

Origins of Enhanced Enantioselectivity in the Pd-Catalyzed Decarboxylative Allylic Alkylation of N-Benzoyl Lactams †


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
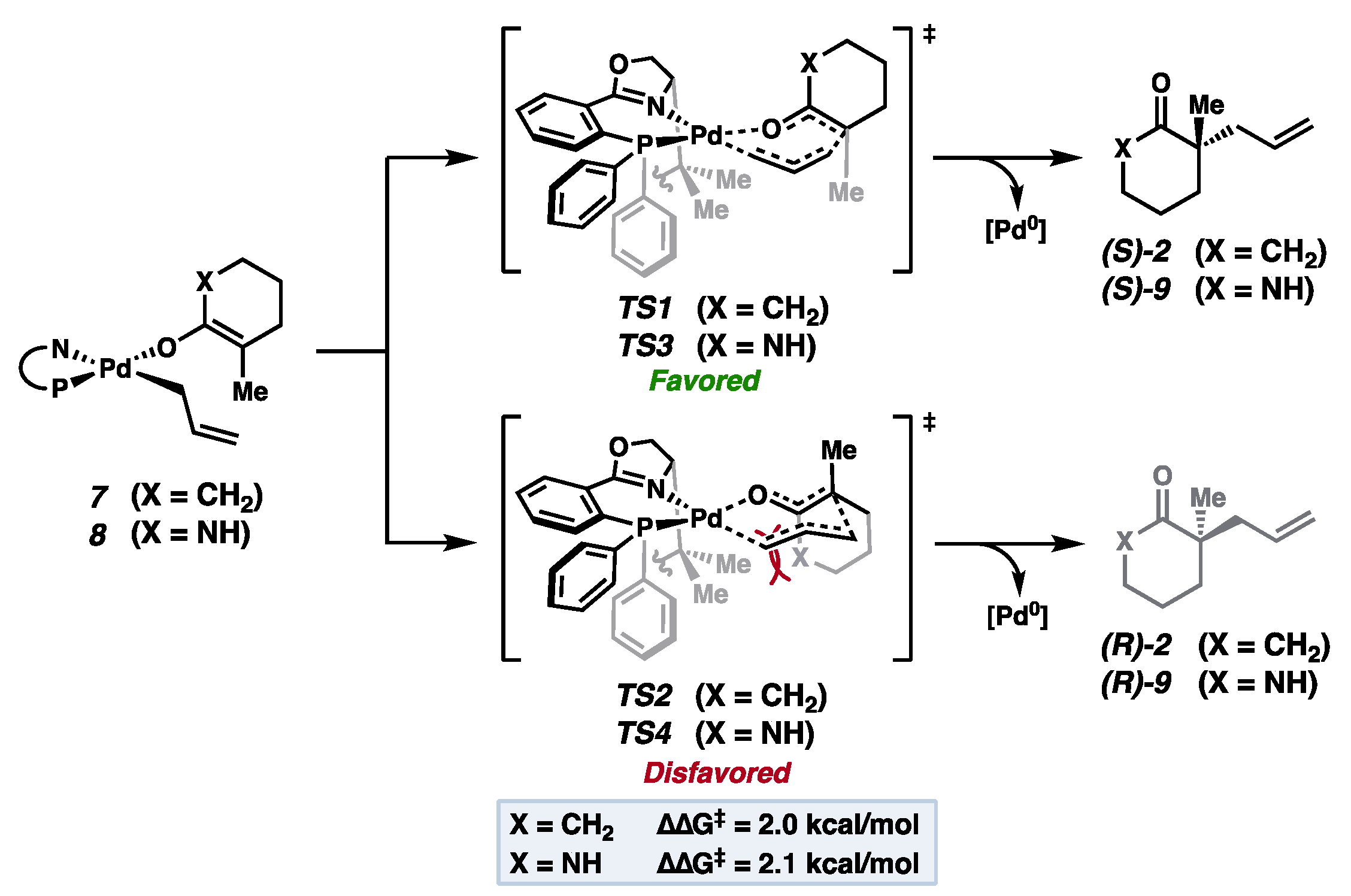
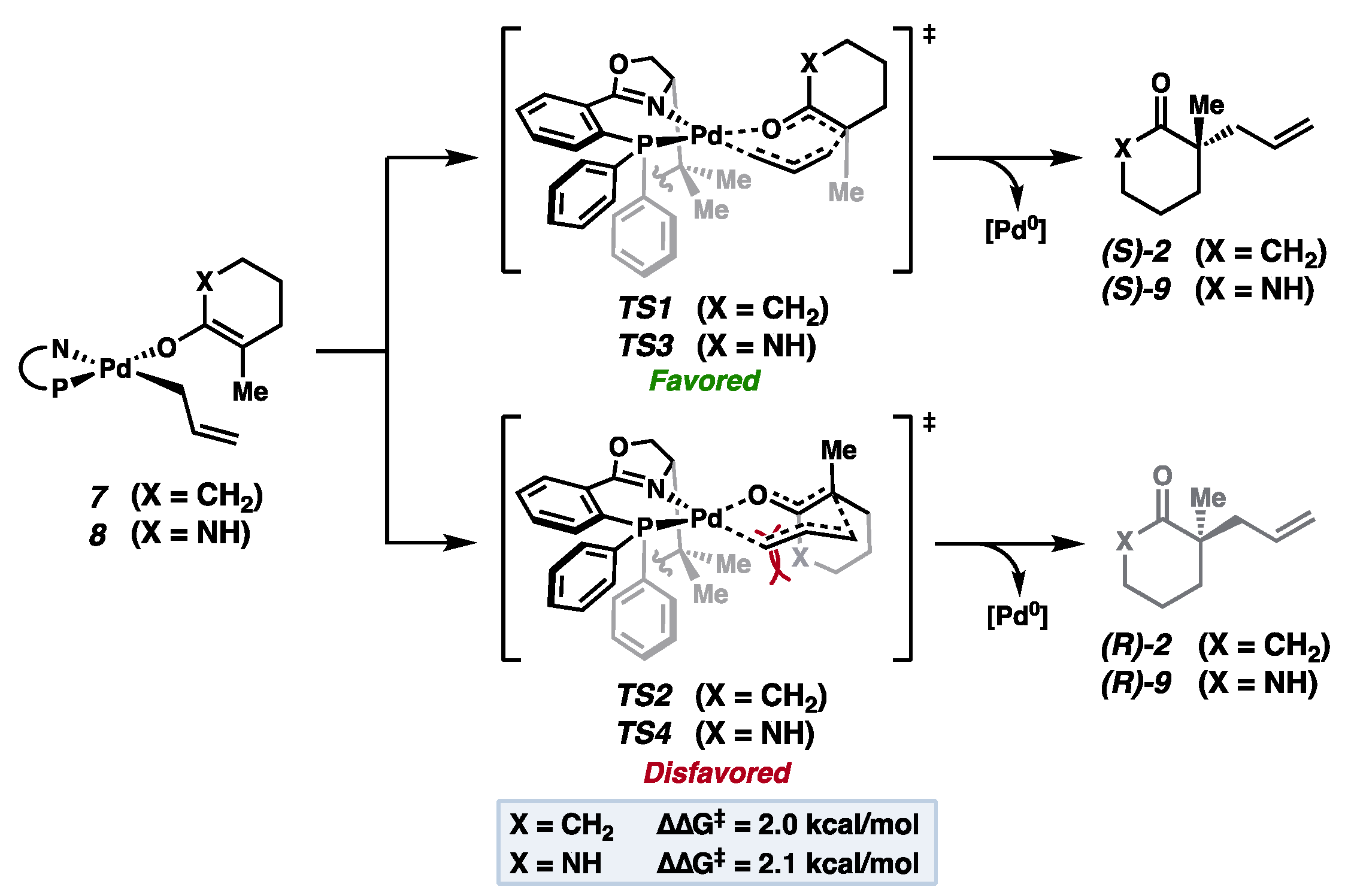
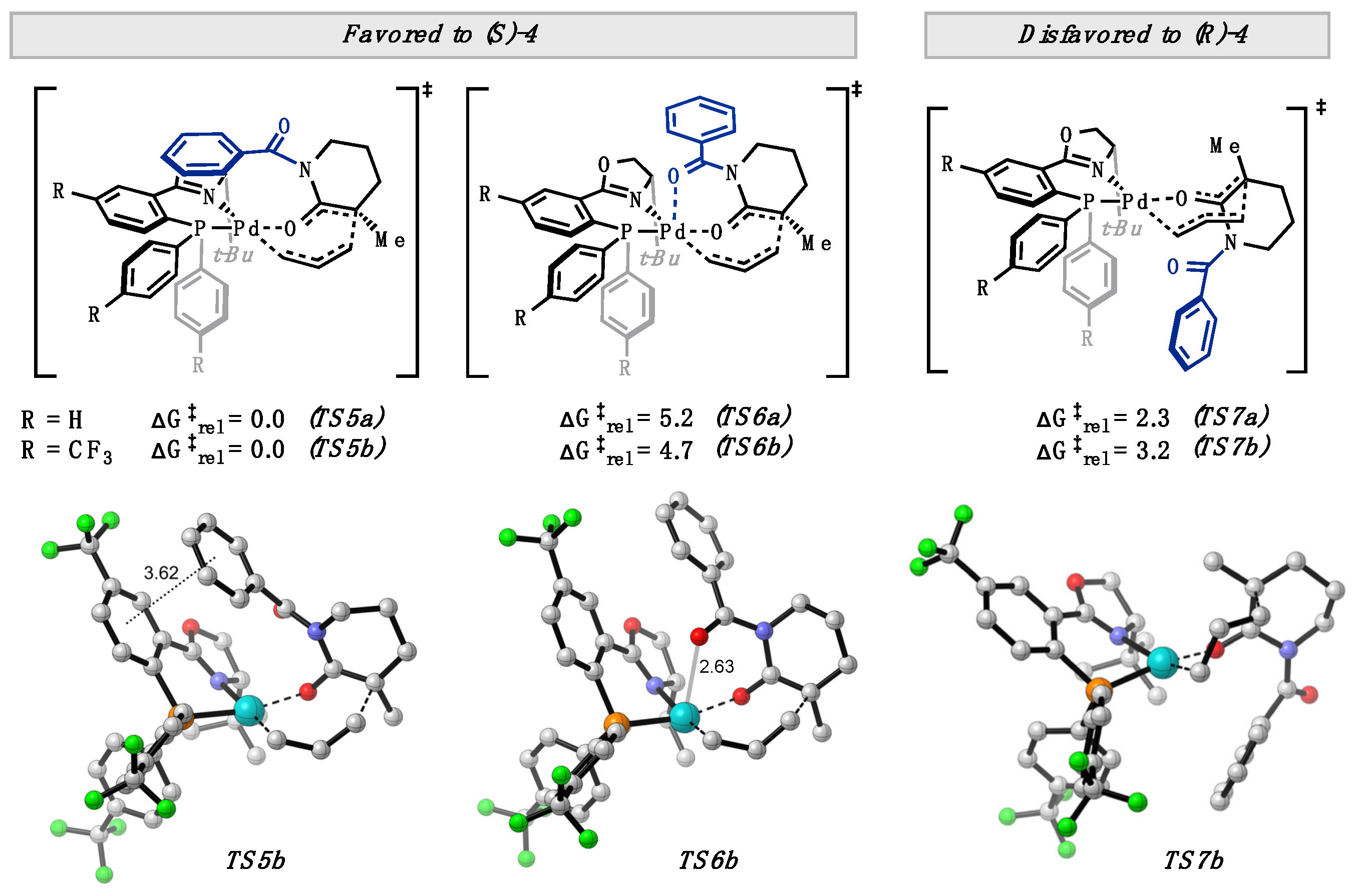
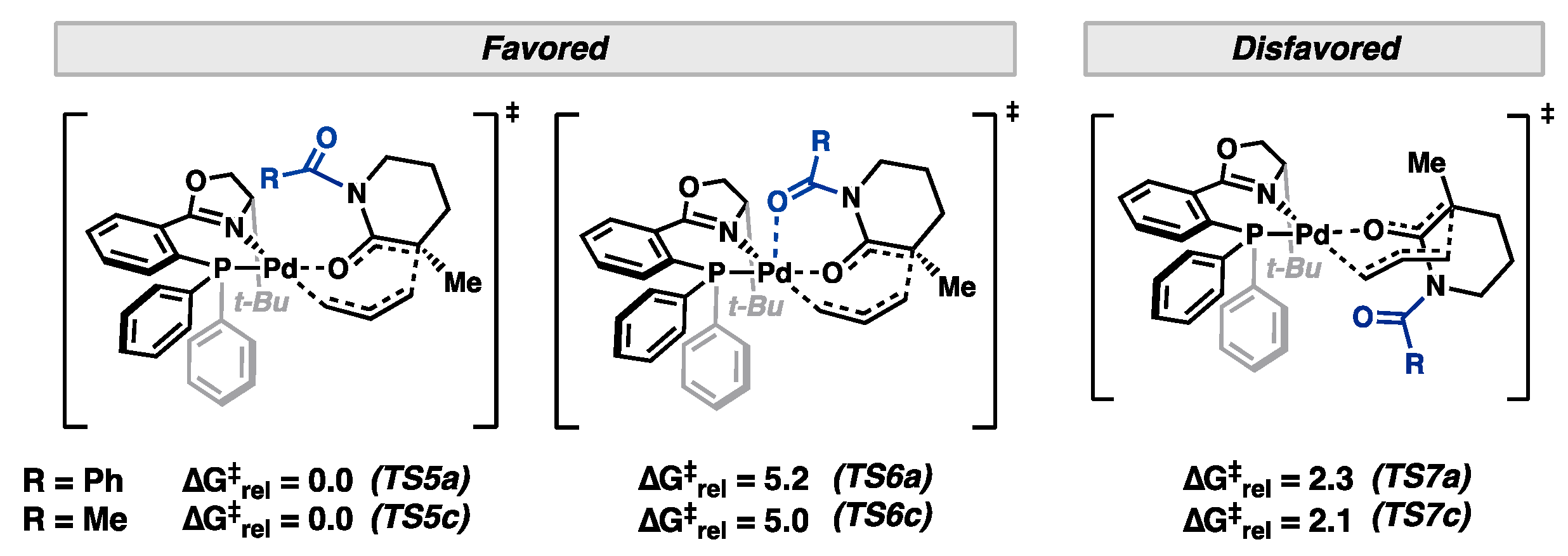
2. Results and Discussion
3. Computational Details
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Han, S.-J.; Liu, W.-B.; Stoltz, B.M. Catalytic Enantioselective Construction of Quaternary Stereocenters: Assembly of Key Building Blocks for the Synthesis of Biologically Active Molecules. Acc. Chem. Res. 2015, 48, 740–751. [Google Scholar] [CrossRef]
- Trost, B.M.; Crawley, M.L. Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev. 2003, 103, 2921–2944. [Google Scholar] [CrossRef]
- Behenna, D.C.; Stoltz, B.M. The Enantioselective Tsuji Allylation. J. Am. Chem. Soc. 2004, 126, 15044–15045. [Google Scholar] [CrossRef]
- Mohr, J.T.; Behenna, D.C.; Harned, A.M.; Stoltz, B.M. Deracemization of Quaternary Stereocenters by Pd-Catalyzed Enantioconvergent Decarboxylative Allylation of Racemic β-Ketoesters. Angew. Chem. Int. Ed. 2005, 44, 6924–6927. [Google Scholar] [CrossRef]
- Keith, J.A.; Behenna, D.C.; Mohr, J.T.; Ma, S.; Marinescu, S.C.; Oxgaard, J.; Stoltz, B.M.; Goddard, W.A. The Inner-Sphere Process in the Enantioselective Tsuji Allylation Reaction with (S)-t-Bu-Phosphinooxazoline Ligands. J. Am. Chem. Soc. 2007, 129, 11876–11877. [Google Scholar] [CrossRef]
- Keith, J.A.; Behenna, D.C.; Sherden, N.; Mohr, J.T.; Ma, S.; Marinescu, S.C.; Nielsen, R.J.; Oxgaard, J.; Stoltz, B.M.; Goddard, W.A. The Reaction Mechanism of the Enantioselective Tsuji Allylation: Inner-Sphere and Outer-Sphere Pathways, Internal Rearrangements, and Asymmetric C–C Bond Formation. J. Am. Chem. Soc. 2012, 134, 19050–19060. [Google Scholar] [CrossRef]
- Cusumano, A.Q.; Stoltz, B.M.; Goddard, W.A. Reaction Mechanism, Origins of Enantioselectivity, and Reactivity Trends in Asymmetric Allylic Alkylation: A Comprehensive Quantum Mechanics Investigation of a C(sp3)–C(sp3) Cross-Coupling. J. Am. Chem. Soc. 2020, 142, 13917–13933. [Google Scholar] [CrossRef]
- McPherson, K.E.; Croatt, M.P.; Morehead, A.T.; Sargent, A.L. DFT Mechanistic Investigation of an Enantioselective Tsuji–Trost Allylation Reaction. Organometallics 2018, 37, 3791–3802. [Google Scholar] [CrossRef]
- Sherden, N.H.; Behenna, D.C.; Virgil, S.C.; Stoltz, B.M. Unusual Allylpalladium Carboxylate Complexes: Identification of the Resting State of Catalytic Enantioselective Decarboxylative Allylic Alkylation Reactions of Ketones. Angew. Chem. Int. Ed. 2009, 48, 6840–6843. [Google Scholar] [CrossRef]
- Behenna, D.C.; Liu, Y.; Yurino, T.; Kim, J.; White, D.E.; Virgil, S.C.; Stoltz, B.M. Enantioselective Construction of Quaternary N-Heterocycles by Palladium-Catalysed Decarboxylative Allylic Alkylation of Lactams. Nat. Chem. 2012, 4, 130–133. [Google Scholar] [CrossRef]
- Duquette, D.C.; Cusumano, A.Q.; Lefoulon, L.; Moore, J.T.; Stoltz, B.M. Probing Trends in Enantioinduction via Substrate Design: Palladium-Catalyzed Decarboxylative Allylic Alkylation of α-Enaminones. Org. Lett. 2020, 22, 4966–4969. [Google Scholar] [CrossRef]
- Bennett, N.B.; Duquette, D.C.; Kim, J.; Liu, W.-B.; Marziale, A.N.; Behenna, D.C.; Virgil, S.C.; Stoltz, B.M. Expanding Insight into Asymmetric Palladium-Catalyzed Allylic Alkylation of N-Heterocyclic Molecules and Cyclic Ketones. Chem. Eur. J. 2013, 19, 4414–4418. [Google Scholar] [CrossRef]
- Cusumano, A.Q.; Goddard, W.A.I.; Stoltz, B.M. The Transition Metal Catalyzed [π2s + π2s + σ2s + σ2s] Pericyclic Reaction: Woodward–Hoffmann Rules, Aromaticity, and Electron Flow. J. Am. Chem. Soc. 2020, 142, 19033–19039. [Google Scholar] [CrossRef]
- Aullón, G.; Alvarez, S. Axial Bonding Capabilities of Square Planar d8-ML4 Complexes. Theoretical Study and Structural Correlations. Inorg. Chem. 1996, 35, 3137–3144. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Ehlert, S.; Huniar, U.; Ning, J.; Furness, J.W.; Sun, J.; Kaplan, A.D.; Perdew, J.P.; Brandenburg, J.G. r2SCAN-D4: Dispersion Corrected Meta-Generalized Gradient Approximation for General Chemical Applications. J. Chem. Phys. 2021, 154, 061101. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A Generally Applicable Atomic-Charge Dependent London Dispersion Correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically Convergent Basis Sets with Relativistic Pseudopotentials. II. Small-Core Pseudopotentials and Correlation Consistent Basis Sets for the Post-d Group 16–18 Elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- Santra, G.; Sylvetsky, N.; Martin, J.M.L. Minimally Empirical Double-Hybrid Functionals Trained against the GMTKN55 Database: revDSD-PBEP86-D4, revDOD-PBE-D4, and DOD-SCAN-D4. J. Phys. Chem. A 2019, 123, 5129–5143. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. wB97M-V: A Combinatorially Optimized, Range-Separated Hybrid, Meta-GGA Density Functional with VV10 Nonlocal Correlation. J. Chem. Phys. 2016, 144, 214110. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem. Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cusumano, A.Q.; Zhang, T.; Goddard, W.A., III; Stoltz, B.M. Origins of Enhanced Enantioselectivity in the Pd-Catalyzed Decarboxylative Allylic Alkylation of N-Benzoyl Lactams. Catalysts 2023, 13, 1258. https://doi.org/10.3390/catal13091258
Cusumano AQ, Zhang T, Goddard WA III, Stoltz BM. Origins of Enhanced Enantioselectivity in the Pd-Catalyzed Decarboxylative Allylic Alkylation of N-Benzoyl Lactams. Catalysts. 2023; 13(9):1258. https://doi.org/10.3390/catal13091258
Chicago/Turabian StyleCusumano, Alexander Q., Tianyi Zhang, William A. Goddard, III, and Brian M. Stoltz. 2023. "Origins of Enhanced Enantioselectivity in the Pd-Catalyzed Decarboxylative Allylic Alkylation of N-Benzoyl Lactams" Catalysts 13, no. 9: 1258. https://doi.org/10.3390/catal13091258
APA StyleCusumano, A. Q., Zhang, T., Goddard, W. A., III, & Stoltz, B. M. (2023). Origins of Enhanced Enantioselectivity in the Pd-Catalyzed Decarboxylative Allylic Alkylation of N-Benzoyl Lactams. Catalysts, 13(9), 1258. https://doi.org/10.3390/catal13091258