Abstract
Achieving selective, direct conversion of methane into value-added chemicals requires catalysts that can navigate the intrinsic trade-off between C–H bond activation and over-dehydrogenation. Transition metal phosphides (TMPs) have emerged as promising catalysts that can tune this selectivity. This work utilizes density functional theory (DFT) to systematically assess how the transition metal’s identity (M = Fe, Co, Ni) in isostructural M2P phosphides governs this balance. The findings reveal that the high reactivity of Fe2P and Co2P, which facilitates initial methane activation, also promotes facile deep dehydrogenation pathways to coke precursors like CH*. In stark contrast, Ni2P exhibits a moderated reactivity that kinetically hinders CH* formation while simultaneously exhibiting the lowest activation barrier for the C–C coupling of CH2* intermediates to form ethylene. This revealed trade-off between the high reactivity of Fe/Co phosphides and the high selectivity of Ni2P offers a guiding principle for the rational design of advanced bimetallic phosphides for efficient methane upgrading.
1. Introduction
Methane (CH4), the main component of natural gas, is both a vast C1 feedstock and a potent greenhouse gas, creating strong motivation for its conversion into value-added chemicals like olefins and aromatics [1,2,3,4,5,6]. Currently, the industrial-scale conversion of methane into liquid fuels and chemicals proceeds primarily through an indirect route. This process involves the initial, energy-intensive reforming of methane to produce synthesis gas (syngas), a mixture of carbon monoxide (CO) and hydrogen (H2), which is subsequently converted into longer-chain hydrocarbons via established technologies like Fischer–Tropsch synthesis [7,8]. However, the syngas production step requires extreme operating conditions, with temperatures often exceeding 700 °C, which renders the overall process economically costly and entails a substantial energy penalty [9]. Consequently, the direct catalytic conversion of methane in a single step represents a paramount goal in heterogeneous catalysis, offering the potential for simplified process schemes and reduced energy consumption [10].
Direct methane conversion strategies are broadly categorized into oxidative and non-oxidative pathways. The oxidative coupling of methane (OCM) has been extensively investigated but is fundamentally constrained by a persistent challenge: the desired C2 products (ethane and ethylene) are significantly more reactive than methane itself under oxidizing conditions [11,12]. This leads to the facile over-oxidation of products to thermodynamically favored but undesirable carbon oxides (COx), severely limiting the achievable selectivity and yield.
The non-oxidative coupling of methane (NOCM), represented by the reaction:
offers a simpler and more carbon-efficient alternative by circumventing the formation of COx byproducts [13,14,15,16]. However, the reaction is highly endothermic due to the thermodynamic stability of the methane molecule and the high dissociation energy of its first C–H bond [17], necessitating extreme temperatures to achieve viable conversion rates. These harsh conditions, in turn, promote rapid catalyst deactivation through the formation of coke. The central challenge in NOCM is therefore kinetic: an ideal catalyst must be active enough to cleave the first C–H bond but selective enough to prevent the subsequent, facile dehydrogenation steps that lead to deeply dehydrogenated coke precursors such as CH* and C*.
2CH4 → C2H6 + H2
In this context, transition metal phosphides (TMPs) have emerged as a highly promising class of catalysts. The incorporation of phosphorus into a transition metal lattice induces significant geometric and electronic modifications [18]. Geometrically, P atoms can act as spacers, breaking up large, highly reactive contiguous metal ensembles that are often responsible for deep dehydrogenation and coking on pure metal surfaces. Electronically, charge transfer between the metal and the more electronegative phosphorus atoms modifies the d-band structure of the metal sites, creating a unique bifunctional surface that can be tuned to control catalytic behavior. TMPs are well-established as superior catalysts for hydroprocessing reactions, where they exhibit exceptional selectivity for cleaving C–O, C–S, and C–N bonds, along with high resistance to coking [19,20,21,22,23,24].
In our previous work, we used density functional theory (DFT) to investigate methane activation on the Ni-rich (001) surface of nickel phosphide (Ni2P) [25]. We found that while Ni2P was less reactive for the initial C–H bond scission (ΔHact = 246 kJ mol−1) than pure Ni, this moderated reactivity was key to its selectivity. The presence of phosphorus kinetically hindered the deep dehydrogenation of methane and favored the accumulation of CH3* and CH2* intermediates on the surface. The CH2* species can then be coupled to form ethylene with a relatively low activation barrier compared to further dehydrogenation. These computational findings were consistent with experimental reports showing high selectivity towards C2 products on silica-supported nickel phosphide catalysts [26,27]. This work established the principle that P-modification can alter reactivity to induce selectivity, but it left open the critical question of how the identity of the transition metal in the M2P framework affects this balance.
Here, we extend our previous study by using DFT to systematically investigate methane activation and coupling on the (001) surfaces of iron phosphide (Fe2P) and cobalt phosphide (Co2P), directly comparing their performance to that of the Ni2P catalyst. The results reveal a clear trend in reactivity and selectivity across these isostructural catalysts. The findings indicate that Co2P is the most reactive catalyst, facilitating the initial C–H bond scission with barriers as low as 85 kJ mol−1; however, this high activity is coupled with kinetically facile pathways toward deep dehydrogenation. Conversely, Ni2P is the least reactive but it exhibits the highest selectivity for ethylene formation by kinetically favoring C–C coupling over C–H scission of the CH2* intermediate. Fe2P presents an intermediate case in both reactivity and selectivity. This work elucidates a fundamental reactivity–selectivity trade-off in metal phosphides and suggests that future catalyst design could focus on creating bimetallic phosphide systems that combine the high activity of one metal with the high selectivity of another to optimize performance for direct methane conversion.
2. Results and Discussion
2.1. Optimized Structures and Binding Energies of C1 Intermediates
This section examines the optimized adsorption geometries and corresponding binding energies (ΔEads) of methane-derived intermediates (CHx*, x = 0–4) and atomic hydrogen (H*) on the isostructural (001) surfaces of Fe2P and Co2P. These results are compared with our previous findings for Ni2P to elucidate trends across the Group 8–10 transition metal phosphides [25].
The DFT-optimized structures and calculated binding energies on Fe2P and Co2P are shown in Figure 1. On all three M2P surfaces, the methane molecule (CH4*) is weakly physisorbed with a binding energy near 0 kJ mol−1 (Figure 1a,g), indicating minimal interaction with the catalyst surface prior to activation. This is in contrast to the highly reactive oxide surfaces like IrO2(110), where methane forms a strong dative bond that pre-activates its C–H bond [2,28,29]. The absence of such pre-activation on phosphide surfaces suggests that the initial C–H bond scission is a surface-mediated event requiring the overcoming of a substantial kinetic barrier, rather than a process facilitated by initial molecular destabilization.
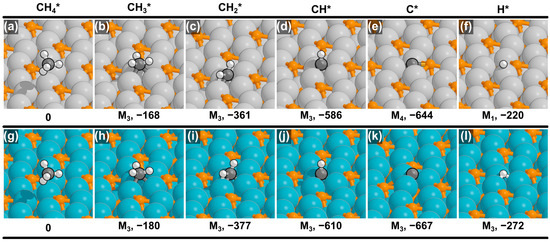
Figure 1.
DFT-optimized structures for CHx* (x = 0–4) and H* on the (a–f) Fe2P(001) and (g–l) Co2P(001) surfaces. The binding energies (in kJ mol−1) and adsorption modes are shown beneath each image (see Section 3 for more details).
Following the initial C–H scission, a notable difference in binding preference emerges compared to our previous work on Ni2P. On both Fe2P and Co2P, all C1 intermediates (CH3*, CH2*, CH*, and C*) preferentially adsorb at high-coordination metallic hollow sites (M3 or M4) rather than interacting with surface phosphorus atoms. On the Ni2P catalyst, however, the less dehydrogenated species CH3* and CH2* favored binding to P-containing sites (P1 and MP, respectively) [25]. These differences in preferred adsorption sites, despite the isostructural nature of the catalyst surfaces examined here, point to a fundamental difference in their electronic properties. The electronic structure of a transition metal phosphide is governed by the interaction between the metal’s d-band and the electron-withdrawing nature of phosphorus. For the more reactive Fe and Co metals, the M3 and M4 hollow sites appear to remain the most energetically favorable binding locations for C1 intermediates, even after modification by phosphorus. In contrast, on the less intrinsically reactive Ni2P surface, the electronic passivation by phosphorus appears to deactivate the Ni hollow sites to a degree that makes the P-containing sites (P1 and MP) competitive for the adsorption of CH3* and CH2*.
A systematic comparison of the binding energies, visualized in the heatmap in Figure 2, highlights the key trends in intermediate stability. On all three catalysts, the binding strength of CHx* intermediates increases monotonically with the degree of dehydrogenation (i.e., as x decreases from 3 to 0). This is an expected result, as species with fewer hydrogen atoms have more available electrons to form stronger covalent bonds with the catalyst surface, a trend widely observed in DFT studies of hydrocarbon chemistry on metal surfaces [30,31,32].
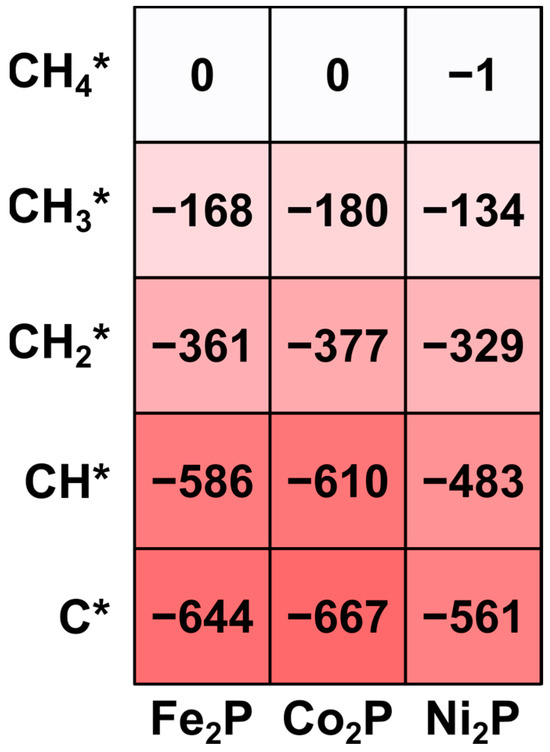
Figure 2.
Heatmap of DFT-predicted binding energies (ΔEads) for CHₓ* (x = 0–4) intermediates on the (001) surfaces of Fe2P, Co2P, and Ni2P. The color scale corresponds to the binding strength in kJ mol−1, with the gradient ranging from dark red for the strongest binding to white for the weakest binding.
More importantly, the results show that the Fe2P and Co2P surfaces are significantly more reactive towards C1 intermediates than the Ni2P surface. This is evidenced by the consistently stronger (more negative) binding energies on Fe2P and Co2P across all CHx* species. For the key CH2* intermediate, for instance, the binding energies on Fe2P (−361 kJ mol−1) and Co2P (−377 kJ mol−1) are more exothermic than on Ni2P (−329 kJ mol−1). This reactivity difference becomes even more pronounced for the deeply dehydrogenated C* species, a common coke precursor. The C* atom binds very strongly to the Fe2P (−644 kJ mol−1) and Co2P (−667 kJ mol−1) surfaces, whereas its interaction with Ni2P (−561 kJ mol−1) is substantially weaker. This indicates that the Ni2P surface is electronically distinct and less reactive, a property that suggests it may be more resistant to coking than Fe2P and Co2P.
To better understand the electronic origins of these differences in adsorption behavior, a Bader charge analysis was performed on the clean M2P(001) surfaces [33,34,35]. The results show that in all cases, the more electronegative phosphorus atoms withdraw electron density from the metal atoms, creating partially positive, Lewis acidic metal sites. The calculated average charge on the surface metal atoms follows the order Fe2P (+0.53 e−) > Co2P (+0.33 e−) > Ni2P (+0.27 e−). This analysis quantifies the electronic modification across the catalysts. The significantly lower positive charge on the Ni atoms indicates a more moderated electronic perturbation by phosphorus compared to Fe and Co. This distinct electronic state likely contributes to the weaker binding of intermediates, the unique preference of CH3* and CH2* for P-containing sites on Ni2P, and ultimately, its superior selectivity for C–C coupling over deep dehydrogenation.
2.2. Methane Dehydrogenation Pathways
Following the analysis of the surface thermodynamics, this section investigates the kinetics of the four successive C–H bond activation steps of methane on the Fe2P, Co2P, and Ni2P surfaces. The activation and reaction enthalpies reported in Table 1 are referenced to the most stable, pre-adsorbed configurations of the reactant and product species on the surface. For each C–H scission event, the resulting hydrogen atom is assumed to recombine and desorb from the surface as ½H2(g). The reactant and product structures illustrated in figures such as Figure 3, however, represent the immediate local minima connected by the transition state, excluding any subsequent diffusion to or from more favorable binding sites.

Table 1.
DFT-predicted forward activation enthalpy (ΔHact = HTS − Hreactants) and enthalpy of reaction (ΔHrxn = Hproducts − Hreactants) for methane dehydrogenation reactions at 1123 K (kJ mol−1).
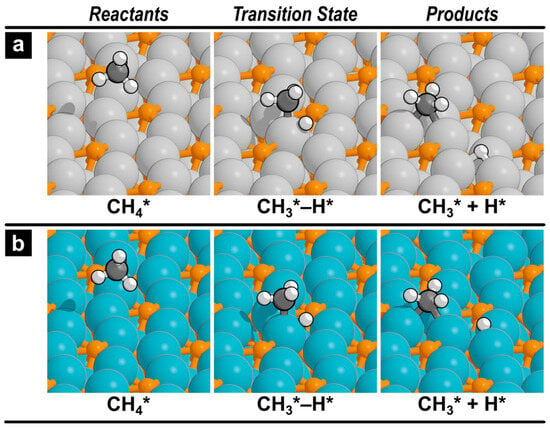
Figure 3.
Reactant, transition state, and product structures for the initial methane activation step on (a) the Fe2P(001) surface and (b) the Co2P(001) surface. Structures for further dehydrogenation reactions are provided in the Supporting Information (Figures S1 and S2).
The initial C–H bond scission in methane is widely recognized as the rate-limiting step for direct methane conversion on most catalyst surfaces, because of the high C–H bond dissociation energy of methane [17,30,31,32]. Our calculations identified highly active pathways on the Fe2P and Co2P surfaces that occur exclusively on multi-metal hollow sites for the first C–H scission. The activation barriers on these sites, presented in Table 1, are remarkably low: 85 kJ mol−1 on Co2P and 101 kJ mol−1 on Fe2P. These values are not only significantly lower than the barrier on Ni2P (246 kJ mol−1) but are also comparable to, or even lower than, those on reactive pure metal surfaces [30,31,32]. Consequently, for Fe2P and Co2P, the initial C–H activation is no longer the most energy-intensive or rate-limiting step. Instead, the phosphorus atoms appear to moderate the subsequent reaction steps rather than simply passivating the initial activity. The structures for this activation step on Fe2P and Co2P, shown in Figure 3, reveal a three-centered transition state involving one metal atom and the breaking C–H bond, a common mechanism observed on various metal surfaces.
Following the initial C–H activation, the kinetics of the subsequent dehydrogenation steps reveal critical differences among these catalysts that govern their selectivity (Table 1). For the second dehydrogenation step to form CH2*, Co2P and Fe2P are again the most active, with barriers of 49 and 52 kJ mol−1, respectively, which are lower than the barrier on Ni2P (65 kJ mol−1). This reactivity trend remains the same for the dehydrogenation of the CH2* species. The formation of CH* is kinetically facile on Fe2P and Co2P (14 and 10 kJ mol−1, respectively), while the same reaction on Ni2P requires a high activation barrier of 84 kJ mol−1. The final dehydrogenation step to atomic carbon (C*) exhibits a high activation barrier on all three surfaces, a finding that is consistent with previous studies on pure metal surfaces [30,31,32].
Figure 4 shows the effective enthalpy reaction coordinate diagram used to assess the methane dehydrogenation pathways on all three catalysts. As discussed previously based on Table 1, the initial C–H activation involves a large energetic barrier on Ni2P compared to Co2P and Fe2P. Following this initial step, a clear reactivity trend emerges for the subsequent dehydrogenation reactions, where the overall reactivity follows the order Co2P > Fe2P > Ni2P.
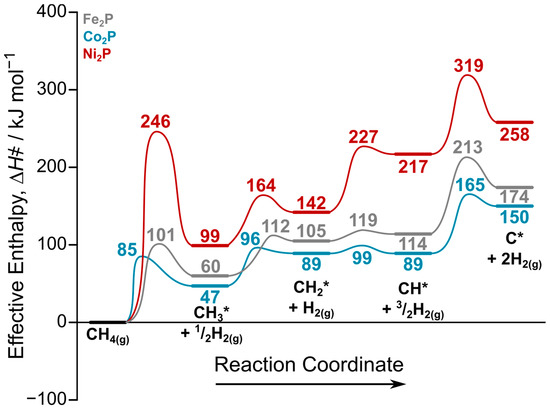
Figure 4.
DFT-predicted reaction coordinate diagram for methane dehydrogenation on Fe2P(001), Co2P(001), and Ni2P(001) surfaces at 1123 K. Effective enthalpies are calculated relative to gas-phase methane and a stoichiometric amount of H2(g) using Equation (3). Each CHx species is located at its most stable adsorption site as discussed in Section 2.1.
The formation of the CH2* intermediate from CH3* proceeds with effective barriers of 96 kJ mol−1 on Co2P, 112 kJ mol−1 on Fe2P, and 164 kJ mol−1 on Ni2P. Once CH2* is formed, its subsequent dehydrogenation is highly catalyst-dependent. On both Fe2P and Co2P, it can readily form CH*, and given that the reverse barriers are also facile while the forward barrier to C* is high, both CH2* and CH* are likely to be the most abundant surface intermediates. In contrast, the profile for Ni2P shows a high effective barrier of 227 kJ mol−1 to form CH*, while the reverse barrier to reform CH2* is only 10 kJ mol−1. This suggests that CH2* and CH3* are the most probable surface intermediates on Ni2P. Furthermore, the effective barrier for C* formation is significantly higher on Ni2P (319 kJ mol−1) compared to Co2P (213 kJ mol−1) and Fe2P (165 kJ mol−1), indicating that it is more resistant to deactivation via coking.
Overall, the reaction coordinate diagram suggests that Ni2P has a unique potential for controlling selectivity towards ethane or ethylene, while the more active Fe2P and Co2P surfaces can readily form CH* species. The possibility of C2 coupling reactions on these surfaces will be discussed next to better assess the selectivity.
2.3. C–C Coupling Reactions and Selectivity Analysis
To assess the potential of each catalyst for producing higher hydrocarbons, the kinetics of C–C coupling reactions were investigated. The forward activation barriers and reaction energies for the formation of ethane, ethylene, and acetylene are listed in Table 2. The results indicate that Ni2P is the most active catalyst for C2 product formation, exhibiting the lowest activation barriers and highest exothermicities, followed by Fe2P and then Co2P. For ethane (*CH3CH3*) formation, the activation barrier on Ni2P is only 98 kJ mol−1, which is significantly lower than on Fe2P (173 kJ mol−1) and Co2P (198 kJ mol−1). A similar reactivity trend is observed for ethylene (*CH2CH2*) formation, though the barriers are considerably lower than for ethane formation on all catalysts. The reaction pathways for ethylene formation on Fe2P and Co2P are shown in Figure 5a,b, where the product is chemisorbed on the surface in a π-bonded configuration. The subsequent desorption of ethylene requires 26 kJ mol−1 on Fe2P and 52 kJ mol−1 on Co2P, while it requires only 7 kJ mol−1 on Ni2P.
Table 2.
DFT-predicted forward activation enthalpy (ΔHact = HTS − Hreactants) and enthalpy of reaction (ΔHrxn = Hproducts − Hreactants) for the C–C coupling reactions at 1123 K (kJ mol−1).
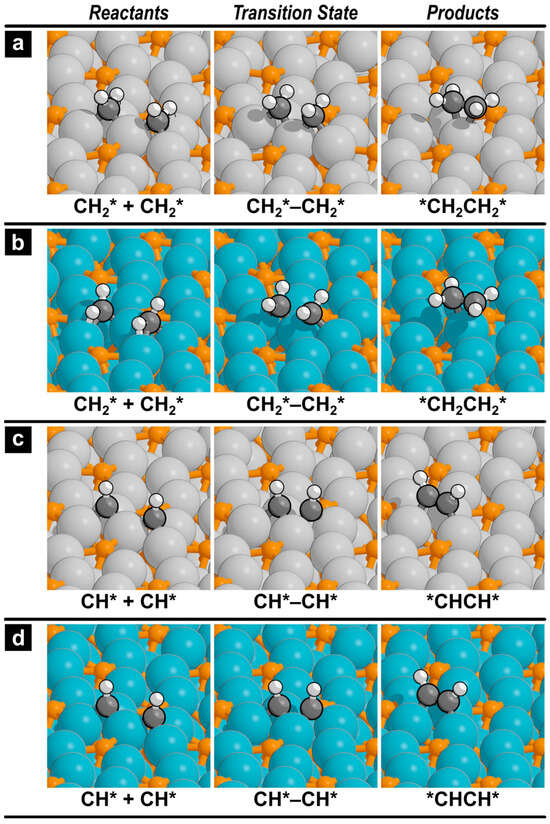
Figure 5.
Reactant, transition state, and product structures for the formation of ethylene on (a) Fe2P(001) and (b) Co2P(001), and acetylene on (c) Fe2P(001) and (d) Co2P(001). Images for the other C–C coupling reactions are shown in the Supporting Information (Figures S3 and S4).
The formation of acetylene (*CHCH*) requires a higher barrier than ethylene formation on both Fe2P (107 kJ mol−1) and Co2P (132 kJ mol−1) but remains facile on Ni2P (56 kJ mol−1). The reaction pathways are shown in Figure 5c,d. The acetylene product adsorbs strongly in a 3-fold hollow site, and its desorption is energetically costly, requiring 175 kJ mol−1 on Fe2P and 192 kJ mol−1 on Co2P, compared to 100 kJ mol−1 on Ni2P. The cross-coupling of CH3* and CH2* to form an ethyl intermediate was also examined, but this pathway was found to be kinetically unfavorable, exhibiting very high activation barriers on all three catalysts (Table 2).
To better examine the overall selectivity, the activation energy for C–C coupling is compared against the competing C–H bond scission step in the parity plot shown in Figure 6. In this plot, points to the right of the diagonal line indicate a kinetic preference for C–C coupling over further dehydrogenation. The results clearly show that Ni2P has the best selectivity for C2 products, as its data points lie in this favorable region. This computational finding is in agreement with experimental studies on silica-supported nickel phosphide catalysts, which have also reported high selectivity towards C2 products [26,27]. In contrast, Co2P and Fe2P are more reactive for C–H activation but exhibit poor selectivity for C2 coupling, with their data points falling to the left of the line. Fe2P comes closest to the selective region, with relatively comparable barriers for acetylene formation (107 kJ mol−1) versus C* formation (99 kJ mol−1). While specific experimental data on Fe2P and Co2P for methane activation is scarce, this predicted trend of high reactivity but poor C2 selectivity is consistent with the well-established behavior of iron- and cobalt-based catalysts in related hydrocarbon reactions. These materials are known to be highly active for C–H bond scission but also have a strong tendency to promote deep dehydrogenation pathways that lead to coking, which limits their selectivity for light olefins in non-oxidative conditions [36,37,38]. These findings suggest that future catalyst design could aim to combine the high reactivity of Fe2P or Co2P with the superior selectivity of Ni2P to enhance overall performance in methane activation and upgrading.
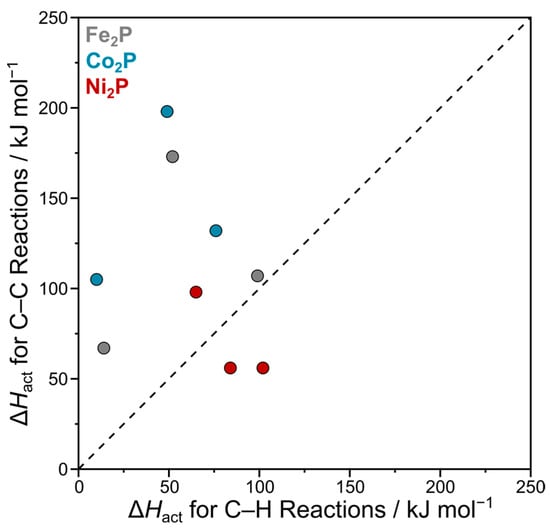
Figure 6.
Parity plot comparing the kinetic selectivity of Fe2P, Co2P, and Ni2P. The plot correlates the activation enthalpy (ΔHact) for C–C coupling (y-axis) with the activation enthalpy for a competing C–H bond scission step (x-axis). Each point compares the competing pathways for a specific C1 intermediate (CH3*, CH2*, or CH*), with all activation enthalpies taken from Table 1 and Table 2. The diagonal dashed line represents equal kinetic favorability; points to the right of the line indicate higher selectivity towards C–C coupling.
3. Computational Methods
All periodic density functional theory (DFT) calculations were performed using the Vienna ab initio simulation package (VASP) as implemented through the Computational Catalysis Interface (CCI) [39,40,41,42,43]. The interactions between ion cores and valence electrons were described using projector augmented-wave (PAW) potentials [44,45]. Exchange and correlation energies were calculated within the generalized gradient approximation (GGA) using the revised Perdew–Burke–Ernzerhof (RPBE) functional [46,47,48]. A plane-wave basis set with an energy cutoff of 396 eV was used for all calculations, and gas-phase molecules were modeled in a periodic 11 × 11 × 11 Å unit cell.
The initial bulk unit cell for Fe2P, which shares the same space group with Ni2P (), was obtained from crystallographic data and then fully optimized using DFT [49]; its calculated lattice parameters were a = b = 5.81 Å and c = 3.42 Å. The stable bulk phase of Co2P has the orthorhombic (Pnma) space group, which differs from the hexagonal () space group of Fe2P and Ni2P. To specifically isolate the role of the metal’s electronic properties, a theoretical Co2P model was constructed using the same () framework as the other catalysts. This approach ensures that any observed trends in catalytic activity and selectivity are a direct consequence of the metal’s identity (Fe, Co, or Ni), thereby decoupling these electronic effects from the geometric effects that would arise from comparing different crystal structures. This methodology has been used in previous studies to deconvolute such effects in transition metal phosphides [18]. Moreover, this isostructural approach is also directly relevant to the study of bimetallic systems, where Co atoms could be incorporated into a () host lattice like Ni2P. The DFT-optimized lattice parameters for this theoretical Co2P model were a = b = 5.72 Å and c = 3.40 Å. Unlike the non-magnetic Ni2P system, spin-polarized calculations were employed for both the Fe2P and Co2P systems.
Based on our previous analysis for Ni2P showing the high thermodynamic stability of the M-rich termination of the (001) facet [22], this surface was used to model both catalysts as shown in Figure 7. To further validate this choice, the surface formation energies for the low-index (001), (100), and (111) facets were calculated. These calculations, detailed in the Supporting Information (Table S1), confirm that the (001) facet is indeed the most thermodynamically stable surface for both Fe2P and Co2P catalysts. The surfaces were modeled as 2 × 2 slabs consisting of four atomic layers, with a 10 Å vacuum region added in the z-direction. During geometric optimization, the bottom two layers were fixed in their bulk positions, while the top two layers and any adsorbates were allowed to fully relax. A 3 × 3 × 1 Monkhorst–Pack k-point mesh was used for sampling the Brillouin zone [50,51]. All structures were relaxed until the forces on each atom were below 0.05 eV Å−1, with the electronic energy converged to within 10−6 eV.
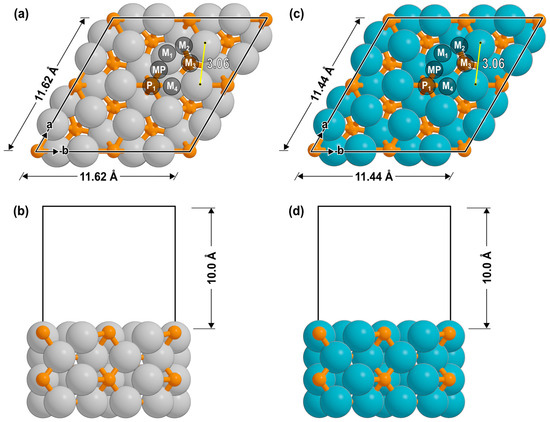
Figure 7.
(a,b) Fe2P(001) and (c,d) Co2P(001) catalyst models as seen from top and side views. Labels denote the binding site (M1: metal atop; M2: metal bridge; M3: 3-fold hollow; M4: 4-fold hollow; P1: phosphorus atop; MP: metal-phosphorus bridge).
Transition state structures were identified using the nudged elastic band (NEB) and dimer methods [52,53,54]. Vibrational frequency calculations were conducted to determine the thermal enthalpy corrections at a temperature of 1123 K. The binding energy of adsorbates (ΔEads) was calculated relative to the clean catalyst surface and the corresponding molecule in the gas phase using Equation (2):
Therefore, a more negative value signifies a more stable, exothermic adsorption. To evaluate the overall reaction kinetics, the effective enthalpy barrier is defined as the enthalpy of forming the transition state and a stoichiometric amount of gas-phase H2 from gas-phase methane:
In this equation, λ corresponds to the number of H2 molecules produced during the dehydrogenation steps. Additional computational details are available in Section S1 of the Supporting Information.
4. Conclusions
In this work, a systematic DFT investigation was conducted to compare methane activation and C–C coupling pathways on the isostructural (001) surfaces of Fe2P, Co2P, and Ni2P. The study aimed to deconvolute the role of the transition metal identity on the catalytic reactivity and selectivity for the non-oxidative coupling of methane.
The results demonstrate a clear trend in thermodynamic surface reactivity towards C1 intermediates, which follows the order Co2P > Fe2P > Ni2P and correlates with the binding strength of the adsorbates. A new, highly active pathway was identified for the initial C–H scission on purely metallic sites of Fe2P and Co2P, with activation barriers (85–101 kJ mol−1) that are comparable to reactive pure metals. The high activity of Fe2P and Co2P for the initial C–H scission also leads to facile pathways for subsequent C–H bond breaking, promoting the formation of a mixture of deeply dehydrogenated intermediates. In contrast, the much higher initial activation barrier on Ni2P makes it significantly less active.
The selectivity of the catalysts is ultimately dictated by the kinetic fate of the CH2* intermediate, the primary precursor for ethylene. On the Ni2P surface, a direct comparison of the competing pathways shows that the activation barrier for C–C coupling to ethylene (56 kJ mol−1) is significantly lower than that for the undesired dehydrogenation to CH* (84 kJ mol−1). This favorable kinetic balance, which is not observed on Fe2P or Co2P, promotes the selective conversion of surface CH2* species to C2 products. Both Fe2P and Co2P heavily favor decomposition, as the barriers for CH2* dehydrogenation (14 and 10 kJ mol−1, respectively) are substantially lower than those for ethylene coupling (67 and 105 kJ mol−1, respectively). These findings are consistent with experimental reports that have demonstrated the high C2 selectivity of Ni2P catalysts.
Overall, this work elucidates a fundamental reactivity–selectivity trade-off in this class of materials. While Fe2P and Co2P are more active for C–H activation, their high reactivity leads to poor selectivity. Ni2P, while significantly less active, is an exceptionally selective catalyst for ethylene formation. These findings provide a guiding principle for the rational design of next-generation catalysts, suggesting that bimetallic phosphide systems could be engineered to combine the activity of one metal with the selectivity of another, offering a promising route towards efficient and selective direct methane conversion.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal15100954/s1, Section S1: Computational Methods Details; Table S1: DFT-predicted surface formation energies; Figure S1: C–H reactions images on the Fe2P(001) surface; Figure S2: C–H reactions images on the Co2P(001) surface; Figure S3: C–C Reactions images on the Fe2P(001) surface; Figure S4: C–C Reactions images on the Co2P(001) surface; Figure S5: DFT-optimized structures for CHx* and H* on the Ni2P catalyst; Figure S6: C–H reactions images on the Ni2P(001) surface; Figure S7: C–C Reactions images on the Ni2P(001) surface.
Funding
This work was supported by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia (Grant No. KFU253254).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Li, T.; Kim, M.; Asthagiri, A.; Weaver, J.F. Low-Temperature Activation of Methane on the IrO2 (110) Surface. Science (1979) 2017, 356, 299–303. [Google Scholar] [CrossRef]
- Kim, D.; Ju, Y.; Kang, D.; Kang, S.B.; Kim, M. Potential of Intrinsic Reactivity toward Value Added Products from Methane Oxidation on RhO2(1 1 0) Surface. Appl. Surf. Sci. 2022, 596, 153499. [Google Scholar] [CrossRef]
- Rosen, A.S.; Notestein, J.M.; Snurr, R.Q. Structure-Activity Relationships That Identify Metal-Organic Framework Catalysts for Methane Activation. ACS Catal. 2019, 9, 3576–3587. [Google Scholar] [CrossRef]
- Sun, X.; Li, X.; Liu, Y.; Yu, Z.; Li, B.; Zhao, Z. The C-H Bond Activation Triggered by Subsurface Mo Dopant on MgO Catalyst in Oxidative Coupling of Methane. Catalysts 2022, 12, 1083. [Google Scholar] [CrossRef]
- Mehmood, A.; Chae, S.Y.; Park, E.D. Photoelectrochemical Conversion of Methane into Value-Added Products. Catalysts 2021, 11, 1387. [Google Scholar] [CrossRef]
- Dry, M.E. The Fischer–Tropsch Process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Schulz, H. Short History and Present Trends of Fischer–Tropsch Synthesis. Appl. Catal. A Gen. 1999, 186, 3–12. [Google Scholar] [CrossRef]
- Salahudeen, N.; Rasheed, A.A.; Babalola, A.; Moses, A.U. Review on Technologies for Conversion of Natural Gas to Methanol. J. Nat. Gas. Sci. Eng. 2022, 108, 104845. [Google Scholar] [CrossRef]
- Latimer, A.A.; Kakekhani, A.; Kulkarni, A.R.; Nørskov, J.K. Direct Methane to Methanol: The Selectivity–Conversion Limit and Design Strategies. ACS Catal. 2018, 8, 6894–6907. [Google Scholar] [CrossRef]
- Desta, B.K.; Araia, A.; Jiang, C.; Robinson, B.; Tewari, K.; Wang, Y.; Sanyal, O.; Hu, J. Direct, Nonoxidative Methane Coupling to Produce Ethylene Using 1M-3Mo/CeO2 Catalysts under Microwave Irradiation: Insights into the Effect of Metal Loading and Promoters. Ind. Eng. Chem. Res. 2025, 64, 10472–10484. [Google Scholar] [CrossRef]
- Lunsford, J.H. The Catalytic Oxidative Coupling of Methane. Angew. Chem. Int. Ed. Engl. 1995, 34, 970–980. [Google Scholar] [CrossRef]
- Zhou, M.; Yang, Z.; Ren, J.; Zhang, T.; Xu, W.; Zhang, J. Non-Oxidative Coupling Reaction of Methane to Hydrogen and Ethene via Plasma-Catalysis Process. Int. J. Hydrogen Energy 2023, 48, 78–89. [Google Scholar] [CrossRef]
- Xiao, Y.; Varma, A. Highly Selective Nonoxidative Coupling of Methane over Pt-Bi Bimetallic Catalysts. ACS Catal. 2018, 8, 2735–2740. [Google Scholar] [CrossRef]
- Talpade, A.D.; Canning, G.; Zhuchen, J.; Arvay, J.; Watt, J.; Miller, J.T.; Datye, A.; Ribeiro, F.H. Catalytic Reactivity of Pt Sites for Non-Oxidative Coupling of Methane (NOCM). Chem. Eng. J. 2024, 481, 148675. [Google Scholar] [CrossRef]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen. Science (1979) 2014, 344, 616–619. [Google Scholar] [CrossRef]
- Blanksby, S.J.; Ellison, G.B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef]
- Waldt, C.; Montalvo-Castro, H.; Almithn, A.; Loaiza-Orduz, Á.; Plaisance, C.; Hibbitts, D. Role of Phosphorous in Transition Metal Phosphides for Selective Hydrogenolysis of Hindered C–O Bonds. J. Catal. 2023, 421, 403–418. [Google Scholar] [CrossRef]
- Guharoy, U.; Ramirez Reina, T.; Gu, S.; Cai, Q. Mechanistic Insights into Selective CO2 Conversion via RWGS on Transition Metal Phosphides: A DFT Study. J. Phys. Chem. C 2019, 123, 22918–22931. [Google Scholar] [CrossRef]
- Duyar, M.S.; Tsai, C.; Snider, J.L.; Singh, J.A.; Gallo, A.; Yoo, J.S.; Medford, A.J.; Abild-Pedersen, F.; Studt, F.; Kibsgaard, J.; et al. A Highly Active Molybdenum Phosphide Catalyst for Methanol Synthesis from CO and CO2. Angew. Chem. 2018, 130, 15265–15270. [Google Scholar] [CrossRef]
- Al-Ali, L.I.; Elmutasim, O.; Al Ali, K.; Singh, N.; Polychronopoulou, K. Transition Metal Phosphides (TMP) as a Versatile Class of Catalysts for the Hydrodeoxygenation Reaction (HDO) of Oil-Derived Compounds. Nanomaterials 2022, 12, 1435. [Google Scholar] [CrossRef]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Hibbitts, D.D.; Flaherty, D.W. Mechanisms and Active Sites for C–O Bond Rupture within 2-Methyltetrahydrofuran over Ni, Ni12P5, and Ni2P Catalysts. ACS Catal. 2018, 8, 7141–7157. [Google Scholar] [CrossRef]
- Li, W.; Dhandapani, B.; Oyama, S.T. Molybdenum Phosphide: A Novel Catalyst for Hydrodenitrogenation. Chem. Lett. 1998, 27, 207–208. [Google Scholar] [CrossRef]
- Almithn, A. Effects of P:Ni Ratio on Methanol Steam Reforming on Nickel Phosphide Catalysts. Molecules 2023, 28, 6079. [Google Scholar] [CrossRef]
- Almithn, A.; Alghanim, S.N.; Mohammed, A.A.; Alghawinim, A.K.; Alomaireen, M.A.; Alhulaybi, Z.; Hossain, S.S. Methane Activation and Coupling Pathways on Ni2P Catalyst. Catalysts 2023, 13, 531. [Google Scholar] [CrossRef]
- Dipu, A.L.; Nishikawa, Y.; Inami, Y.; Iguchi, S.; Yamanaka, I. Development of Highly Active Silica-Supported Nickel Phosphide Catalysts for Direct Dehydrogenative Conversion of Methane to Higher Hydrocarbons. Catal. Lett. 2022, 152, 199–212. [Google Scholar] [CrossRef]
- Dipu, A.L.; Ohbuchi, S.; Nishikawa, Y.; Iguchi, S.; Ogihara, H.; Yamanaka, I. Direct Nonoxidative Conversion of Methane to Higher Hydrocarbons over Silica-Supported Nickel Phosphide Catalyst. ACS Catal. 2020, 10, 375–379. [Google Scholar] [CrossRef]
- Kim, M.; Franklin, A.D.; Martin, R.; Bian, Y.; Weaver, J.F.; Asthagiri, A. Kinetics of Low-Temperature Methane Activation on IrO2(1 1 0): Role of Local Surface Hydroxide Species. J. Catal. 2020, 383, 181–192. [Google Scholar] [CrossRef]
- Martin, R.; Kim, M.; Asthagiri, A.; Weaver, J.F. Alkane Activation and Oxidation on Late-Transition-Metal Oxides: Challenges and Opportunities. ACS Catal. 2021, 11, 4682–4703. [Google Scholar] [CrossRef]
- Han, Z.; Yang, Z.; Han, M. Comprehensive Investigation of Methane Conversion over Ni(111) Surface under a Consistent DFT Framework: Implications for Anti-Coking of SOFC Anodes. Appl. Surf. Sci. 2019, 480, 243–255. [Google Scholar] [CrossRef]
- Li, T.; Wen, X.; Li, Y.-W.; Jiao, H. Successive Dissociation of CO, CH4, C2H6, and CH3CHO on Fe(110): Retrosynthetic Understanding of FTS Mechanism. J. Phys. Chem. C 2018, 122, 28846–28855. [Google Scholar] [CrossRef]
- Zuo, Z.; Huang, W.; Han, P.; Li, Z. A Density Functional Theory Study of CH4 Dehydrogenation on Co(111). Appl. Surf. Sci. 2010, 256, 5929–5934. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A Fast and Robust Algorithm for Bader Decomposition of Charge Density. Comput. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A Grid-Based Bader Analysis Algorithm without Lattice Bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.R.C.M.; Oliveira, H.A.; Guarino, A.C.P.F.; Toledo, B.B.; Moura, M.B.T.; Oliveira, B.T.M.; Passos, F.B. Effect of Support on Methane Decomposition for Hydrogen Production over Cobalt Catalysts. Int. J. Hydrogen Energy 2016, 41, 6763–6772. [Google Scholar] [CrossRef]
- Horn, R.; Schlögl, R. Methane Activation by Heterogeneous Catalysis. Catal. Lett. 2015, 145, 23–39. [Google Scholar] [CrossRef]
- Tan, P. Active Phase, Catalytic Activity, and Induction Period of Fe/Zeolite Material in Nonoxidative Aromatization of Methane. J. Catal. 2016, 338, 21–29. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kravchenko, P.; Plaisance, C.; Hibbitts, D. A New Computational Interface for Catalysis. ChemRxiv 2019. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, W. Comment on “Generalized Gradient Approximation Made Simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Koumina, A.; Bacmann, M.; Fruchart, D.; Soubeyroux, J.-L.; Wolfers, P.; Tobola, J.; Kaprzyk, S.; Niziol, S.; Mesnaoui, M.; Zach, R. Crystallographic and Magnetic Properties of Fe2P. In Annales de Chimie Science des Materiaux; Elsevier: Amsterdam, The Netherlands, 1998; Volume 23, pp. 177–180. [Google Scholar]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special Points for Brillouin-Zone Integrations”—A Reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1998; pp. 385–404. [Google Scholar]
- Henkelman, G.; Jónsson, H. A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).