

New Platinum Complexes from Salen- and Hydroxy-Substituted Salpn-Naphthalene Ligands with CO2 Reduction Activity

 , , ,
, , ,
Abstract
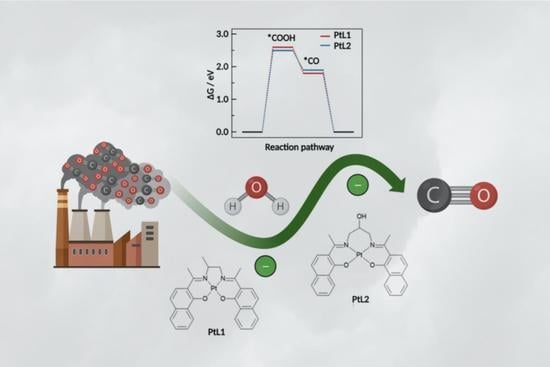
1. Introduction
2. Results and Discussion
2.1. Spectroscopic Properties
2.2. Structural Characterization
2.3. DFT Calculations
2.4. Electrochemical Studies
2.5. CO2 Reduction
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis and Characterization
3.3. DFT Calculations
3.4. Electrochemical Setup
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Du, C.; Wang, X.; Chen, W.; Feng, S.; Wen, J.; Wu, Y.A. CO2 Transformation to Multicarbon Products by Photocatalysis and Electrocatalysis. Mater Today Adv. 2020, 6, 100071. [Google Scholar] [CrossRef]
- Roy, S.C.; Varghese, O.K.; Paulose, M.; Grimes, C.A. Toward Solar Fuels: Photocatalytic Conversion of Carbon Dioxide to Hydrocarbons. ACS Nano 2010, 4, 1259–1278. [Google Scholar] [CrossRef]
- Cramer, H.H.; Chatterjee, B.; Weyhermüller, T.; Werlé, C.; Leitner, W. Controlling the Product Platform of Carbon Dioxide Reduction: Adaptive Catalytic Hydrosilylation of CO2 Using a Molecular Cobalt(II) Triazine Complex. Angew. Chem.-Int. Ed. 2020, 15681, 15674–15681. [Google Scholar] [CrossRef]
- Chiong, M.R.; Paraan, F.N.C. Controlling the Nucleophilic Properties of Cobalt Salen Complexes for Carbon Dioxide Capture. RSC Adv. 2019, 9, 23254–23260. [Google Scholar] [CrossRef]
- Sun, L.; Reddu, V.; Fisher, A.C.; Wang, X. Electrocatalytic Reduction of Carbon Dioxide: Opportunities with Heterogeneous Molecular Catalysts. Energy Environ. Sci. 2020, 13, 374–403. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.-M. Catalysis of the Electrochemical Reduction of Carbon Dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef]
- Jiang, C.; Nichols, A.W.; Machan, C.W. A Look at Periodic Trends in D-Block Molecular Electrocatalysts for CO2 Reduction. Dalton Trans. 2019, 48, 9454–9468. [Google Scholar] [CrossRef]
- Francke, R.; Schille, B.; Roemelt, M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide—Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118, 4631–4701. [Google Scholar] [CrossRef]
- Nichols, A.W.; Hooe, S.L.; Kuehner, J.S.; Dickie, D.A.; Machan, C.W. Electrocatalytic CO2 Reduction to Formate with Molecular Fe(III) Complexes Containing Pendent Proton Relays. Inorg. Chem. 2020, 59, 5854–5864. [Google Scholar] [CrossRef]
- Zhu, G.; Li, Y.; Zhu, H.; Su, H.; Chan, S.H.; Sun, Q. Curvature-Dependent Selectivity of CO2 Electrocatalytic Reduction on Cobalt Porphyrin Nanotubes. ACS Catal. 2016, 6, 6294–6301. [Google Scholar] [CrossRef]
- Inglis, J.L.; MacLean, B.J.; Pryce, M.T.; Vos, J.G. Electrocatalytic Pathways towards Sustainable Fuel Production from Water and CO2. Coord. Chem. Rev. 2012, 256, 2571–2600. [Google Scholar] [CrossRef]
- Yoshida, T.; Kamato, K.; Tsukamoto, M.; Iida, T.; Schlettwein, D.; Wöhrle, D.; Kaneko, M. Selective Electroacatalysis for CO2 Reduction in the Aqueous Phase Using Cobalt Phthalocyanine/Poly-4-Vinylpyridine Modified Electrodes. J. Electroanal. Chem. 1995, 385, 209–225. [Google Scholar] [CrossRef]
- Nie, W.; Tarnopol, D.E.; McCrory, C.C.L. Enhancing a Molecular Electrocatalyst’s Activity for CO2 Reduction by Simultaneously Modulating Three Substituent Effects. J. Am. Chem. Soc. 2021, 143, 3764–3778. [Google Scholar] [CrossRef]
- Abe, T.; Yoshida, T.; Tokita, S.; Taguchi, F.; Imaya, H.; Kaneko, M. Factors Affecting Selective Electrocatalytic CO2 Reduction with Cobalt Phthalocyanine Incorporated in a Polyvinylpyridine Membrane Coated on a Graphite Electrode. J. Electroanal. Chem. 1996, 412, 125–132. [Google Scholar] [CrossRef]
- Costentin, C.; Passard, G.; Robert, M.; Savéant, J.-M. Pendant Acid–Base Groups in Molecular Catalysts: H-Bond Promoters or Proton Relays? Mechanisms of the Conversion of CO2 to CO by Electrogenerated Iron(0) Porphyrins Bearing Prepositioned Phenol Functionalities. J. Am. Chem. Soc. 2014, 136, 11821–11829. [Google Scholar] [CrossRef]
- Singh, S.; Phukan, B.; Mukherjee, C.; Verma, A. Salen Ligand Complexes as Electrocatalysts for Direct Electrochemical Reduction of Gaseous Carbon Dioxide to Value Added Products. RSC Adv. 2015, 5, 3581–3589. [Google Scholar] [CrossRef]
- Bose, P.; Mukherjee, C.; Golder, A.K. A NiII Complex of the Tetradentate Salen Ligand H2LNH2 Comprising an Anchoring -NH2 Group: Synthesis, Characterization and Electrocatalytic CO2 Reduction to Alcohols. Inorg. Chem. Front. 2019, 6, 1721–1728. [Google Scholar] [CrossRef]
- Bose, P.; Mukherjee, C.; Kumar Golder, A. Reduction of CO2 to Value-Added Products on a Cu(II)-Salen Complex Coated Graphite Electrocatalyst. ChemistrySelect 2020, 5, 9281–9287. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, J.H.; Dao, X.Y.; Zhang, X.D.; Zhao, Y.; Sun, W.Y. Coordination Polymers with a Pyridyl-Salen Ligand for Photocatalytic Carbon Dioxide Reduction. Chem. Comm. 2020, 56, 4110–4113. [Google Scholar] [CrossRef]
- Kong, X.; Liu, Y.; Li, P.; Ke, J.; Liu, Z.; Ahmad, F.; Yan, W.; Li, Z.; Geng, Z.; Zeng, J. Coordinate Activation in Heterogeneous Carbon Dioxide Reduction on Co-Based Molecular Catalysts. Appl. Catal. B 2020, 268, 118452. [Google Scholar] [CrossRef]
- Ulusoy, M.; Birel, Ö.; Şahin, O.; Büyükgüngör, O.; Cetinkaya, B. Structural, Spectral, Electrochemical and Catalytic Reactivity Studies of a Series of N2O2 Chelated Palladium(II) Complexes. Polyhedron 2012, 38, 141–148. [Google Scholar] [CrossRef]
- Oliveira, Y.A.; Olean-Oliveira, A.; Teixeira, M.F.S. Short Communication: Molecular Architecture Based on Palladium-Salen Complex/Graphene for Low Potential Water Oxidation. J. Electroanal. Chem. 2021, 880, 114928. [Google Scholar] [CrossRef]
- Ghabdian, M.; Nasseri, M.A.; Allahresani, A.; Motavallizadehkakhky, A. Pd Salen Complex@CPGO as a Convenient, Effective Heterogeneous Catalyst for Suzuki–Miyaura and Heck–Mizoroki Cross-Coupling Reactions. Res. Chem. Intermed. 2021, 47, 1713–1728. [Google Scholar] [CrossRef]
- Movassagh, B.; Parvis, F.S.; Navidi, M. Pd(II) Salen Complex Covalently Anchored to Multi-Walled Carbon Nanotubes as a Heterogeneous and Reusable Precatalyst for Mizoroki-Heck and Hiyama Cross-Coupling Reactions. Appl. Organomet. Chem. 2015, 29, 40–44. [Google Scholar] [CrossRef]
- Lwin, S.; Wachs, I.E. Olefin Metathesis by Supported Metal Oxide Catalysts. ACS Catal. 2014, 4, 2505–2520. [Google Scholar] [CrossRef]
- Gutierrez, K.; Corchado, J.; Lin, S.; Chen, Z.; Piñero Cruz, D.M. A Non-Innocent Salen Naphthalene Ligand and Its Co2+, Ni2+ and Cu2+ Metal Complexes: Structural, Electrochemical, and Spectroscopic Characterization and Computational Studies. Inorg. Chim. Acta 2018, 474, 118–127. [Google Scholar] [CrossRef]
- Kamounah, F.S.; Salman, S.R.; Mahmoud, A.A.K. Substitution and Solvent Effect of Some Substituted Hydroxy Schiff Bases. Spectrosc. Lett. 1998, 31, 1557–1567. [Google Scholar] [CrossRef]
- Gutiérrez, K.J.; Pedreira, W.M.; Piñero Cruz, D.M. Synthesis, Structural, Electrochemical and Spectroscopic Characterization, and Theoretical Calculations of Two New Cu(II) and Ni(II) Complexes from 2-(1-((Pyridine-2-Ylmethyl)Imino)Ethyl)Naphtalen-1-ol. J. Coord. Chem. 2019, 72, 2654–2668. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, F.; Angeles, L.; Zhu, X.; Wong, W. New Phosphorescent Platinum (II) Schiff Base Complexes for PHOLED. J. Mater. Chem. 2012, 22, 16448–16457. [Google Scholar] [CrossRef]
- Bosnich, B. An Interpretation of the Circular Dichroism and Electronic Spectra of Salicylaldimine Complexes of Square-Coplanar Diamagnetic Nickel (II). J. Am. Chem. Soc. 1968, 90, 627–632. [Google Scholar] [CrossRef]
- Houjou, H.; Hoga, Y.; Ma, Y.-L.; Achira, H.; Yoshikawa, I.; Mutai, T.; Matsumura, K. Dinuclear Fused Salen Complexes of Group-10 Metals: Peculiarity of the Crystal Structure and near-Infrared Luminescence of a Bis(Pt-Salen) Complex. Inorg. Chim. Acta 2017, 461, 27–34. [Google Scholar] [CrossRef]
- Tauc, J. States in the Gap. J. Non-Cryst. Solids 1972, 10, 569–585. [Google Scholar] [CrossRef]
- Nichols, A.W.; Chatterjee, S.; Sabat, M.; Machan, C.W. Electrocatalytic Reduction of CO2 to Formate by an Iron Schiff Base Complex. Inorg. Chem. 2018, 57, 2111–2121. [Google Scholar] [CrossRef]
- Solomon, M.B.; Chan, B.; Kubiak, C.P.; Jolliffe, K.A.; D’Alessandro, D.M. The Spectroelectrochemical Behaviour of Redox-Active Manganese Salen Complexes. Dalton Trans. 2019, 48, 3704–3713. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Delley, B. From Molecules to Solids with the DMol3 Approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Sathrum, A.J.; Kubiak, C.P. Kinetics and limiting current densities of homogeneous and heterogeneous electrocatalysts. J. Phys. Chem. Lett. 2011, 2, 2372–2379. [Google Scholar] [CrossRef]
- Hooe, S.L.; Dressel, J.M.; Dickie, D.A.; Machan, C.W. Highly efficient electrocatalytic reduction of CO2 to CO by a molecular chromium complex. ACS Catal. 2019, 10, 1146–1151. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Experimental Eg (eV) a |
|---|---|
| L1 | 2.73 |
| L2 | 2.76 |
| PtL1 | 2.67 |
| PtL2 | 2.66 |
| Metal Complex | Atmosphere | Voltage (V vs. Fc/Fc+) | H2O (M) | CO FE (%) |
|---|---|---|---|---|
| PtL1 | CO2 | −2.7 | 0 | 5.5 |
| CO2 | −2.7 | 3 | 5.4 | |
| CO2 | −2.5 | 3 | 0.8 | |
| CO2 | −2.9 | 3 | 0.9 | |
| N2 | −2.7 | 0 | N.D. | |
| N2 | −2.7 | 3 | N.D. | |
| PtL2 | CO2 | −2.5 | 0 | 4.4 |
| CO2 | −2.5 | 3 | 42.0 | |
| CO2 | −2.3 | 3 | 4.15 | |
| CO2 | −2.7 | 3 | 0.8 | |
| N2 | −2.5 | 0 | N.D. | |
| N2 | −2.5 | 3 | N.D. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera-Reyes, J.O.; Soto-Pérez, J.; Sepulveda-Pagán, M.; Lu, L.; Borrero-Negrón, J.; Luna-Ramírez, A.V.; Trinidad-Pérez, P.; Pagán-Torres, Y.; Chen, Z.; Cabrera, C.R.; et al. New Platinum Complexes from Salen- and Hydroxy-Substituted Salpn-Naphthalene Ligands with CO2 Reduction Activity. Catalysts 2023, 13, 911. https://doi.org/10.3390/catal13050911
Rivera-Reyes JO, Soto-Pérez J, Sepulveda-Pagán M, Lu L, Borrero-Negrón J, Luna-Ramírez AV, Trinidad-Pérez P, Pagán-Torres Y, Chen Z, Cabrera CR, et al. New Platinum Complexes from Salen- and Hydroxy-Substituted Salpn-Naphthalene Ligands with CO2 Reduction Activity. Catalysts. 2023; 13(5):911. https://doi.org/10.3390/catal13050911
Chicago/Turabian StyleRivera-Reyes, Javier O., Joesene Soto-Pérez, Miguel Sepulveda-Pagán, Linguo Lu, Justin Borrero-Negrón, Alanys V. Luna-Ramírez, Pedro Trinidad-Pérez, Yomaira Pagán-Torres, Zhongfang Chen, Carlos R. Cabrera, and et al. 2023. "New Platinum Complexes from Salen- and Hydroxy-Substituted Salpn-Naphthalene Ligands with CO2 Reduction Activity" Catalysts 13, no. 5: 911. https://doi.org/10.3390/catal13050911
APA StyleRivera-Reyes, J. O., Soto-Pérez, J., Sepulveda-Pagán, M., Lu, L., Borrero-Negrón, J., Luna-Ramírez, A. V., Trinidad-Pérez, P., Pagán-Torres, Y., Chen, Z., Cabrera, C. R., West, W. C., Jones, J.-P., & Piñero Cruz, D. M. (2023). New Platinum Complexes from Salen- and Hydroxy-Substituted Salpn-Naphthalene Ligands with CO2 Reduction Activity. Catalysts, 13(5), 911. https://doi.org/10.3390/catal13050911