
Electrochemical Promotion of CO2 Hydrogenation Using Rh Catalysts Supported on O2− Conducting Solid Electrolyte




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
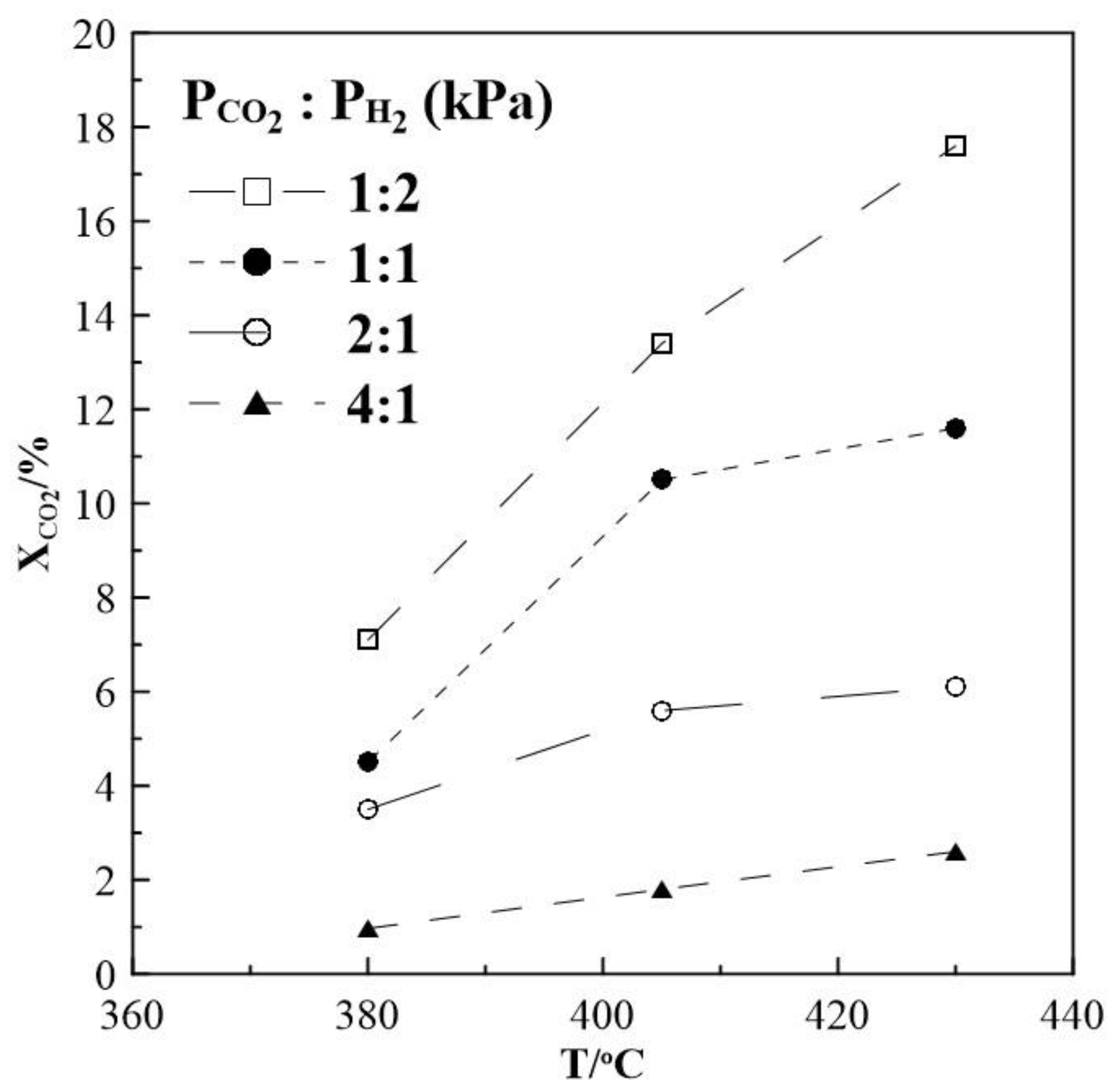
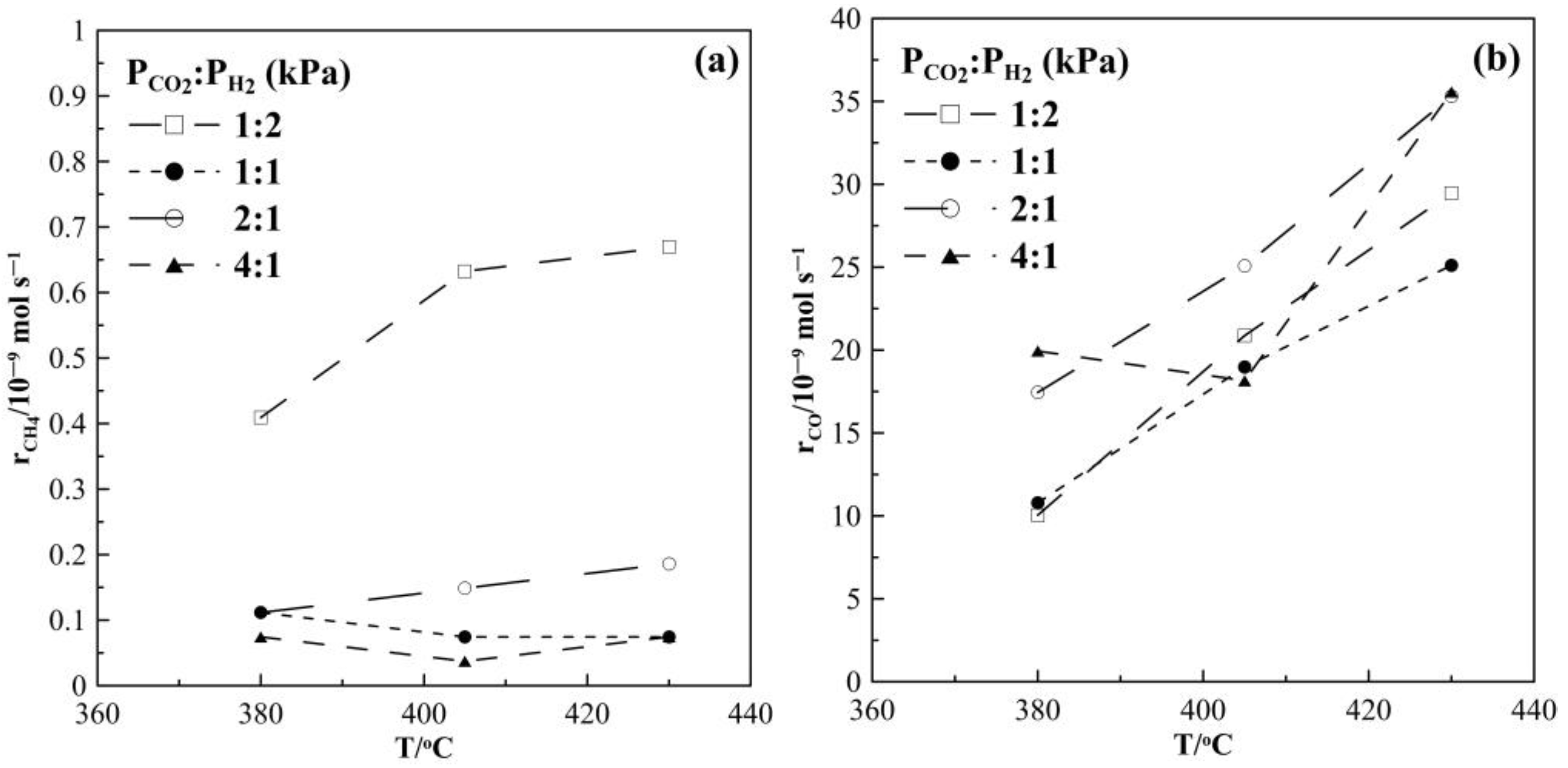
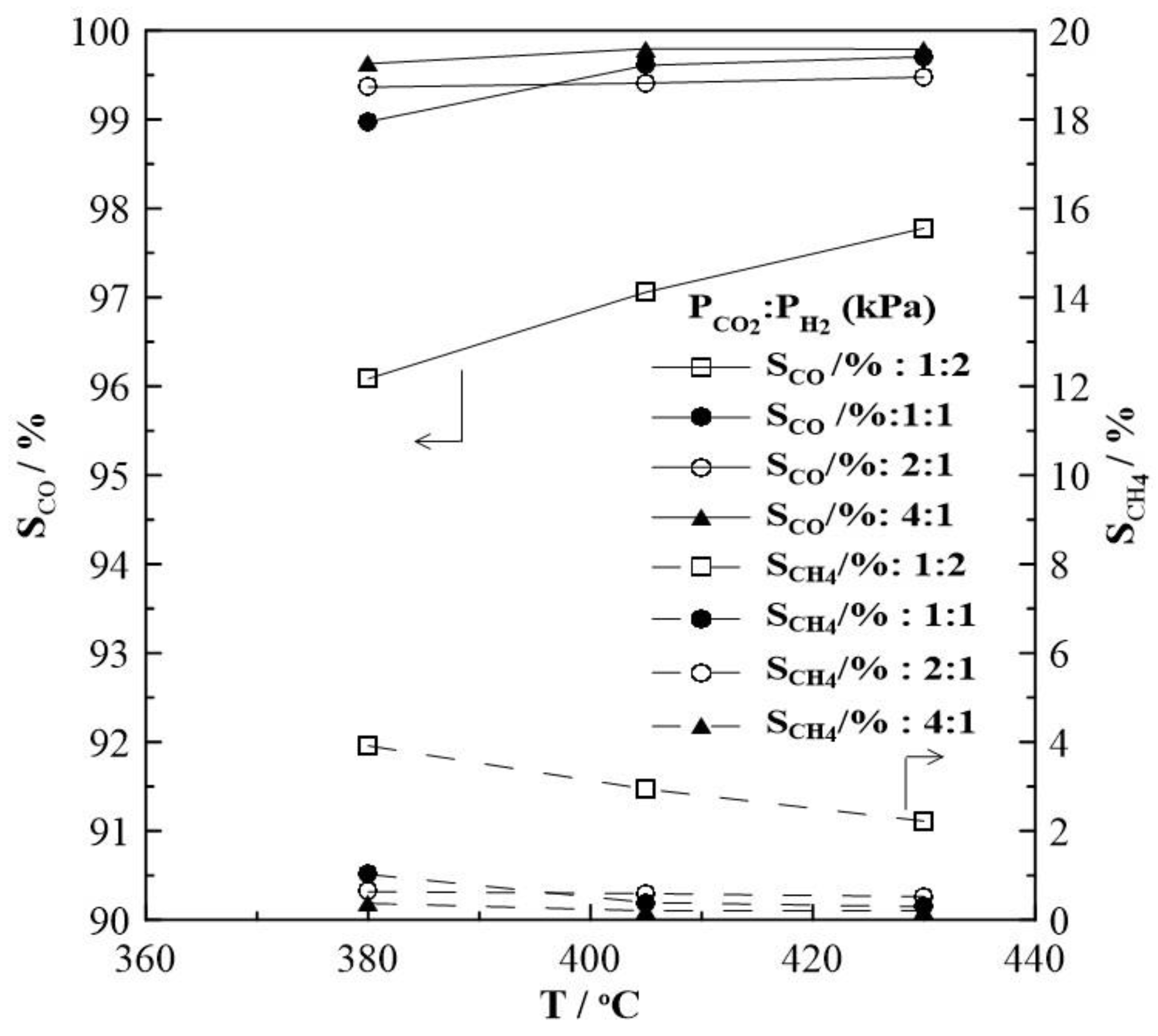
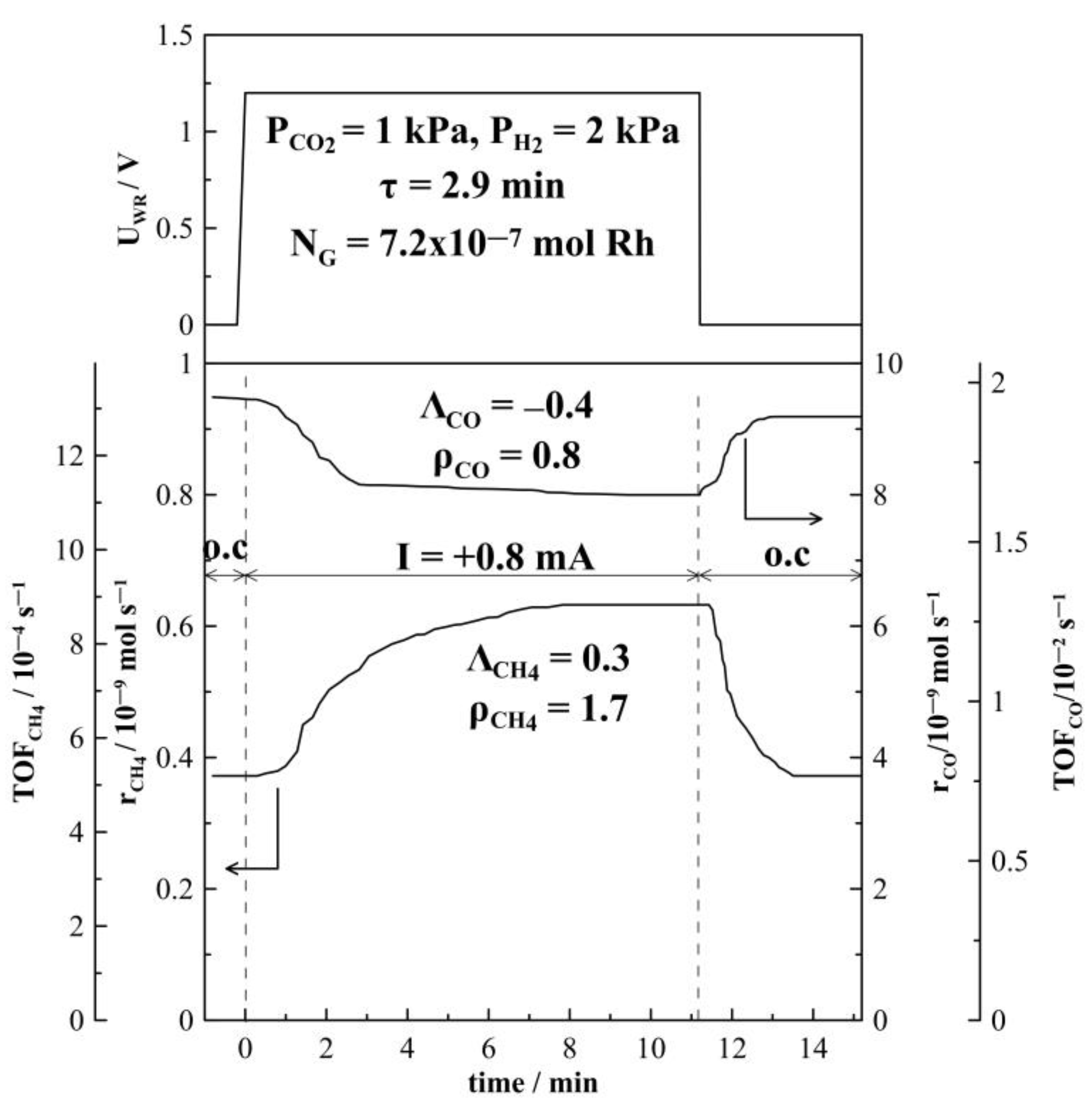
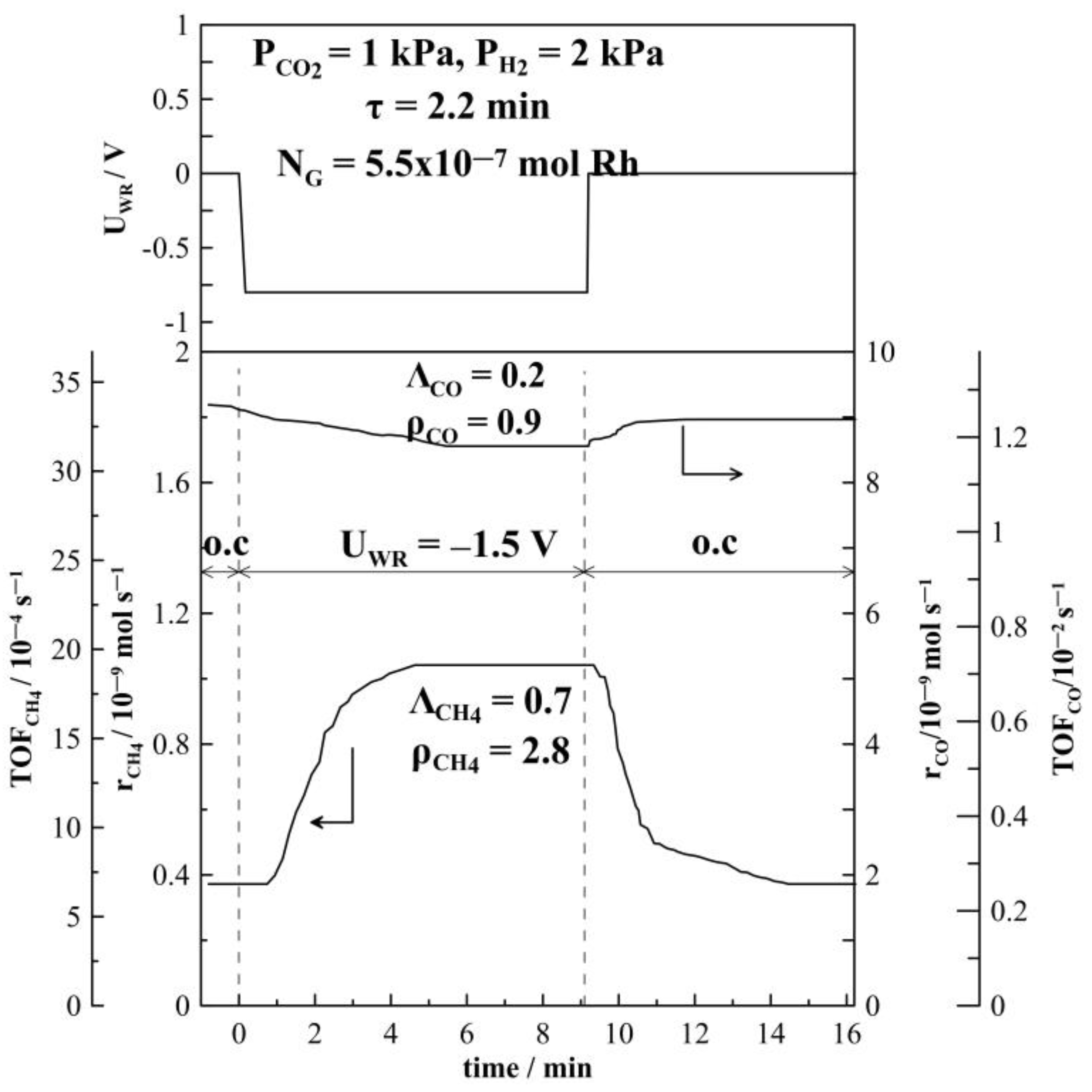
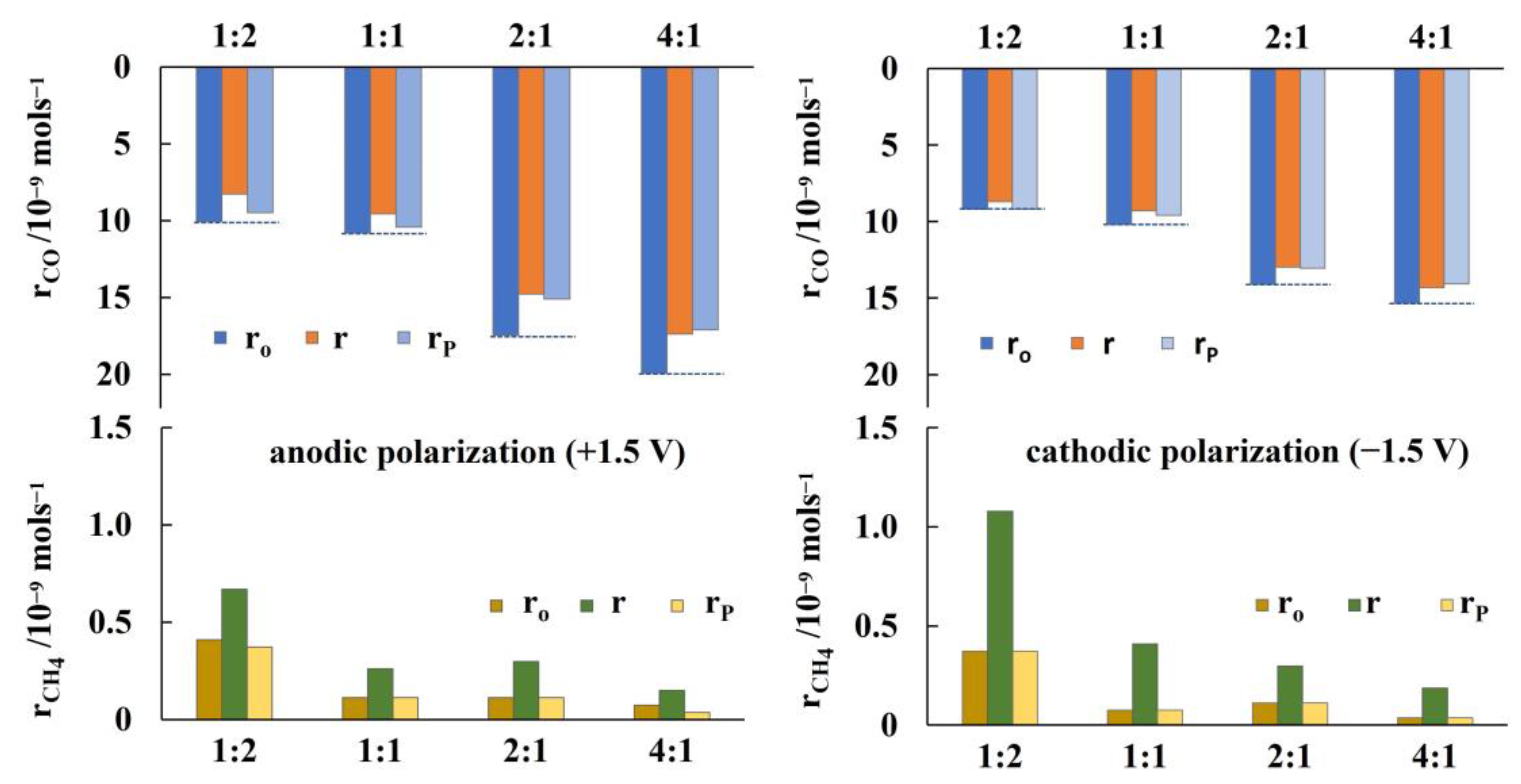
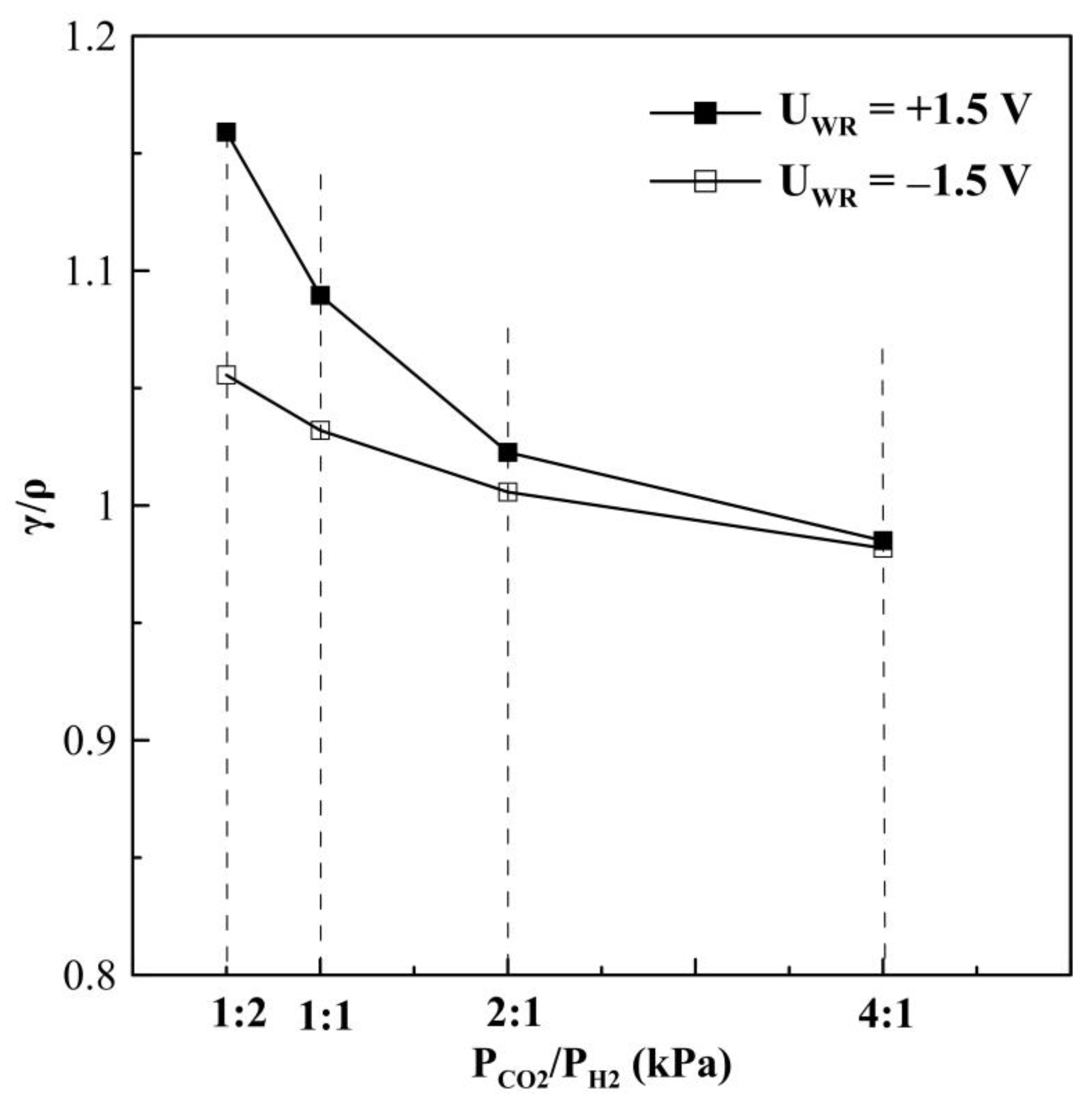
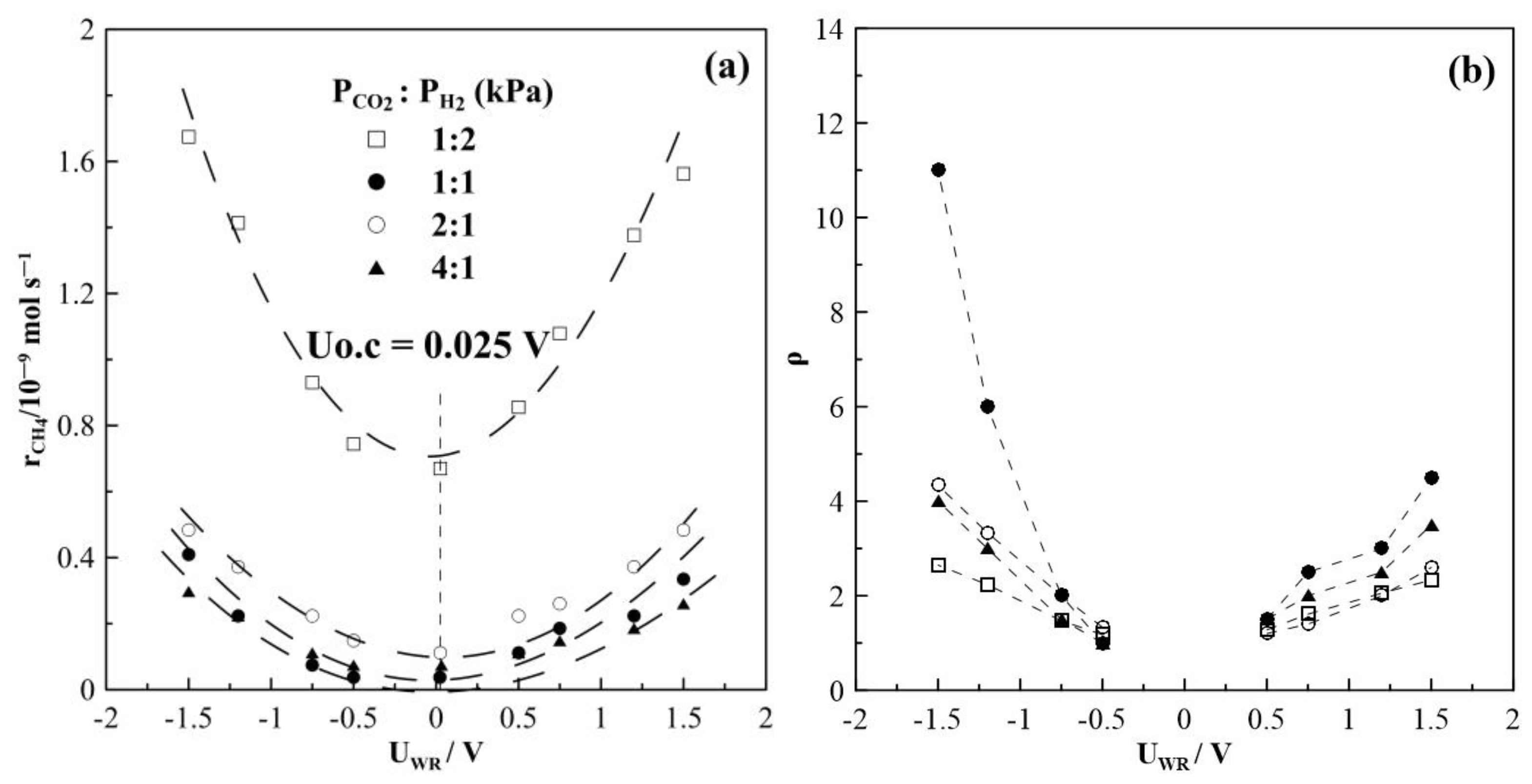
2. Results and Discussion
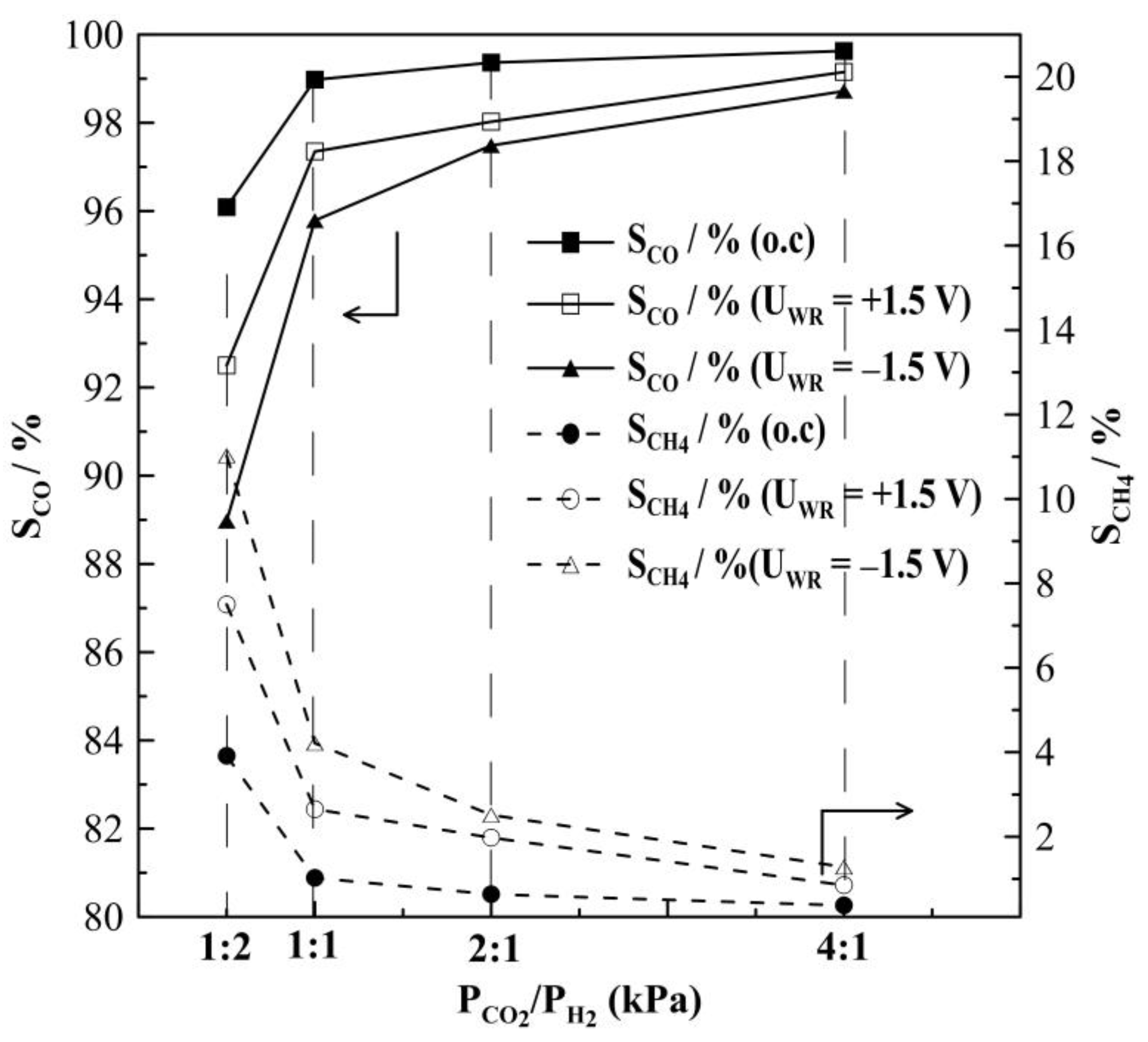
2.1. Hydrogenation Activity Measurements
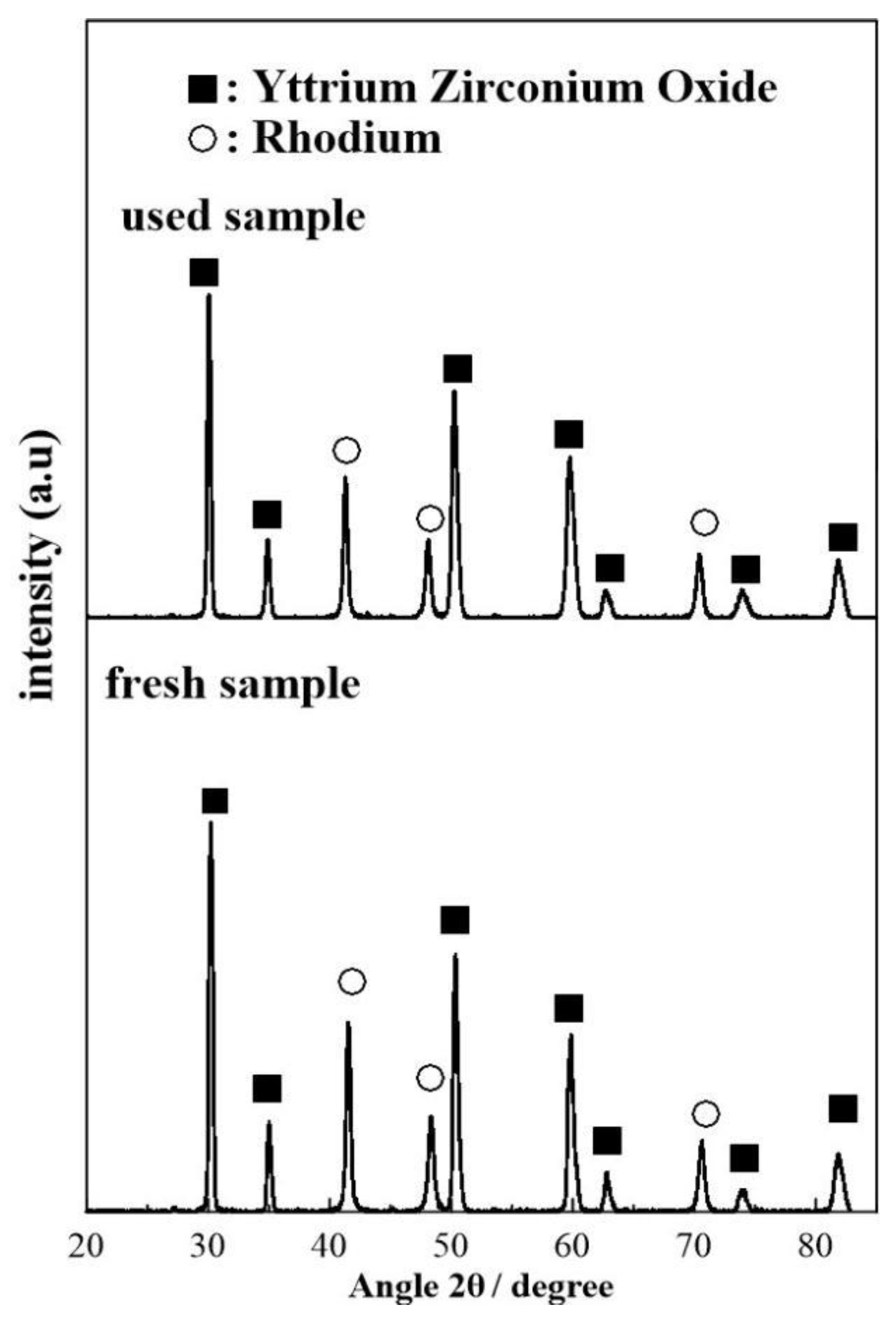
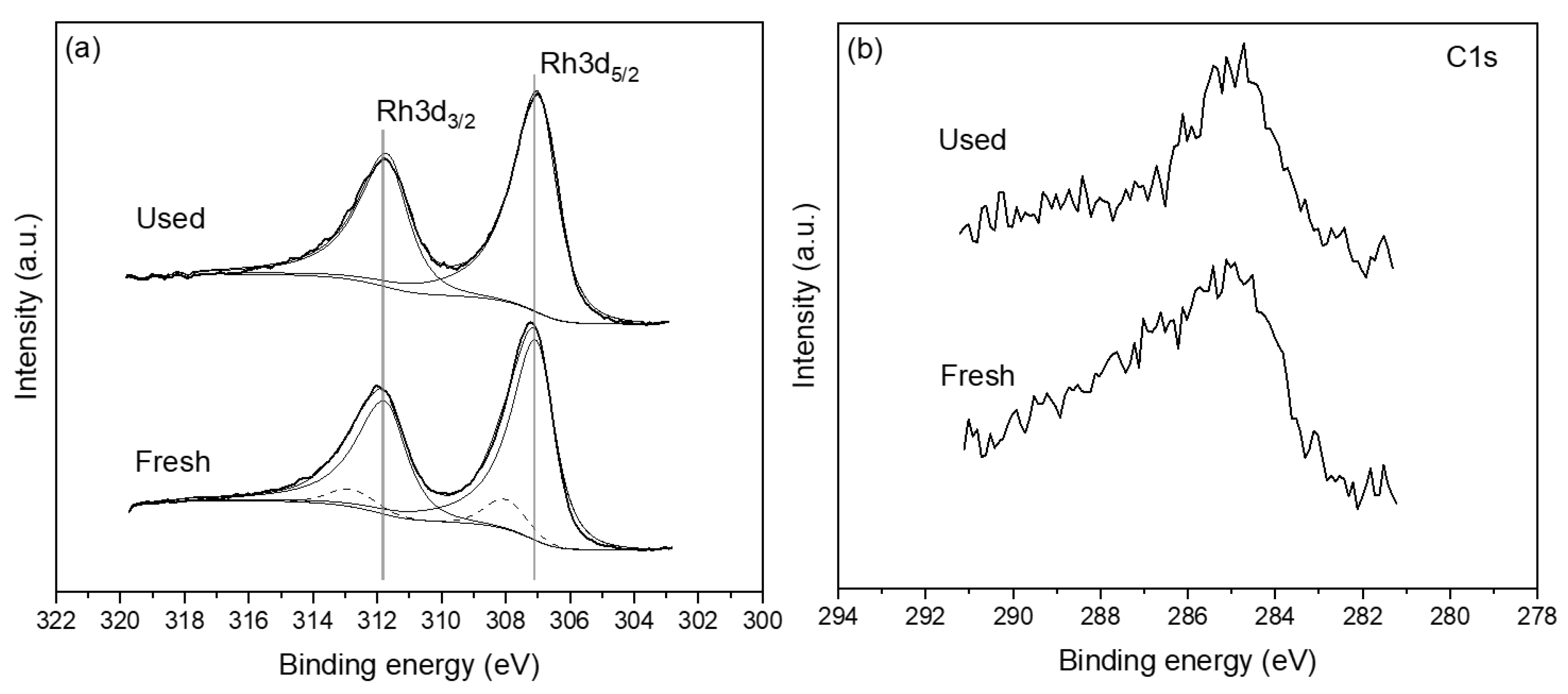
2.2. Catalyst Characterization
3. Experimental
3.1. Catalyst Preparation
3.2. Reactor Operation
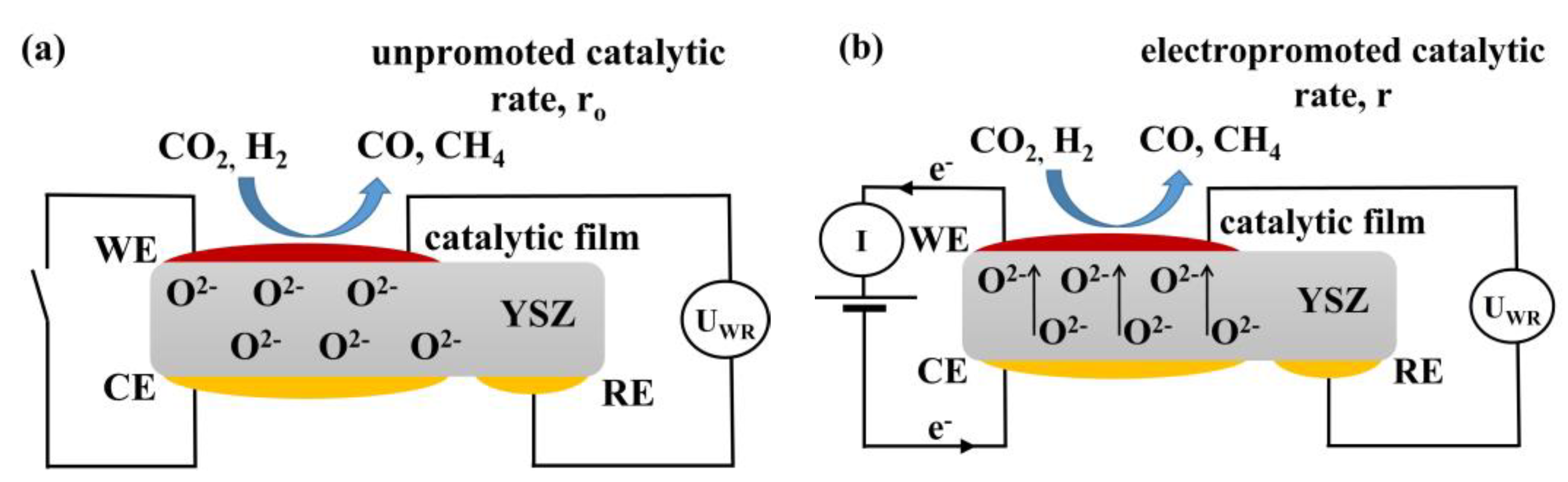
3.3. Electrochemical Promotion Parameters Computation
3.4. Catalyst Characterization
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kinney, P.L. Interactions of Climate Change, Air Pollution, and Human Health. Curr. Environ. Health Rep. 2018, 5, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Berry, H.L.; Waite, T.D.; Dear, K.B.G.; Capon, A.G.; Murray, V. The Case for Systems Thinking about Climate Change and Mental Health. Nat. Clim. Chang. 2018, 8, 282–290. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning Selectivity of CO2 Hydrogenation Reactions at the Metal/Oxide Interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef] [PubMed]
- Knutson, T.R.; Tuleya, R.E. Impact of CO2-Induced Warming on Simulated Hurricane Intensity and Precipitation: Sensitivity to the Choice of Climate Model and Convective Parameterization. J. Clim. 2004, 17, 3477–3495. [Google Scholar] [CrossRef]
- Schwiderowski, P.; Ruland, H.; Muhler, M. Current Developments in CO2 Hydrogenation towards Methanol: A Review Related to Industrial Application. Curr. Opin. Green Sustain. Chem. 2022, 38, 100688. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Y.; Ding, M.; Hong, X.; Liu, G.; Tsang, S.C.E. Advances in Higher Alcohol Synthesis from CO2 Hydrogenation. Chem 2021, 7, 849–881. [Google Scholar] [CrossRef]
- Chatzilias, C.; Martino, E.; Zagoraios, D.; Kyriakou, G.; Katsaounis, A. Electrochemical Promotion of Catalysis for CO2 Valorization. In Recent Advances in Electrochemical Promotion of Catalysis; Modern Aspects of Electrochemistry; Vernoux, P., Vayenas, C.G., Eds.; Springer: Cham, Switzerland, 2022; Volume 61. [Google Scholar]
- Nozaki, F.; Sodesawa, T.; Satoh, S.; Kimura, K. Hydrogenation of Carbon Dioxide into Light Hydrocarbons at Atmospheric Pressure over Rh/Nb205 or Cu/Si02-Rh/Nb205. J. Catal. 1987, 104, 339–346. [Google Scholar] [CrossRef]
- Solymosi, F.; Pasztor, M. Analysis of the R-Spectral Behavior of Adsorbed CO Formed in H2 + CO2 Surface Interaction over Supported Rhodium. J. Catal. 1987, 104, 312–322. [Google Scholar] [CrossRef]
- Lapidus, A.L.; Gaidai, N.A.; Nekrasov, N.V.; Tishkova, L.A.; Agafonov, Y.A.; Myshenkova, T.N. The Mechanism of Carbon Dioxide Hydrogenation on Copper and Nickel Catalysts. Pet. Chem. 2007, 47, 91–98. [Google Scholar] [CrossRef]
- Borodko, Y.; Somorjai, G.A. Catalytic Hydrogenation of Carbon Oxides-a 10-Year Perspective. Appl. Catal. A Gen. 1999, 186, 355–362. [Google Scholar] [CrossRef]
- Kusama, H.; Kitamura Bando, K.; Okabe, K.; Arakawa, H. Effect of Metal Loading on CO2 Hydrogenation Reactivity over Rh/SiO2 Catalysts. Appl. Catal. 2000, 197, 255–268. [Google Scholar] [CrossRef]
- Gasser, D.; Baiker, A. Hydrogenation of Carbon Dioxide over Copper-Zirconia Catalysts Prepared by In-Situ Activation of Amorphous Copper-Zirconium Alloy. Appl. Catal. 1989, 48, 279–294. [Google Scholar] [CrossRef]
- Amenomiya, Y. Methanol synthesis from CO + H2. II Copper-based binary and ternary catalysts. Appl. Catal. 1987, 30, 57–68. [Google Scholar] [CrossRef]
- Sahibzada, M.; Chadwick, D.; Metcalfe, I.S. Hydrogenation of Carbon Dioxide to Methanol over Palladium-Promoted Cu/ZnO/A12O3 Catalysts. Catal. Today 1996, 29, 367–372. [Google Scholar] [CrossRef]
- Solymosi, F.; Erdhelyi, A.; Bansagi, T. Methanation of CO, on Supported Rhodium Catalyst. J. Catal. 1981, 68, 371–382. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, Y.; Tsubaki, N. Dual Catalysis Mechanism of Alcohol Solvent and Cu Catalyst for a New Methanol Synthesis Method. Catal. Commun. 2005, 6, 275–279. [Google Scholar] [CrossRef]
- Schild, C.; Wokaun, A.; Baiker, A. On the Mechanism of CO and CO2 Hydrogenation Reactions on Zirconia-Supported Catalysts: A Diffuse Reflectance FTIR Study Part II. Surface Species on Copper/Zirconia Catalysts: Implications for Methanol Synthesis Selectivity. J. Mol. Catal. 1990, 63, 243–254. [Google Scholar] [CrossRef]
- Vannice, M.A. The Catalytic Synthesis of Hydrocarbons from HJCO Mixtures over the Group VIII Metals I. The Specific Activities and Product Distributions of Supported Metals. J. Catal. 1975, 31, 449–461. [Google Scholar] [CrossRef]
- Araki, R.; Ponec, V. Methanation of Carbon Monoxide on Nickel and Nickel-Copper Alloys. J. Catal. 1976, 44, 439–448. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Selective Methanation of CO over Supported Noble Metal Catalysts: Effects of the Nature of the Metallic Phase on Catalytic Performance. Appl. Catal. A Gen. 2008, 344, 45–54. [Google Scholar] [CrossRef]
- Henderson, A.; Worley, S.D.; Peebles, D.E. An Infrared Study of the Hydrogenation of Carbon Dioxide on Supported Rhodium Catalys An inverse spillover effect. J. Phys. Chem. 1985, 89, 392–394. [Google Scholar] [CrossRef]
- Solymosi, F.; Tombacz, I.; Koszta, J. Effects of Variation of Electric Properties of TiOp Support on Hydrogenation of CO and CO2 over Rh Catalysts. J. Catal. 1985, 95, 578–586. [Google Scholar] [CrossRef]
- Brown Bourzutschky, J.A.; Homs, N.; Bell, A.A.T. Hydrogenation of 002 and 002/00 Mixtures over Copper-Containing Catalysts. J. Catal. 1990, 124, 73–85. [Google Scholar] [CrossRef]
- Chanchlani, K.G.; Hudgins, R.R.; Silveston, P.L. Methanol Synthesis from H2, CO, and 002 over Cu/ZnO Catalysts. J. Catal. 1992, 136, 59–75. [Google Scholar] [CrossRef]
- Nitta, Y.; Suwata, O.; Ikeda, Y.; Okamoto, Y.; Imanaka, T. Copper-Zirconia Catalysts for Methanol Synthesis from Carbon Dioxide: Effect of ZnO Addition to Cu-ZrO2 Catalysts. Catal. Lett. 1994, 26, 345–354. [Google Scholar] [CrossRef]
- Arena, F.; Italiano, G.; Barbera, K.; Bordiga, S.; Bonura, G.; Spadaro, L.; Frusteri, F. Solid-State Interactions, Adsorption Sites and Functionality of Cu-ZnO/ZrO2 Catalysts in the CO2 Hydrogenation to CH3OH. Appl. Catal. A Gen. 2008, 350, 16–23. [Google Scholar] [CrossRef]
- Reubroycharoen, P.; Vitidsant, T.; Yoneyama, Y.; Tsubaki, N. Development of a New Low-Temperature Methanol Synthesis Process. Catal. Today 2004, 89, 447–454. [Google Scholar] [CrossRef]
- Hori, Y.; Wakebe, H.; Tsukamoto, T.; Koga, O. Adsorption of CO Accompanied with Simultaneous Charge Transfer on Copper Single Crystal Electrodes Related with Electrochemical Reduction of CO2 to Hydrocarbons. Surf. Sci. 1995, 335, 258–260. [Google Scholar] [CrossRef]
- Ando, H.; Xu, Q.; Fujiwara, M.; Matsumura, Y.; Tanaka, M.; Souma, Y. Hydrocarbon Synthesis from CO2 over Fe ± Cu. Catal. Today 1998, 45, 229–234. [Google Scholar] [CrossRef]
- Trovarelli, A.; Mustazza, C.; Dolcetti, G.; Kagpar, J.; Graziani, M. Carbon dioxide hydrogenation on rhodium supported on transition metal oxides. Effect of reduction temperature on product distribution. Appl. Catal. 1990, 65, 129–142. [Google Scholar] [CrossRef]
- Marwood, M.; Doepper, R.; Renken, A. In-Situ Surface and Gas Phase Analysis for Kinetic Studies under Transient Conditions: The Catalytic Hydrogenation of CO2. Appl. Catal. 1997, 151, 223–246. [Google Scholar] [CrossRef]
- Falconer, J.L.; Zagli, A.E. Adsorption and Methanation of Carbon Dioxide on a Nickel/Silica Catalyst. J. Catal. 1980, 62, 280–285. [Google Scholar] [CrossRef]
- Biloen, P.; Helle, J.N.; Van Den Berg, F.G.A.; Sachtler, W.M.H. On the Activity of Fischer-Tropsch and Methanation Catalysts: A Study Utilizing Isotopic Transients. J. Catal. 1983, 81, 450–463. [Google Scholar] [CrossRef]
- Coenen, J.W.E.; Van Nisselrooy, P.F.M.T.; De Croon, M.H.J.M.; Van Dooren, P.F.H.A.; Van Meerten, R.Z.C. The dynamics of methanation of carbon modoxide on Nickel catalysts. Appl. Catal. 1986, 25, 1–8. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, J.; Ma, Z.; Zhang, G.; Zhang, H.; Fu, X.; Ma, Y.; Liu, F.; Liu, M.; Huang, H. Seeded Growth of Gold–Copper Janus Nanostructures as a Tandem Catalyst for Efficient Electroreduction of CO2 to C2+ Products. Small 2022, 18, 2201695. [Google Scholar] [CrossRef]
- Zhong, Y.; Kong, X.; Song, Z.; Liu, Y.; Peng, L.; Zhang, L.; Luo, X.; Zeng, J.; Geng, Z. Adjusting Local CO Confinement in Porous-Shell Ag@Cu Catalysts for Enhancing C-C Coupling toward CO2 Eletroreduction. Nano Lett. 2022, 22, 2554–2560. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Pliangos, C.; Brosda, S.; Tsiplakides, D. Electrochemical Activation of Catalysis Promotion, Electrochemical Promotion, and Metal-Support Interactions; Springer: New York, NY, USA, 2002. [Google Scholar]
- Theleritis, D.; Souentie, S.; Siokou, A.; Katsaounis, A.; Vayenas, C.G. Hydrogenation of CO2 over Ru/YSZ Electropromoted Catalysts. ACS Catal. 2012, 2, 770–780. [Google Scholar] [CrossRef]
- Kalaitzidou, I.; Makri, M.; Theleritis, D.; Katsaounis, A.; Vayenas, C.G. Comparative Study of the Electrochemical Promotion of CO2 Hydrogenation on Ru Using Na+, K+, H+ and O2− Conducting Solid Electrolytes. Surf. Sci. 2016, 646, 194–203. [Google Scholar] [CrossRef]
- Panaritis, C.; Michel, C.; Couillard, M.; Baranova, E.A.; Steinmann, S.N. Elucidating the Role of Electrochemical Polarization on the Selectivity of the CO2 Hydrogenation Reaction over Ru. Electrochim. Acta 2020, 350, 136405. [Google Scholar] [CrossRef]
- Chatzilias, C.; Martino, E.; Tsatsos, S.; Kyriakou, G.; Katsaounis, A.; Vayenas, C.G. Kinetic study of CO2 hydrogenation on Ru/ YSZ catalyst using a monolithic electropromoted reactor (MEPR). Chem. Eng. J. 2022, 430, 132967. [Google Scholar] [CrossRef]
- Zagoraios, D.; Panaritis, C.; Krassakopoulou, A.; Baranova, E.A.; Katsaounis, A.; Vayenas, C.G. Electrochemical Promotion of Ru Nanoparticles Deposited on a Proton Conductor Electrolyte during CO2 Hydrogenation. Appl. Catal. B 2020, 276, 119148. [Google Scholar] [CrossRef]
- Bebelis, S.; Karasali, H.; Vayenas, C.G. Electrochemical Promotion of CO2 Hydrogenation on Rh/YSZ Electrodes. J. Appl. Electrochem. 2008, 38, 1127–1133. [Google Scholar] [CrossRef]
- Papaioannou, E.I.; Souentie, S.; Hammad, A.; Vayenas, C.G. Electrochemical Promotion of the CO2 Hydrogenation Reaction Using Thin Rh, Pt and Cu Films in a Monolithic Reactor at Atmospheric Pressure. Catal. Today 2009, 146, 336–344. [Google Scholar] [CrossRef]
- Kotsiras, A.; Kalaitzidou, I.; Grigoriou, D.; Symillidis, A.; Makri, M.; Katsaounis, A.; Vayenas, C.G. Electrochemical Promotion of Nanodispersed Ru-Co Catalysts for the Hydrogenation of CO2. Appl. Catal. B 2018, 232, 60–68. [Google Scholar] [CrossRef]
- Panaritis, C.; Zgheib, J.; Ebrahim, S.A.H.; Couillard, M.; Baranova, E.A. Electrochemical In-Situ Activation of Fe-Oxide Nanowires for the Reverse Water Gas Shift Reaction. Appl. Catal. B 2020, 269, 118826. [Google Scholar] [CrossRef]
- Nicole, J.; Comninellis, C.; Tsiplakides, D.; Pliangos, C.; Verykios, X.E.; Vayenas, C.G. Electrochemical Promotion and Metal-Support Interactions. J. Catal. 2001, 204, 23–34. [Google Scholar] [CrossRef]
- Stoukides, M.; Vayenas, C.G. The Effect of Electrochemical Oxygen Pumping on the Rate and Selectivity of Ethylene Oxidation on Polycrystalline Silver. J. Catal. 1981, 70, 137–146. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Koutsodontis, C.G. Non-Faradaic Electrochemical Activation of Catalysis. J. Chem. Phys. 2008, 128, 182506. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Ladas, S. Dependence of Catalytic Rates on Catalyst Work Function. Nature 1990, 343, 625–627. [Google Scholar] [CrossRef]
- Bebelis, S.; Vayenas, C.G. Non-Faradaic Electrochemical Modification of Catalytic Activity 1. The Case of Ethylene Oxidation on Pt. J. Catal. 1989, 118, 125–146. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Despotopoulou, M. Non-Faradaic Electrochemical Modification of Catalytic Activity 4. The Use of Β″-Al2O3 as the Solid Electrolyte. J. Catal. 1991, 128, 415–435. [Google Scholar] [CrossRef]
- Ladas, S.; Bebelis, S.; Vayenas, C.G. Work Function Measurements on Catalyst Films Subject to in Situ Electrochemical Promotion. Surf. Sci. 1991, 251–252, 1062–1068. [Google Scholar] [CrossRef]
- Neophytides, S.G.; Tsiplakides, D.; Stonehart, P.; Jaksic, M.M.; Vayenas, C.G. Electrochemical Enhancement of a Catalytic Reaction in Aqueous Solution. Nature 1994, 370, 45–47. [Google Scholar] [CrossRef]
- Pliangos, C.; Raptis, C.; Badas, T.; Tsiplakides, D.; Vayenas, C.G. Electrochemical Promotion of a Classically Promoted Rh Catalyst for the Reduction of NO. Electrochim. Acta 2000, 46, 331–339. [Google Scholar] [CrossRef]
- Vernoux, P.; Gaillard, F.; Bultel, L.; Siebert, E.; Primet, M. Electrochemical Promotion of Propane and Propene Oxidation on Pt/YSZ. J. Catal. 2002, 208, 412–421. [Google Scholar] [CrossRef]
- Katsaounis, A.; Teschner, D.; Zafeiratos, S. The Effect of Polarization and Reaction Mixture on the Rh/YSZ Oxidation State During Ethylene Oxidation Studied by Near Ambient Pressure XPS. Top. Catal. 2018, 61, 2142–2151. [Google Scholar] [CrossRef]
- Constantinou, I.; Archonta, D.; Brosda, S.; Lepage, M.; Sakamoto, Y.; Vayenas, C.G. Electrochemical Promotion of NO Reduction by C3H6 on Rh Catalyst-Electrode Films Supported on YSZ and on Dispersed Rh/YSZ Catalysts. J. Catal. 2007, 251, 400–409. [Google Scholar] [CrossRef]
- Souentie, S.; Xia, C.; Falgairette, C.; Li, Y.D.; Comninellis, C. Investigation of the “Permanent” Electrochemical Promotion of Catalysis (P-EPOC) by Electrochemical Mass Spectrometry (EMS) Measurements. Electrochem. Commun. 2010, 12, 323–326. [Google Scholar] [CrossRef]
- Falgairette, C.; Jaccoud, A.; Fóti, G.; Comninellis, C. The Phenomenon of “Permanent” Electrochemical Promotion of Catalysis (P-EPOC). J. Appl. Electrochem. 2008, 38, 1075–1082. [Google Scholar] [CrossRef]
- Kokka, A.; Petala, A.; Panagiotopoulou, P. Support Effects on the Activity of Ni Catalysts for the Propane Steam Reforming Reaction. Nanomaterials 2021, 11, 1948. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Goula, G.; Panagiotopoulou, P.; Kampouri, S.; Taylor, M.J.; Kyriakou, G.; Lambert, R.M. Stabilization of Catalyst Particles against Sintering on Oxide Supports with High Oxygen Ion Lability Exemplified by Ir-Catalyzed Decomposition of N2O. Appl. Catal. B 2016, 192, 357–364. [Google Scholar] [CrossRef]
- Blomberg, S.; Lundgren, E.; Westerström, R.; Erdogan, E.; Martin, N.M.; Mikkelsen, A.; Andersen, J.N.; Mittendorfer, F.; Gustafson, J. Structure of the Rh2O3(0001) Surface. Surf. Sci. 2012, 606, 1416–1421. [Google Scholar] [CrossRef]
- Holzwarth, U.; Gibson, N. The Scherrer Equation versus the “Debye-Scherrer Equation”. Nat. Nanotechnol. 2011, 6, 534. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Borja, C.; De Lucas-Consuegra, A.; Sapountzi, F.; Dorado, F.; Katsaounis, A.; Valverde, J.L. Oscillatory Behavior of Rh/YSZ under Electropromoted Conditions. Chem. Phys. Lett. 2012, 519–520, 89–92. [Google Scholar] [CrossRef]
- Hamdy, M.S.; Alhanash, A.M.; Benaissa, M.; Alsalme, A.; Alharthi, F.A.; Al-Zaqri, N. Rhodium Nanoparticles Incorporated Mesoporous Silica as an Active Catalyst for Cyclohexene Hydrogenation under Ambient Conditions. Catalysts 2020, 10, 925. [Google Scholar] [CrossRef]
- Tsatsos, S.; Kyriakou, G. Copper Growth on a Stepped Nickel Surface: Electronic and Geometric Effects on CO Reactivity. J. Phys. Chem. 2023, 127, 6337–6346. [Google Scholar] [CrossRef]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokkinou, N.; Xydas, F.; Brosda, S.; Kyriakou, G.; Katsaounis, A. Electrochemical Promotion of CO2 Hydrogenation Using Rh Catalysts Supported on O2− Conducting Solid Electrolyte. Catalysts 2023, 13, 1014. https://doi.org/10.3390/catal13061014
Kokkinou N, Xydas F, Brosda S, Kyriakou G, Katsaounis A. Electrochemical Promotion of CO2 Hydrogenation Using Rh Catalysts Supported on O2− Conducting Solid Electrolyte. Catalysts. 2023; 13(6):1014. https://doi.org/10.3390/catal13061014
Chicago/Turabian StyleKokkinou, Nikoleta, Fotios Xydas, Susanne Brosda, Georgios Kyriakou, and Alexandros Katsaounis. 2023. "Electrochemical Promotion of CO2 Hydrogenation Using Rh Catalysts Supported on O2− Conducting Solid Electrolyte" Catalysts 13, no. 6: 1014. https://doi.org/10.3390/catal13061014
APA StyleKokkinou, N., Xydas, F., Brosda, S., Kyriakou, G., & Katsaounis, A. (2023). Electrochemical Promotion of CO2 Hydrogenation Using Rh Catalysts Supported on O2− Conducting Solid Electrolyte. Catalysts, 13(6), 1014. https://doi.org/10.3390/catal13061014