
Glycine Substitution of Residues with Unfavored Dihedral Angles Improves Protein Thermostability


 ,
, 
Abstract
:
1. Introduction
2. Results
2.1. Predicting of Thermostable Mutants
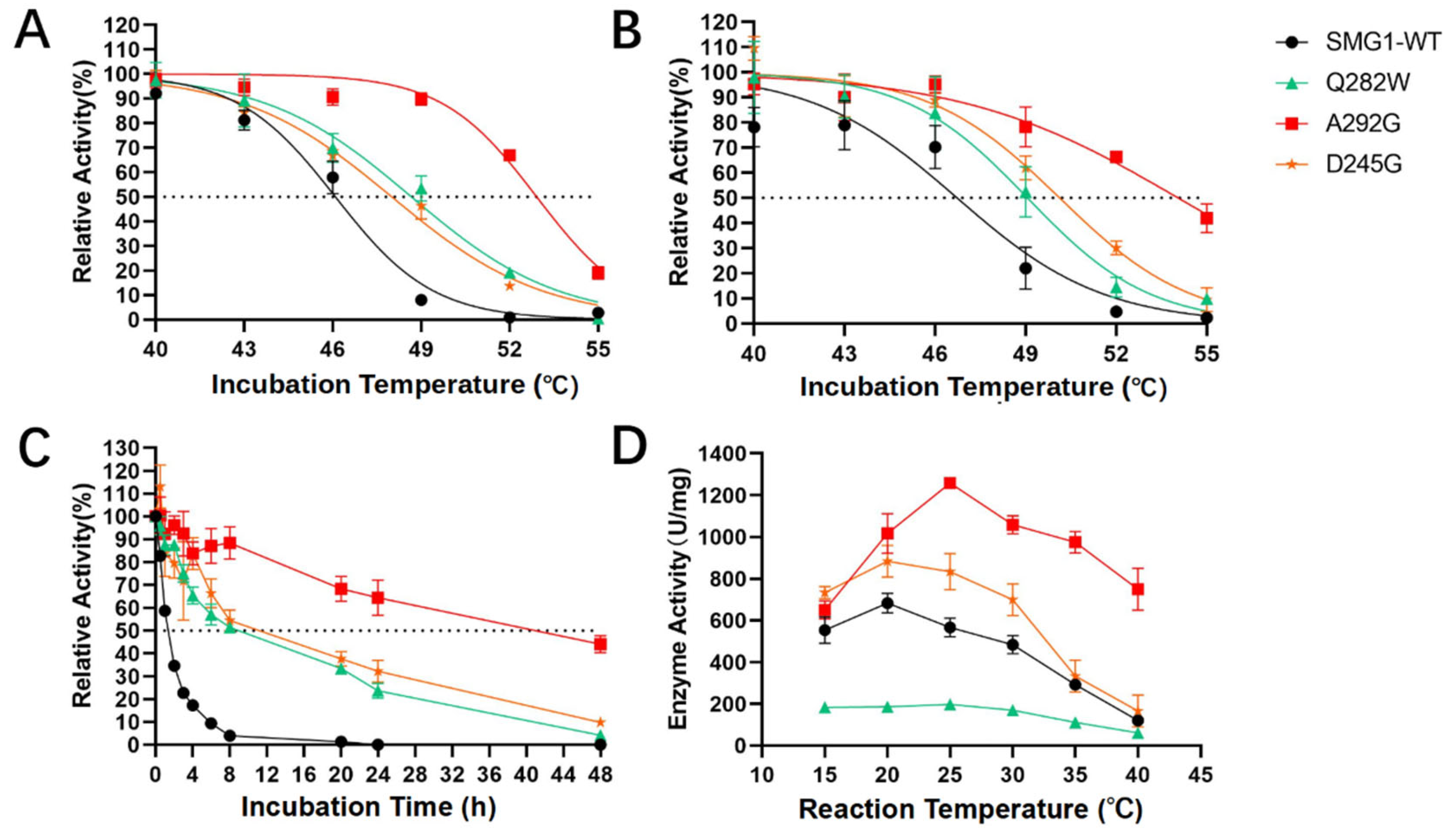
2.2. Characterization of Thermostability-Enhanced Mutants
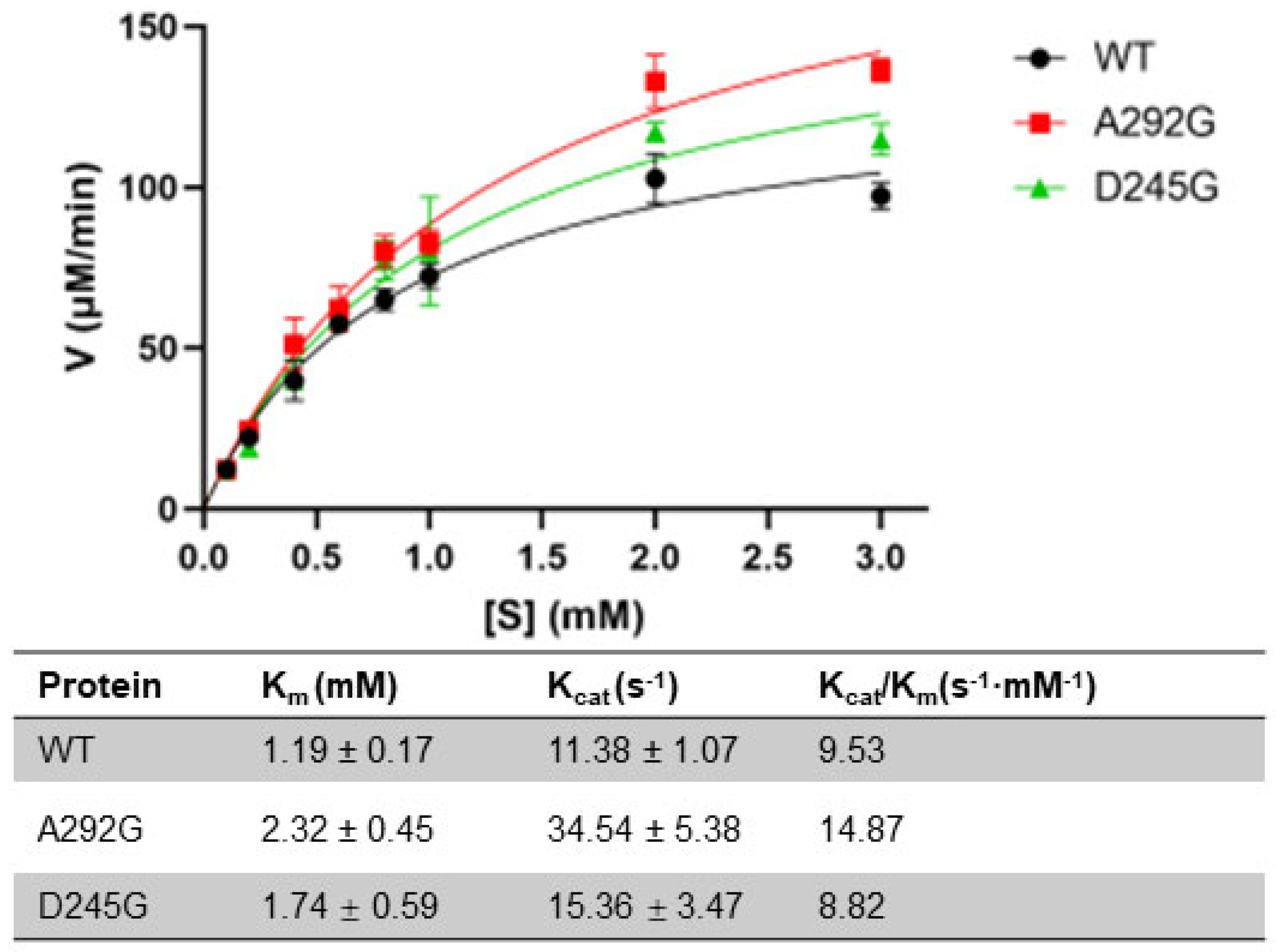
2.3. Enzyme Kinetics of the WT and Mutants
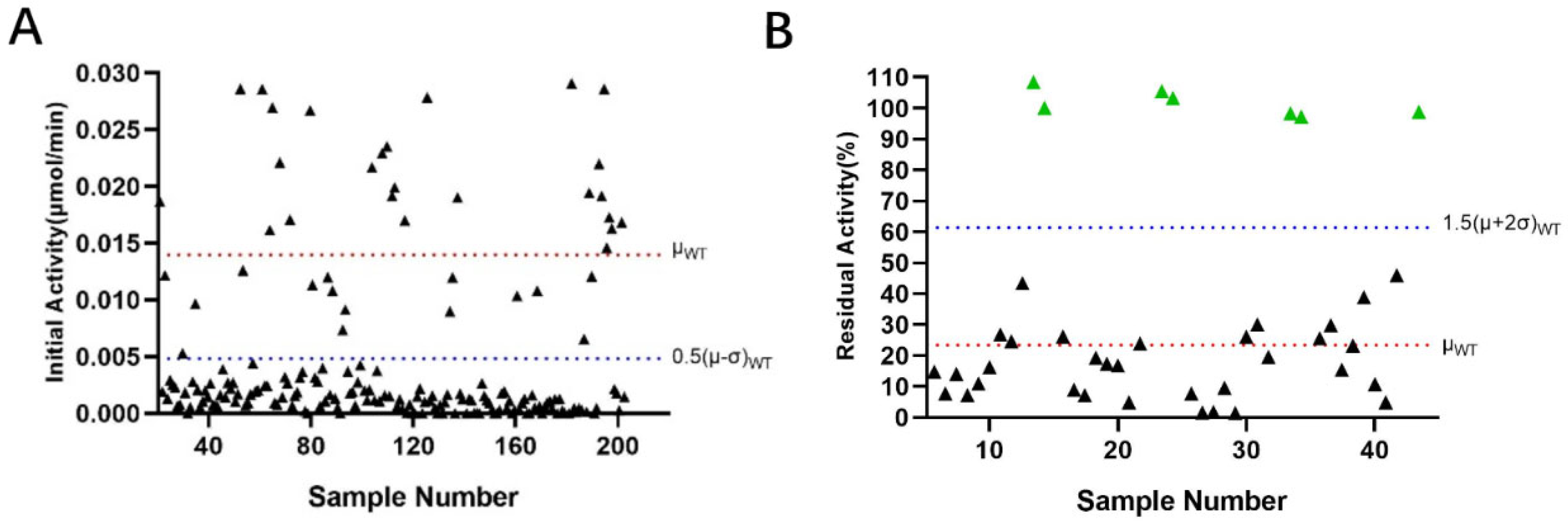
2.4. Site-Directed Saturation Library Analysis
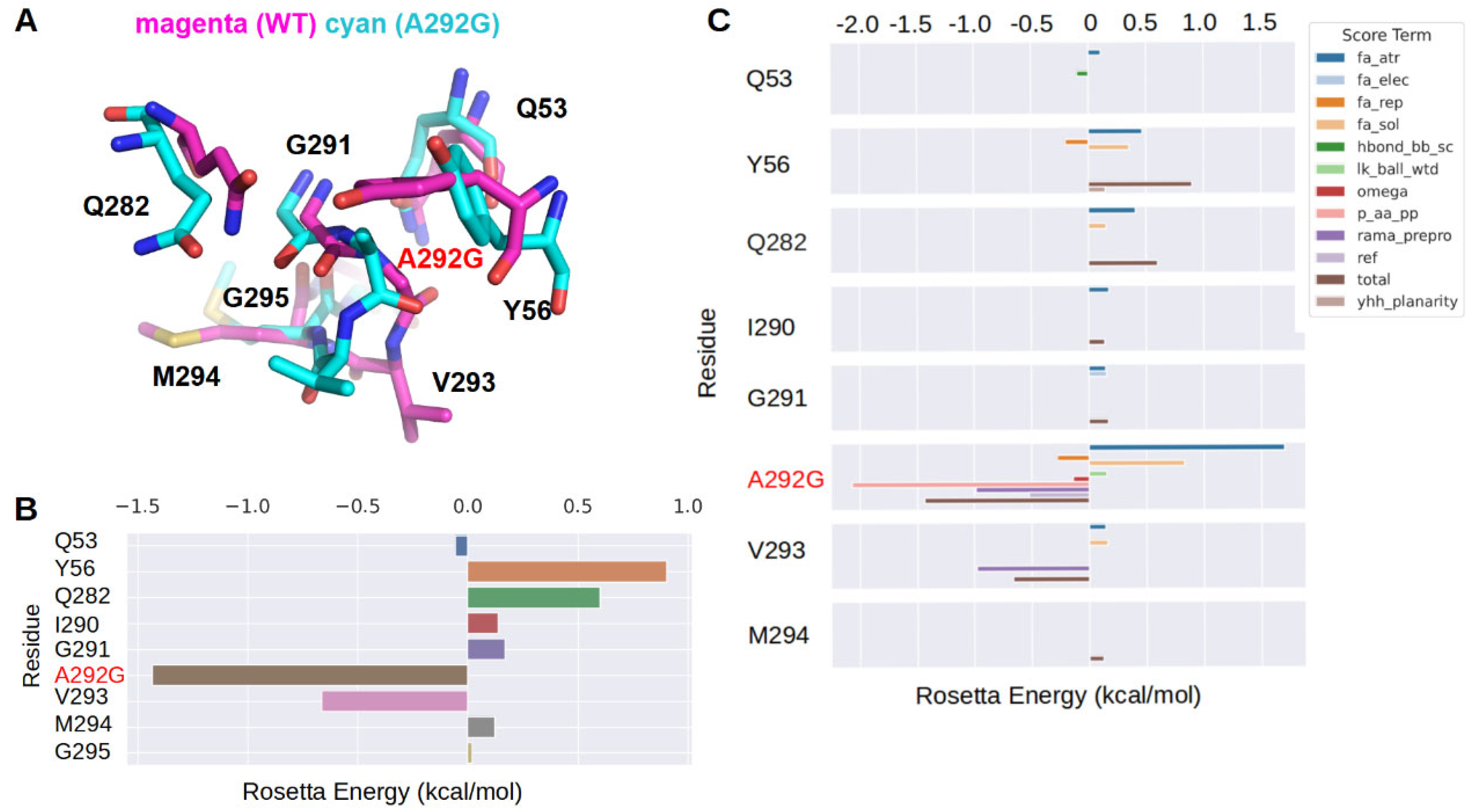
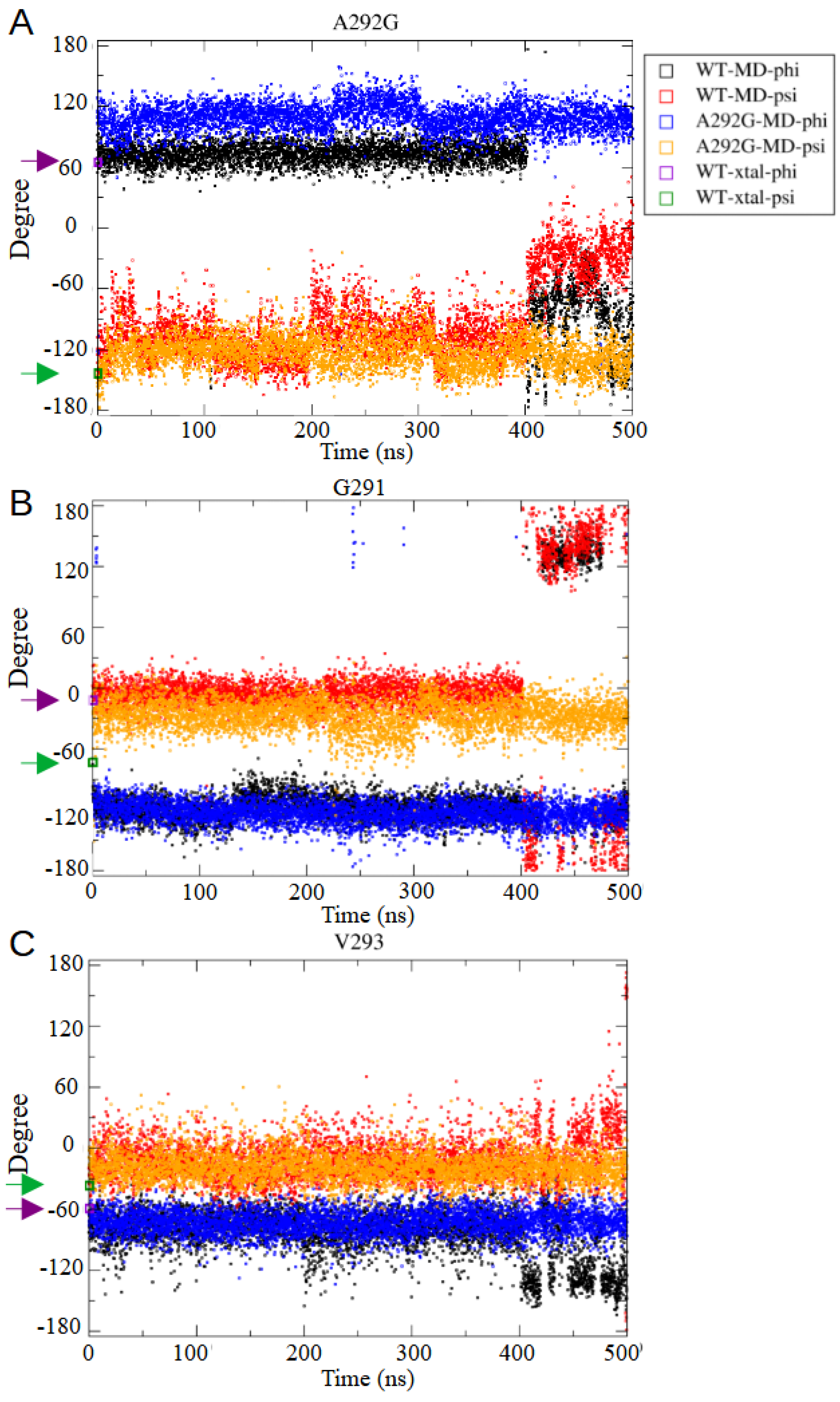
2.5. Molecular Mechanism for Thermostability Enhancement
3. Discussion
4. Materials and Methods
4.1. In Silico Screening of Thermostable Mutations
4.2. Protein Expression and Purification
4.3. Crude Lysate Preparation
4.4. Determination of T5015 Using 4-Nitrophenyl Caprylate
4.5. The Half-Life and Optimal Reaction Temperature of the SMG1 WT and Mutants
4.6. Determination of Melting Temperature from the Thermal Shift Assay
4.7. Enzyme Kinetic Measurement
4.8. Site-Directed Saturation Mutagenesis and Screening
4.9. Protein Preparation for Modeling
4.10. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chandra, P.; Singh, R.; Arora, P.K. Microbial lipases and their industrial applications: A comprehensive review. Microb. Cell Factories 2020, 19, 1–42. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, Y.M.; Saunders, C.W.; Johnstone, K.R.; Reeder, N.L.; Coleman, C.G.; Kaczvinsky, J.R., Jr.; Gale, C.; Walter, R.; Mekel, M.; Lacey, M.P.; et al. Isolation and expression of a Malassezia globosa lipase gene, LIP1. J. Investig. Dermatol. 2007, 127, 2138–2146. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.F.; Li, T.; Ning, Z.X.; Wang, Y.H.; Yang, B.; Yang, X.Q. Production of extremely pure diacylglycerol from soybean oil by lipase-catalyzed glycerolysis. Enzym. Microb. Technol. 2011, 49, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.J.; Yang, Z.; Guan, F.F.; Zhang, S.S.; Cui, D.; Guan, G.H.; Li, Y. A novel mono- and diacylglycerol lipase highly expressed in Pichia pastoris and its application for food emulsifier preparation. Process Biochem. 2013, 48, 1899–1904. [Google Scholar] [CrossRef]
- Li, D.; Faiza, M.; Ali, S.; Wang, W.; Tan, C.P.; Yang, B.; Wang, Y. Highly Efficient Deacidification of High-Acid Rice Bran Oil Using Methanol as a Novel Acyl Acceptor. Appl. Biochem. Biotechnol. 2018, 184, 1061–1072. [Google Scholar] [CrossRef]
- Xing, Y.-N.; Tan, J.; Wang, Y.; Wang, J. Enhancing the thermostability of a mono-and diacylglycerol lipase from Malassizia globose by stabilizing a flexible loop in the catalytic pocket. Enzym. Microb. Technol. 2021, 149, 109849. [Google Scholar] [CrossRef]
- Xu, T.; Liu, L.; Hou, S.; Xu, J.; Yang, B.; Wang, Y.; Liu, J. Crystal structure of a mono- and diacylglycerol lipase from Malassezia globosa reveals a novel lid conformation and insights into the substrate specificity. J. Struct. Biol. 2012, 178, 363–369. [Google Scholar] [CrossRef]
- Guo, S.; Xu, J.; Pavlidis, I.V.; Lan, D.; Bornscheuer, U.T.; Liu, J.; Wang, Y. Structure of product-bound SMG1 lipase: Active site gating implications. FEBS J. 2015, 282, 4538–4547. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Cao, H.; Wang, J.; He, L.; Qi, Y.; Zhang, J.Z. DeepDDG: Predicting the Stability Change of Protein Point Mutations Using Neural Networks. J. Chem. Inf. Modeling 2019, 59, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinform. 2006, 62, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33 (Suppl. 2), W306–W310. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, E.H.; Leaver-Fay, A.; Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins Struct. Funct. Bioinform. 2011, 79, 830–838. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33 (Suppl. 2), W382–W388. [Google Scholar] [CrossRef]
- Sumbalova, L.; Stourac, J.; Martinek, T.; Bednar, D.; Damborsky, J. HotSpot Wizard 3.0: Web server for automated design of mutations and smart libraries based on sequence input information. Nucleic Acids Res. 2018, 46, W356–W362. [Google Scholar] [CrossRef]
- Bednar, D.; Beerens, K.; Sebestova, E.; Bendl, J.; Khare, S.; Chaloupkova, R.; Prokop, Z.; Brezovsky, J.; Baker, D.; Damborsky, J. FireProt: Energy-and evolution-based computational design of thermostable multiple-point mutants. PLoS Comput. Biol. 2015, 11, e1004556. [Google Scholar] [CrossRef]
- Weinstein, J.J.; Goldenzweig, A.; Hoch, S.; Fleishman, S.J. PROSS 2: A new server for the design of stable and highly expressed protein variants. Bioinformatics 2021, 37, 123–125. [Google Scholar] [CrossRef]
- Huang, P.; Chu, S.K.S.; Frizzo, H.N.; Connolly, M.P.; Caster, R.W.; Siegel, J.B. Evaluating Protein Engineering Thermostability Prediction Tools Using an Independently Generated Dataset. ACS Omega 2020, 5, 6487–6493. [Google Scholar] [CrossRef]
- Stites, W.E.; Meeker, A.K.; Shortle, D. Evidence for strained interactions between side-chains and the polypeptide backbone. J. Mol. Biol. 1994, 235, 27–32. [Google Scholar] [CrossRef]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef] [PubMed]
- Bendl, J.; Stourac, J.; Sebestova, E.; Vavra, O.; Musil, M.; Brezovsky, J.; Damborsky, J. HotSpot Wizard 2.0: Automated design of site-specific mutations and smart libraries in protein engineering. Nucleic Acids Res. 2016, 44, W479–W487. [Google Scholar] [CrossRef] [PubMed]
- Rathi, P.C.; Fulton, A.; Jaeger, K.E.; Gohlke, H. Application of Rigidity Theory to the Thermostabilization of Lipase A from Bacillus subtilis. PLoS Comput. Biol. 2016, 12, e1004754. [Google Scholar] [CrossRef] [PubMed]
- Contreras, F.; Nutschel, C.; Beust, L.; Davari, M.D.; Gohlke, H.; Schwaneberg, U. Can constraint network analysis guide the identification phase of KnowVolution? A case study on improved thermostability of an endo-β-glucanase. Comput. Struct. Biotechnol. J. 2021, 19, 743–751. [Google Scholar] [CrossRef]
- Frenz, B.; Lewis, S.M.; King, I.; DiMaio, F.; Park, H.; Song, Y. Prediction of protein mutational free energy: Benchmark and sampling improvements increase classification accuracy. Front. Bioeng. Biotechnol. 2020, 8, 1175. [Google Scholar] [CrossRef]
- Pancotti, C.; Benevenuta, S.; Birolo, G.; Alberini, V.; Repetto, V.; Sanavia, T.; Capriotti, E.; Fariselli, P. Predicting protein stability changes upon single-point mutation: A thorough comparison of the available tools on a new dataset. Brief. Bioinform. 2022, 23, bbab555. [Google Scholar] [CrossRef]
- Masumoto, K.; Ueda, T.; Motoshima, H.; Imoto, T. Relationship between local structure and stability in hen egg white lysozyme mutant with alanine substituted for glycine. Protein Eng. 2000, 13, 691–695. [Google Scholar] [CrossRef]
- Trevino, S.R.; Schaefer, S.; Scholtz, J.M.; Pace, C.N. Increasing protein conformational stability by optimizing β-turn sequence. J. Mol. Biol. 2007, 373, 211–218. [Google Scholar] [CrossRef]
- Stojanoski, V.; Adamski, C.J.; Hu, L.; Mehta, S.C.; Sankaran, B.; Zwart, P.; Prasad, B.V.; Palzkill, T. Removal of the side chain at the active-site serine by a glycine substitution increases the stability of a wide range of serine β-lactamases by relieving steric strain. Biochemistry 2016, 55, 2479–2490. [Google Scholar] [CrossRef]
- Lan, D.; Zhao, G.; Holzmann, N.; Yuan, S.; Wang, J.; Wang, Y. Structure-Guided Rational Design of a Mono- and Diacylglycerol Lipase from Aspergillus oryzae: A Single Residue Mutant Increases the Hydrolysis Ability. J. Agric. Food Chem. 2021, 69, 5344–5352. [Google Scholar] [CrossRef]
- Osuna, S.; Jimenez-Oses, G.; Noey, E.L.; Houk, K. Molecular dynamics explorations of active site structure in designed and evolved enzymes. Acc. Chem. Res. 2015, 48, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT or Mutants | Half-Life (h) | Optimal Reaction Temperature (°C) | Enzymatic Activity toward DAG at Optimal Reaction Temperature (U/mg) | T5015 (°C, Substrate 4-Nitrophenyl Caprylate) | T5015 (°C, Substrate DAG) | Tm |
|---|---|---|---|---|---|---|
| WT | 1.7 | 20 | 683.3 | 46.23 | 46.73 | 51.34 |
| D245G | 15.4 | 20 | 883.3 | 48.01 (+1.8) * | 50.13 (+3.4) | 60.05 (+8.71) |
| Q282W | 12.5 | 25 | 197.2 | 48.67 (+2.5) | 49.06 (+2.4) | ND |
| A292G | 39.9 | 25 | 1258.3 | 52.85 (+6.7) | 54.40 (+7.7) | 61.70 (+10.36) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Z.; Zhong, Q.; Li, J.; Zhou, B.; Xing, Y.-N.; Liu, K.; Cao, K.; Lan, D.; Zhou, T.; Wang, Y.; et al. Glycine Substitution of Residues with Unfavored Dihedral Angles Improves Protein Thermostability. Catalysts 2022, 12, 898. https://doi.org/10.3390/catal12080898
Lu Z, Zhong Q, Li J, Zhou B, Xing Y-N, Liu K, Cao K, Lan D, Zhou T, Wang Y, et al. Glycine Substitution of Residues with Unfavored Dihedral Angles Improves Protein Thermostability. Catalysts. 2022; 12(8):898. https://doi.org/10.3390/catal12080898
Chicago/Turabian StyleLu, Zhili, Qiaoxian Zhong, Jingxian Li, Bingjie Zhou, Yan-Ni Xing, Kaien Liu, Kexin Cao, Dongming Lan, Teng Zhou, Yonghua Wang, and et al. 2022. "Glycine Substitution of Residues with Unfavored Dihedral Angles Improves Protein Thermostability" Catalysts 12, no. 8: 898. https://doi.org/10.3390/catal12080898
APA StyleLu, Z., Zhong, Q., Li, J., Zhou, B., Xing, Y.-N., Liu, K., Cao, K., Lan, D., Zhou, T., Wang, Y., & Wang, J. (2022). Glycine Substitution of Residues with Unfavored Dihedral Angles Improves Protein Thermostability. Catalysts, 12(8), 898. https://doi.org/10.3390/catal12080898