al. Simple Design of an Enzyme-Inspired Supported Catalyst Based on a Catalytic Triad. Chem 2017, 2, 732–745. [Google Scholar] [CrossRef]
- Hammes, G.G.; Benkovic, S.J.; Hammes-Schiffer, S. Flexibility, Diversity, and Cooperativity: Pillars of Enzyme Catalysis. Biochemistry 2011, 50, 10422–10430. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.K.M. Supramolecular Catalysis in Transition. Chem.—A Eur. J. 1998, 4, 1378–1383. [Google Scholar] [CrossRef]
- Kirby, A.J. Enzyme Mechanisms, Models, and Mimics. Angew. Chem. Int. Ed. Engl. 1996, 35, 706–724. [Google Scholar] [CrossRef]
- Szőllősi, G. Asymmetric one-pot reactions using heterogeneous chemical catalysis: Recent steps towards sustainable processes. Catal. Sci. Technol. 2018, 8, 389–422. [Google Scholar] [CrossRef]
- Martínez, S.; Veth, L.; Lainer, B.; Dydio, P. Challenges and Opportunities in Multicatalysis. ACS Catal. 2021, 11, 3891–3915. [Google Scholar] [CrossRef]
- Chen, T.; Xu, Z. Recent advances in co-immobilization of organic acids and bases for cooperative and tandem catalysis. J. Catal. 2024, 435, 115577. [Google Scholar] [CrossRef]
- Chen, T.; Qiu, M.; Peng, Y.; Yi, C.; Xu, Z. Engineering synergistic effects of immobilized cooperative catalysts. Coord. Chem. Rev. 2023, 474, 214863. [Google Scholar] [CrossRef]
- Motokura, K.; Ding, S.; Usui, K.; Kong, Y. Enhanced Catalysis Based on the Surface Environment of the Silica-Supported Metal Complex. ACS Catal. 2021, 11, 11985–12018. [Google Scholar] [CrossRef]
- Fernandes, A.E.; Jonas, A.M. Design and engineering of multifunctional silica-supported cooperative catalysts. Catal. Today 2019, 334, 173–186. [Google Scholar] [CrossRef]
- Jin, R.; Zheng, D.; Liu, R.; Liu, G. Silica-Supported Molecular Catalysts for Tandem Reactions. ChemCatChem 2018, 10, 1739–1752. [Google Scholar] [CrossRef]
- Kasinathan, P.; Lang, C.; Gaigneaux, E.M.; Jonas, A.M.; Fernandes, A.E.; Fernandes, A.E. Influence of Site Pairing in Hydrophobic Silica-Supported Sulfonic Acid Bifunctional Catalysts. Langmuir 2020, 36, 13743–13751. [Google Scholar] [CrossRef] [PubMed]
- Kasinathan, P.; Lang, C.; Radhakrishnan, S.; Schnee, J.; D’Haese, C.; Breynaert, E.; Martens, J.A.; Gaigneaux, E.M.; Jonas, A.M.; Fernandes, A.E. “Click” Silica-Supported Sulfonic Acid Catalysts with Variable Acid Strength and Surface Polarity. Chemistry 2019, 25, 6753–6762. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qin, Q.; Kardas, S.; Fossépré, M.; Surin, M.; Fernandes, A.E.; Glinel, K.; Jonas, A.M. Sequence Rules the Functional Connections and Efficiency of Catalytic Precision Oligomers. ACS Catal. 2022, 12, 2126–2131. [Google Scholar] [CrossRef]
- Chandra, P.; Jonas, A.M.; Fernandes, A.E. Synthesis of discrete catalytic oligomers and their potential in silica-supported cooperative catalysis. RSC Adv. 2019, 9, 14194–14197. [Google Scholar] [CrossRef]
- Chandra, P.; Jonas, A.M.; Fernandes, A.E. Sequence and Surface Confinement Direct Cooperativity in Catalytic Precision Oligomers. J. Am. Chem. Soc. 2018, 140, 5179–5184. [Google Scholar] [CrossRef]
- Chandra, P.; Jonas, A.M.; Fernandes, A.E. Spatial Coordination of Cooperativity in Silica-Supported Cu/TEMPO/Imidazole Catalytic Triad. ACS Catal. 2018, 8, 6006–6011. [Google Scholar] [CrossRef]
- Fernandes, A.E.; Riant, O.; Jonas, A.M.; Jensen, K.F. One “Click” to controlled bifunctional supported catalysts for the Cu/TEMPO-catalyzed aerobic oxidation of alcohols. RSC Adv. 2016, 6, 36602–36605. [Google Scholar] [CrossRef]
- Fernandes, A.E.; Riant, O.; Jensen, K.F.; Jonas, A.M. Molecular Engineering of Trifunctional Supported Catalysts for the Aerobic Oxidation of Alcohols. Angew. Chem. Int. Ed. 2016, 55, 11044–11048. [Google Scholar] [CrossRef]
- Olivo, G.; Capocasa, G.; Del Giudice, D.; Lanzalunga, O.; Di Stefano, S. New horizons for catalysis disclosed by supramolecular chemistry. Chem. Soc. Rev. 2021, 50, 7681–7724. [Google Scholar] [CrossRef]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W. Supramolecular catalysis. Part 1: Non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W. Supramolecular catalysis. Part 2: Artificial enzyme mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Luo, Q.; Liu, J. Artificial enzymes based on supramolecular scaffolds. Chem. Soc. Rev. 2012, 41, 7890–7908. [Google Scholar] [CrossRef] [PubMed]
- Wiester, M.J.; Ulmann, P.A.; Mirkin, C.A. Enzyme mimics based upon supramolecular coordination chemistry. Angew. Chem. Int. Ed. Engl. 2011, 50, 114–137. [Google Scholar] [CrossRef]
- Dong, Z.; Yongguo, W.; Yin, Y.; Liu, J. Supramolecular enzyme mimics by self-assembly. Curr. Opin. Colloid. Interface Sci. 2011, 16, 451–458. [Google Scholar] [CrossRef]
- Meeuwissen, J.; Reek, J.N. Supramolecular catalysis beyond enzyme mimics. Nat. Chem. 2010, 2, 615–621. [Google Scholar] [CrossRef]
- Zhang, B.; Reek, J.N.H. Supramolecular Strategies for the Recycling of Homogeneous Catalysts. Chem.—Asian J. 2021, 16, 3851–3863. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Noto, R. “Release and catch” catalytic systems. Green Chem. 2013, 15, 2608–2618. [Google Scholar] [CrossRef]
- Zhang, L.; Luo, S.; Cheng, J.-P. Non-covalent immobilization of asymmetric organocatalysts. Catal. Sci. Technol. 2011, 1, 507–516. [Google Scholar] [CrossRef]
- Fraile, J.M.; García, J.I.; Mayoral, J.A. Noncovalent Immobilization of Enantioselective Catalysts. Chem. Rev. 2009, 109, 360–417. [Google Scholar] [CrossRef]
- Liu, F.; Qu, P.; Weiss, J.; Guo, K.; Weck, M. Substrate Channeling in Compartmentalized Nanoreactors. Macromolecules 2024, 57, 6805–6815. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.J.; Lee, Y.S.; Yuan, M.; Alkotaini, B.; Zhao, J.; Blumenthal, E.; Minteer, S.D. Substrate Channeling by a Rationally Designed Fusion Protein in a Biocatalytic Cascade. JACS Au 2021, 1, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Chavan, K.S.; Calabrese Barton, S. Simulation of Intermediate Channeling by Nanoscale Confinement. J. Phys. Chem. C 2018, 122, 14474–14480. [Google Scholar] [CrossRef]
- Ke, G.; Liu, M.; Jiang, S.; Qi, X.; Yang, Y.R.; Wootten, S.; Zhang, F.; Zhu, Z.; Liu, Y.; Yang, C.J.; et al. Directional Regulation of Enzyme Pathways through the Control of Substrate Channeling on a DNA Origami Scaffold. Angew. Chem. Int. Ed. 2016, 55, 7483–7486. [Google Scholar] [CrossRef]
- Chen, T.; Peng, Y.; Qiu, M.; Yi, C.; Xu, Z. Heterogenization of homogeneous catalysts in polymer nanoparticles: From easier recovery and reuse to more efficient catalysis. Coord. Chem. Rev. 2023, 489, 215195. [Google Scholar] [CrossRef]
- Nestl, B.M.; Hauer, B. Engineering of Flexible Loops in Enzymes. ACS Catal. 2014, 4, 3201–3211. [Google Scholar] [CrossRef]
- Kokkinidis, M.; Glykos, N.M.; Fadouloglou, V.E. Protein flexibility and enzymatic catalysis. Adv. Protein Chem. Struct. Biol. 2012, 87, 181–218. [Google Scholar] [CrossRef]
- Pabis, A.; Risso, V.A.; Sanchez-Ruiz, J.M.; Kamerlin, S.C.L. Cooperativity and flexibility in enzyme evolution. Curr. Opin. Struct. Biol. 2018, 48, 83–92. [Google Scholar] [CrossRef]
- Xu, S.; Liu, Y.; Zhang, B.; Li, S.; Ye, X.; Wang, Z.-G. Self-Assembly of Multimolecular Components for Engineering Enzyme-Mimetic Materials. Acc. Mater. Res. 2024, 5, 1072–1086. [Google Scholar] [CrossRef]
- Nicosia, C.; Huskens, J. Reactive self-assembled monolayers: From surface functionalization to gradient formation. Mater. Horiz. 2014, 1, 32–45. [Google Scholar] [CrossRef]
- Wang, L.; Polyansky, D.E.; Concepcion, J.J. Self-Assembled Bilayers as an Anchoring Strategy: Catalysts, Chromophores, and Chromophore-Catalyst Assemblies. J. Am. Chem. Soc. 2019, 141, 8020–8024. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, L.; Brady, M.D.; Dares, C.J.; Meyer, G.J.; Meyer, T.J.; Concepcion, J.J. Self-Assembled Chromophore–Catalyst Bilayer for Water Oxidation in a Dye-Sensitized Photoelectrosynthesis Cell. J. Phys. Chem. C 2019, 123, 30039–30045. [Google Scholar] [CrossRef]
- Dwars, T.; Paetzold, E.; Oehme, G. Reactions in micellar systems. Angew. Chem. Int. Ed. Engl. 2005, 44, 7174–7199. [Google Scholar] [CrossRef]
- Tasçioǧlu, S. Micellar solutions as reaction media. Tetrahedron 1996, 52, 11113–11152. [Google Scholar] [CrossRef]
- Lorenzetto, T.; Fabris, F.; Scarso, A. Recent metallosurfactants for sustainable catalysis in water. Curr. Opin. Colloid Interface Sci. 2023, 64, 101689. [Google Scholar] [CrossRef]
- Nothling, M.D.; Xiao, Z.; Hill, N.S.; Blyth, M.T.; Bhaskaran, A.; Sani, M.A.; Espinosa-Gomez, A.; Ngov, K.; White, J.; Buscher, T.; et al. A multifunctional surfactant catalyst inspired by hydrolases. Sci. Adv. 2020, 6, eaaz0404. [Google Scholar] [CrossRef]
- Ren, C.Z.J.; Solís-Muñana, P.; Warr, G.G.; Chen, J.L.Y. Dynamic and Modular Formation of a Synergistic Transphosphorylation Catalyst. ACS Catal. 2020, 10, 8395–8401. [Google Scholar] [CrossRef]
- Matich, O.; Naiya, M.M.; Salam, J.; Tiban Anrango, B.A.; Chen, J.L.Y. Modular Assembly and Optimization of an Artificial Esterase from Functionalised Surfactants. ChemCatChem 2024, 16, e202400945. [Google Scholar] [CrossRef]
- Solís-Muñana, P.; Salam, J.; Ren, C.Z.J.; Carr, B.; Whitten, A.E.; Warr, G.G.; Chen, J.L.Y. An Amphiphilic (salen)Co Complex—Utilizing Hydrophobic Interactions to Enhance the Efficiency of a Cooperative Catalyst. Adv. Synth. Catal. 2021, 363, 3207–3213. [Google Scholar] [CrossRef]
- Mandal, T.; Zhao, C.G. Modularly designed organocatalytic assemblies for direct nitro-Michael addition reactions. Angew. Chem. Int. Ed. Engl. 2008, 47, 7714–7717. [Google Scholar] [CrossRef]
- Xia, A.B.; Xu, D.Q.; Luo, S.P.; Jiang, J.R.; Tang, J.; Wang, Y.F.; Xu, Z.Y. Dual organocatalytic ion-pair assemblies: A highly efficient approach for the enantioselective oxa-Michael-mannich reaction of salicylic aldehydes with cyclohexenones. Chemistry 2010, 16, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Ohmatsu, K.; Hara, Y.; Ooi, T. In situ generation of ion-paired chiral ligands: Rapid identification of the optimal ligand for palladium-catalyzed asymmetric allylation. Chem. Sci. 2014, 5, 3645–3650. [Google Scholar] [CrossRef]
- Corrêa, G.A.; de Castro, B.; Rebelo, S.L.H. Binuclear Mn(III) and Fe(III) porphyrin nanostructured materials in catalytic reduction of 4-nitrophenol. Catal. Today 2023, 418, 114149. [Google Scholar] [CrossRef]
- Martins, M.B.M.S.; Corrêa, G.A.; Moniz, T.; Medforth, C.J.; de Castro, B.; Rebelo, S.L.H. Nanostructured binuclear Fe(III) and Mn(III) porphyrin materials: Tuning the mimics of catalase and peroxidase activity. J. Catal. 2023, 419, 125–136. [Google Scholar] [CrossRef]
- Correa, G.A.; Kuzniarska-Biernacka, I.; Fernandes, D.M.; Rebelo, S.L.H. Polarized Bimetallic Site Synergy in Ionic Structures of Cu(II), Fe(III), and Mn(III) Porphyrins: Electrochemistry and Catalytic Hydrogenation of Nitroaromatics. Inorg. Chem. 2024, 63, 22865–22879. [Google Scholar] [CrossRef]
- Weis, M.; Waloch, C.; Seiche, W.; Breit, B. Self-Assembly of Bidentate Ligands for Combinatorial Homogeneous Catalysis: Asymmetric Rhodium-Catalyzed Hydrogenation. JACS 2006, 128, 4188–4189. [Google Scholar] [CrossRef]
- Chevallier, F.; Breit, B. Self-assembled bidentate ligands for Ru-catalyzed anti-Markovnikov hydration of terminal alkynes. Angew. Chem. Int. Ed. Engl. 2006, 45, 1599–1602. [Google Scholar] [CrossRef]
- Breit, B.; Seiche, W. Self-assembly of bidentate ligands for combinatorial homogeneous catalysis based on an A-T base-pair model. Angew. Chem. Int. Ed. Engl. 2005, 44, 1640–1643. [Google Scholar] [CrossRef]
- Breit, B.; Seiche, W. Hydrogen bonding as a construction element for bidentate donor ligands in homogeneous catalysis: Regioselective hydroformylation of terminal alkenes. J. Am. Chem. Soc. 2003, 125, 6608–6609. [Google Scholar] [CrossRef]
- Waloch, C.; Wieland, J.; Keller, M.; Breit, B. Self-assembly of bidentate ligands for combinatorial homogeneous catalysis: Methanol-stable platforms analogous to the adenine-thymine base pair. Angew. Chem. Int. Ed. Engl. 2007, 46, 3037–3039. [Google Scholar] [CrossRef]
- Reetz, M.T. Combinatorial transition-metal catalysis: Mixing monodentate ligands to control enantio-, diastereo-, and regioselectivity. Angew. Chem. Int. Ed. Engl. 2008, 47, 2556–2588. [Google Scholar] [CrossRef] [PubMed]
- de Greef, M.; Breit, B. Self-assembled bidentate ligands for the nickel-catalyzed hydrocyanation of alkenes. Angew. Chem. Int. Ed. Engl. 2009, 48, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Wieland, J.; Breit, B. A combinatorial approach to the identification of self-assembled ligands for rhodium-catalysed asymmetric hydrogenation. Nat. Chem. 2010, 2, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Laungani, A.C.; Slattery, J.M.; Krossing, I.; Breit, B. Supramolecular bidentate ligands by metal-directed in situ formation of antiparallel beta-sheet structures and application in asymmetric catalysis. Chemistry 2008, 14, 4488–4502. [Google Scholar] [CrossRef]
- Clarke, M.L.; Fuentes, J.A. Self-assembly of organocatalysts: Fine-tuning organocatalytic reactions. Angew. Chem. Int. Ed. Engl. 2007, 46, 930–933. [Google Scholar] [CrossRef]
- Park, J.; Lang, K.; Abboud, K.A.; Hong, S. Self-assembled dinuclear cobalt(II)-salen catalyst through hydrogen-bonding and its application to enantioselective nitro-aldol (Henry) reaction. J. Am. Chem. Soc. 2008, 130, 16484–16485. [Google Scholar] [CrossRef]
- Ma, D.Y.; Norouzi-Arasi, H.; Sheibani, E.; Wärnmark, K. Dynamic Supramolecular [(Salen)CrCl] Complexes as Efficient Catalysts for Ring Opening of Epoxides. ChemCatChem 2010, 2, 629–632. [Google Scholar] [CrossRef]
- Park, J.; Lang, K.; Abboud, K.A.; Hong, S. Self-assembly approach toward chiral bimetallic catalysts: Bis-urea-functionalized (salen)cobalt complexes for the hydrolytic kinetic resolution of epoxides. Chemistry 2011, 17, 2236–2245. [Google Scholar] [CrossRef]
- Qin, Q.; Li, J.; Dellemme, D.; Fossepre, M.; Barozzino-Consiglio, G.; Nekkaa, I.; Boborodea, A.; Fernandes, A.E.; Glinel, K.; Surin, M.; et al. Dynamic self-assembly of supramolecular catalysts from precision macromolecules. Chem. Sci. 2023, 14, 9283–9292. [Google Scholar] [CrossRef]
- Pimentel, E.B.; Peters-Clarke, T.M.; Coon, J.J.; Martell, J.D. DNA-Scaffolded Synergistic Catalysis. J. Am. Chem. Soc. 2021, 143, 21402–21409. [Google Scholar] [CrossRef]
- Zheng, T.; Tang, Q.; Wan, L.; Zhao, Y.; Xu, R.; Xu, X.; Li, H.; Han, D. Controlled Self-Assembly of the Catalytic Core of Hydrolases Using DNA Scaffolds. Nano Lett. 2023, 23, 2081–2086. [Google Scholar] [CrossRef] [PubMed]
- Chuzel, O.; Magnier-Bouvier, C.; Schulz, E. Assembly of chiral monodentate ligands to chelates by donor–acceptor interactions. Tetrahedron Asymmetry 2008, 19, 1010–1019. [Google Scholar] [CrossRef]
- Blechschmidt, D.R.; Woodhouse, M.D.; Inagaki, S.; Whitfield, M.; Ogunsanya, A.; Yoder, A.; Lilly, D.; Heim, E.W.; Soucie, L.N.; Liang, J.; et al. Aromatic donor-acceptor interaction promoted catalyst assemblies for hydrolytic kinetic resolution of epichlorohydrin. Org. Biomol. Chem. 2018, 17, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Soucie, L.N.; Blechschmidt, D.R.; Yoder, A.; Gustafson, A.; Liu, Y. Aromatic Donor-Acceptor Interaction-Based Co(III)-salen Self-Assemblies and Their Applications in Asymmetric Ring Opening of Epoxides. Org. Lett. 2019, 21, 513–518. [Google Scholar] [CrossRef]
- Meso, G.; Gregorich, D.; Waite, A.J.; Smith, S.; Liu, L.; Cortez, J.; Harris, E.; Baluyut, R.D.; Correa, N.; Liang, J.; et al. Organocatalytic Assembly Based on Aromatic Donor–Acceptor Interactions for the Asymmetric Aldol Reaction on Water. ChemCatChem 2023, 15, e202300012. [Google Scholar] [CrossRef]
- Chen, T.; Xiao, W.; Wang, Z.; Gan, P.; Yi, C.; Xu, Z. Pyrene-Containing Polymer-Supported Cu/TEMPO Catalytic Systems: Aromatic Stacking-Enhanced Cooperative Catalysis. J. Phys. Chem. C 2021, 126, 309–316. [Google Scholar] [CrossRef]
- Chen, T.; Xiao, W.; Yang, J.; Qiu, M.; Yi, C.; Xu, Z. Polyimide-supported Cu/2,2,6,6-tetramethyl-1-piperidine-N-oxyl catalytic systems: Aromatic donor-acceptor interaction-directed cooperative catalysis. J. Colloid. Interface Sci. 2022, 622, 202–208. [Google Scholar] [CrossRef]
- Chen, L.A.; Xu, W.; Huang, B.; Ma, J.; Wang, L.; Xi, J.; Harms, K.; Gong, L.; Meggers, E. Asymmetric catalysis with an inert chiral-at-metal iridium complex. J. Am. Chem. Soc. 2013, 135, 10598–10601. [Google Scholar] [CrossRef]
- Xu, W.; Arieno, M.; Low, H.; Huang, K.; Xie, X.; Cruchter, T.; Ma, Q.; Xi, J.; Huang, B.; Wiest, O.; et al. Metal-Templated Design: Enantioselective Hydrogen-Bond-Driven Catalysis Requiring Only Parts-per-Million Catalyst Loading. J. Am. Chem. Soc. 2016, 138, 8774–8780. [Google Scholar] [CrossRef]
- Chen, L.A.; Tang, X.; Xi, J.; Xu, W.; Gong, L.; Meggers, E. Chiral-at-metal octahedral iridium catalyst for the asymmetric construction of an all-carbon quaternary stereocenter. Angew. Chem. Int. Ed. Engl. 2013, 52, 14021–14025. [Google Scholar] [CrossRef]
- Ma, J.; Ding, X.; Hu, Y.; Huang, Y.; Gong, L.; Meggers, E. Metal-templated chiral Bronsted base organocatalysis. Nat. Commun. 2014, 5, 4531. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Lin, H.; Gong, L.; Meggers, E. Enantioselective Sulfa-Michael Addition to α,β-Unsaturated γ-Oxoesters Catalyzed by a Metal-Templated Chiral Brønsted Base. Asian J. Org. Chem. 2015, 4, 434–437. [Google Scholar] [CrossRef]
- Hu, Y.; Zhou, Z.; Gong, L.; Meggers, E. Asymmetric aza-Henry reaction to provide oxindoles with quaternary carbon stereocenter catalyzed by a metal-templated chiral Brønsted base. Org. Chem. Front. 2015, 2, 968–972. [Google Scholar] [CrossRef]
- Liu, J.; Gong, L.; Meggers, E. Asymmetric Friedel–Crafts alkylation of indoles with 2-nitro-3-arylacrylates catalyzed by a metal-templated hydrogen bonding catalyst. Tetrahedron Lett. 2015, 56, 4653–4656. [Google Scholar] [CrossRef]
- Ding, X.; Tian, C.; Hu, Y.; Gong, L.; Meggers, E. Tuning the Basicity of a Metal-Templated Brønsted Base to Facilitate the Enantioselective Sulfa-Michael Addition of Aliphatic Thiols to α,β-Unsaturated N-Acylpyrazoles. Eur. J. Org. Chem. 2016, 2016, 887–890. [Google Scholar] [CrossRef]
- Huang, K.; Ma, Q.; Shen, X.; Gong, L.; Meggers, E. Metal-Templated Asymmetric Catalysis: (Z)-1-Bromo-1-Nitrostyrenes as Versatile Substrates for Friedel–Crafts Alkylation of Indoles. Asian J. Org. Chem. 2016, 5, 1198–1203. [Google Scholar] [CrossRef]
- Ma, Q.; Gong, L.; Meggers, E. Enantioselective β-alkylation of pyrroles with the formation of an all-carbon quaternary stereocenter. Org. Chem. Front. 2016, 3, 1319–1325. [Google Scholar] [CrossRef]
- Fanourakis, A.; Docherty, P.J.; Chuentragool, P.; Phipps, R.J. Recent Developments in Enantioselective Transition Metal Catalysis Featuring Attractive Noncovalent Interactions between Ligand and Substrate. ACS Catal. 2020, 10, 10672–10714. [Google Scholar] [CrossRef]
- Serra-Pont, A.; Alfonso, I.; Jimeno, C.; Sola, J. Dynamic assembly of a zinc-templated bifunctional organocatalyst in the presence of water for the asymmetric aldol reaction. Chem. Commun. 2015, 51, 17386–17389. [Google Scholar] [CrossRef]
- Serra-Pont, A.; Alfonso, I.; Sola, J.; Jimeno, C. A copper-templated, bifunctional organocatalyst: A strongly cooperative dynamic system for the aldol reaction. Org. Biomol. Chem. 2017, 15, 6584–6591. [Google Scholar] [CrossRef]
- Serra-Pont, A.; Alfonso, I.; Sola, J.; Jimeno, C. An efficient dynamic asymmetric catalytic system within a zinc-templated network. Chem. Commun. 2019, 55, 7970–7973. [Google Scholar] [CrossRef] [PubMed]
- Sors-Vendrell, A.; Ortiz, A.; Meneses, D.; Alfonso, I.; Sola, J.; Jimeno, C. A Degenerate Metal-Templated Catalytic System with Redundant Functional Groups for the Asymmetric Aldol Reaction. J. Org. Chem. 2022, 87, 7509–7513. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, C.; Solà, J.; Alfonso, I. In Situ Assembly of a Metal-Templated Catalyst for Asymmetric Aldol Reactions: From the Metal Complexes Equilibria to a Practical System. Eur. J. Org. Chem. 2024, 27, e202301298. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, A.E.; Jonas, A.M. Design of Multicatalytic Systems Through Self-Assembly. Catalysts 2025, 15, 265. https://doi.org/10.3390/catal15030265
Fernandes AE, Jonas AM. Design of Multicatalytic Systems Through Self-Assembly. Catalysts. 2025; 15(3):265. https://doi.org/10.3390/catal15030265
Chicago/Turabian StyleFernandes, Antony E., and Alain M. Jonas. 2025. "Design of Multicatalytic Systems Through Self-Assembly" Catalysts 15, no. 3: 265. https://doi.org/10.3390/catal15030265
APA StyleFernandes, A. E., & Jonas, A. M. (2025). Design of Multicatalytic Systems Through Self-Assembly. Catalysts, 15(3), 265. https://doi.org/10.3390/catal15030265