Abstract
The β-d-mannopyranoside linkage is found in a number of biological structures, in particular, in the core trisaccharide of N-linked glycoproteins, as well as within the antigenic polysaccharides of Salmonella, yeasts, and glycolipids. The construction of this glycosydic bond by chemical approach is very challenging and requires cumbersome protection and activation steps prior to glycosylation. In this context, β-mannosidase from Cellulomonas fimi (Cf-β-Man) was immobilized for the first time, and it was employed in the synthesis of β-mannosides. Cf-β-Man immobilized on IDA-Co2+-agarose allows the synthesis of the disaccharide, cyanomethyl β-d-mannopyranosyl-(1→6)-2-acetamido-2-deoxy-1-thio-β-d-glucopyranoside, with a higher conversion compared to the soluble enzyme (20% vs. 5%) after 6 h under best conditions. This explorative work opens new scenarios concerning the design of engineered Cf-β-Man mutants and their immobilization in order to obtain a robust and recyclable biocatalyst for applications in chemoenzymatic glycan synthesis.
1. Introduction
Oligosaccharides are involved in a multitude of biological events in all living organisms, and are often found in conjunction with other macromolecules, such as lipids or proteins [1,2]. Alterations in cell surface oligosaccharides are associated with various pathological conditions, including malignant transformation [3]. There are two main classes of glycoproteins, namely N-linked and O-linked glycoproteins. The N-linked glycoproteins are a biologically important class of structures containing a β-linked mannopyranoside unit. The β-d-mannopyranoside linkage is probably the most difficult glycosidic bond to synthesize chemically and all synthetic methods require a number of protection and activation steps prior to glycosylation [4,5].
Glycoside hydrolases (glycosidases) are enzymes that hydrolyze glycosidic bonds between two or more carbohydrate modules or sugar and a non-sugar moiety within oligosaccharides. In this context, mannosidases (mannoside mannohydrolases, EC 3.2.1.x), and mannanases (mannan mannohydrolases, EC 3.2.1.x) are involved in the processing of mannosides. β-Mannosidases (EC 3.2.1.25) are exo-acting enzymes that catalyse the hydrolysis of β-1,4-linked mannosides, releasing mannose from the non-reducing end of mannans and mannan oligosaccharides, while β-mannanases (EC 3.2.1.78) are the pivotal enzymes that initiate degradation of mannans by randomly cleaving the backbone to liberate short β-1,4-manno-oligomers, producing new chain ends [6]. These biocatalysts can also act in the opposite direction, leading to synthesis of novel oligosaccharides by reverse hydrolysis reaction. In particular, mannosidases were exploited for the synthesis of manno-oligosaccharides [4].
Despite the versatility of mannosidases, the use of these enzymes in soluble form has some limitations, including low stability, unmanageable recovery and reuse, short shelf life, difficulty in handling, and loss of activity at prolonged operational conditions [7,8]. However, immobilization of enzymes on or into solid matrixes can address these issues. Few examples of immobilization protocols of mannosidases and mannanases were reported so far. Most of them are based on non-covalent protocols using different immobilization matrixes, which include chitosan magnetic nanocomposites [9], sodium alginate-grafted β-cyclodextrin [10], cross-linked enzyme aggregates [11], and calcium alginate beads [12]. Recently, β-mannanase from Thermotoga maritima was covalently immobilized on glutaraldehyde cross-linked chitosan beads. The immobilized enzyme showed an improved activity and stability compared to the soluble enzyme [8].
In this work, an immobilization study of β-mannosidase from Cellulomonas fimi (Cf-β-Man) was performed for the chemoenzymatic synthesis of β-mannosides. For this purpose, different support matrixes and binding chemistries were investigated. Combining the (proper) selection of the matrix and operative immobilization conditions, itwas possible to select an immobilized catalyst (Co-IDA-agarose-Cf-β-Man), suitable for the synthesis of mannose-based disaccharides.
We screened the enzyme activity using different sugar substrates, such as d-mannose (2a), d-mannose-SCH2CN (2b), N-acetyl-d-glucosamine (2c), and N-acetyl-d-glucosamine-SCH2CN (2d). The compounds 2b and 2d carried a thiocyanomethyl group at the C1 position as precursor of the reactive IME (2-iminomethoxyethyl), a suitable linker useful for the conjugation to the proteins (Scheme 1).
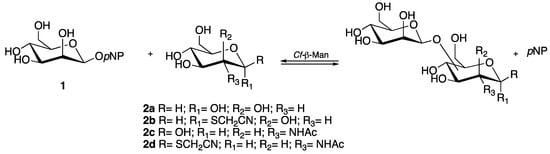
Scheme 1.
Transglycosylation reaction: screening of acceptors. pNP = para-nitrophenol.
2. Results and Discussion
2.1. Transglycosylation: Screening of Acceptors
With the aim of preparing disaccharides completely in water, we decided to explore an enzyme-catalyzed transmannosylation reaction employing para-nitrophenyl-β-mannoside (pNP-β-Man) (1) as donor and an array of mannose- and N-acetylglucosamine-based acceptors, using the β1,4-mannosidase from Cellulomonas fimi as biocatalyst (Scheme 1). As thiocyanomethyl glycosides were widely used for attaching carbohydrates to proteins under mild reaction conditions, both mannose (Man) and N-acetylglucosamine (GlcNAc) were also functionalized with this group and employed as acceptors in the enzymatic synthesis of the disaccharides of interest.
The activity and regioselectivity of soluble Cf-β-Man was studied in the transglycosylation reaction using different sugar acceptors, such as d-mannose (2a), d-mannose-SCH2CN (2b), N-acetyl-d-glucosamine (2c), and N-acetyl-d-glucosamine-SCH2CN (2d). The formation of the transglycosylation disaccharide products was monitored by LC-MS.
In particular, with compounds 2a and 2b as acceptors, the formation of two different transglycosylation products was observed at the expected m/z (Figure S1A,B). When compound 2c was used as an acceptor, a great number of products were observed with the expected m/z, thus suggesting a lower regioselectivity for this acceptor (Figure S1C). Finally, when compound 2d was used as an acceptor, the formation of a single peak with the expected m/z was observed (Figure S1D).
Thus, given the high regioselectivity of the enzyme in the presence of 2d acceptor, and considering that in the N-linked glycoproteins of both eukaryotes and several viral pathogens (e.g., HIV, Ebola, and some coronaviruses), the oligosaccharide chains are linked to the asparagine residue of the proteins through a conserved pentasaccharide core, which contains an N-acetylglucosamine unit linked via β1,4 bond to a mannose unit [2]; we decided to study this reaction more in detail in order to optimize the reaction conditions with the final aim of identifying the transglycosylation product.
2.2. Transglycosylation: Reaction Conditions Optimization
In order to try to optimize the formation of the transglycosylation product using compound 2d as glycosyl acceptor, various parameters were evaluated: (i) use of different organic (green) solvents (acetonitrile; glycerol formal; tetrahydrofuran; and tert-butanol) and one ionic liquid (Bmim[BF4]) at 30% v/v; (ii) donor/acceptor ratio variations; and (iii) addition of donor and enzyme in aliquots over time.
The reference reaction was set up in maleate buffer 100 mM pH 6.5 (optimal conditions suggested by supplier) and 1:6 donor/acceptor ratio using 2.5 UI/mL of soluble enzyme. From LC-MS analysis, the formation of the transglycosylation product (m/z 277) is already observed after 1 h reaction time, reaching its maximum formation around 3 h and remaining almost constant up to 24 h (Figure S2).
Previous work from our laboratory showed that the use of certain (green) organic solvents or ionic liquids as cosolvents in the reaction media could increase the enzyme activity and also the regioselectivity of glucosidases, allowing for an increase in reaction yield [13]. None of the solvents, nor the ionic liquid tested, allowed for an increase in the conversion; on the contrary, their use negatively influenced enzyme activity as the glycosyl donor pNP-β-Man (1) was not consumed during incubation compared to the standard reaction, for which, after 3 h reaction time, the complete consumption of the sugar donor could be observed (Figure S3). The low activity is probably due to low enzyme stability (see below).
Moreover, we also tried to add more glycosyl donor (1) to the reaction media once we observed its complete disappearance by TLC in order to improve the yield. After 1 h reaction with the standard donor acceptor ratio (1:6), we added an additional 1 equivalent of the glycosyl donor (1). However, monitoring the reaction up to 4 h after the addition did not allow the complete consumption of the donor, probably also in this case due to low enzyme stability in reaction conditions (see below). Finally, due to stability issues, we also tried to add both the glycosyl donor pNP-β-Man (1) and the enzyme in four different aliquots over time; however, also in this case, we did not observe the complete consumption of the glycosyl donor.
Based on these results, the best conditions for the transglycosylation reaction are maleate buffer 100 mM pH 6.5, 1:6 donor/acceptor ratio, and 2.5 UI/mL of soluble enzyme at 37 °C for 3 h.
2.3. Cf-β-Man Stability
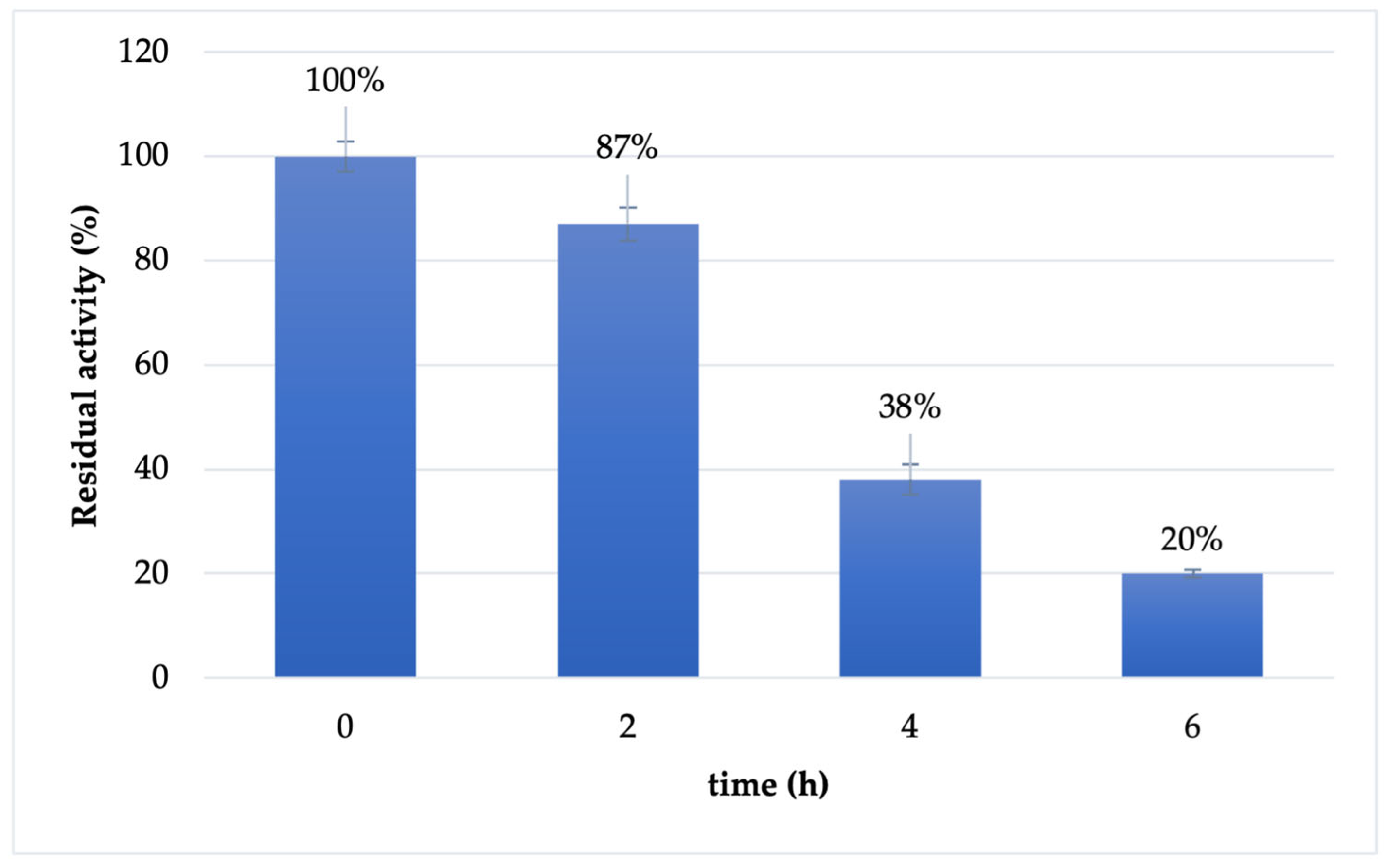
The stability of soluble Cf-β-Man was investigated in the optimal conditions reported for enzyme activity (maleate buffer 100 mM pH 6.5). As reported in Figure 1, the enzyme is almost stable in the first two hours of the transglycosylation reaction (87% retained activity). However, already after 4 h of incubation, the retained activity of the biocatalyst drops at 38%, reaching 20% of retained activity after 6 h of incubation.
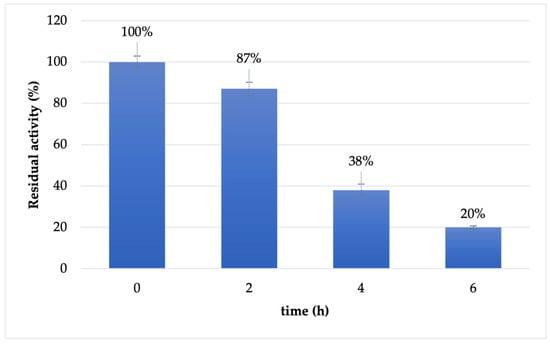
Figure 1.
Stability of soluble Cf-β-Man in maleate buffer 100 mM pH 6.5 and 37 °C.
Due to the low stability of the soluble enzyme in reaction conditions, we decided to try to stabilize it through immobilization in order to obtain a robust biocatalyst that could be used for synthetic applications.
2.4. Immobilization of Cf-β-Man on Different Immobilization Carriers
Cf-β-Man was first covalently immobilized on two different carriers: SepabeadsTM, which is a hydrophobic matrix [14], and agarose, which is a sharply hydrophilic carrier [15]. The results of the immobilization screening are shown in Table 1.

Table 1.
Immobilization of Cf-β-Man on Sepabeads and agarose-based carriers.
First, both carriers were easily functionalized with aldehyde groups for imine-bond formation with the ε-amino groups of the lysine residues of the biocatalyst. The immobilization of Cf-β-Man on glyoxyl-Sepabeads (GLX-Sepabeads) was evaluated using two pH values: (i) pH 10.0, which is the pH value at which this type of immobilization is generally carried out, allowing a multipoint interaction between the enzyme and the carrier (entry 1) and (ii) pH 8.0, which allows for a milder interaction between the aldehyde groups of the support and the ε-amino groups of lysins present on the surface of the enzyme (entry 2). Furthermore, the addition of d-mannose (1% w/v) as an additive was also evaluated in the immobilization carried out at pH 10.0, with the aim to improve the output of the immobilization process by maintaining the correct three-dimensional conformation of the active site and therefore preserving the catalytic pocket from distortions. As reported in Table 1, a quantitative yield of immobilization in terms of bound protein, and an excellent yield (around 90%) in terms of activity were obtained in both pH conditions. Both final enzymatic derivatives were active (0.94 IU/g and 0.96 IU/g, respectively, at pH 10.0 and pH 8.0); however, a modest activity recovery (13.8% and 15.0%, respectively) was obtained. The modest activity recovery obtained could be due to the distortion of the three-dimensional structure of the protein during the binding with the carrier. Using a mild immobilization pH (pH 8.0; entry 2) or lowering the immobilization temperature from 25 °C to 4 °C (pH 10.0; entry 1) to allow for slower interaction between the enzyme and the carrier do not appear to protect the enzyme from inactivation. Furthermore, the addition of d-mannose as an additive does not seem to improve the immobilization outcome; however, in all the other immobilization protocols, we decided to add it, unless otherwise specified.
At this point, exploiting the same binding chemistry between the carrier and the enzyme, we decided to evaluate glyoxyl-agarose (GLX-AG) as a carrier for Cf-β-Man immobilization. GLX-AG is a highly hydrophilic carrier widely exploited for the covalent immobilization of enzymes [15].
Immobilization of Cf-β-Man on GLX-AG (Table 1, entry 3) gave poor yields both in terms of immobilized protein (49%) and in terms of activity (77%) compared to GLX-Sepabeads (entry 1) even if the immobilization time was extended up to 24 h in order to try to push the reaction to completion. Furthermore, the final derivative obtained was almost inactive (0.08 UI/g), showing a very poor activity recovery (<5%), suggesting better immobilization outcomes when using hydrophobic carriers.
Given the low activity of the derivatives obtained both with GLX-Sepabeads (0.94 UI/g) and with GLX-AG (0.08 UI/g), the subsequent chemical reduction of the imine bonds to stable C-N bonds, which is generally carried out to avoid enzyme leaching from the carrier was not evaluated also because the reduction step, generally, reduces drastically the activity of the enzymatic derivative. However, imine bonds are not very stable at acid pH [16], which is the working pH of the enzyme used in this work. Therefore, in order to obtain an active and already stable derivative without the reduction step, in the screening, two other Sepabeads- and agarose-based carriers, respectively, were derivatized with furan-2,5-dicarbaldehyde for the covalent immobilization of Cf-β-Man. DFF-Sepabeads (Table 1, entry 4) and DFF-EDA-AG (Table 1, entry 5) have the same binding chemistry as their corresponding glyoxyl-carriers, but these carriers are characterized by a spacer arm that could confer greater flexibility to the immobilized enzyme. Furan-2,5-dicarbaldehyde is a bio-based molecule that was recently proposed as an alternative to glutaraldehyde, which is normally used for the insertion of a spacer on the carrier surface [16]. Furthermore, thanks to its furan ring structure, once the imine bond between the enzyme and the carrier is formed, an aromatic system is formed, stabilizing the imine bond even at acidic pH [16]. Both supports showed good immobilization yields in terms of activity (76.5% and 74%, for Sepabeads and agarose, respectively) and in terms of protein (51.3% and 88.8%), but the final derivatives were almost completely inactive (0.06 IU/g), with a poor activity recovery (<5%).
Since the multipoint immobilization protocols exploiting aldehyde groups led to enzyme derivatives with low activity, we decided to screen a different binding chemistry. The covalent immobilization of Cf-β-Man on cyanogen bromide-activated agarose (CNBr-AG) was investigated (Table 1, entry 6), since it allows for obtaining single bonds with the primary NH2 groups of the protein and providing for greater flexibility to the enzyme once bound to the carrier. In this case, a quantitative immobilization yield was obtained both in terms of protein and in terms of activity, and also a good activity recovery (70.6%). The final derivative showed good retained activity (1.75 IU/g); however, already after a few hours stored at 4 °C, it showed an exponential decrease in activity, up to complete inactivation after 7 days.
Thus, as the covalent immobilization protocols screened have not allowed us to obtain an active and stable enzyme derivative over time, we decided, also on the basis of the literature [9,10,12], to evaluate immobilization protocols based on milder non-covalent interactions. As reported in Table 1, three immobilization trials were performed by ionic interaction between the carboxyl groups of the enzyme and the amino groups of the Sepabeads carrier derivatized with polyethyleneimine under different conditions (entries 7–9). Good immobilization yields, both in terms of protein and in terms of activity, were detected in all conditions analyzed (60–99%), significantly reducing the immobilization time (1–2 h) even at 4 °C compared to covalent protocols. In this case, however, the decrease in temperature from 25 °C to 4 °C and the addition of 1% w/v sucrose significantly increased the activity recovery (from 5.2 to 33.5%), allowing for the obtaining of an enzyme derivative with higher final activity (1.74 IU/g) (Table 1, entry 8) than that obtained at 25 °C and without the addition of sucrose (0.26 IU/g) (Table 1, entry 7). However, also in this case, the derivative with the highest activity at the end of the immobilization process (Table 1, entry 8), already after a few hours of storage at 4 °C, showed a decrease in activity, up to complete inactivation after 7 days. In order to overcome this issue, given that the immobilization protocol used was not covalent, we tried to trap Cf-β-Man, previously adsorbed by ionic interaction, by crosslinking with aldehyde dextran. After the crosslinking, the activity of the final derivative was lower (0.63 IU/g) (Table 1, entry 9), and consequently, also the activity recovery dropped (13.5%). Additionally in this case, given the low activity of the derivative obtained, the subsequent chemical reduction of the imine bonds between dextran and the protein to stable C-N bonds, which is generally carried out, was not performed. However, despite the cross-linking, even if without reduction, also this derivative showed a decrease in activity after 40 h of storage at 4 °C.
At this point, in order to obtain an enzymatic derivative with a good activity maintained over time, the last immobilization strategy studied exploited the formation of a coordination bond between the enzyme, in particular, the His-Tag sequence, and a metal by using an agarose-iminodiacetic acid-Co2+ -based carrier (IDA-Co2+-AG). Initially, optimal yields were obtained both in terms of activity (82.2%) and in terms of protein (100%) (Table 1, entry 10), and the enzymatic derivative maintained very high activity (3.10 IU/g); therefore, a good activity recovery was obtained (72.8%). Moreover, in this case, the final derivative maintained its activity over time up to 30 days after the immobilization when stored at 4 °C.
Based on these results, IDA-Co2+-AG was selected as the best carrier for Cf-β-Man immobilization (high immobilization yield and activity recovery). A further strength point of Cf-β-Man immobilized on IDA-Co2+-AG was its shelf life: this immobilized preparation retained its starting activity after 30 days of storage at 4 °C.
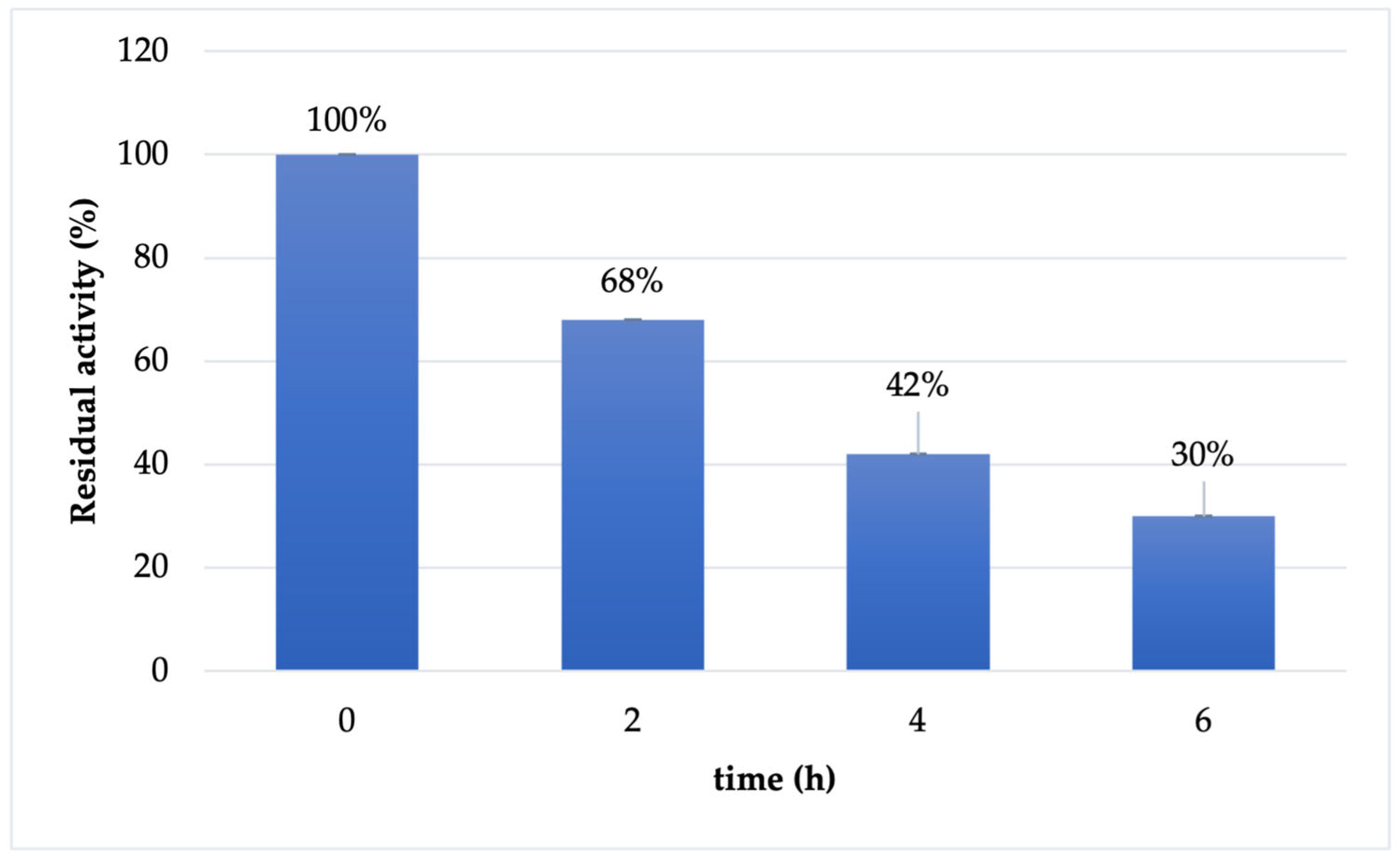
The stability of immobilized Cf-β-Man derivative was tested in reaction conditions (maleate buffer 100 mM pH 6.5). As reported in Figure 2, the immobilized derivative has a similar behavior to the soluble enzyme: the enzyme is almost stable in the first two hours of the reaction (68% retained activity). However, already after 4 h of incubation, the retained activity of the biocatalysts drops at 42%, maintaining 30% of activity after 6 h of incubation.
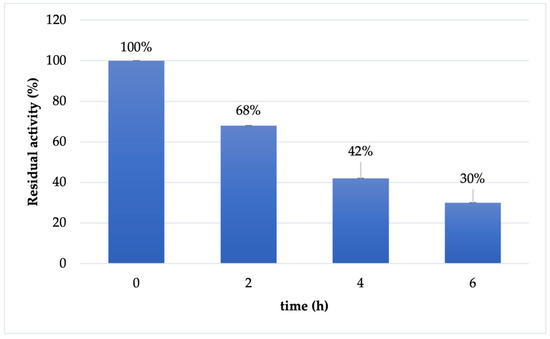
Figure 2.
Stability of immobilized Cf-β-Man on IDA-Co2+-AG in maleate buffer 100 mM pH 6.5 and 37 °C.
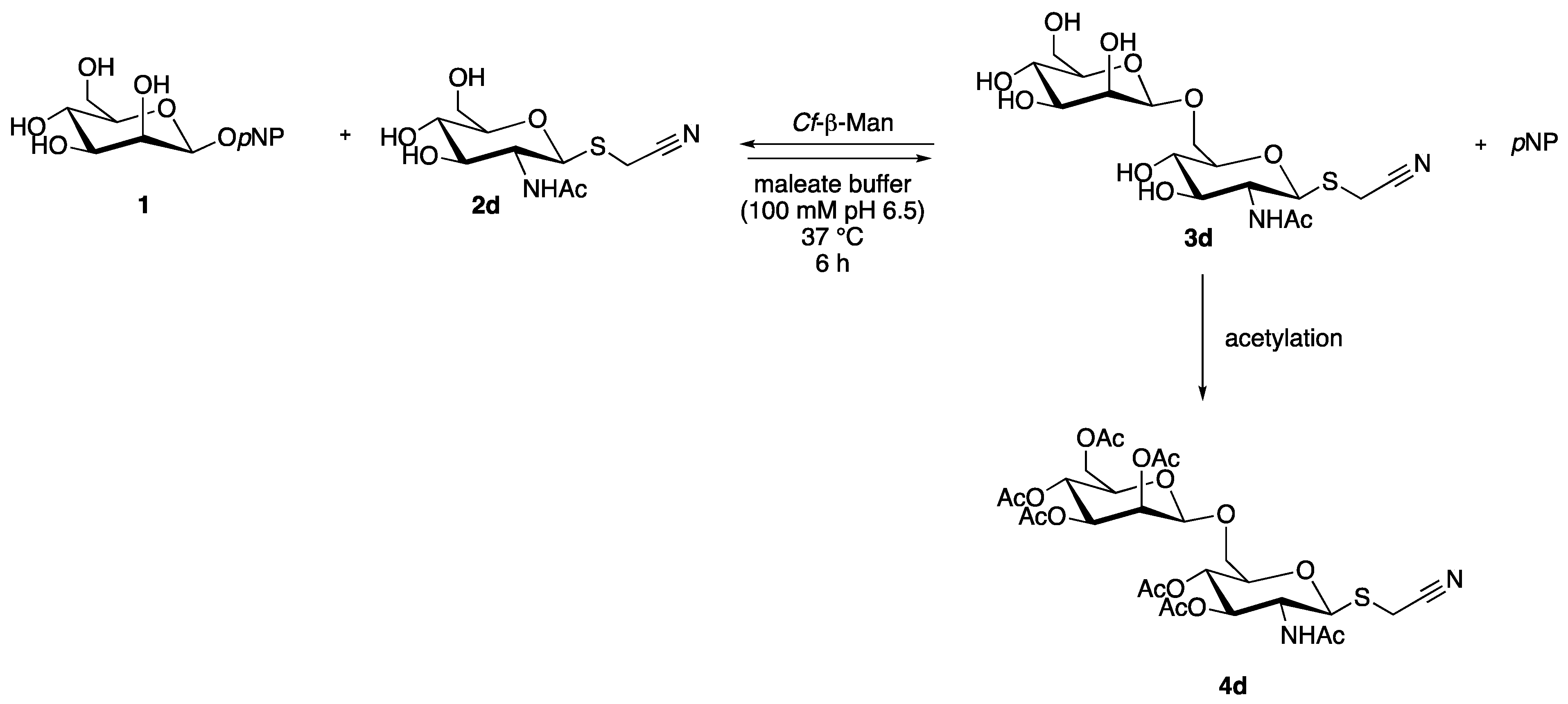
2.5. Synthesis and Identification of Cyanomethyl β-d-Mannopyranosyl-(1→6)-2-Acetamido-2-Deoxy-1-thio-β-d-Glucopyranoside (3d)
The transglycosylation reaction was performed both with the soluble and immobilized enzyme in order to determine reaction conversion in HPLC based on calibration curve and to identify the disaccharide product (4d) after chemical acetylation through complete NMR analysis (Scheme 2).
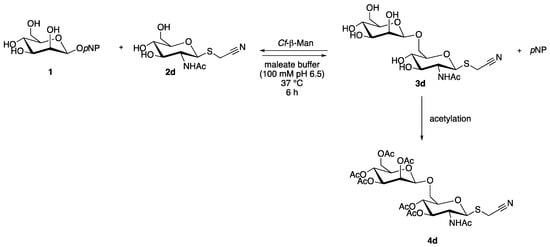
Scheme 2.
Transglycosylation reaction with acceptor 2d for the synthesis of 3d, isolated and identified after chemical acetylation (4d). pNP = para-nitrophenol.
The reaction with the immobilized enzyme was performed in the standard reaction conditions, lowering the enzymatic units from 1.25 IU to 0.9 IU in order to allow a good dispersion of the immobilized enzyme in reaction media. Using the immobilized derivative, a complete consumption of the glycosyl donor (1) was observed already after 4 h of reaction (Figure S4A) and after acetylation, a conversion of 20% was registered. While using the soluble enzyme, in the same reaction conditions, the glycosyl donor (1) was not completely consumed after 6 h of reaction (Figure S4B). After acetylation, a conversion of 5% was registered, demonstrating better performance for the immobilized enzyme.
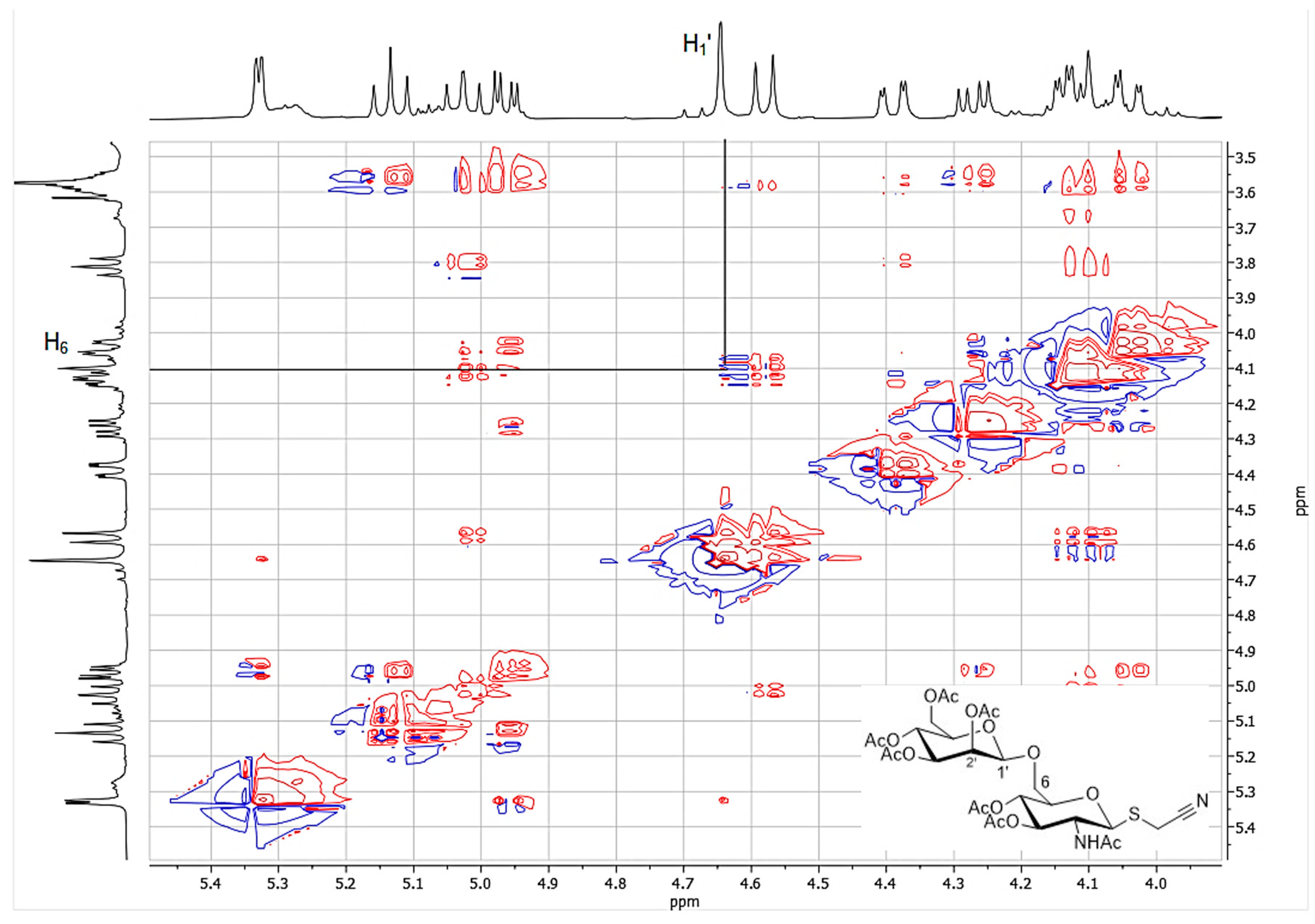
The reaction catalyzed by immobilized Cf-β-Man was further scaled up in 2 mL final volume in order to isolate and characterize the product. The acetylated disaccharide (4d) was purified by flash chromatography (17% isolated yield; 10 mg) and characterized by ESI-MS (Figure S5). The identity of the product was determined by complete NMR analysis (Figures S6–S9). In particular, NMR analysis allowed the determination of both the β configuration of the glycosidic bond and the type of linkage (1→6) between the two sugar units. Indeed, the H1′ and H6 signals were found to be correlated in the TOCSY spectrum (Figure 3). On the other hand, no useful structural information could be drawn from the value of the coupling constant between the anomeric proton H1′ at 4.65 ppm and H2′ at 5.33 ppm (J = 1.2 Hz). Indeed, as opposed to d-glucosides, typical J1,2 values for d-mannose derivatives are 1.6 Hz for coupling between diequatorial protons (α anomer) and 0.8 Hz for the axial-equatorial coupling of the β anomer [17]. However, the configuration at C1 of 4d could be inferred to be β by direct comparison of its 1H NMR spectrum with that of the α anomer, recently obtained by reacting 2,3,4,6-tetra-O-acetyl-α-d-mannopyranosyl trichloracetimidate with cyanomethyl 2-acetamido-3,4-di-O-acetyl-2-deoxy-1-thio-β-d-glucopyranoside. In fact, while the α anomer shows a series of multiplets between 5.39 and 4.80 ppm (including H1′ and H1 at 4.86 and 4.80 ppm, respectively), the corresponding signals in 4d are significantly upfield shifted (specifically, in the 5.33–4.58 ppm region). Remarkable differences in δ values also affect the remaining areas of the spectrum [18]. Moreover, based on literature data [19] Cf-β-Man is a retaining glycosidase, thus its natural β stereoselectivity should be conserved while the regioselectivity can differ based on the type of glycosyl acceptor used in the reaction.
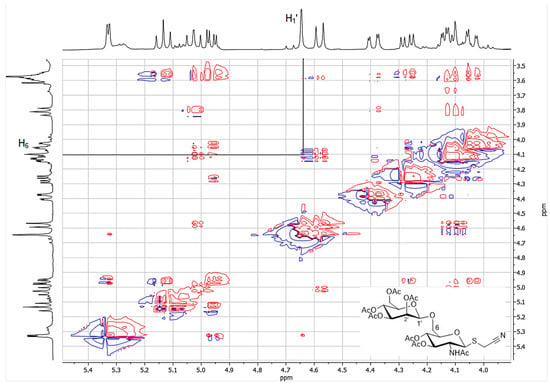
Figure 3.
Expansion of the TOCSY spectrum of 4d showing the correlation between H1′ and H6.
3. Materials and Methods
β-Mannosidase from C. fimi (Cf-β-Man) was from Megazyme (Bray, Ireland). 4-Nitrophenyl-β-d-mannopyranoside (pNP-β-Man) was from Apollo Scientific (Breadbury, United Kingdom). Bradford reagent, glycidol, sodium periodate (NaIO4), sodium borohydride (NaBH4), ethylenediamine (EDA), and cyanogen bromide-activated Sepharose® 4B (CNBr-agarose) were purchased from Sigma-Aldrich (Milano, Italy). Sepharose CL-6B (agarose) was from GE Healthcare (Uppsala, Sweden). Agarose IDA-Co2+ was from Jena Bioscience (Jena, Germany).
Sepabeads® EC-EP/S and ReliZyme EA112/S were a gift from Resindion (Binasco, Italy).
Spectrophotometric assays were performed using a Shimadzu UV-1601 UV-visible spectrophotometer equipped with magnetic stirring. HPLC analyses were run with a L-7100 HPLC (Merck Hitachi, Tokyo, Japan) equipped with Merck Hitachi D-7000 HPLC Multi HSM Manager, a L-7400 detector, an L-7300 oven and an Adamas RP-C18 extreme column (SepaChrom, 250 mm × 4.6 mm, 5 μm particles) (Rho, Italy).
LC-MS analyses were performed on a Dionex UltiMate 3000 HPLC system (Thermo Scientific, San Jose, CA, USA) equipped with a mobile-phase online degasser, ternary pump, autosampler, column thermostated compartment, and variable wavelength detector, and controlled by Chromeleon software (6.8 version). MS detection was achieved by using a linear ion trap mass spectrometer (LTQ) equipped with an electrospray ion source (ESI) (Thermo Scientific, San Jose, CA, USA) and controlled by X-calibur software (2.0.7 version).
1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively, on a Bruker AVANCE 400 spectrometer equipped with a TOPSPIN software package (Bruker, Karlsruhe, Germany) (Available online: https://www.bruker.com/en/products-and-solutions/mr/nmr-software/topspin.html, accessed on 28 August 2023) at 300 K. CDCl3 (Merck) was used as solvent. 1H and 13C chemical shifts (δ) are given in parts per million (ppm) and referenced to the solvent signals (δH 7.26 and δC 77.16) from tetramethylsilane (TMS). Coupling constants (J) are reported in Hertz (Hz).
3.1. Enzyme Characterization
3.1.1. Standard Activity Assay
The standard activity assay (2 mL) was performed at 37 °C in sodium maleate buffer (pH 6.5, 100 mM) containing pNP-β-Man (1 mM) and an appropriate amount of enzyme (6 μL of 1/10 dilution in sodium maleate buffer 100 mM pH 6.5). Three reactions were incubated in parallel for 1, 2, and 3 min under mechanical stirring (1000 rpm). At the established endpoints, each reaction was quenched by the addition of NaOH 10 M (20 μL). After quenching, the formation of para-nitro-phenol (pNP) was monitored spectrophotometrically at 410 nm using an extinction coefficient of 18.3 mM−1 cm−1. One unit of Cf-β-Man was defined as the amount of enzyme that produces 1 μmole of pNP per minute from pNP-β-Man. The volumetric activity of Cf-β-Man was 30 IU/mL.
3.1.2. Bradford Assay
The protein content was determined by Bradford assay using bovine serum albumin (BSA) as standard. The protein content was 20.5 mg/mL.
3.2. Transglycosylation Reaction for the Synthesis of Mannose- and N-Acetylglucosamine-Based Disaccharides
The transglycosylation reaction was set up following a procedure reported in the literature [13]. First, 4 different acceptors: d-mannose (2a), d-mannose-SCH2CN (2b), N-acetyl-d-glucosamine (2c), and N-acetyl-d-glucosamine-SCH2CN (2d) were tested to investigate the specificity of the enzyme (Scheme 1). Subsequently, N-acetyl-d-glucosamine-SCH2CN (2d) was chosen as a model acceptor and the reaction conditions were investigated deeply.
3.2.1. Transglycosylation Reaction for the Synthesis of Mannose- and N-Acetylglucosamine-Based Disaccharides: Acceptor Screening
The donor (1, pNP-β-Man, 0.020 mmol, 6.5 mg) and the different acceptors (0.125 mmol) were solubilized in maleate buffer (100 mM, pH 6.5, 500 mL) at 37 °C until an opalescent mixture was obtained. The reaction was started with the addition of the enzyme (83 μL, 1.25 IU). The reaction was allowed to stir at 37 °C for 24 h. At different endpoints, samples were withdrawn (40 µL) from the reaction mixture, filtered with Microcon® filter (cutoff 10 kDa) for 10 min at 13,000 rpm and 25 °C to eliminate the enzyme, and samples were monitored in LC-MS.
3.2.2. Transglycosylation Reaction for the Synthesis of Cyanomethyl β-d-Mannopyranosyl-(1→6)-2-Acetamido-2-Deoxy-1-thio-β-d-Glucopyranoside (3d): Reaction Conditions Screening
The donor pNP-β-Man (0.020 mmol) and the acceptor N-acetyl-d-glucosamine-SCH2CN (2d) (0.125 mmol) were solubilized in maleate buffer (100 mM, pH 6.5, 500 μL) at 37 °C. The reaction was started through the addition of 1.25 IU of enzyme (reference reaction). In an attempt to optimize the reaction, various parameters were evaluated: (i) the addition of various organic solvents at 30% v/v in the reaction buffer: acetonitrile (ACN), glycerol formal (GL), tetrahydrofuran (THF), and tert-butanol (t-BuOH) as well as the ionic liquid 1-butyl-2,3-dimethylimidazoliumtetrafluoroborate (Bmim[BF4]); (ii) higher concentration of glycosyl donor; as well as (iii) the addition of the donor and fresh enzyme fractionated into 4 aliquots. For reactions 2–5, 150 μL of the corresponding organic co-solvent were added in 350 μL of maleate buffer 100 mM pH 6.5 containing the glycosyl donor pNP-β-Man (0.020 mmol) and the acceptor N-acetyl-d-glucosamine-SCH2CN (2d) (0.125 mmol). The reactions were started through the addition of the enzyme (1.25 IU; 42 μL). For reaction 6, the standard reaction was set up, but after 1 h additional glycosyl donor pNP-β-Man (0.020 mmol) was added and incubated further for 5 additional hours. Finally, reaction 7 was set up by adding fresh aliquots both of the glycosyl donor pNP-β-Man (0.010 mmol × 4 times: t0, t = 30 min, t = 1.5 h, t = 3 h) and of fresh enzyme (0.32 IU × 4 times: t0, t = 30 min, t = 1.5 h, t = 3 h).
The reactions were monitored both by TLC (CHCl3/MeOH, 5:1) and LC-MS.
3.3. Cf-β-Man Stability
The stability of the soluble enzyme was evaluated in the sodium maleate buffer (100 mM, pH 6.5). Soluble enzyme (1.25 IU, 42 μL) was solubilized in sodium maleate buffer (100 mM, pH 6.5, 500 μL) and incubated at 37 °C on a rolling shaker (1000 rpm) for 24 h. At different endpoints, samples were withdrawn (40 μL) and the residual activity was determined by the standard activity assay at least in duplicate.
The stability of the immobilized enzyme on Co2+-IDA-AG was evaluated in sodium maleate buffer (100 mM, pH 6.5). For each endpoint studied, an independent reaction was set up containing immobilized derivative (50 mg) suspended in sodium maleate buffer (100 mM, pH 6.5, 500 μL). The reactions were incubated at 37 °C on a rolling shaker (1000 rpm) for 24 h. At the desired endpoints, the reactions were filtered with Microcon® filter (cutoff 10 kDa) for 10 min at 13,000 rpm and 25 °C. The residual activity of the immobilized derivative (10–15 mg) was determined by the standard activity assay at least in duplicate.
3.4. Enzyme Immobilization
Enzyme immobilization on different carriers was performed using an enzyme loading of 5 IU per gram of carrier. The enzyme is sold as a 3.2 M ammonium sulphate suspension; thus, before being used in the immobilization protocols, the enzyme was pretreated by centrifugation at 13,000 rpm for 10 min at 4 °C, the supernatant was eliminated, and the pellet was resuspended in the appropriate buffer for each immobilization protocol (see below). A 10:1 ratio volume of immobilization reaction/volume of the carrier was used.
During immobilization, the supernatant was monitored by measuring the amount of protein in the solution (Bradford assay) (10–50 μL), and the residual activity of the supernatant (5–30 μL) was checked by the standard activity assay described before.
3.4.1. Immobilization of Cf-β-Man on Glyoxyl-Sepabeads® (GLX-Sepabeads)
GLX-Sepabeads was prepared as reported in the literature [20]. Briefly, Sepabeads® EC-EP/S (20 g) was suspended in H2SO4 (0.5 M, 260 mL) under mechanical stirring for 2 h at r.t., then filtered under a vacuum and washed thoroughly with distilled H2O until achieving neutral pH. The resin was then oxidized by incubation with NaIO4 (100 mM, 80 mL) for 2 h at r.t., filtered, washed thoroughly with distilled H2O, and stored at 4 °C till use.
Immobilization of Cf-β-Man on GLX-Sepabeads was performed testing two different pH values: (i) pH 8, which allows for a milder interaction between the enzyme and the carrier, and (ii) pH 10, which allows for multipoint interactions. Briefly, GLX-Sepabeads was washed abundantly either with KH2PO4 (50 mM, pH 8) or NaHCO3 buffer (50 mM, pH 10) and then filtered under reduced pressure until dryness. Soluble enzyme (2.5 IU, 83 μL), previously pretreated as reported before, was solubilized into the appropriate buffer (7 mL). In the immobilization protocol performed at pH 10, the reaction media was added with d-mannopyranoside (1% w/v) as additive. Then, the carrier (0.5 g) was added, and the suspensions were allowed to stir at 4 °C or 25 °C for 3–6 h. The immobilized enzyme was then filtered, rinsed thoroughly with distilled H2O, and stored at 4 °C till use.
3.4.2. Immobilization of Cf-β-Man on Glyoxyl-Agarose (GLX-AG)
GLX-AG was prepared as reported in literature [21]. Briefly, SepharoseTM CL-6B (agarose, 15 g) was suspended in deionized H2O (3.6 mL) and NaOH (1.7 M, 7.1 mL) containing NaBH4 (142.5 mg/mL). Subsequently, glycidol (5.1 mL) was added dropwise, keeping the vessel at 4 °C in an ice bath. The reaction was kept under gently stirring overnight at r.t. After the incubation period, the suspension was filtered, and the carrier was washed abundantly with deionized H2O. Oxidation was initiated by adding NaIO4 (100 mM, 102 mL). The reaction was carried out for 2 h at r.t., and then the carrier was filtered under reduced pressure and washed abundantly with deionized H2O and stored at 4 °C until use.
Immobilization of Cf-β-Man on GLX-AG was performed following a standard protocol [21]. Briefly, GLX-AG was washed abundantly with NaHCO3 buffer (50 mM, pH 10) and then filtered under reduced pressure until dryness. Soluble enzyme (2.5 IU, 83 μL), previously pretreated as reported before, was solubilized into NaHCO3 buffer (50 mM, pH 10, 7 mL) containing d-mannopyranoside (1% w/v) as additive. Then, the carrier (0.5 g) was added, and the suspension was allowed to stir at 4 °C for 24 h. The immobilized enzyme was then filtered, rinsed thoroughly with distilled H2O, and stored at 4 °C till use.
3.4.3. Immobilization of Cf-β-Man on Furan-2,5-Dicarbaldehyde-ReliZyme® (DFF-ReliZyme)
DFF-ReliZyme was prepared following a standard protocol [16]. ReliZyme™ EA112/S (1 g) was suspended in KH2PO4 buffer (25 mM, pH 7, 1.6 mL) and DFF (0.025 g) was added to the mixture. The mixture was allowed to stir at r.t. for 2 h. The supernatant was then removed, and the activated carrier was rinsed thoroughly with distilled H2O, and stored at 4 °C till use.
Immobilization of Cf-β-Man on DFF-ReliZyme was performed as reported before for GLX-AG (see Section 3.4.2).
3.4.4. Immobilization of Cf-β-Man on Furan-2,5-Dicarbaldehyde-Ethylendiamine-Agarose (DFF-EDA-AG)
DFF-EDA-AG was prepared, modifying a standard protocol [16]. First, GLX-AG (1 g) was aminated using EDA (2 M, pH 10, 5.7 mL) for 2 h at r.t. and subsequently reduced for 2 h at r.t. with NaBH4 (5.7 mg) [21]. The carrier was then filtered and rinsed thoroughly with distilled H2O. Subsequently, EDA-AG (1 g) was suspended in KH2PO4 buffer (25 mM, pH 7, 1.6 mL) and DFF (0.025 g) was added to the mixture. The mixture was allowed to stir at r.t. for 2 h. The supernatant was then removed, and the activated carrier was rinsed thoroughly with distilled H2O, and stored at 4 °C till use.
Immobilization of Cf-β-Man on DFF-EDA-AG was performed as reported before for GLX-AG (see Section 3.4.2)
3.4.5. Immobilization of Cf-β-Man on Cyanogen Bromide Agarose (CNBr-AG)
Commercial CNBr-agarose (0.25 g) was treated with HCl (1 mM, 50 mL) under mild mechanical stirring at r.t. for 1 h. The mixture was then filtered, washed with distilled H2O, and dried under vacuum.
Immobilization of Cf-β-Man on CNBr-AG was performed following a standard protocol [21]. Briefly, CNBr-AG was washed abundantly with KH2PO4 buffer (50 mM, pH 7.5) and then filtered under reduced pressure until dryness. Soluble enzyme (1.25 IU, 42 μL), previously pretreated as reported before, was solubilized into KH2PO4 buffer (50 mM, pH 7.5, 3.5 mL) containing d-mannopyranoside (1% w/v) as additive. Then, the carrier (0.25 g) was added, and the suspension was allowed to stir at 4 °C for 2 h. The immobilized enzyme was then filtered, rinsed thoroughly with distilled H2O, and stored at 4 °C till use.
3.4.6. Immobilization of Cf-β-Man on Sepabeads-Polyethylenimine (Sepabeads-PEI)
Sepabeads®-PEI and aldehyde dextran were prepared following a standard protocol [22]. Briefly, polyethylenimine (PEI, MW = 600 Da) (6.6 g) and NaCl (3.76 g) were dissolved in water (64.3 mL). The pH was adjusted to 11 using NaOH and then Sepabeads® EC-EP/S (5 g) was added. The reaction was carried out for 24 h at r.t. under mechanical stirring. The suspension was then filtered under reduced pressure and the carrier was first washed with NaCl (1 M, pH 11) and then with deionized H2O. The carrier was stored at 4 °C until use. Dextran (MW = 20 kDa) (1.67 g) was suspended in deionized H2O (50 mL) containing NaIO4 (0.435 g) to obtain an oxidation degree of 10% [22]. The reaction was carried out for 2 h at r.t. and then the solution was dialyzed (3500 Da molecular weight cut-off, MWCO) against deionized H2O.
Immobilization of Cf-β-Man on Sepabeads®-PEI was performed following a standard protocol [22]. Briefly, Sepabeads®-PEI was washed abundantly with KH2PO4 buffer (5 mM, pH 7.5) and then filtered under reduced pressure until dryness. The soluble enzyme (2.5 IU, 83 μL), previously pretreated as reported before, was solubilized into KH2PO4 buffer (5 mM, pH 7.5, 7 mL) with or without sucrose (1% w/v) as additive. Then, the carrier (0.5 g) was added, and the suspension was allowed to stir at 25 °C for 1 h (without sucrose as additive) and at 4 °C for 2 h (with sucrose as additive). Moreover, an additional cross-linking step with aldehyde dextran was performed on the derivative obtained with sucrose as additive. After 2 h, aldehyde dextran (0.7 mL) was added and allowed to stir for 1 additional hour at 4 °C.
The immobilized enzyme was then filtered, rinsed thoroughly with distilled H2O, and stored at 4 °C till use.
3.4.7. Immobilization of Cf-β-Man on Cobalt-Iminodiacetic Acid-Agarose (Co2+-IDA-AG)
Immobilization of Cf-β-Man on Co2+-IDA-AG was performed following supplier indications. Briefly, Co2+-IDA-AG was washed abundantly with KH2PO4 buffer (5 mM, pH 7.5) and then filtered under reduced pressure until dryness. Soluble enzyme (2.5 IU, 83 μL), previously pretreated as reported before, was solubilized into KH2PO4 buffer (5 mM, pH 7.5, 7 mL) containing d-mannopyranoside (1% w/v) as additive. Then, the carrier (0.5 g) was added, and the suspension was allowed to stir at 4 °C for 4 h. The final derivative was filtered, rinsed thoroughly with distilled H2O, and stored at 4 °C till use.
3.5. Synthesis of Cyanomethyl β-d-Mannopyranosyl-(1→6)-2-Acetamido-2-Deoxy-1-thio-β-d-Glucopyranoside (3d) and Identification
3.5.1. Transglycosylation Reaction
The donor pNP-β-Man (0.020 mmol) and the acceptor N-acetyl-d-glucosamine-SCH2CN (2d) (0.125 mmol) were solubilized in maleate buffer (100 mM, pH 6.5, 500 μL) at 37 °C. The reaction was started through the addition of 0.9 IU of soluble or immobilized enzyme. The reactions were maintained at 1000 rpm at 37 °C until exhaustion of the glycosyl donor (1) by TLC monitoring (CHCl3/MeOH, 5:1). The reactions were stopped by centrifugation for 10 min at 13,000 rpm at 25 °C with Microcon® filter (cutoff 10 kDa), and subsequently, the supernatants were dried under N2 flow.
3.5.2. Acetylation Reaction
The reaction crudes (9–15 mg, 1 eq., 0.1 M) were solubilized in dichloromethane (DCM). Acetic anhydride (6 eq.), Et3N (7 eq.) and DMAP (a spatula tip) were added to this solution. The reactions were incubated at r.t. under magnetic stirring for 6 h. The reaction mixtures were monitored in TLC (AcOEt/n-hexane, 7:3). At the endpoint, the mixtures were diluted with water and the aqueous phases were separated from the organic phase and extracted 3 times with DCM.
The organic phases were dried with anhydrous Na2SO4, filtered, and dried under vacuum. The samples were solubilized in 200 μL acetonitrile and analyzed by HPLC. Conversion was determined by using a calibration curve.
3.5.3. Scale-Up and Identification
The scale-up of the transglycosylation was performed in order to isolate and identify the product. The donor (1, 26 mg, 0.085 mmol) and the acceptor (2d, 138 mg, 0.500 mmol) were dissolved in maletae buffer (100 mM, pH 6.5, 2 mL) under magnetic stirring at 37 °C in a water bath. The reaction was started through the addition of the immobilized enzyme (5 IU). The reaction was maintained under magnetic stirring at 37 °C until complete exhaustion of the donor by TLC monitoring (CHCl3/MeOH, 5:1) (6 h). The reaction was stopped by centrifugation for 10 min at 13,000 rpm at 25 °C with Microcon® filter (cutoff 10 kDa) and subsequently dried under N2 flow. The crude was subsequently acetylated and purified by flash column chromatography (DCM/acetone, 8:2, Rf = 0.29).
The identity of the product (4d) was determined by ESI-MS (Figure S5) and complete NMR (Figures S6–S9) analysis.
MS: m/z = 713.67 [M+Na]+ (calculated 713.19).
1H NMR (CDCl3, 400 MHz): δ (ppm) 5.67 (d, J = 9.4 Hz, 1H), 5.33 (d, J = 3.4 Hz, 1H), 5.13 (t, J = 9.8 Hz, 1H), 5.03 (dd, J = 10.3, 9.0 Hz, 1H), 4.96 (dd, J = 9.9, 3.4 Hz, 1H), 4.65 (br s, 1H), 4.58 (d, J = 10.4 Hz, 1H), 4.39 (dd, J = 12.3, 2.5 Hz, 1H), 4.27 (dd, J = 12.3, 5.2 Hz, 1H), 4-17-4.01 (m, 4H), 3.81 (t, J = 9.4 Hz, 1H), 3.60 (m, J = 16.9 Hz, 1H), 3.21 (m, J = 17.0 Hz, 1H), 3.08 (q, J = 7.3 Hz, 1H), 2.08 (s, 3H), 2.06 (s, 3H), 2.05 (s, 3H), 2.03 (s, 3H), 1.98 (s, 3H), 1.92 (s, 3H), 1.90 (s, 3H).
13C NMR (CDCl3, 100 MHz): δ (ppm) 171.2, 170.5, 170.3, 169.9, 169.6, 169.1, 158.8, 116.1, 97.7, 83.2, 77.0, 74.5, 73.2, 72.7, 70.7, 68.3, 65.8, 62.3, 62.0, 52.5, 23.1, 20.8, 20.7, 20.7, 20.7, 20.6, 20.5, 14.6.
3.6. Calibration Curve of Cyanomethyl (2′,3′,4′,6′-Tetra-O-Acetyl-β-d-Mannopyranosyl)-(1→6)-2-Acetamido-3,4-di-O-Acetyl-2-Deoxy-1-thio-β-d-Glucopyranoside (4d)
Conversion was determined by using a calibration curve (Figure S10). The purified disaccharide produced in the scale-up reaction (5 mg) was solubilized in 300 μL of acetonitrile and further diluted to obtain the concentration interval of the calibration curve (5–25 μM). The samples were analyzed by HPLC.
3.7. Analytical Methods
3.7.1. LC-MS
LC-MS analyses were carried out on a Waters X-Bridge Amide column, 2.5 μm × 150 mm × 3 mm, using acetonitrile + 0.1% formic acid (A) and H2O + 0.1% formic acid (B) as mobile phases. The elution program entailed 2 min of isocratic condition at 15% B, followed by a linear gradient from 15% to 35% B in 18 min, and a wash step at 50% B for 1 min. The column was thermostated at 50 °C, flow rate was set at 0.35 mL/min, and the injection volume was 2 μL. The following MS parameters were applied: positive ion mode, scan range 150–1000 m/z in full scan mode, source voltage 4.6 kV, capillary voltage 30 V, sheath gas flow rate 10 (arbitrary units), auxiliary gas flow rate 4 (arbitrary units), capillary temperature 250 °C, and tube lens voltage 80 V.
3.7.2. HPLC
HPLC analyses were carried out on a SepaChrom Adamas C18-Extreme 5 μm, 250 mm × 4.6 mm column using KH2PO4 10 mM pH 4.0 (A), and acetonitrile (B) as mobile phases. The elution program was set as follows: 0 → 10 min 100% A-0% B (isocratic), 10.1 → 15 min 90% A-10% B (linear gradient), 15.1 → 30 min 80% A-20% B (linear gradient), 30.1 → 35 min 80% A-20% B (isocratic), 35.1 → 45 min 100% A-10% B (isocratic), flow rate 1 mL/min, and injection volume = 10 μL, λ = 220 nm.
3.8. Building Block Synthesis
Cyanomethyl 1-thio-α-d-mannopyranoside (2b)
Cyanomethyl 1-thio-α-d-mannopyranoside (2b) was synthesized as depicted in Scheme S1 following standard protocols [18,23].
1-Chloro-2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside (5b)
Briefly, titanium tetrachloride (1.1 mL, 16.77 mmol) was added to a solution of commercial α-d-mannose pentaacetate (5a, 5 g, 12.82 mmol) in anhydrous 1,2-dichloroethane (30 mL) at 35 °C, and the mixture was stirred under nitrogen overnight. The reaction was monitored by TLC (dichloromethane/methanol, 95:5), Rf = 0.64. The solution was diluted with dichloromethane and poured into ice-cold water (50 mL). The aqueous phase was extracted 3 times with dichloromethane and the organic layers were combined and washed with saturated sodium bicarbonate, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude solid was purified by flash chromatography (ethyl acetate/hexane, 1:1) to give 5b with a yield of 90%.
2-S-(2,3,4,6-Tetra-O-acetyl-α-d-mannopyranosyl)-2-thiopseudourea hydrochloride (5c)
Chloro-2,3,4,6-tetra-O-acetyl-α-d-mannopyranoside (5b, 4.2 g, 11.48 mmol) and thiourea (1.9 g, 25 mmol) were dissolved in anhydrous acetonitrile (20 mL) under an atmosphere of N2 in the presence of activated molecular sieves 4 Å. The mixture was refluxed for 6 h at 80 °C, batch cooled to room temperature, and crystallised (5c), as a white crystalline solid, with the aid of petrol as anti-solvent with a yield of 80%.
Cyanomethyl 2,3,4,6-tetra-O-acetyl-1-thio-α-d-mannopyranoside (5d)
Tetra-O-acetyl-α-d-mannopyranosyl-2-thiopseudourea hydrochloride (5c, 4.06 g, 9.19 mmol) was dissolved in 1:1 water:acetone mixture (40 mL) and sodium metabisulphite (3.44 g, 18.11 mmol), potassium carbonate (1.5 g, 10.84 mmol), and chloroacetonitrile (11.4 mL, 20 eq) were added. The mixture was stirred at room temperature and the reaction was monitored by TLC (ethyl acetate/hexane, 1:1), Rf = 0.46. Upon completion, 130 mL of ice water were added to the solution that was stirred for 2 h. The reaction was extracted with dichloromethane and the combined organics extracts were washed with brine and dried over Na2SO4 and concentrated in vacuo. Crystallization from hot methanol affording 5d, as a white crystalline foamy solid, with a yield of 60%. 1H-NMR was in agreement with that previously reported [23].
Cyanomethyl 1-thio-α-d-mannopyranoside (2b)
Cyanomethyl-2,3,4,6-tetra-O-acetyl-1-thio-α-d-mannopyranoside (5d) (1.0 g, 2.48 mmol) was solubilized in anhydrous MeOH (50 mL) in atmosphere of N2 at r.t. Then, MeONa (0.005 g, 0.100 mmol) was added. The reaction mixture was kept under magnetic stirring for 6 h at r.t. Deacetylation was monitored in TLC (AcOEt/MeOH, 9:1, Rf = 0.13) and ESI-MS until deacetylation was complete. At the end of the reaction, the product (2d) was obtained as a white solid by drying under vacuum. The product was characterized by ESI-MS.
Cyanomethyl 2-acetamido -2-deoxy-1-thio-β-d-glucopyranoside (2d)
Cyanomethyl 2-acetamido -2-deoxy-1-thio-β-d-glucopyranoside (2d) was synthesized as depicted in Scheme S1 following standard protocols [18,23].
1-Chloro-2-acetamido-3,4,6,-tri-O-acetyl-2-deoxy-α-d-glucopyranoside (6b)
N-Acetyl glucosamine (6a, 2 g, 9.041 mmol) was suspended in acetyl chloride (20 mL) in the presence of activated molecular sieves 4 Å, and was stirred for 48 h at 30 °C and the reaction was monitored by TLC (dichloromethane/methanol, 95:5). The solution was diluted with dichloromethane and poured into ice-cold water (25 mL). The aqueous phase was extracted 3 times with dichloromethane and the organic layers were combined and washed with saturated sodium bicarbonate, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude solid was purified by flash chromatography (ethyl acetate/hexane, 7:3) and gave a white solid of 6b with a yield of 68%. Rf = 0.71 (dichloromethane/methanol, 95:5).
1-Thiourea-2-acetamido-3,4,6,-tri-O-acetyl-2-deoxy-α-d-glucopyranoside (6c)
1-Chloro-2-acetamido-3,4,6,-tri-O-acetyl-2-deoxy-α-d-glucopyranoside (6b, 1 g, 2.734 mmol) and thiourea (0.38 g, 5 mmol) were dissolved in anhydrous acetone (10 mL) under an atmosphere of N2 in the presence of activated molecular sieves 4 Å. The mixture was refluxed for 3 h at room temperature. A white precipitate was formed and compound 6c was isolated by filtration, washed with ethanol, and concentrated in vacuo gave pure product with a yield of 90%.
Cyanomethyl 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-1-thio-β-d-glucopyranoside (6d)
1-Thiourea-2-acetamido-3,4,6,-tri-O-acetyl-2-deoxy-α-d-glucopyranoside (6c, 0.245 g, 0.554 mmol) was dissolved in 1:1 water:acetone mixture (2.6 mL) and sodium metabisulphite (0.212 g, 1.115 mmol), potassium carbonate (0.093 g, 0.672 mmol), and chloroacetonitrile (0.712 mL, 20 eq.) were added. The mixture was stirred at room temperature and the reaction was monitored by TLC. Upon completion, 8 mL of ice water was added to the solution that was stirred for 45 min. The reaction was extracted with dichloromethane and the combined organics extracts were washed with brine and dried over anhydrous Na2SO4 and concentrated in vacuo, affording 6d with a yield of 98%. Rf = 0.71 (dichloromethane/methanol, 95:5, v/v).
Cyanomethyl 2-acetamido-2-deoxy-1-thio-β-d-glucopyranoside (2d)
Cyanomethyl 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-1-thio-β-d-glucopyranoside (6d) (1.0 g, 2486 mmol) was solubilized in anhydrous MeOH (50 mL), in atmosphere of N2 at r.t., then, MeONa (0.005 g, 0.100 mmol) was added. The reaction mixture was kept under magnetic stirring for 6 h at r.t. Deacetylation was monitored in TLC (AcOEt/MeOH, 9:1, Rf = 0.13) and ESI-MS until deacetylation was complete. At the end of the reaction, the product (2d) was obtained as a white solid by drying under vacuum. The product was characterized by ESI-MS.
4. Conclusions
In this work, β-mannosidase from Cellulomonas fimi was immobilized for the first time, and it was employed in the synthesis of β-mannosides. In the enzymatic transglycosylation reaction, different free monosaccharides (d-mannose 2a, d-mannose-SCH2CN 2b, N-acetyl-d-glucosamine 2c, and N-acetyl-d-glucosamine-SCH2CN 2d) were used as acceptors and para-nitrophenyl-β-mannoside (1) as a donor. Only the compound 2d allowed for the formation of one regioisomer disaccharide 3d functionalized with a thiocyanomethyl group at the C1 position useful for preparation of glycoproteins. The use of Cf-β-Man immobilized on IDA-Co2+-agarose allows the synthesis of 4d with a higher conversion compared to the soluble enzyme (20% vs. 5%) after 6 h. This explorative work opens the door to new studies concerning the design of engineered Cf-β-Man mutants and their immobilization in order to obtain a robust and recyclable biocatalyst for large-scale applications.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13111399/s1, Figure S1: LC-MS chromatograms: acceptor screening; Figure S2: LC-MS monitoring transglycosylation in 100 mM maleate buffer pH 6.5; Figure S3: Co-solvents screening; Figure S4: Transglycosylation reaction catalyzed by immobilized Cf-β-Man (A) and soluble Cf-β-Man (B); Figure S5: ESI-MS spectrum of 4d; Figure S6: 1H NMR spectrum of 4d; Figure S7: 13C NMR spectrum of 4d; Figure S8: COSY spectrum of 4d; Figure S9: TOCSY spectrum of 4d; Figure S10: HPLC calibration curve of 4d; Scheme S1: Synthesis of cyanomethyl 1-thio-α-d-mannopyranoside (2b) and β-d-N-acetylglucosamine (2d).
Author Contributions
Conceptualization, T.B.; investigation M.S.R. (immobilization study, transglycosylation reaction and isolation of product 4d), S.T. (study of transglycosylation reaction conditions by LC-MS) and M.R. (identification of product 4d by NMR spectroscopy); writing—original draft preparation, M.S.R. and T.B.; writing—review and editing, G.S. and T.B.; supervision, G.S. (identification of product 4d) and T.B.; funding acquisition, M.T. All co-authors participated equally and substantially to the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Italian Ministry of Health (Project Immunoterapia: cura e prevenzione di malattie infettive e tumorali (Immuno-HUB), project number T4-CN-02).
Data Availability Statement
Data is contained within the article or Supplementary Materials.
Acknowledgments
Resindion S.r.l. (Binasco, Italy) is gratefully acknowledged for the supply of SepabeadsTM EC-EP.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Varki, A. Biological Roles of Glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed]
- Shivatare, S.S.; Shivatare, V.S.; Wong, C.-H. Glycoconjugates: Synthesis, Functional Studies, and Therapeutic Developments. Chem. Rev. 2022, 122, 15603–15671. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in Health and Disease. Nat. Rev. Nephrol. 2019, 15, 347–366. [Google Scholar] [CrossRef] [PubMed]
- Gridley, J.J.; Osborn, H.M.I. Recent Advances in the Construction of β-D-Mannose and β-D-Mannosamine Linkages. J. Chem. Soc. Perkin Transform. 2000, 10, 1471–1491. [Google Scholar] [CrossRef]
- El Ashry, E.S.H.; Rashed, N.; Ibrahim, E.S.I. Strategies of Synthetic Methodologies for Constructing β-Mannosidic Linkage. Curr. Org. Synth. 2005, 2, 175–213. [Google Scholar] [CrossRef]
- Malgas, S.; van Dyk, J.S.; Pletschke, B.I. A Review of the Enzymatic Hydrolysis of Mannans and Synergistic Interactions between β-Mannanase, β-Mannosidase and α-Galactosidase. World J. Microbiol. Biotechnol. 2015, 31, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Karav, S.; Cohen, J.L.; Barile, D.; Nobrega de Moura Bell, J.M.L. Recent Advances in Immobilization Strategies for Glycosidases. Biotechnol. Prog. 2017, 33, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Sadaqat, B.; Sha, C.; Ahmad Dar, M.; Dhanavade, M.J.; Sonawane, K.D.; Mohamed, H.; Shao, W.; Song, Y. Modifying Thermostability and Reusability of Hyperthermophilic Mannanase by Immobilization on Glutaraldehyde Cross-Linked Chitosan Beads. Biomolecules 2022, 12, 999. [Google Scholar] [CrossRef]
- Panwar, D.; Kaira, G.S.; Kapoor, M. Cross-Linked Enzyme Aggregates (CLEAs) and Magnetic Nanocomposite Grafted CLEAs of GH26 Endo-β-1,4-Mannanase: Improved Activity, Stability and Reusability. Int. J. Biol. Macromol. 2017, 105, 1289–1299. [Google Scholar] [CrossRef]
- Dhiman, S.; Srivastava, B.; Singh, G.; Khatri, M.; Arya, S.K. Immobilization of Mannanase on Sodium Alginate-Grafted-β-Cyclodextrin: An Easy and Cost Effective Approach for the Improvement of Enzyme Properties. Int. J. Biol. Macromol. 2020, 156, 1347–1358. [Google Scholar] [CrossRef]
- Behera, S.; Dev, M.J.; Singhal, R.S. Cross-Linked β-Mannanase Aggregates: Preparation, Characterization, and Application for Producing Partially Hydrolyzed Guar Gum. Appl. Biochem. Biotechnol. 2022, 194, 1981–2004. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, M.Y.; El-Assar, S.A.; Youssef, A.S.; El-Sersy, N.A.; Beltagy, E.A. Extracellular β-Mannanase Production by the Immobilization of the Locally Isolated Aspergillus niger. Int. J. Agric. Biol. 2006, 8, 57–62. [Google Scholar]
- Hoyos, P.; Bavaro, T.; Perona, A.; Rumbero, A.; Tengattini, S.; Terreni, M.; Hernáiz, M.J. Highly Efficient and Sustainable Synthesis of Neoglycoproteins Using Galactosidases. ACS Sustain. Chem. Eng. 2020, 8, 6282–6292. [Google Scholar] [CrossRef]
- Benítez-Mateos, A.I.; Contente, M.L. Agarose vs. Methacrylate as Material Supports for Enzyme Immobilization and Continuous Processing. Catalysts 2021, 11, 814. [Google Scholar] [CrossRef]
- Zucca, P.; Fernandez-Lafuente, R.; Sanjust, E. Agarose and Its Derivatives as Supports for Enzyme Immobilization. Molecules 2016, 21, 1577. [Google Scholar] [CrossRef] [PubMed]
- Danielli, C.; van Langen, L.; Boes, D.; Asaro, F.; Anselmi, S.; Provenza, F.; Renzi, M.; Gardossi, L. 2,5-Furandicarboxaldehyde as a Bio-Based Crosslinking Agent Replacing Glutaraldehyde for Covalent Enzyme Immobilization. RSC Adv. 2022, 12, 35676. [Google Scholar] [CrossRef] [PubMed]
- Bubb, W.A. NMR Spectroscopy in the Study of Carbohydrates: Characterizing the Structural Complexity. Concepts Magn. Reson. 2003, 19, 1–19. [Google Scholar] [CrossRef]
- Tanzi, L.; Robescu, M.S.; Marzatico, S.; Recca, T.; Zhang, Y.; Terreni, M.; Bavaro, T. Developing a Library of Mannose-Based Mono- and Disaccharides: A General Chemoenzymatic Approach to Monohydroxylated Building Blocks. Molecules 2020, 25, 5764. [Google Scholar] [CrossRef]
- Nashiru, O.; Zechel, D.L.; Stoll, D.; Mohammadzadeh, T.; Warren, R.A.J.; Withers, S.G. β-Mannosynthase: Synthesis of β-Mannosides with a Mutant β-Mannosidase. Angew. Chem. Int. Ed. 2001, 40, 417–420. [Google Scholar] [CrossRef]
- Semproli, R.; Robescu, M.S.; Sangiorgio, S.; Pargoletti, E.; Bavaro, T.; Rabuffetti, M.; Cappelletti, G.; Speranza, G.; Ubiali, D. From Lactose to Alkyl Galactoside Fatty Acid Esters as Non-ionic Biosurfactants: A Two-step Enzymatic Approach to Cheese Whey Valorization. ChemPlusChem 2023, 88, e202200331. [Google Scholar] [CrossRef]
- Bruni, M.; Robescu, M.S.; Ubiali, D.; Marrubini, G.; Vanna, R.; Morasso, C.; Benucci, I.; Speranza, G.; Bavaro, T. Immobilization of γ-Glutamyl Transpeptidase from Equine Kidney for the Synthesis of Kokumi Compounds. ChemCatChem 2020, 12, 210–218. [Google Scholar] [CrossRef]
- Robescu, M.S.; Serra, I.; Terreni, M.; Ubiali, D.; Bavaro, T. A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine 5′-Monophosphate. Catalysts 2020, 10, 60. [Google Scholar] [CrossRef]
- Bavaro, T.; Filice, M.; Temporini, C.; Tengattini, S.; Serra, I.; Morelli, C.F.; Massolini, G.; Terreni, M. Chemoenzymatic Synthesis of Neoglycoproteins Driven by the Assessment of Protein Surface Reactivity. RSC Adv. 2014, 4, 56455–56466. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).