
Low-Temperature Electrochemical Oxidation of Methane into Alcohols



Abstract
1. Introduction
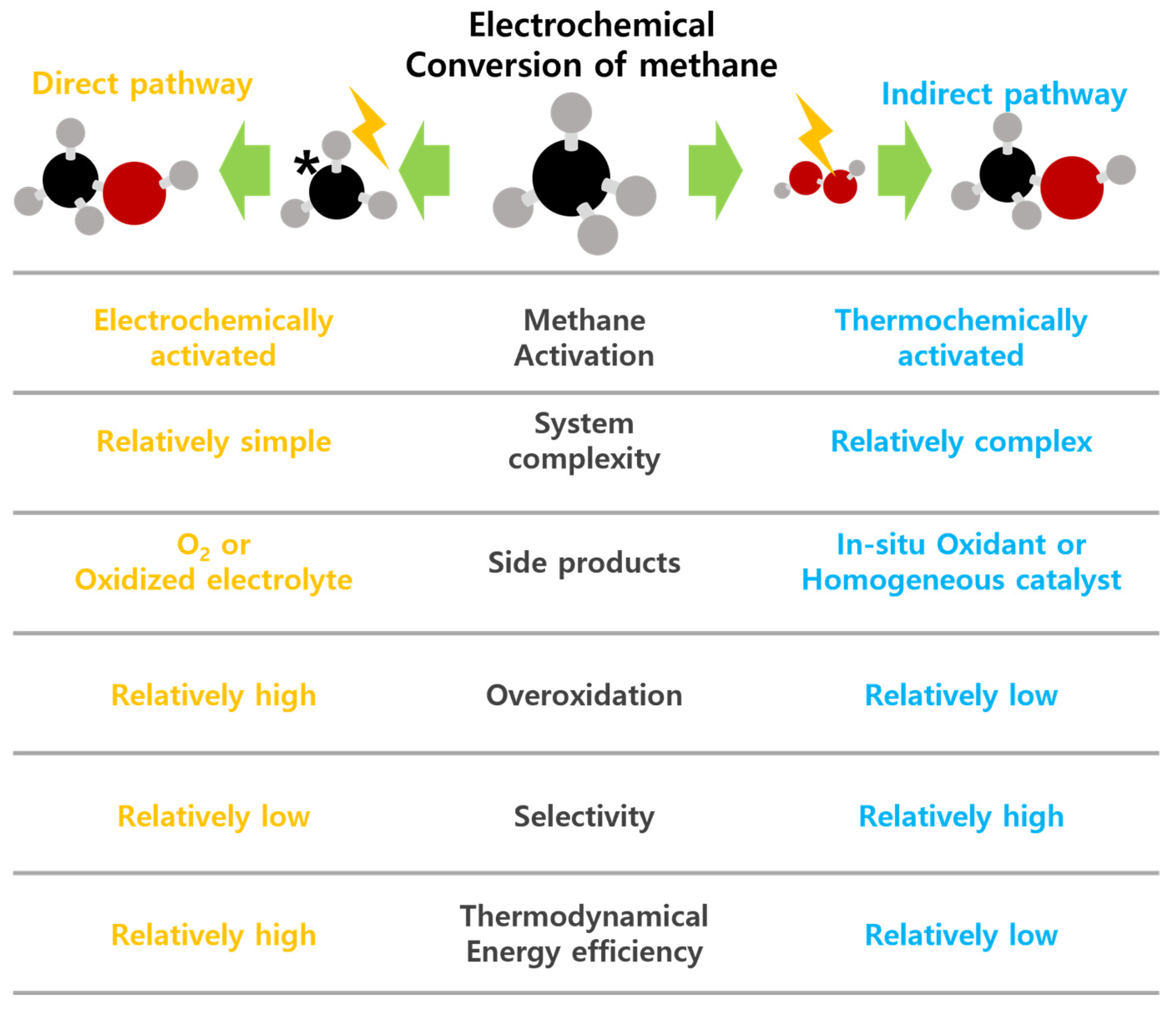
2. Direct Electrochemical Conversion of Methane
2.1. Methane to C2+ Alcohols
2.1.1. Carbonate Electrolyte
2.1.2. Alkaline Electrolyte
2.1.3. Other Electrolytes
2.2. Methane to Methanol
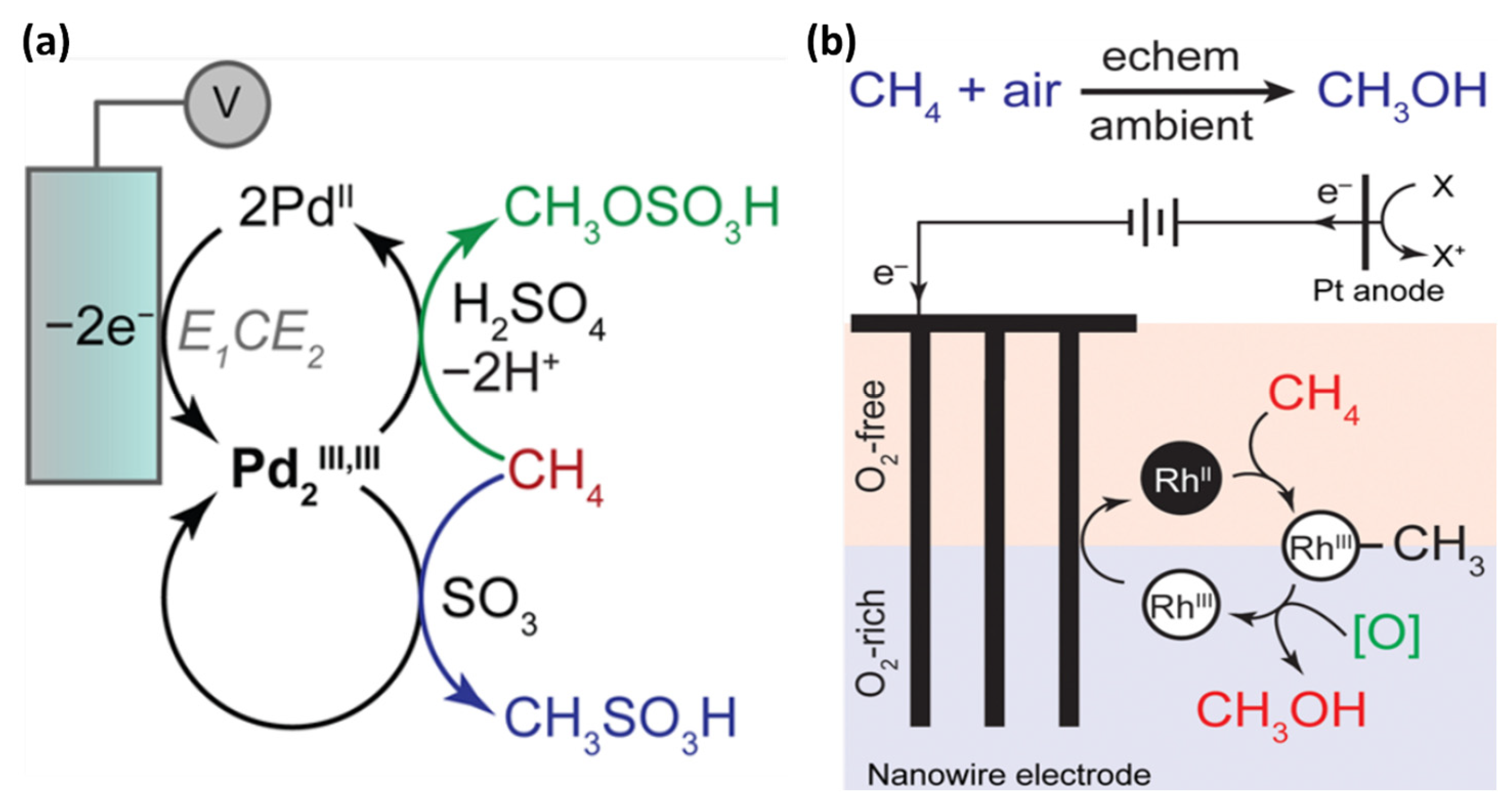
3. Indirect Electrochemical Conversion of Methane
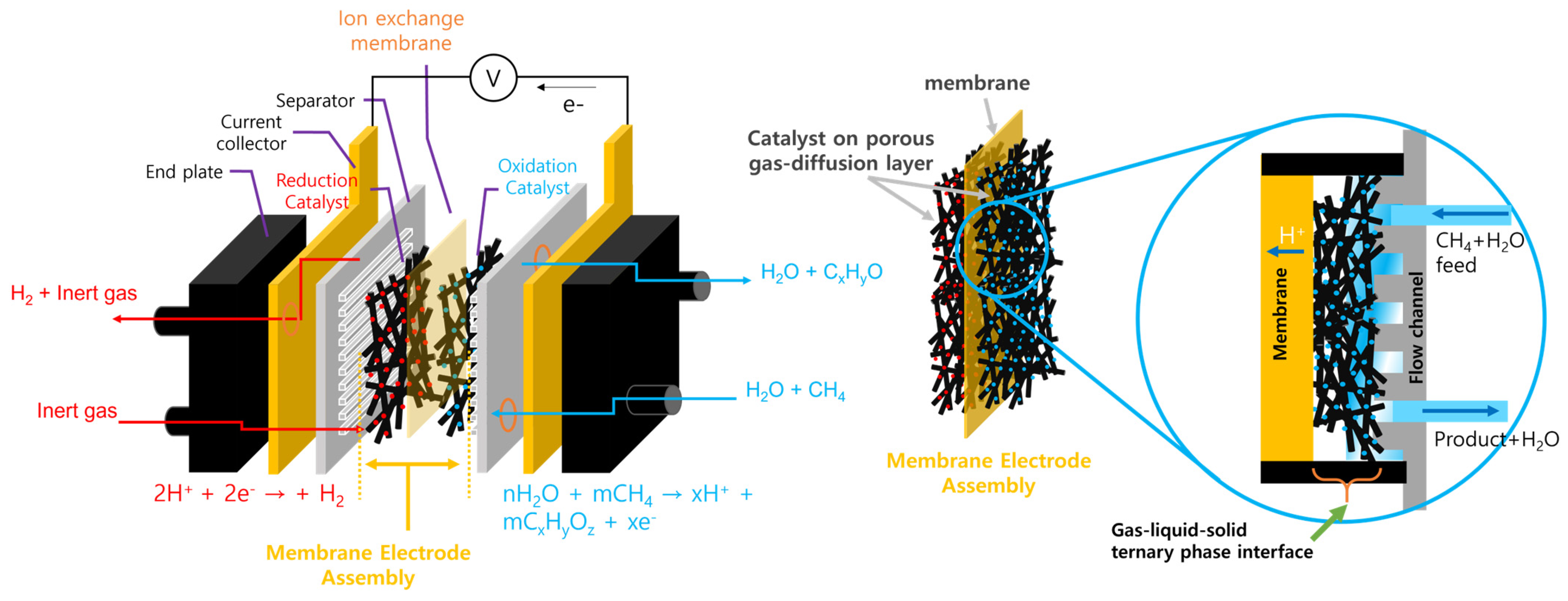
4. Electrochemical Conversion of Methane via a MEA Structure Reactor
Performance-Limiting Factor of the MEA Electrochemical Reactor
5. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-, Electro-, or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- Richard, D.; Huang, Y.-C.; Morales-Guio, C.G. Recent advances in the electrochemical production of chemicals from methane. Curr. Opin. Electrochem. 2021, 30, 100793. [Google Scholar] [CrossRef]
- Soucie, H.; Elam, M.; Mustain, W.E. Practical Assessment for At-Scale Electrochemical Conversion of Methane to Methanol. ACS Energy Lett. 2023, 8, 1218–1229. [Google Scholar] [CrossRef]
- Zakaria, Z.; Kamarudin, S.K. Direct conversion technologies of methane to methanol: An overview. Renew. Sustain. Energy Rev. 2016, 65, 250–261. [Google Scholar] [CrossRef]
- Huang, K.; Miller, J.B.; Huber, G.W.; Dumesic, J.A.; Maravelias, C.T. A General Framework for the Evaluation of Direct Nonoxidative Methane Conversion Strategies. Joule 2018, 2, 349–365. [Google Scholar] [CrossRef]
- Jackson, R.B.; Saunois, M.; Bousquet, P.; Canadell, J.G.; Poulter, B.; Stavert, A.R.; Bergamaschi, P.; Niwa, Y.; Segers, A.; Tsuruta, A. Increasing anthropogenic methane emissions arise equally from agricultural and fossil fuel sources. Environ. Res. Lett. 2020, 15, 071002. [Google Scholar] [CrossRef]
- Hussin, F.; Aroua, M.K. Recent development in the electrochemical conversion of carbon dioxide: Short review. AIP Conf. Proc. 2019, 2124, 030017. [Google Scholar] [CrossRef]
- Arminio-Ravelo, J.A.; Escudero-Escribano, M. Strategies toward the sustainable electrochemical oxidation of methane to methanol. Curr. Opin. Green Sustain. Chem. 2021, 30, 100489. [Google Scholar] [CrossRef]
- Abdelkader Mohamed, A.G.; Zahra Naqviab, S.A.; Wang, Y. Advances and Fundamental Understanding of Electrocatalytic Methane Oxidation. ChemCatChem 2021, 13, 787–805. [Google Scholar] [CrossRef]
- Wang, Q.; Kan, M.; Han, Q.; Zheng, G. Electrochemical Methane Conversion. Small Struct. 2021, 2, 2100037. [Google Scholar] [CrossRef]
- Bagherzadeh Mostaghimi, A.H.; Al-Attas, T.A.; Kibria, M.G.; Siahrostami, S. A review on electrocatalytic oxidation of methane to oxygenates. J. Mater. Chem. A 2020, 8, 15575–15590. [Google Scholar] [CrossRef]
- Dhandole, L.K.; Kim, S.H.; Moon, G.-h. Understanding (photo)electrocatalysis for the conversion of methane to valuable chemicals through partial oxidation processes. J. Mater. Chem. A 2022, 10, 19107–19128. [Google Scholar] [CrossRef]
- Jang, J.; Shen, K.; Morales-Guio, C.G. Electrochemical Direct Partial Oxidation of Methane to Methanol. Joule 2019, 3, 2589–2593. [Google Scholar] [CrossRef]
- Shi, T.; Sridhar, D.; Zeng, L.; Chen, A. Recent advances in catalyst design for the electrochemical and photoelectrochemical conversion of methane to value-added products. Electrochem. Commun. 2022, 135, 107220. [Google Scholar] [CrossRef]
- Upham, D.C.; Kristoffersen, H.H.; Snodgrass, Z.R.; Gordon, M.J.; Metiu, H.; McFarland, E.W. Bromine and iodine for selective partial oxidation of propane and methane. Appl. Catal. A 2019, 580, 102–110. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.; Bao, X. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, B.; Jiang, Y.-J.; Zhou, Y.; Sun, X.; Wang, Y.; Zhu, W. Photocatalytic Conversion of Methane: Current State of the Art, Challenges, and Future Perspectives. ACS Environ. Au 2023, 3, 252–276. [Google Scholar] [CrossRef]
- Xie, S.; Lin, S.; Zhang, Q.; Tian, Z.; Wang, Y. Selective electrocatalytic conversion of methane to fuels and chemicals. J. Energy Chem. 2018, 27, 1629–1636. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef]
- Arnarson, L.; Schmidt, P.S.; Pandey, M.; Bagger, A.; Thygesen, K.S.; Stephens, I.E.L.; Rossmeisl, J. Fundamental limitation of electrocatalytic methane conversion to methanol. Phys. Chem. Chem. Phys. 2018, 20, 11152–11159. [Google Scholar] [CrossRef]
- Sher Shah, M.S.A.; Oh, C.; Park, H.; Hwang, Y.J.; Ma, M.; Park, J.H. Catalytic Oxidation of Methane to Oxygenated Products: Recent Advancements and Prospects for Electrocatalytic and Photocatalytic Conversion at Low Temperatures. Adv. Sci. 2020, 7, 2001946. [Google Scholar] [CrossRef]
- O’Reilly, M.E.; Kim, R.S.; Oh, S.; Surendranath, Y. Catalytic Methane Monofunctionalization by an Electrogenerated High-Valent Pd Intermediate. ACS Cent. Sci. 2017, 3, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Stoukides, M.; Eng, D.; Chiang, P.H.; Alqahtany, H. Electrochemical Modification of the Activity and Selectivity of Metal Catalysts. In Studies in Surface Science and Catalysis; Guczi, L., Solymosi, F., TÉTÉNyi, P., Eds.; Elsevier: Amsterdam, The Netherlands, 1993; Volume 75, pp. 2131–2134. [Google Scholar]
- Patterson, B.D.; Mo, F.; Borgschulte, A.; Hillestad, M.; Joos, F.; Kristiansen, T.; Sunde, S.; van Bokhoven, J.A. Renewable CO2 recycling and synthetic fuel production in a marine environment. Proc. Natl. Acad. Sci. USA 2019, 116, 12212–12219. [Google Scholar] [CrossRef] [PubMed]
- Tomita, A.; Nakajima, J.; Hibino, T. Direct Oxidation of Methane to Methanol at Low Temperature and Pressure in an Electrochemical Fuel Cell. Angew. Chem. Int. Ed. 2008, 47, 1462–1464. [Google Scholar] [CrossRef]
- Lee, B.; Hibino, T. Efficient and selective formation of methanol from methane in a fuel cell-type reactor. J. Catal. 2011, 279, 233–240. [Google Scholar] [CrossRef]
- Sobyanin, V.A.; Belyaev, V.D. Gas-phase electrocatalysis: Methane oxidation to syngas in a solid oxide fuel cell reactor. Solid State Ionics 2000, 136–137, 747–752. [Google Scholar] [CrossRef]
- Fornaciari, J.C.; Primc, D.; Kawashima, K.; Wygant, B.R.; Verma, S.; Spanu, L.; Mullins, C.B.; Bell, A.T.; Weber, A.Z. A Perspective on the Electrochemical Oxidation of Methane to Methanol in Membrane Electrode Assemblies. ACS Energy Lett. 2020, 5, 2954–2963. [Google Scholar] [CrossRef]
- Promoppatum, P.; Viswanathan, V. Identifying Material and Device Targets for a Flare Gas Recovery System Utilizing Electrochemical Conversion of Methane to Methanol. ACS Sustain. Chem. Eng. 2016, 4, 1736–1745. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, L.; Han, Z.; Tang, Y.; Sun, Y.; Wan, P.; Chen, Y.; Argyle, M.D.; Fan, M. Direct conversion of methane to methanol by electrochemical methods. Green Energy Environ. 2022, 7, 1132–1142. [Google Scholar] [CrossRef]
- Kishore, M.R.A.; Lee, S.; Yoo, J.S. Fundamental Limitation in Electrochemical Methane Oxidation to Alcohol: A Review and Theoretical Perspective on Overcoming It. Adv. Sci. 2023, 10, 2301912. [Google Scholar] [CrossRef]
- Amenomiya, Y.; Birrs, V.I.; Goledzinowski, M.; Galuszka, J.; Sanger, A.R. Conversion of Methane by Oxidative Coupling. Catal. Rev.-Sci. Eng. 1990, 32, 163–227. [Google Scholar] [CrossRef]
- Spinner, N.; Mustain, W.E. Electrochemical Methane Activation and Conversion to Oxygenates at Room Temperature. J. Electrochem. Soc. 2013, 53, 1. [Google Scholar] [CrossRef]
- Ma, M.; Jin, B.J.; Li, P.; Jung, M.S.; Kim, J.I.; Cho, Y.; Kim, S.; Moon, J.H.; Park, J.H. Ultrahigh Electrocatalytic Conversion of Methane at Room Temperature. Adv. Sci. 2017, 4, 1700379. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Oh, C.; Kim, J.; Moon, J.H.; Park, J.H. Electrochemical CH4 oxidation into acids and ketones on ZrO2:NiCo2O4 quasi-solid solution nanowire catalyst. Appl. Catal. B 2019, 259, 118095. [Google Scholar] [CrossRef]
- Kaur, M.; Li, Z.; Meng, S.; Li, W.; Song, H. Electrocatalytic methane conversion to high value chemicals at ambient conditions. Energy Convers. Manag. 2023, 285, 117029. [Google Scholar] [CrossRef]
- Oh, C.; Kim, J.; Hwang, Y.J.; Ma, M.; Park, J.H. Electrocatalytic methane oxidation on Co3O4- incorporated ZrO2 nanotube powder. Appl. Catal. B 2021, 283, 119653. [Google Scholar] [CrossRef]
- Qiao, J.; Li, H.F.; Chang, Y.; Guan, L.S.; Shuang, S.; Dong, C. Voltammetric Study and Detection of Methane on Nickel Hydroxide–Modified Nickel Electrode. Anal. Lett. 2008, 41, 593–598. [Google Scholar] [CrossRef]
- Omasta, T.J.; Rigdon, W.A.; Lewis, C.A.; Stanis, R.J.; Liu, R.; Fan, C.Q.; Mustain, W.E. Two Pathways for Near Room Temperature Electrochemical Conversion of Methane to Methanol. ECS Trans. 2015, 66, 129. [Google Scholar] [CrossRef]
- Kadosh, Y.; Ben-Eliyahu, Y.; Bochlin, Y.; Ezuz, L.; Iflah, Y.; Halevy, S.; Kozuch, S.; Korin, E.; Bettelheim, A. A bilayer coating as an oxygen-transfer cascade for the electrochemical ambient conversion of methane to oxygenates. Chem. Commun. 2022, 58, 3154–3157. [Google Scholar] [CrossRef]
- Xu, N.; Coco, C.A.; Wang, Y.; Su, T.; Wang, Y.; Peng, L.; Zhang, Y.; Liu, Y.; Qiao, J.; Zhou, X.-D. Electro-conversion of methane to alcohols on “capsule-like” binary metal oxide catalysts. Appl. Catal. B 2021, 282, 119572. [Google Scholar] [CrossRef]
- Luo, M.; Li, J.; Wang, M.; Ma, Y.; Zheng, G.; Wang, M.; Zhou, Y. Electrooxidation of methane to acetic acid over ZnO nanosheets: Defect-sites engineering. J. Environ. Chem. Eng. 2023, 11, 109539. [Google Scholar] [CrossRef]
- Ponticorvo, E.; Iuliano, M.; Cirillo, C.; Sarno, M. Selective C2 electrochemical synthesis from methane on modified alumina supporting single atom catalysts. Chem. Eng. J. 2023, 451, 139074. [Google Scholar] [CrossRef]
- Kim, C.; Min, H.; Kim, J.; Sul, J.; Yang, J.; Moon, J.H. NiO/ZnO heterojunction nanorod catalyst for high-efficiency electrochemical conversion of methane. Appl. Catal. B 2023, 323, 122129. [Google Scholar] [CrossRef]
- Song, Y.; Zhao, Y.; Nan, G.; Chen, W.; Guo, Z.; Li, S.; Tang, Z.; Wei, W.; Sun, Y. Electrocatalytic oxidation of methane to ethanol via NiO/Ni interface. Appl. Catal. B 2020, 270, 118888. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, W.; Song, Y.; Dong, X.; Li, G.; Wei, W.; Sun, Y. Efficient methane electrocatalytic conversion over a Ni-based hollow fiber electrode. Chin. J. Catal. 2020, 41, 1067–1072. [Google Scholar] [CrossRef]
- Xie, Z.; Chen, M.; Chen, Y.; Guan, A.; Han, Q.; Zheng, G. Electrocatalytic Methane Oxidation to Ethanol via Rh/ZnO Nanosheets. J. Phys. Chem. C 2021, 125, 13324–13330. [Google Scholar] [CrossRef]
- Kim, C.; Min, H.; Kim, J.; Moon, J.H. Boosting electrochemical methane conversion by oxygen evolution reactions on Fe–N–C single atom catalysts. Energy Environ. Sci. 2023, 16, 3158–3165. [Google Scholar] [CrossRef]
- Ramos, A.S.; Santos, M.C.L.; Godoi, C.M.; Oliveira Neto, A.; Fernando Brambilla De Souza, R. Obtaining C2 and C3 Products from Methane Using Pd/C as Anode in a Solid Fuel Cell-type Electrolyte Reactor. ChemCatChem 2020, 12, 4517–4521. [Google Scholar] [CrossRef]
- Hibino, T.; Kobayashi, K.; Nagao, M.; Zhou, D.; Chen, S.; Yamamoto, Y. Alternating Current Electrolysis for Individual Synthesis of Methanol and Ethane from Methane in a Thermo-electrochemical Cell. ACS Catal. 2023, 13, 8890–8901. [Google Scholar] [CrossRef]
- Li, J.; Luo, M.; Wang, M.; Ma, Y.; Zheng, G.; Wang, M.; Zhou, Y.; Lu, Y.; Zhu, C.; Chen, B. Electrocatalytic conversion of methane to ethanol by ultrathin WO3 nanosheets: Oxygen vacancy engineering. Appl. Mater. Today 2023, 32, 101855. [Google Scholar] [CrossRef]
- Santos, M.C.L.; Godoi, C.M.; Kang, H.S.; de Souza, R.F.B.; Ramos, A.S.; Antolini, E.; Neto, A.O. Effect of Ni content in PdNi/C anode catalysts on power and methanol co-generation in alkaline direct methane fuel cell type. J. Colloid Interface Sci. 2020, 578, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Sarno, M.; Ponticorvo, E.; Funicello, N.; De Pasquale, S. Methane electrochemical oxidation at low temperature on Rh single atom/NiO/V2O5 nanocomposite. Appl. Catal. A 2020, 603, 117746. [Google Scholar] [CrossRef]
- Godoi, C.M.; Santos, M.C.L.; Silva, A.J.; Tagomori, T.L.; Ramos, A.S.; de Souza, R.F.B.; Neto, A.O. Methane conversion to higher value-added product and energy co-generation using anodes OF PdCu/C in a solid electrolyte reactor: Alkaline fuel cell type monitored by differential mass spectroscopy. Res. Chem. Intermed. 2021, 47, 743–757. [Google Scholar] [CrossRef]
- Hsieh, C.-T.; Tang, Y.-T.; Ho, Y.-S.; Shao, W.-K.; Cheng, M.-J. Methane to Methanol Conversion over N-Doped Graphene Facilitated by Electrochemical Oxygen Evolution: A First-Principles Study. J. Phys. Chem. C 2023, 127, 308–318. [Google Scholar] [CrossRef]
- Kang, Y.; Li, Z.; Lv, X.; Song, W.; Wei, Y.; Zhang, X.; Liu, J.; Zhao, Z. Active oxygen promoted electrochemical conversion of methane on two-dimensional carbide (MXenes): From stability, reactivity and selectivity. J. Catal. 2021, 393, 20–29. [Google Scholar] [CrossRef]
- Prajapati, A.; Collins, B.A.; Goodpaster, J.D.; Singh, M.R. Fundamental insight into electrochemical oxidation of methane towards methanol on transition metal oxides. Proc. Natl. Acad. Sci. USA 2021, 118, e2023233118. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.-Y.; Jiang, H.-M.; Han, Z.-W.; Zhang, L.-T.; Tang, Y.; Wan, P.-Y.; Khan, Z.U.H.; Chen, Y.-M. Direct Conversion of Methane to Methanol on LaCo0.5Fe0.5O3 Anode in Aqueous Ionic Liquid. Int. J. Electrochem. Sci. 2022, 17, 221161. [Google Scholar] [CrossRef]
- Zeng, L.; Thiruppathi, A.R.; van der Zalm, J.; Shi, T.; Chen, A. Tailoring trimetallic CoNiFe oxide nanostructured catalysts for the efficient electrochemical conversion of methane to methanol. J. Mater. Chem. A 2022, 10, 15012–15025. [Google Scholar] [CrossRef]
- Shen, K.; Kumari, S.; Huang, Y.-C.; Jang, J.; Sautet, P.; Morales-Guio, C.G. Electrochemical Oxidation of Methane to Methanol on Electrodeposited Transition Metal Oxides. J. Am. Chem. Soc. 2023, 145, 6927–6943. [Google Scholar] [CrossRef]
- Lee, J.; Yang, J.; Moon, J.H. Solar Cell-Powered Electrochemical Methane-to-Methanol Conversion with CuO/CeO2 Catalysts. ACS Energy Lett. 2021, 6, 893–899. [Google Scholar] [CrossRef]
- Kim, R.S.; Surendranath, Y. Electrochemical Reoxidation Enables Continuous Methane-to-Methanol Catalysis with Aqueous Pt Salts. ACS Cent. Sci. 2019, 5, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.S.; Reis, R.M.; Lanza, M.R.V.; Bertazzoli, R. Electrosynthesis of methanol from methane: The role of V2O5 in the reaction selectivity for methanol of a TiO2/RuO2/V2O5 gas diffusion electrode. Electrochim. Acta 2013, 87, 606–610. [Google Scholar] [CrossRef]
- Cook, R.L.; Sammells, A.F. Ambient Temperature Methane Activation to Condensed Species under Cathodic Conditions. J. Electrochem. Soc. 1990, 137, 2007. [Google Scholar] [CrossRef]
- Latimer, A.A.; Kakekhani, A.; Kulkarni, A.R.; Nørskov, J.K. Direct Methane to Methanol: The Selectivity–Conversion Limit and Design Strategies. ACS Catal. 2018, 8, 6894–6907. [Google Scholar] [CrossRef]
- Ogura, K.; Migita, C.T.; Ito, Y. Combined Photochemical and Electrochemical Oxidation of Methane. J. Electrochem. Soc. 1990, 137, 500. [Google Scholar] [CrossRef]
- Frese, K.W., Jr. Partial electrochemical oxidation of methane under mild conditions. Langmuir 1991, 7, 13–15. [Google Scholar] [CrossRef]
- Gunsalus, N.J.; Koppaka, A.; Park, S.H.; Bischof, S.M.; Hashiguchi, B.G.; Periana, R.A. Homogeneous Functionalization of Methane. Chem. Rev. 2017, 117, 8521–8573. [Google Scholar] [CrossRef]
- Deng, J.; Lin, S.-C.; Fuller, J.; Iñiguez, J.A.; Xiang, D.; Yang, D.; Chan, G.; Chen, H.M.; Alexandrova, A.N.; Liu, C. Ambient methane functionalization initiated by electrochemical oxidation of a vanadium (V)-oxo dimer. Nat. Commun. 2020, 11, 3686. [Google Scholar] [CrossRef]
- Kim, R.S.; Nazemi, A.; Cundari, T.R.; Surendranath, Y. A PdIII Sulfate Dimer Initiates Rapid Methane Monofunctionalization by H Atom Abstraction. ACS Catal. 2020, 10, 14782–14792. [Google Scholar] [CrossRef]
- Natinsky, B.S.; Lu, S.; Copeland, E.D.; Quintana, J.C.; Liu, C. Solution Catalytic Cycle of Incompatible Steps for Ambient Air Oxidation of Methane to Methanol. ACS Cent. Sci. 2019, 5, 1584–1590. [Google Scholar] [CrossRef]
- Dang, H.T.; Lee, H.W.; Lee, J.; Choo, H.; Hong, S.H.; Cheong, M.; Lee, H. Enhanced Catalytic Activity of (DMSO)2PtCl2 for the Methane Oxidation in the SO3–H2SO4 System. ACS Catal. 2018, 8, 11854–11862. [Google Scholar] [CrossRef]
- Zimmermann, T.; Soorholtz, M.; Bilke, M.; Schüth, F. Selective Methane Oxidation Catalyzed by Platinum Salts in Oleum at Turnover Frequencies of Large-Scale Industrial Processes. J. Am. Chem. Soc. 2016, 138, 12395–12400. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, T.; Yang, C.; Chen, M.; Guan, A.; Yang, L.; Li, S.; Lv, X.; Wang, Y.; Zheng, G. Electrocatalytic Methane Oxidation Greatly Promoted by Chlorine Intermediates. Angew. Chem. Int. Ed. 2021, 60, 17398–17403. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, A.; Sartape, R.; Kani, N.C.; Gauthier, J.A.; Singh, M.R. Chloride-Promoted High-Rate Ambient Electrooxidation of Methane to Methanol on Patterned Cu–Ti Bimetallic Oxides. ACS Catal. 2022, 12, 14321–14329. [Google Scholar] [CrossRef]
- Higgins, D.; Hahn, C.; Xiang, C.; Jaramillo, T.F.; Weber, A.Z. Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New Paradigm. ACS Energy Lett. 2019, 4, 317–324. [Google Scholar] [CrossRef]
- Weng, L.-C.; Bell, A.T.; Weber, A.Z. Towards membrane-electrode assembly systems for CO2 reduction: A modeling study. Energy Environ. Sci. 2019, 12, 1950–1968. [Google Scholar] [CrossRef]
- Seidel, Y.E.; Schneider, A.; Jusys, Z.; Wickman, B.; Kasemo, B.; Behm, R.J. Transport Effects in the Electrooxidation of Methanol Studied on Nanostructured Pt/Glassy Carbon Electrodes. Langmuir 2010, 26, 3569–3578. [Google Scholar] [CrossRef]
- Márquez-Montes, R.A.; Collins-Martínez, V.H.; Pérez-Reyes, I.; Chávez-Flores, D.; Graeve, O.A.; Ramos-Sánchez, V.H. Electrochemical Engineering Assessment of a Novel 3D-Printed Filter-Press Electrochemical Reactor for Multipurpose Laboratory Applications. ACS Sustain. Chem. Eng. 2020, 8, 3896–3905. [Google Scholar] [CrossRef]
- Heo, P.; Kajiyama, N.; Kobayashi, K.; Nagao, M.; Sano, M.; Hibino, T. Proton Conduction in Sn0.95Al0.05P2O7 – PBI – PTFE Composite Membrane. Electrochem. Solid-State Lett. 2008, 11, B91. [Google Scholar] [CrossRef]
- Heo, P.; Ito, K.; Tomita, A.; Hibino, T. A Proton-Conducting Fuel Cell Operating with Hydrocarbon Fuels. Angew. Chem. Int. Ed. 2008, 47, 7841–7844. [Google Scholar] [CrossRef]
- Paschos, O.; Kunze, J.; Stimming, U.; Maglia, F. A review on phosphate based, solid state, protonic conductors for intermediate temperature fuel cells. J. Phys. Condens. Matter 2011, 23, 234110. [Google Scholar] [CrossRef]
- Kusoglu, A.; Weber, A.Z. New Insights into Perfluorinated Sulfonic-Acid Ionomers. Chem. Rev. 2017, 117, 987–1104. [Google Scholar] [CrossRef]
- Li, W.; Manthiram, A.; Guiver, M.D. Acid–base blend membranes consisting of sulfonated poly(ether ether ketone) and 5-amino-benzotriazole tethered polysulfone for DMFC. J. Membr. Sci. 2010, 362, 289–297. [Google Scholar] [CrossRef]
- Ma, M.; Clark, E.L.; Therkildsen, K.T.; Dalsgaard, S.; Chorkendorff, I.; Seger, B. Insights into the carbon balance for CO2 electroreduction on Cu using gas diffusion electrode reactor designs. Energy Environ. Sci. 2020, 13, 977–985. [Google Scholar] [CrossRef]
- Lee, B.; Sakamoto, Y.; Hirabayashi, D.; Suzuki, K.; Hibino, T. Direct oxidation of methane to methanol over proton conductor/metal mixed catalysts. J. Catal. 2010, 271, 195–200. [Google Scholar] [CrossRef]
- Santos, M.C.L.; Nunes, L.C.; Silva, L.M.G.; Ramos, A.S.; Fonseca, F.C.; de Souza, R.F.B.; Neto, A.O. Direct Alkaline Anion Exchange Membrane Fuel Cell to Converting Methane into Methanol. ChemistrySelect 2019, 4, 11430–11434. [Google Scholar] [CrossRef]
- Nandenha, J.; Piasentin, R.M.; Silva, L.M.G.; Fontes, E.H.; Neto, A.O.; de Souza, R.F.B. Partial oxidation of methane and generation of electricity using a PEMFC. Ionics 2019, 25, 5077–5082. [Google Scholar] [CrossRef]
- Rocha, R.S.; Camargo, L.M.; Lanza, M.R.V.; Bertazzoli, R. A Feasibility Study of the Electro-recycling of Greenhouse Gases: Design and Characterization of a (TiO2/RuO2)/PTFE Gas Diffusion Electrode for the Electrosynthesis of Methanol from Methane. Electrocatalysis 2010, 1, 224–229. [Google Scholar] [CrossRef]
- Nandenha, J.; Fontes, E.H.; Piasentin, R.M.; Fonseca, F.C.; Neto, A.O. Direct oxidation of methane at low temperature using Pt/C, Pd/C, Pt/C–ATO and Pd/C–ATO electrocatalysts prepared by sodium borohydride reduction process. J. Fuel Chem. Technol. 2018, 46, 1137–1145. [Google Scholar] [CrossRef]
- Nandenha, J.; Nagahama, I.H.F.; Yamashita, J.Y.; Fontes, E.H.; Ayoub, J.M.S.; de Souza, R.F.B.; Fonseca, F.C.; Neto, A.O. Activation of Methane on PdZn/C Electrocatalysts in an Acidic Electrolyte at Low Temperatures. Int. J. Electrochem. Sci. 2019, 14, 10819–10834. [Google Scholar] [CrossRef]
- de Moura Souza, F.; de Souza, R.F.B.; Batista, B.L.; dos Santos, M.C.; Fonseca, F.C.; Neto, A.O.; Nandenha, J. Methane activation at low temperature in an acidic electrolyte using PdAu/C, PdCu/C, and PdTiO2/C electrocatalysts for PEMFC. Res. Chem. Intermed. 2020, 46, 2481–2496. [Google Scholar] [CrossRef]
- Fan, Q. Method for Producing Methanol from Methane. U.S. Patent 2014/0124381 A1, 20 October 2015. [Google Scholar]
- Fan, Q. Non-Faradaic Electrochemical Promotion of Catalytic Methane Reforming for Methanol Production. U.S. Patent 9499917B2, 22 November 2016. [Google Scholar]
- Garcia, L.M.S.; Rajak, S.; Chair, K.; Godoy, C.M.; Silva, A.J.; Gomes, P.V.R.; Sanches, E.A.; Ramos, A.S.; De Souza, R.F.B.; Duong, A.; et al. Conversion of Methane into Methanol Using the [6,6′-(2,2′-Bipyridine-6,6′-Diyl)bis(1,3,5-Triazine-2,4-Diamine)](Nitrato-O)Copper(II) Complex in a Solid Electrolyte Reactor Fuel Cell Type. ACS Omega 2020, 5, 16003–16009. [Google Scholar] [CrossRef]
- Liu, F.; Yan, Y.; Chen, G.; Wang, D. Recent advances in ambient electrochemical methane conversion to oxygenates using metal oxide electrocatalysts. Green Chem. 2024. Advance Article. [Google Scholar] [CrossRef]
- Verma, S.; Lu, S.; Kenis, P.J.A. Co-electrolysis of CO2 and glycerol as a pathway to carbon chemicals with improved technoeconomics due to low electricity consumption. Nat. Energy 2019, 4, 466–474. [Google Scholar] [CrossRef]
- Koolen, C.D.; Luo, W.; Züttel, A. From Single Crystal to Single Atom Catalysts: Structural Factors Influencing the Performance of Metal Catalysts for CO2 Electroreduction. ACS Catal. 2023, 13, 948–973. [Google Scholar] [CrossRef]
- Zhang, J.; Pham, T.H.M.; Ko, Y.; Li, M.; Yang, S.; Koolen, C.D.; Zhong, L.; Luo, W.; Züttel, A. Tandem effect of Ag@C@Cu catalysts enhances ethanol selectivity for electrochemical CO2 reduction in flow reactors. Cell Rep. Phys. Sci. 2022, 3, 100949. [Google Scholar] [CrossRef]
- Maiyalagan, T.; Saji, V.S. Electrocatalysts for Low Temperature Fuel Cells: Fundamentals and Recent Trends; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
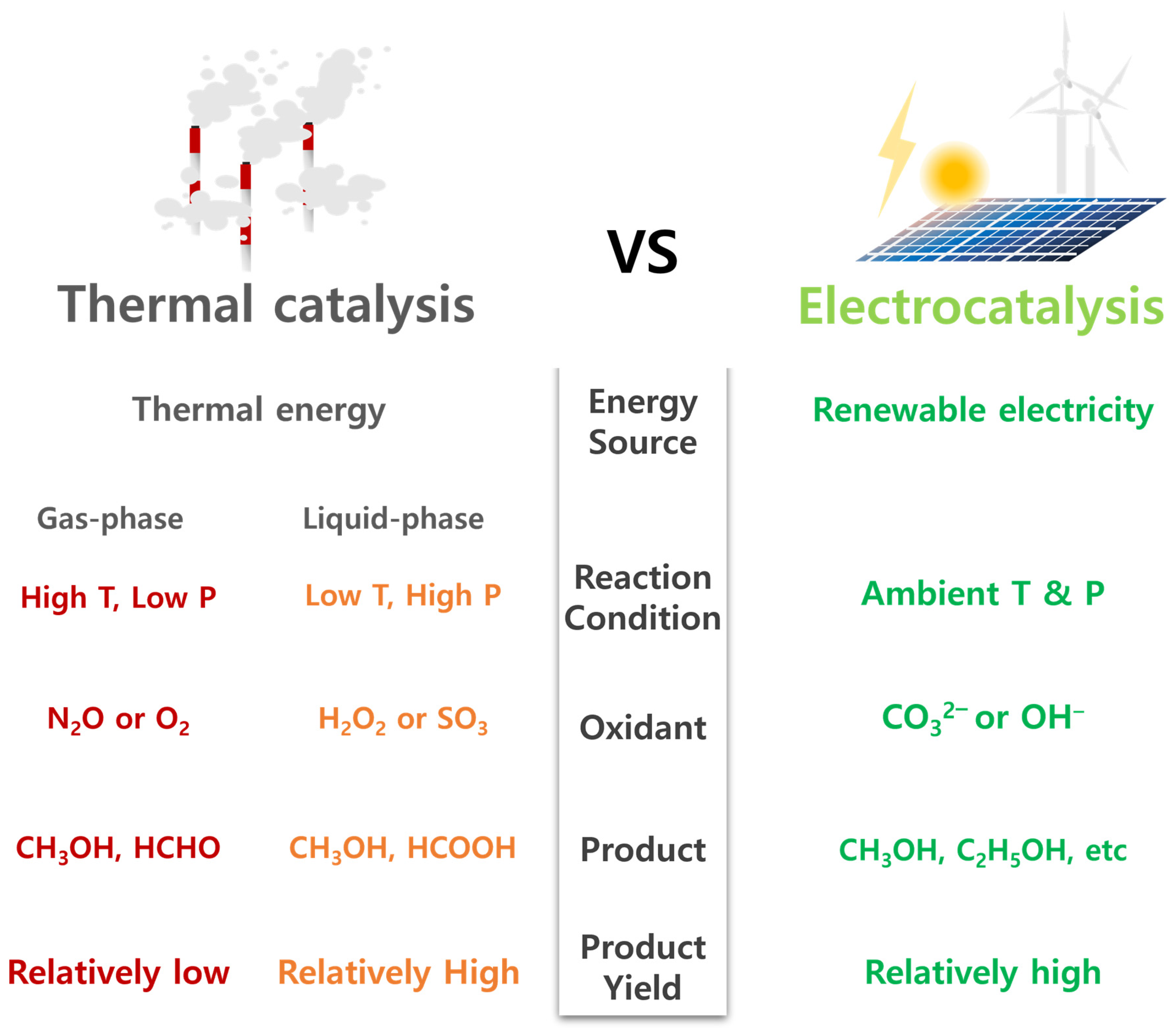

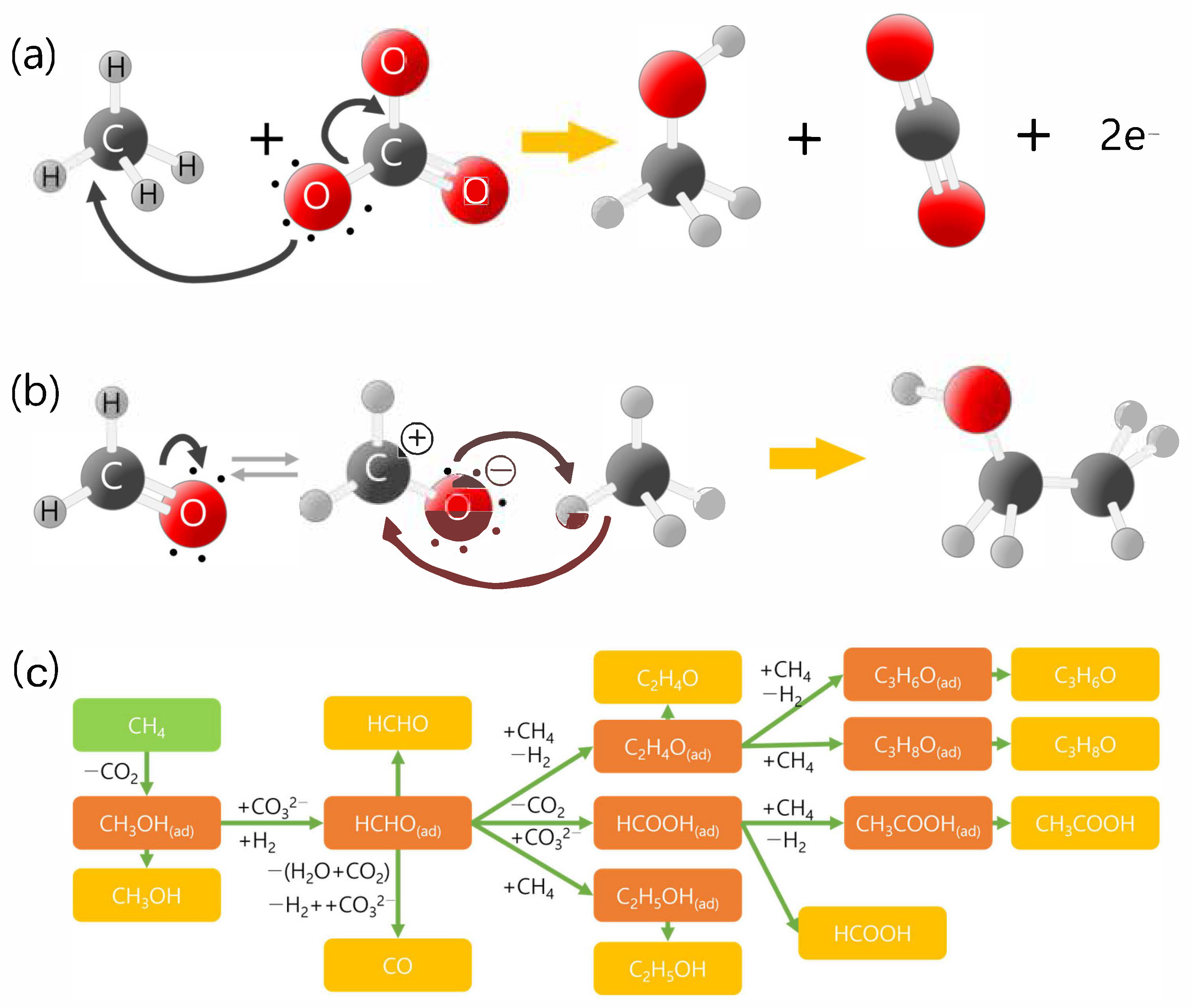




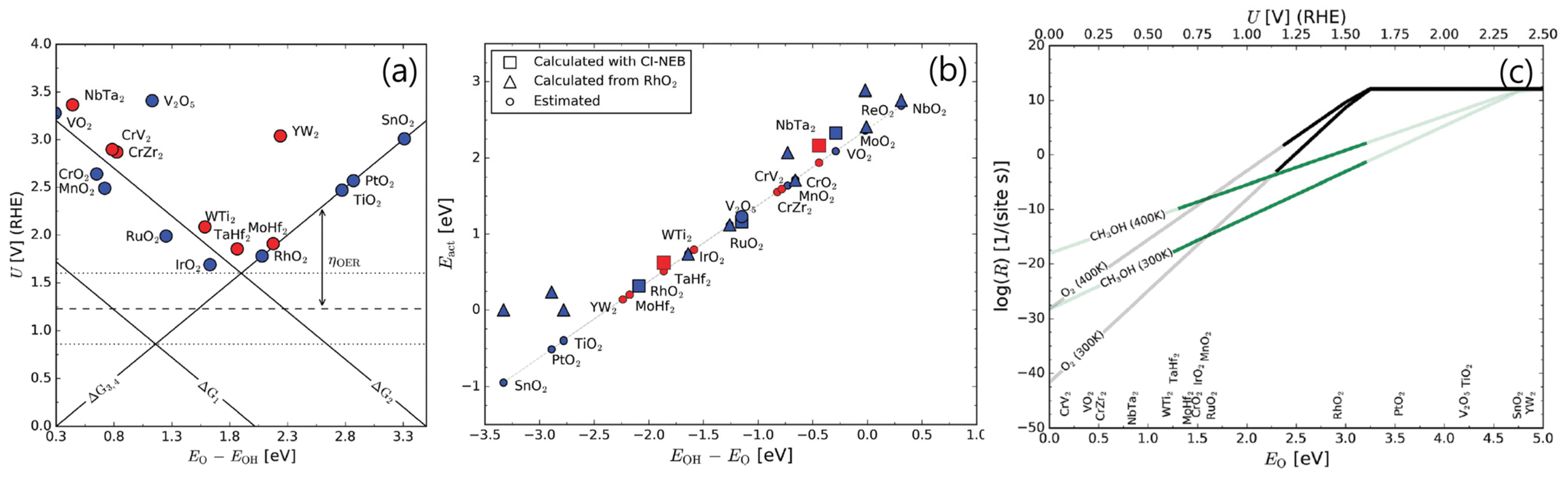




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Half-Cell Reactions | Proton Exchange Membrane | V vs. SHE |
|---|---|---|
| Anode | CH4 + H2O → CH3OH + 2H+ + 2e− 2H2O → O2 + 4H+ + 4e− | 0.58 1.23 |
| Cathode | O2 + 4H+ + 4e− → 2H2O 2H+ + 2e− → 2H2 | 1.23 0.00 |
| Anion Exchange Membrane | ||
| Anode | CH4 + 2OH− → CH3OH + H2O + 2e− 4OH− → O2 + 2H2O + 4e− | −0.18 −0.40 |
| Cathode | O2 + 2H2O + 4e− → 4OH− 2H2O + 2e− → H2 + 2OH− | −0.40 −0.83 |
| Electrocatalyst | Temperature (°C) | Product | Potential (V) | Electrolyte | FE (%) | Yield (mmol g−1 h−1) | Selectivity (%) | Ref |
|---|---|---|---|---|---|---|---|---|
| NiO/Ni | RT | CH3CH2OH | 1.40 vs. RHE | NaOH | 89 | 2.5 × 10−2 | 87 | [45] |
| Co3O4/ZrO2 | RT | CH3CH2CH2OH, CH3CH(OH)CH3 | 1.6 vs. RHE | Na2CO3 | --- | 2.4 | 92 | [37] |
| NiCo2O4/ZrO2 | RT | CH3CH2CH2OH, CH3CH(OH)CH3 | 2.0 vs. Pt | Na2CO3 | --- | 1.2 | --- | [35] |
| 0.6% Rh/ZnO CuO/CeO2 Rh/NiO/V2O5 | RT RT 100 | CH3CH2OH CH3OH CH3OH | 1.4 vs. RHE 0.8 vs. SCE 0.7 vs. RHE | KOH Na2CO3 Nafion | 23 91 | 0.79 0.75 6.5 × 102 | 85 79 98 | [47] [61] [53] |
| Pt(IV) | 130 | CH3OH | 0.68 vs. SHE | NaCl, H2SO4 | 100 | 2.9 × 10−4 | 70 | [62] |
| NiO/Ni | RT | CH3OH, CH3CH2OH | 1.46 vs. SHE | NaOH | 54, 85 | --- | 78, 95 | [46] |
| NiO/ZrO2 | 40 | CH3OH, HCHO, HCOOH, C2H5OH, CH3COOH, C3H8O, C3H6O | 2.0 vs. Cathode | Na2CO3 | --- | --- | HCHO: 44 | [33] |
| TiO2/RuO2/V2O5 | RT | CH3OH, HCHO, HCOOH | 2.0 vs. SCE | Na2SO4 | 57 | --- | CH3OH: 97.7 | [63] |
| ZrO2/Co3O4 | RT | CH3CH2CH2OH, (CH3)2CHOH, CH3CHO | 2.0 vs. Pt | Na2CO3 | >100 | --- | >60 | [34] |
| Electrode | Temperature (°C) | Potential (V) | Electrolyte/Membrane | Oxidant | Product (Production Rate) | Current Density (mA/cm2) | Ref. |
|---|---|---|---|---|---|---|---|
| V2O5/SnO2-PTFE anode | 100 | 0.9 | Sn0.9In0.1P2O7 | H2O | CH3OH | 4 | [26] |
| PdAu/C cathode | 50 | Sn0.9In0.1P2O7 | O2 | CH3OH (0.4 µmol h−1 cm−2), | 400 | [25] | |
| Pt/C cathode | 85 | 0.4 | Nafion 117 | H2O2 | CH3OH (0.14 mol L–1) | [88] | |
| NiO-ZrO2 anode | 40 | 2.0 | 1.0 M Na2CO3 + DMF/ralex AM-PAD | CO32− | CH3OH, HCHO, CO, HCOOH, C2H5OH, CH3COOH, CH3COCH3, CH3CHOHCH3 | 21 | [33] |
| Co3O4-ZrO2/CP-nafion 117 anode | 25 | 2.0 VPt | 0.5 M Na2CO3/Nafion 117 | CO32− | CH3OH (9.9 µg L−1 h−1), CH2O, C2H5OH, C2H4O, C3H8O, C3H6O | ~10 | [34,35] |
| Pd/C Anode | 80 | 0.05–1.2 V | Nafion | H2O | HCOOH | ~10 | [90] |
| PdZn/C Anode | 80 | 0.05–1.2 V | Nafion | H2O | CH3OH, HCOOH | ~10 | [91] |
| PdAu/C Anode | 80 | 0.05–1.2 V | Nafion | H2O | CH3OH, HCOOH | ~10 | [92] |
| Pd/C, Pt/C, Ni/C anode | 25 (anode) 85 (cathode) | 0.3 | 6.0 M KOH/Nafion 117 | OH− | CH3OH (0.9 mol L–1) | ~1 | [87] |
| MOx anode (M = Mn, Fe, Ni, Os, Pt) | 160 | -- | KOH + H2O (catholyte)/ion exchange membrane | OH− | CH3OH | -- | [93] |
| MOx anode (M = Ni, Co, Cu, Ag, Pt, Au, Ce, Pb, Fe, Mn, Zn Os, Pt) | 25–160 | -- | KOH + H2O (catholyte)/Daramic anion exchange membrane | OH− | CH3OH, CO2 | -- | [94] |
| [6,6′-(2,2′-Bipyridine-6,6′-Diyl)bis(1,3,5-Triazine-2,4-Diamine)](Nitrato-O)Copper(II) Complex/C Anode | 25 | 0.2–0.3 | KOH-doped Nafion membrane | OH− | CH3OH (1.85 mol L–1 h–1), HCOOH | ~1 | [95] |
| TiO2/RuO2/V2O5/anode | 25 | 2.0 VSCE | 0.1 M Na2SO4/PTFE | H2O (OH−) | CH3OH (300 mg L−1 h−1), HCHO | 57 | [63] |
| TiO2/RuO2 anode | 25 | 2.1 VSCE | 0.1 M Na2SO4/PTFE | H2O (OH−) | CH3OH (220 mg L−1 cm2), HCHO, HCOOH | 30 | [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehmood, A.; Chae, S.Y.; Park, E.D. Low-Temperature Electrochemical Oxidation of Methane into Alcohols. Catalysts 2024, 14, 58. https://doi.org/10.3390/catal14010058
Mehmood A, Chae SY, Park ED. Low-Temperature Electrochemical Oxidation of Methane into Alcohols. Catalysts. 2024; 14(1):58. https://doi.org/10.3390/catal14010058
Chicago/Turabian StyleMehmood, Adeel, Sang Youn Chae, and Eun Duck Park. 2024. "Low-Temperature Electrochemical Oxidation of Methane into Alcohols" Catalysts 14, no. 1: 58. https://doi.org/10.3390/catal14010058
APA StyleMehmood, A., Chae, S. Y., & Park, E. D. (2024). Low-Temperature Electrochemical Oxidation of Methane into Alcohols. Catalysts, 14(1), 58. https://doi.org/10.3390/catal14010058