Abstract
D-serine plays an essential role in the field of medicine and cosmetics. Diaminopimelate dehydrogenase (DAPDH) is a kind of oxidoreductase that can reduce keto acid into the corresponding D-amino acid. Because of its high stereoselectivity and lack of by-product production, DAPDH has become the preferred enzyme for the efficient one-step synthesis of D-amino acids. However, the types of DAPDH with a reductive amination function reported so far are limited. Although the DAPDH from Symbiobacterium thermophilum (StDAPDH) demonstrates reductive amination activity toward a series of macromolecular keto acids, activity toward hydroxypyruvate (HPPA) for D-serine synthesis has not been reported. In this study, we investigated the activity of the available StDAPDH/H227V toward HPPA by measuring the desired product D-serine. After homologous structure modeling and docking analysis concerning the substrate-binding pocket, four residues, D92, D122, M152, and N253, in the active pocket were predicted for catalyzing HPPA. Through single-point saturation mutation and iterative mutation, a mutant D92E/D122W/M152S was obtained with an 8.64-fold increase in enzyme activity, exhibiting a specific activity of 0.19 U/mg and kcat value of 3.96 s−1 toward HPPA. Using molecular dynamics simulation, it was speculated that the increase in enzyme activity might be related to the change in substrate pocket size and the enhancement of the interactions between the substrate and key residues.
1. Introduction
D-serine is an unnatural amino acid with a high concentration in the hippocampus, forebrain, striatum, and other parts of mammals [1,2]. This amino acid also plays an essential role in cosmetics and medicine, such as as the drug intermediate for synthesizing D-cycloserine for treating pulmonary tuberculosis caused by infection [3,4,5,6]. The methods of synthesizing D-serine mainly include chemical synthesis and biosynthesis. The chemical method mainly synthesizes D-serine by splitting the racemate of D, L-amino acid [7,8]. However, the disadvantage of chemical synthesis is that the process produces a large amount of waste, polluting the environment. In addition, the reaction process requires a specific temperature and pressure environment, increasing production costs, and the optical purity of the product is low. With the development of biotechnology, the preparation of D-amino acids using the biological method has become a research hotspot. Presently, the preparation of D-serine using the biological method is mainly through the selective reaction and degradation of D,L-amino acid or derivatives of its racemate with enzymes to separate a single D-amino acid [9]. Although this method is environmentally friendly, low yield and cumbersome operation steps are disadvantages. Therefore, finding a highly selective, efficient, and safe method to produce D-serine is necessary.
The asymmetric catalysis of prochiral keto acids to chiral amino acids with selective amino acid dehydrogenase is a potential method. For example, D-serine or L-serine can be synthesized via reductive amination of the corresponding keto acid with amino acid dehydrogenase of a desired selectivity. Once the appropriate enzyme is found, the prochiral substrate can be catalyzed to synthesize the corresponding D-amino acid in one step. The advantages include a simple operation, high selectivity, and no by-product generation. Diaminopimelate dehydrogenase (DAPDH) is a kind of NADPH-dependent D-amino acid dehydrogenase mainly distributed in bacteria and plants [10,11] and can be divided into two types. Type I DAPDH shows a high degree of specificity for the natural substrate meso-2,6-diaminopimelate (meso-DAP), such as the DAPDHs from Corynebacterium glutamicum (CgDAPDH) and Ureibacillus thermosphaericus [12]. Most DAPDHs belong to type I. Type II DAPDH can catalyze natural substrates and shows apparent ammonification activity. Compared with other enzymes, DAPDH can directly catalyze the asymmetric reductive amination of α-keto acids to synthesize D-amino acids. In the reaction process, no other amino acids are introduced as ammonia donors, which benefits the separation of the products. Therefore, the catalytic route for synthesizing D-amino acids from DAPDH has received extensive attention.
In recent years, the research on DAPDH mainly focuses on Symbiobacterium thermophilum (StDAPDH), the first member of the meso-DAPDH family found to catalyze the asymmetric reductive amination of α-keto acids to D-amino acids. In addition to the natural substrate, it can catalyze the reductive amination of keto acids such as pyruvate and 4-methyl-2-oxovalerate to D-alanine and D-leucine, respectively [13,14,15]. Zhu et al. explored the molecular mechanism of the unique properties of StDAPDH through structural analysis and site mutation [14]. Based on this, the amino acid residues in the substrate pocket of StDAPDH were mutated, and a mutant H227V was obtained. The substrate-binding pocket of the H227V mutant was expanded, and the enzyme activity toward large hydrophobic substrates with steric hindrance, such as phenylpyruvate and tert-leucine, was improved, which provided a basis for the synthesis of D-phenylalanine and D-tert-leucine [14]. Subsequently, mutation of StDAPDH was carried out to obtain a mutant W121L/H227I, which showed high activity against a variety of large steric keto acids for synthesizing D-phenylglycine, D-tryptophan, and D-tert-leucine [16]. In addition to StDAPDH, mutations of CgDAPDH also expended their activity toward certain keto acids for the synthesis of D-valine, D-leucine, D-isoleucine, and D-cyclohexylalanine [17,18,19,20,21].
Through molecular modification, the substrate spectrum of DAPDH was broadened, and its activity toward a series of macromolecular keto acids was improved. However, limited studies have reported that these available DAPDH mutants exhibit activity and selectivity toward hydroxypyruvate (HPPA) with small steric hindrance for D-serine synthesis.
In order to develop a D-amino acid dehydrogenase that can catalyze the reductive amination of HPPA to D-serine, with the promising variant StDAPDH/H227V as the initial object, its catalytic activity for the asymmetric reductive amination of HPPA was investigated in this study. Structural analysis concerning the substrate-binding pocket was carried out further to improve the activity for D-serine synthesis via molecular modification. The HPPA substrate was docked into the catalytic pocket of the StDAPDH/H227V structural model, and the potential residues were predicted and selected for site-directed saturation mutagenesis and iterative mutation. Finally, the positive variants with increased activity were obtained. The increase in enzyme activity was explained through conformational analysis and molecular dynamics simulation.
2. Results and Discussion
2.1. Enzyme Activity Determination of StDAPDH/H227V
In order to determine the catalytic activity of StDAPDH/H227V toward D-serine, HPPA, D-serine, and L-serine were separately used as the substrate to react in a Tris-HCl buffer solution.
The experimental results show that when HPPA was used as the substrate, HPPA was consumed. Only D-serine appeared as a product, indicating that StDAPDH/H227V has stereoisomerization selectivity (Figure 1A). When D-serine was used as the substrate, no HPPA was produced in the reaction system, which ensures that the D-serine final product of the catalytic cascade system would not be consumed (Figure 1B). Similarly, When L-serine was used as the substrate, no HPPA and D-serine were produced in the reaction system. Therefore, the single-step catalytic reaction had stereoisomeric selectivity, and no reverse reaction occurred (Figure 1C). The reaction direction of the single-step reaction catalyzed by StDAPDH/H227V was consistent with the direction of generating D-serine from HPPA.
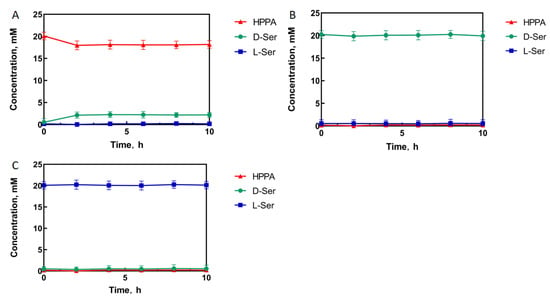
Figure 1.
StDAPDH/H227V catalytic process with hydroxypyruvate (A), D-serine (D-Ser) (B), and L-serine (L-Ser) (C) as substrates, respectively.
2.2. Prediction of Key Residues for Saturation Mutagenesis
The above experiments found that StDAPDH/H227V has activity toward HPPA, but the conversion rate was only 8.5% (Figure 1A). Therefore, structure-based engineering was used to improve the activity of StDAPDH/H227V. The homologous structural model of StDAPDH/H227V was constructed based on the crystal structure of StDAPDH (PDB ID: 3WBB). Furthermore, to have a detailed view of substrate-involved molecular interactions, HPPA was connected to the catalytic pocket of the StDAPDH/H227V model with AutoDock. A total of nine docking results were obtained, and the highest docking result score was selected to predict key residues. As shown in Figure 2, the 4 residues within 4 Å from the ligand, D92, D122, M152, and N253, were identified as potential key sites for substrate binding. Of these, M152 and N253 form hydrogen bonds with HPPA. These four residues were selected as the key residue sites for saturation mutation.
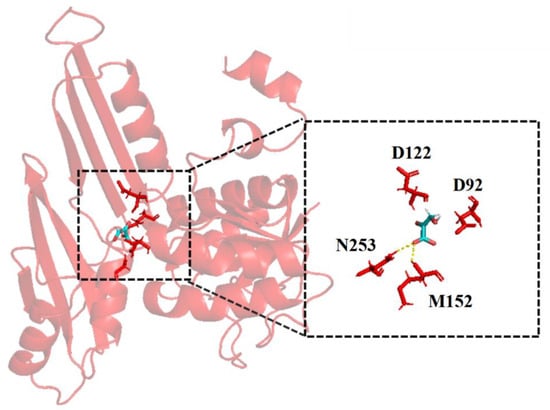
Figure 2.
The docking results of hydroxypyruvate with the StDAPDH/H227V model.
2.3. Screening of Positive Single Mutants
In order to screen mutants efficiently, it is necessary to find a suitable high-throughput screening method. Xu et al. constructed a high-throughput method to rapidly screen leucine dehydrogenase [22]. To verify that this method can also be used to screen StDAPDH mutants, different D-serine concentrations were added to the Cu(II) calcein complex in a Tris buffer. After 30 min of reaction, the fluorescence intensity of the excitation and emission spectra were read. The results show that the Cu(II) calcein complex had the highest peaks at 485 nm and 525 nm. Therefore, 485 nm and 525 nm were selected as the excitation and emission wavelengths of the Cu(II) calcein complex. With the increase in the D-serine concentration, the fluorescence intensity also increased (Figure 3A), which indicates that D-serine affected the fluorescence intensity of excitation and emission spectra.
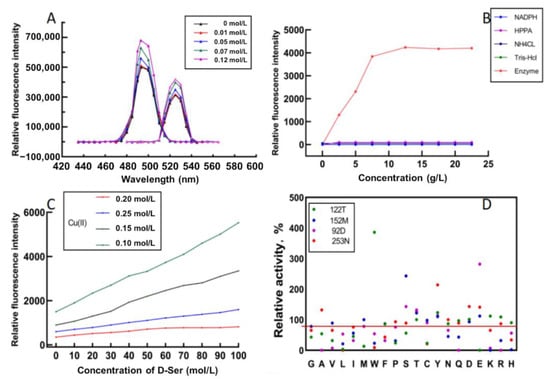
Figure 3.
High-throughput screening of the mutants with Cu(II) calcein complex. (A) Excitation (left) and emission (right) wavelengths of Cu(II) calcein complex in the absence and presence of D-serine. (B) Fluorescence intensities for different concentrations of HPPA, NADPH, NH4CL, Tris-HCl, and enzyme. (C) Fluorescence intensities of different concentrations of D-serine and Cu(II) at the same enzyme concentration (2 g/L) and calcein concentration. (D) Relative activities of the screened mutants.
A crude enzyme solution was used to catalyze the reaction to screen mutants efficiently. The reaction system for evaluating the capability of mutants catalyzing the D-serine synthesis included HPPA, NH4Cl, NADPH, and a Tris-HCl buffer. In order to eliminate the interference of irrelevant factors on the fluorescence intensity, different concentrations of HPPA, NH4Cl, NAPDH, and Tris-HCL were added to the Cu(II) calcein complex. The results show that they did not interfere with the detection of fluorescence intensity. In contrast, the fluorescence intensity increased with the increase in the crude enzyme concentration within a specific range. When the enzyme concentration exceeded 6 g/L, the fluorescence intensity no longer increased (Figure 3B), presumably due to the limitation on enzyme activity. Then, the reaction solution was centrifuged, and the fluorescence intensity of the supernatant was measured. It was found that the supernatant had the same fluorescence intensity as the reaction solution, indicating that the crude enzyme system also did not affect the determination of fluorescence intensity.
The concentration ratio of Cu(II) and calcein greatly influenced the fluorescence intensity. In order to achieve the best linear fit between the fluorescence intensity and the concentration of D-serine, different concentrations of Cu(II) and D-serine were used to construct the curve, and the substrate concentration and enzyme concentration were fixed. The results show that the best linear fitting degree (R2 = 0.9979) was obtained at Cu(II) 0.25 mM and calcein 0.5 mM, and with a detection limit of between 0 and 100 mM (Figure 3C).
Then, site-directed saturation mutagenesis was performed on the four key residues, D92, D122, M152, and N253. The above high-throughput screening method screened the mutant library with HPPA as the substrate. The fluorescence intensity showed the concentration of D-serine produced by StDAPDH/H227V catalyzed HPPA. The higher concentration of D-serine, the higher the fluorescence intensity. The specific concentration of D-serine was calculated according to the standard curve under the conditions of Cu(II) 0.25 mM and calcein 0.5 mM. The results showed that the enzyme activities of 17 mutants were improved, among which D92E, D122W, M152S, and N253Y were the most considerably improved (Figure 3D). Then, the specific activity and kinetic parameters were measured. The results indicate that the specific activity of D92E, D122W, M152S, and N253Y increased by 2.61-, 3.92-, 2.27-, and 2.01-fold, respectively, compared with the wild type (Table 1).

Table 1.
Specific activities of StDAPDH/H227V and the mutants toward hydroxypyruvate.
2.4. Iterative Saturation Mutagenesis
In order to further improve the catalytic activity, D92E, D122W, M152S, and N253Y were used as templates for the separate saturation mutagenesis of the other three sites (Figure 4). It was found that when D122W or M152S was used as the template, the enzyme activity of the D122W/M152S (M2) mutant increased markedly. Then, D122W/M152S was used as the template, and the other two sites were saturated. The screening library obtained the mutant D92E/D122W/M152S (M3) with an obvious increase in enzymatic activity. Finally, using the M3 mutant as the template, saturation mutation was performed on the N253 site.
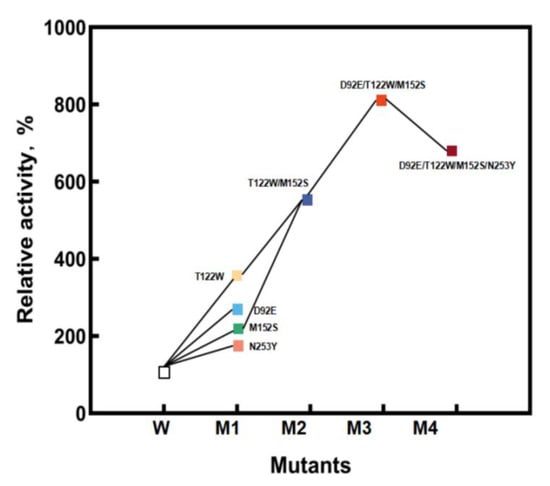
Figure 4.
The process of iterative saturation mutagenesis.
The kinetic parameters of the above mutants are shown in Table 1. During further mutation and screening, the mutant D92E/D122W/M152S was found to have 8.64-fold specific activity improved specific activity toward HPPA compared with StDAPDH/H227V.
To measure the conversion of HPPA catalyzed by the D92E/D122W/M152S mutant, HPPA, D-serine, and L-serine were used separately as the substrate to react in a Tris-HCl buffer solution. The experimental results showed that when 20 mM of HPPA was used as the substrate, HPPA was consumed, and the final product was only D-serine, with a conversion rate of 65% and an optical purity of >99% e.e. (Figure 5A). No reaction occurred when L-serine or D-serine were used as the substrate (Figure 5B,C). This indicates that the mutations did not affect stereoselectivity but improved activity efficiently. In order to further investigate the capability of the D92E/D122W/M152S mutant for D-serine synthesis, 40 mM of HPPA was used as the substrate under the same catalytic conditions as those for 20 mM of the substrate. Consequently, the final product was only D-serine, with a conversion rate of 61% and an optical purity of >99% e.e. (Figure 5D), indicating that the conversion rate of the M3 mutant was almost constant, even for a two-fold increase in the substrate concentration. Thus, the catalytic efficiency of this positive mutant would be promising for D-serine synthesis and the productivity of HPPA reductive amination would be further improved by the optimization of the catalytic system and reaction conditions.
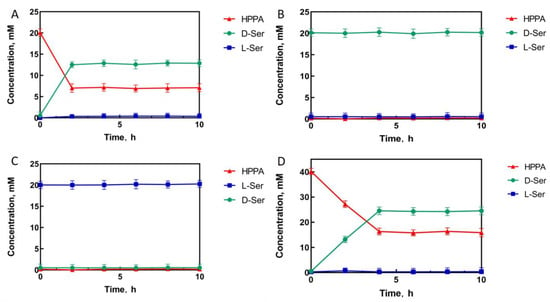
Figure 5.
The mutant D92E/D122W/M152S (M3) catalytic process with HPPA ((A): 20 mM; (D): 40 mM), D-serine (B), and L-serine (C) as substrates, respectively.
2.5. Role of the Mutation Sites for Enhanced Activity
According to Figure 3D, most of the mutants with increased enzyme activity have the corresponding amino acid residues containing a hydroxyl group at D92, M152, and N253. The kinetic parameters of D92E, M152S, and N253Y with highly increased enzyme activity were measured. The specific activity of D92E, M152S, and N253Y increased by 2.61-, 2.27-, and 2.01-fold, respectively, compared with StDAPDH/H227V (Table 1). This result shows that the increase in the enzyme activities of D92E, M152S, and N253Y was due to the improvement of their affinity toward HPPA. Most of the mutants with increased enzyme activity at D122 have the corresponding amino acid residues containing bulky groups (Figure 3D). The kinetic parameters of D122W with highly increased enzyme activity were measured. The specific activity of D122W increased by 3.92-fold compared with StDAPDH/H227V, and the kcat value increased from 1.09 ± 0.02 s−1 to 3.44 ± 0.05 s−1 (Table 1), which was the major contributor to the improved catalytic efficiency. This indicates that the increase in the enzyme activity of D122W was due to the improvement of the conversion rate from HPPA to D-serine.
The increased enzyme activity of D92E/D122W/M152S was higher than the sum of the enzyme activity of the single mutants D92E, D122W, and M152S (Table 1). This result shows that there would be a synergistic effect among D92E, D122W, and M152S. By measuring the kinetic parameters, it was found that, compared with the initial object StDAPDH/H227V (control), the kcat values generally increased and Km values decreased for the positive single mutants and their combined mutants, indicating that these mutations led to the improvement in catalytic efficiency (Table 1). The increase in enzyme activity increased the conversion rate from the substrate to D-serine, and the affinity between the substrate and active pocket of the enzyme.
In order to further analyze the mechanism for the increased catalytic activity of the mutant, 50-ns molecular dynamics simulations of StDAPDH/H227V and the mutant M3 were performed using Gromacs. According to the root-mean-square displacement (RMSD) data (Figure 6C), the low activity of StDAPDH/H227V resulted from the substrate HPPA being unstabilized in the catalytic pocket. This phenomenon can be explained as follows: (i) HPPA has a weak interaction with the key residues, and (ii) HPPA is relatively small compared with the catalytic pocket. However, the RMSD data from the 50-ns molecular dynamics simulation of the M3 showed that the mutant complex became dynamically equilibrated after 40 ns of simulation (Figure 6C). When HPPA was docked into the active site of the homologous model of the M3 (Figure 6A,B), the mutation of D122W from a small molecular residue to a bulky residue reduced the active pocket of the enzyme. Therefore, the chance of HPPA contacting the catalytic center was increased, thereby improving the catalytic efficiency. In addition, the increased affinity of the substrate for the active pocket was also presumed by calculating the hydrogen bond occupancy (Table S4). It was found that the ligand formed two stable hydrogen bonds with M152S and one stable hydrogen bond with D92E, while only one hydrogen bond with M152 before the mutation (Figure 6D,E). On the other hand, the activity of the combined mutant M4 (D92E/D122W/M152S/N253Y) was lower than that of M3 (D92E/D122W/M152S) (Figure 4). Although the single mutation N253Y in addition to H227V (control) increased the enzymatic activity, when the mutation N253Y was iterated to M3 by replacing asparagine with the relatively large tyrosine, the substrate-binding pocket space might have been too small to accommodate HPPA with a catalysis-favored conformation. Therefore, the M3 mutation giving a noticeable improvement in enzymatic activity reduced the size of the active pocket and enhanced the molecular interaction between the substrate and the catalytic residues.
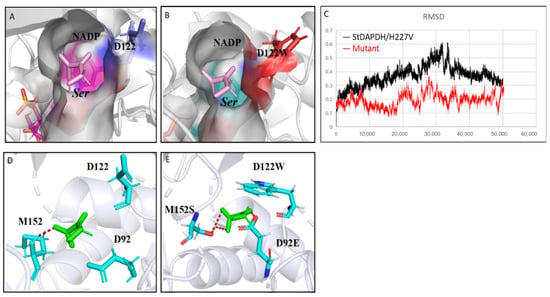
Figure 6.
Structural analysis of the mutation sites. (A) The docking structure of the pocket of StDAPDH/H227V with HPPA; (B) the docking structure of the pocket of M3 mutant with HPPA; (C) root-mean-square displacement of StDAPDH/H227V and M3 mutant; and (D) interaction between hydroxypyruvate (HPPA) and key sites of StDAPDH/H227V. Red: hydrogen bond; blue: key site; and green: HPPA. (E) Interaction between HPPA and key sites of M3 mutant. Red: hydrogen bond; blue: key site red as the substrate.
3. Materials and Methods
3.1. Molecular Modeling and Docking Calculations
The homologous structural models of StDAPDH/H227V and its mutant were constructed using the SWISS-MODEL online server (https://swissmodel.expasy.org/interactive, accessed on 20 December 2022), based on the protein structure of StDAPDH (PDB ID: 3WBB) [16]. The structures of HPPA and NADPH were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov, accessed on 20 December 2022). The HPPA was docked to the StDAPDH receptor using AutoDock. The ranking of the docking results for ligands is listed in Table S2. The structures were analyzed, and potential binding sites were identified using PyMOL. All molecular dynamics simulations were performed using the AMBER 11 simulation package. The standard AMBER force field (ff03.r1) and the general AMBER force field (gaff) were used to establish the topologies and parameters of the enzyme and substrate, respectively. The program Antechamber was used to handle the force field parameters of the substrate, and the NADPH parameters reported by Ryde were used. The whole system was solvated in an octahedral periodic box of TIP3P water molecules. Before the molecular dynamics simulations, minimizations were carried out via three steps. First, water/ions were optimized by restraining the protein. Second, the side chains of the protein were relaxed by restraining the backbone atoms. Third, the whole system was optimized without any restraints. Following minimizations, the systems were gradually heated from 0 to 303 K over 60 ps, and then 50 ns MD simulations with a 2.0 fs time step were performed under a constant temperature of 303 K. The SHAKE algorithm was employed to constrain bonds involving hydrogen atoms. The hydrogen bond occupancy was calculated via VMD.
3.2. Site-Directed Saturation Mutagenesis
Site-directed saturation mutants were constructed using a previously published method [23]. Primer synthesis and DNA sequencing were completed by Shenggong Co., Ltd. (Shanghai, China). The primers used in this study are shown in Table S1.
3.3. Protein Expression of StDAPDH Mutants
The site-directed saturation mutants of StDAPDH expressed in E. coli BL21(DE3) were inoculated into 4 mL of Luria–Bertani medium containing 4 mg·mL−1 of kanamycin and cultured at 37 °C with shaking at 200 rpm for 8–10 h. The cultured bacterial solution was transferred to a 250 mL Erlenmeyer flask containing 50 mL of Luria–Bertani medium at a 2% inoculation amount and cultured at 37 °C with shaking at 200 rpm for 2–3 h. When OD600 was 0.6–1.0, isopropyl β-d-1-thiogalactopyranoside with a final concentration of 0.5 mM was added for induction. The induction temperature was 17 °C, and the shaking culture was performed at 200 rpm for 17 h.
3.4. Cell Fragmentation
The fermentation broth of 20 mL was centrifuged at 4 °C and 10,000 rpm for 10 min, and the supernatant was discarded. The bacterial precipitate was resuspended in normal saline, washed twice under the same conditions, and resuspended in 50 mM of the Tris-HCl buffer (pH 9.5; 50 mM). The precipitate was then re-volumed to 20 mL and ultrasonically broken in an ice water bath. The disrupted cell solution was centrifuged at 4 °C and 10,000 rpm for 30 min to separate the supernatant and cell debris. The supernatant was used as a crude enzyme solution.
3.5. Mutants Screening
HPPA, NADPH, and NH4CL were added to 2 mL of the crude enzyme solution to final concentrations of 20 mM, 10 mM, and 40 mM, respectively, in each well of 96 MTPs. Then, 100 μL of the mixture was taken, and the fluorescence intensity was recorded as the initial fluorescence. The mixture was reacted in a rotary oscillator at 37 °C and 200 rpm for 12 h. A total of 100 μL of the reaction solution was taken out from each well and added to a black 96 MTP well containing 100 μL of the Tris-Cl (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) buffer (pH 9.5; 50 mM) and 60 μL of the Cu(II) calcein (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) complex. After 30 min, the fluorescence intensity of the mixture in each well was detected at an excitation wavelength of 485 nm and an emission wavelength of 525 nm.
3.6. Purification of Proteins
Purification was performed using the AKTA purifier protein purification instrument. A GE HisTrap HP affinity chromatography column was first balanced with solution A (Tris-HCl 20 mM; NaCl 150 mM; pH 7.5) to 4 column volumes. After the crude enzyme solution was loaded, the impurities unbound to the nickel column were washed with solution A. After equilibrium was obtained, the protein with the histidine tag was linearly eluted with 42% solution A and 58% solution B (Tris-HCl 20 mM; NaCl 150 mM; imidazole 500 mM; pH 7.5), and the elution peaks were collected. The purity of the target protein was determined via SDS-PAGE. Chemicals used for protein purification were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).
3.7. Determination of Kinetic Parameters
An absorbance change mainly determines diaminopimelate dehydrogenase activity at 340 nm. Therefore, the 200 μL reaction system comprised 200 mM of NH4Cl, 0.5 mM of NADPH, and 50 mM of Na2CO3-NaHCO3 buffer (pH 9.5), and the substrate HPPA concentration was approximately 2–40 mM. After incubation at 30 °C for 3 min, 20 μL of the enzyme solution was added, and the absorbance change at 340 nm was immediately detected using the Spectramax M2e microplate reader. The enzyme activity unit U was defined as the amount of enzyme required to consume one μmol of NADPH per minute under test conditions. The Km and Vmax values of the enzyme-catalyzed HPPA reductive amination were calculated with a Lineweaver–Burk plot. The enzyme activity calculation formula was as follows: enzyme activity (U) = EW × V × 103/(6220 × 0.3). The specific activity was calculated using this formula: specific activity (U·mg−1) = enzyme activity (U)/protein (mg). Chemicals used for activity assay were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).
3.8. Enzymatic Reactions and Detection of L-serine, D-serine, and HPPA
For the reductive amination, HPPA was used as the substrate (with a final concentration of 20 mM) to react in a 2 mL Tris-HCl buffer (20 mM; pH 9.5) solution containing 4 mg/mL of the purified enzyme of StDAPDH/H227V or its mutants, 40 mM of NH4Cl, and 10 mM of NADPH for 10 h. For the control reaction, HPPA was added to 2 mL of the reaction solution to a final concentration of 20 mM, without the addition of the purified enzyme. To test the reverse reaction, D-serine or L-serine was used as the substrate (with a final concentration of 20 mM) to react in a 2 mL Tris-HCl buffer (20 mM; pH 9.5) solution containing 4 mg/mL of the purified enzyme of StDAPDH/H227V or its mutants, and 10 mM of NADP+ for 10 h. For the control reaction, D-serine or L-serine was added to a 2 mL reaction solution to a final concentration of 20 mM, without the addition of the purified enzyme. Three parallel experiments were set up for each reaction. The reaction temperature was 42 °C, and 50 μL of the reaction solution was taken as the sample for further analysis every 2 h. Samples were derivatized with an FDAA derivatization agent and then detected using HPLC (Chromaster CM5110, Hitachi, Tokyo, Japan) to detect L-serine and D-serine. A 10 μL sample was added to a 1.5 mL Eppendorf tube, and then 8 μL of a 1 M NaHCO3 solution and 40 μL of 1% (w/v) FDAA acetone solution were added. The reaction solution was heated in a metal bath at 40 °C for 1 h. The sample was taken out and cooled to room temperature. A total of 8 μL of 1 M HCl was added and mixed well. Finally, 934 μL of a 40% acetonitrile aqueous solution was added for dilution. The HPLC was performed after filtration with 0.22 μm of an organic microporous membrane. The samples were detected using a Develosil ODS-UG-5 column (150 mm × 4.6 mm). The detection conditions were as follows: mobile phase A was 0.05% aqueous solution, and mobile phase B was 0.05% acetonitrile solution. The pump program was calibrated for 0–40 min, and the proportion of mobile phase B increased from 30% to 45%. After 40–45 min, the proportion of mobile phase B increased from 40% to 50% with a constant pump flow rate of 1 mL/min. The detection wavelength was 340 nm, and the column temperature was 40 °C. The injection volume was 20 μL. Samples were derivatized with 2,4-Dinitrophenylhydrazine to detect HPPA. A 10 μL sample was added to a 1.5 mL Eppendorf tube, then 40 μL of a 10 mM 2,4-Dinitrophenylhydrazine solution and 25 μL if a 0.2 mol/L NaOH solution were added. The reaction solution was heated in a metal bath at 30 °C for 20 min. Then, HPLC was performed after filtration with a 0.22 μm organic microporous membrane. Lastly, the samples were detected using a Develosil ODS-UG-5 column (150 mm × 4.6 mm). The detection conditions were as follows: mobile phase A was 0.05% aqueous solution, and mobile phase B was 0.05% acetonitrile solution. The pump program was 0–40 min, and the proportion of mobile phase B increased from 20% to 35%, after 40–45 min. The proportion of mobile phase B increased from 35% to 50%, and the pump flow rate was constant at 1 mL/min with a detection wavelength of 324 nm and column temperature of 40 °C. The injection volume was 20 μL. The retention times of L-serine, D-serine, and HPPA are listed in Table S3. Chemicals used for enzymatic reactions and products analysis were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).
4. Conclusions
In this study, molecular docking predicted the key residues involved in the substrate binding to improve the activity of StDAPDH toward HPPA. The four residues, D92, D122, M152, and N253, were observed to have obviously close connections with HPPA and were selected for further site-directed saturation mutagenesis. Consequently, the single mutations at these sites generated positive variants, D92E, D122W, M152S, and N253Y, with noticeable improvements in catalytic activity. After iterative mutation, a D92E/D122W/M152S mutant was finally achieved. It was speculated that the increase in enzyme activity could be related to the change in active pocket size and the enhancement of molecular interactions between the substrate and key residues. Compared with previously reported research on engineering StDAPDH for catalyzing macromolecular keto acid substrates, this study engineered StDAPDH for enhanced activity toward small-molecule HPPA. These results broaden the substrate spectrum of the StDAPDH family and provided a basis for tuning the enzyme with the potential for the one-step catalytic synthesis of D-serine.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13030576/s1, Table S1: Primers used in this study; Table S2: The ranking of docking results of HPPA; Table S3: The retention times of high-performance liquid chromatography (HPLC); Table S4: Hydrogen bond occupancy.
Author Contributions
Conceptualization, Y.N. and Y.X.; investigation, Z.W., H.Q. and W.L.; formal analysis, Z.W. and H.Q.; validation, Z.W., H.Q. and W.L.; writing—original draft preparation, Z.W.; writing—review and editing, Y.N.; supervision, Y.N. and Y.X.; funding acquisition, Y.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the National Key R&D Program of China (grant number 2021YFC2102000), the National Natural Science Foundation of China (grant numbers 22178147, 31872891), the Program of Introducing Talents of Discipline to Universities (grant number 111-2-06), the High-End Foreign Experts Recruitment Program (grant number G2021144005L), the National Program for Support of Top-notch Young Professionals, the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Top-notch Academic Programs Project of Jiangsu Higher Education Institutions, the Jiangsu Province “Collaborative Innovation Center for Advanced Industrial Fermentation” Industry Development Program, and the National First-Class Discipline Program of Light Industry Technology and Engineering (grant number LITE2018-09).
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Materials. Derived data supporting the findings of this study are available upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Genchi, G. An overview on D-amino acids. Amino Acids 2017, 49, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Z.; Ma, Q.Y.; Zhu, H.L. Distribution, industrial applications, and enzymatic synthesis of D-amino acids. Appl. Microbiol. Biotechnol. 2015, 99, 3341–3349. [Google Scholar] [CrossRef]
- Bommarius, A.S.; Schwarm, M.; Drauz, K. Biocatalysis to amino acid-based chiral pharmaceuticals—Examples and perspectives. J. Mol. Catal. B Enzym. 1998, 5, 1–11. [Google Scholar] [CrossRef]
- Yang, H.; Zheng, G.; Peng, X. D-Amino acids and D-Tyr-tRNATyr deacylase: Stereospecificity of the translation machine revisited. FEBS Lett. 2003, 552, 95–98. [Google Scholar] [CrossRef]
- Ma, J.S. Unnatural amino acids in drug discovery. Chim. Oggi. 2003, 21, 65–68. [Google Scholar]
- Friedman, M.; Levin, C. Nutritional and medicinal aspects of D amino acids. Amino Acids 2012, 42, 1553–1582. [Google Scholar] [CrossRef]
- Bruckner, H.; Westhauser, T. Chromatographic determination of L- and D-amino acids in plants. Amino Acids 2003, 24, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Wehofsky, N.; Pech, A.; Liebscher, S.; Schmidt, S.; Komeda, H.; Asano, Y.; Bordusa, F. D-amino acid specific proteases and native all-L-proteins: A convenient combination for semisynthesis. Angew. Chem. Int. Ed. 2008, 47, 5456–5460. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.D.; Grogan, G.; Bommarius, A.; Hyungdon, Y. Oxidoreductase-Catalyzed Synthesis of Chiral Amines. ACS Catal. 2018, 8, 10985–11015. [Google Scholar] [CrossRef]
- Misono, H.; Togawa, H.; Yamamoto, T.; Soda, K. Meso-alpha, epsilondiaminopimelate D-dehydrogenase: Distribution and the reaction product. J. Bacteriol. 1979, 137, 22–27. [Google Scholar] [CrossRef]
- Pollegioni, L.; Rosini, E.; Molla, G. Advances in enzymatic synthesis of D-amino acids. Int. J. Mol. Sci. 2020, 21, 3206. [Google Scholar] [CrossRef] [PubMed]
- Bornadel, A.; Bisagni, S.; Pushpanath, A.; Montgomery, S.; Turner, N.; Dominguez, B. Technical considerations for scale-up of imine-reductase-catalyzed reductive amination: A case study. Org. Process Res. Dev. 2019, 23, 1262–1268. [Google Scholar] [CrossRef]
- Gao, X.Z.; Chen, X.; Liu, W.D.; Feng, J.H.; Wu, Q.; Hua, L.; Zhu, D.M. A novel meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum: Overexpression, characterization, and potential for D-amino acid synthesis. Appl. Environ. Microbiol. 2012, 78, 8595–8600. [Google Scholar] [CrossRef]
- Gao, X.Z.; Huang, F.; Feng, J.H.; Chen, X.; Zhang, H.L.; Wang, Z.X.; Wu, Q.; Zhu, D.M. Engineering the meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum by site saturation mutagenesis for D-phenylalanine synthesis. Appl. Environ. Microbiol. 2013, 79, 5078–5081. [Google Scholar] [CrossRef]
- Liu, W.; Li, Z.; Huang, C.H.; Guo, R.T.; Zhao, L.M.; Zhang, D.L.; Chen, X.; Wu, Q.; Zhu, D.M. Structural and mutational studies on the unusual substrate specificity of meso-diaminopimelate dehydrogenase from Symbiobacterium thermophilum. Chembiochem 2014, 15, 217–222. [Google Scholar] [CrossRef]
- Cheng, X.K.; Chen, X.; Feng, J.H.; Wu, Q.; Zhu, D.M. Structure-guided engineering of meso-diaminopimelate dehydrogenase for enantioselective reductive amination of sterically bulky 2-keto acids. Catal. Sci. Technol. 2018, 8, 4994–5002. [Google Scholar] [CrossRef]
- Vedha-Peters, K.; Gunawardana, M.; Rozzell, J.D. Creation of a broad-range and highly stereoselective D-amino acid dehydrogenase for the one-step synthesis of D-amino acids. J. Am. Chem. Soc. 2006, 128, 10923–10929. [Google Scholar] [CrossRef] [PubMed]
- Akita, H.; Doi, K.; Kawarabayasi, Y. Creation of a thermostable NADP+-dependent D-amino acid dehydrogenase from Ureibacillus thermosphaericus strain A1 meso-diaminopimelate dehydrogenase by site-directed mutagenesis. Biotechnol. Lett. 2012, 34, 1693–1699. [Google Scholar] [CrossRef]
- Hayashi, J.; Seto, T.; Akita, H.; Watanabe, M.; Hoshino, T.; Yoneda, K.; Ohshima, T.; Sakuraba, H. Structure-based engineering of an artificially generated NADP+-dependent D-amino acid dehydrogenase. Appl. Environ. Microbiol. 2017, 83, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Akita, H.; Suzuki, H.; Doi, K. Efficient synthesis of D-branched-chain amino acids and their labeled compounds with stable isotopes using D-amino acid dehydrogenase. Appl. Microbiol. Biotechnol. 2014, 98, 1135–1143. [Google Scholar] [CrossRef]
- Hanson, R.L.; Davis, B.L.; Goldberg, S.L.; Johnston, R.M.; Parker, W.L.; Tully, T.P.; Montana, M.A.; Patel, R.N. Enzymatic preparation of a D-amino acid from a racemic amino acid or keto acid. Org. Process Res. Dev. 2008, 12, 1119–1129. [Google Scholar] [CrossRef]
- Xu, J.M.; Fu, F.T.; Hu, H.F.; Zheng, Y.G. A high-throughput screening method for amino acid dehydrogenas. Anal. Biochem. 2016, 495, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yao, P.; Gong, R.; Li, J.; Wu, Q.; Zhu, D.M.; Liu, P.; Li, G.Y.; Yao, P.Y.; Lonsdale, R.; et al. Simultaneous engineering of an enzyme’s entrance tunnel and active site: The case of monoamine oxidase MAO-N. Chem. Sci. 2017, 8, 4093–4099. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).