
2D Nanomaterial—Based Electrocatalyst for Water Soluble Hydroperoxide Reduction



{kind=link}
{kind=link}
{kind=link}
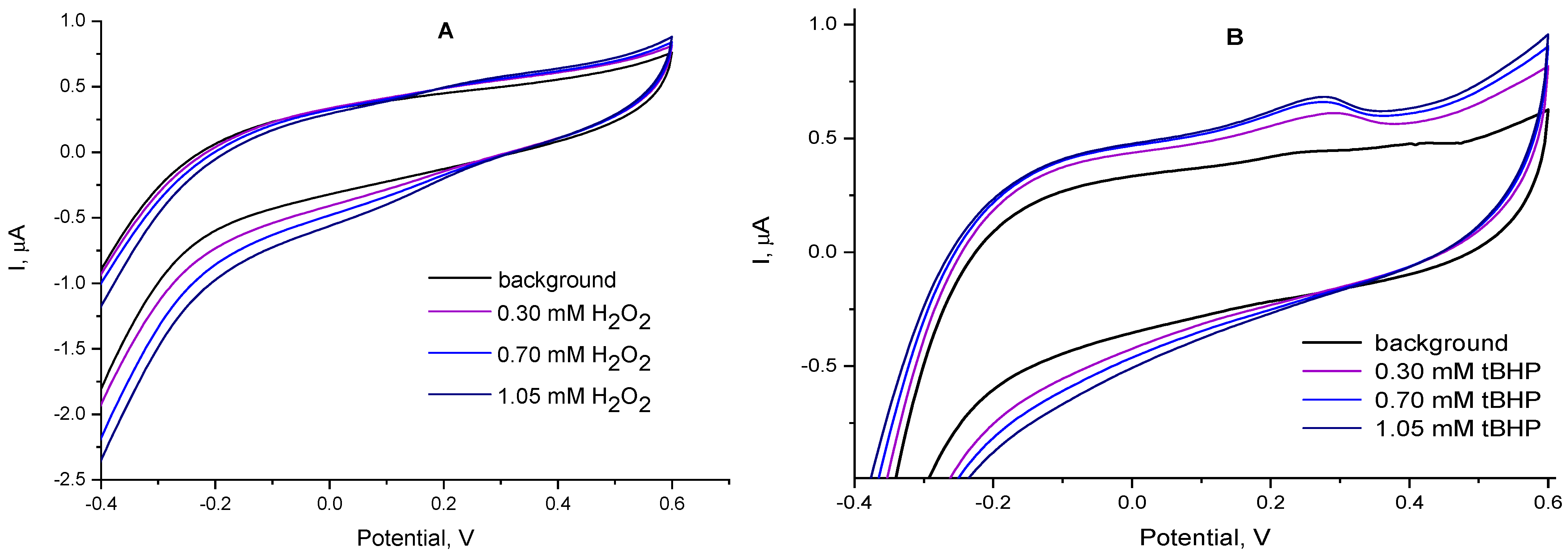{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
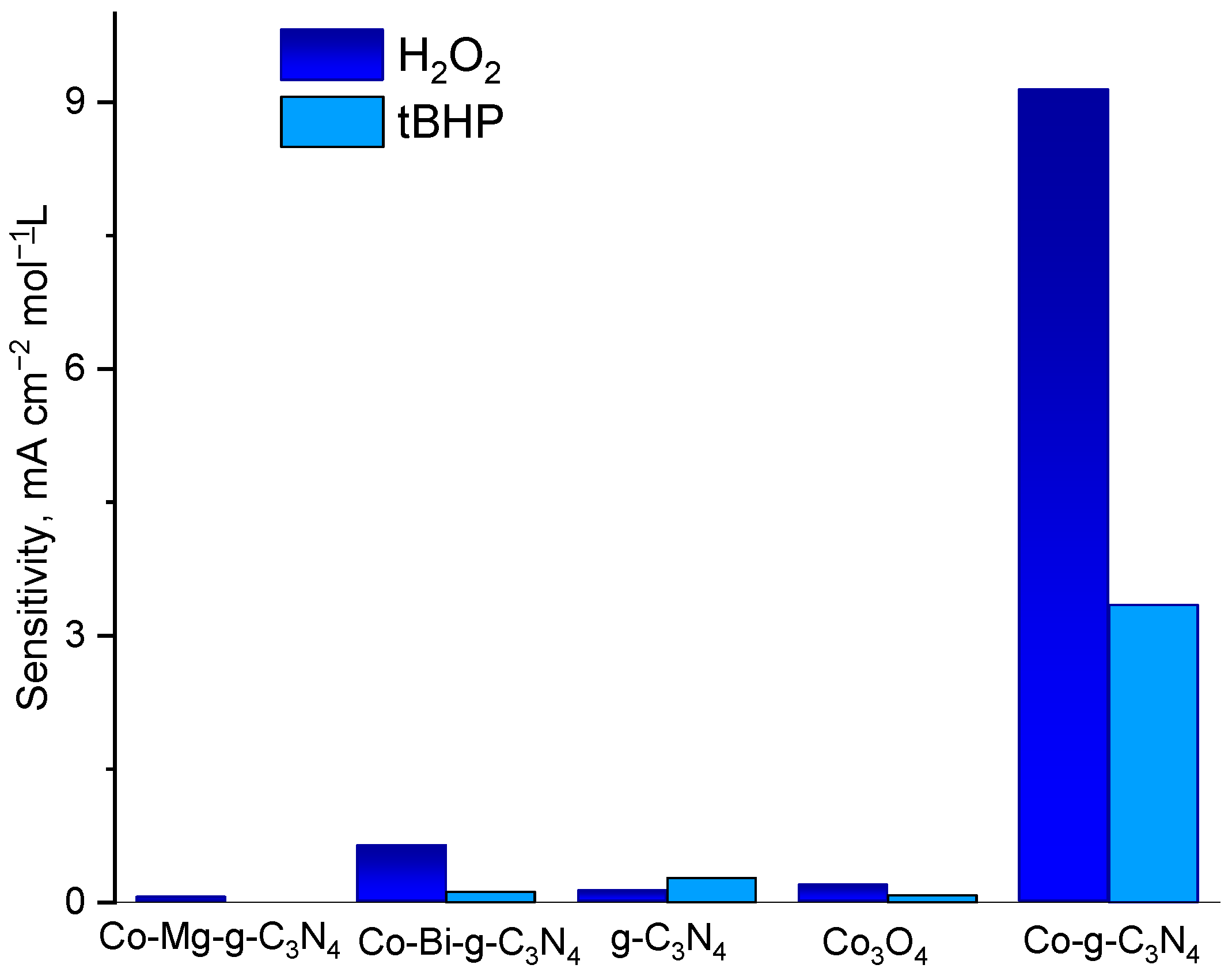
2.1. Electrocatalytic Activity of Pristine and Metal Oxide—Doped g-C3N4
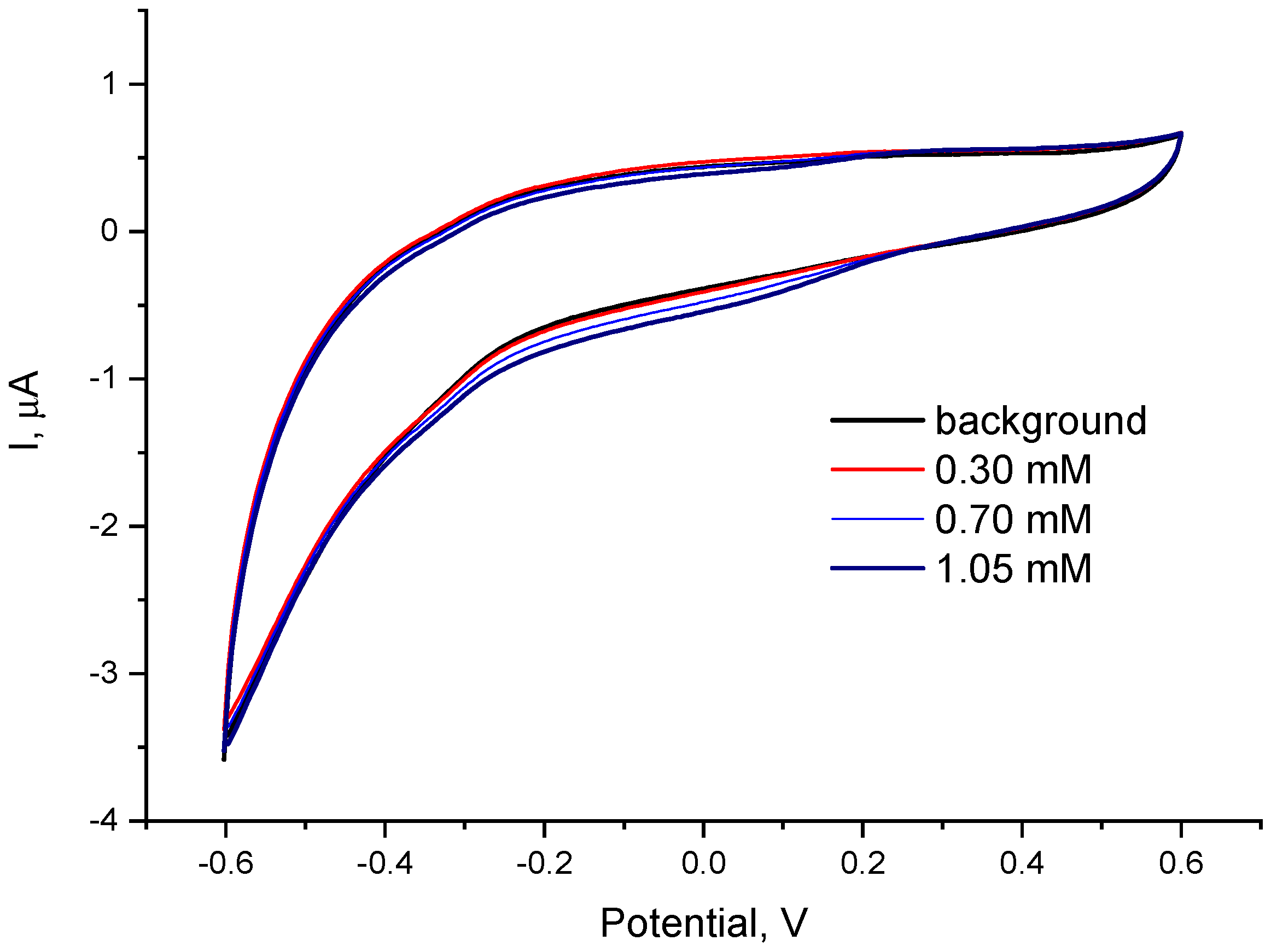
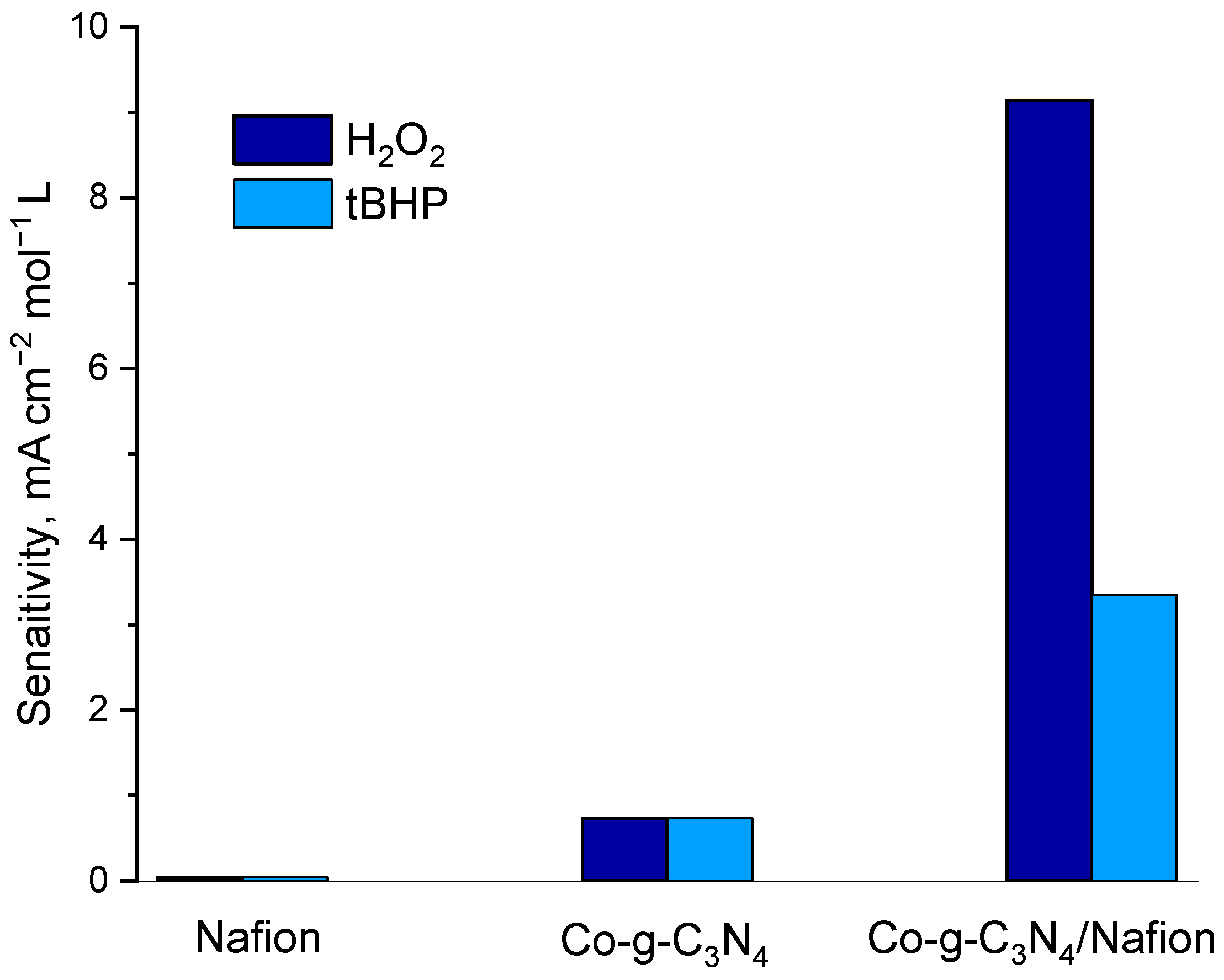
2.2. Optimization of the Polymer-Catalyst Ratio
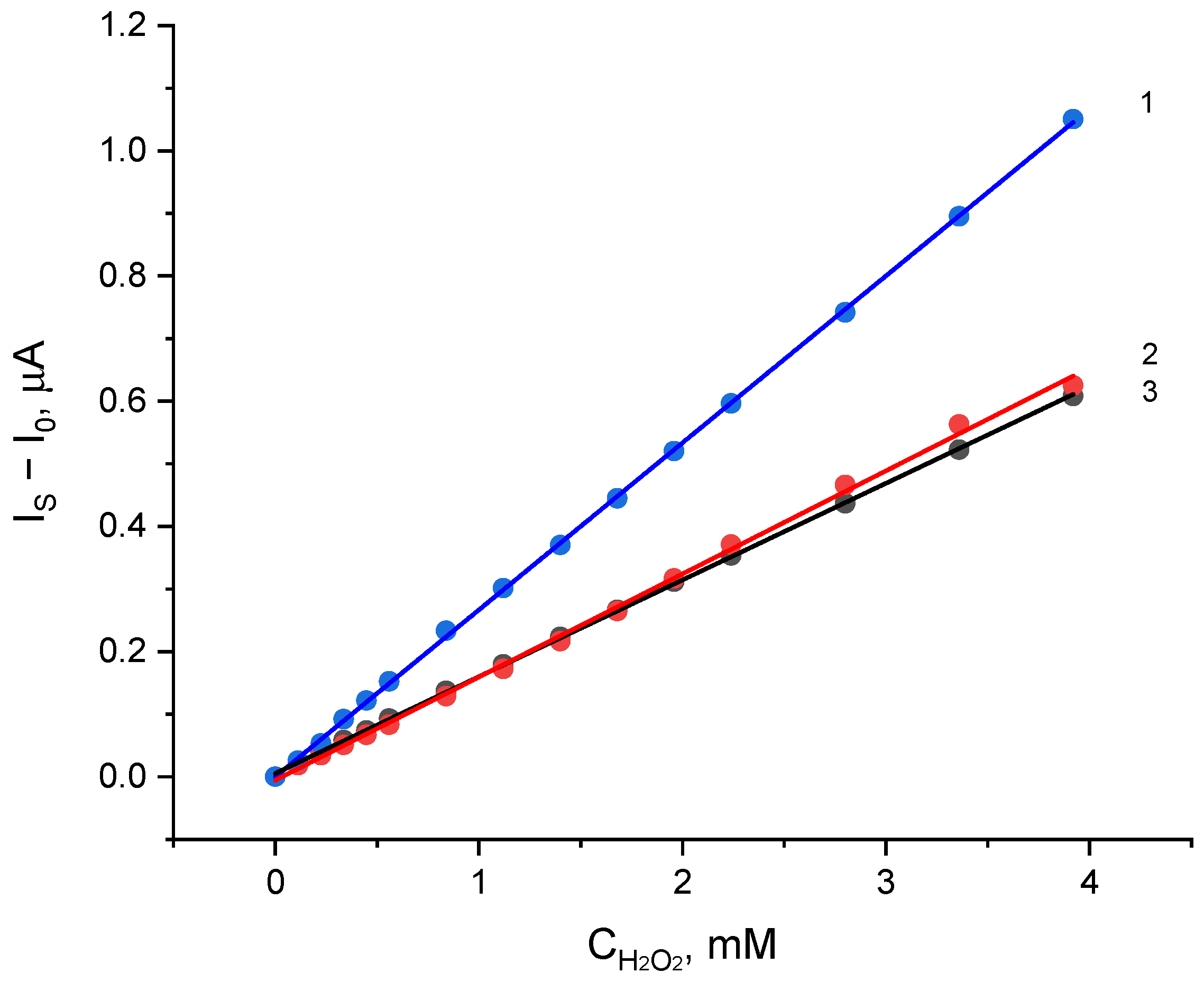
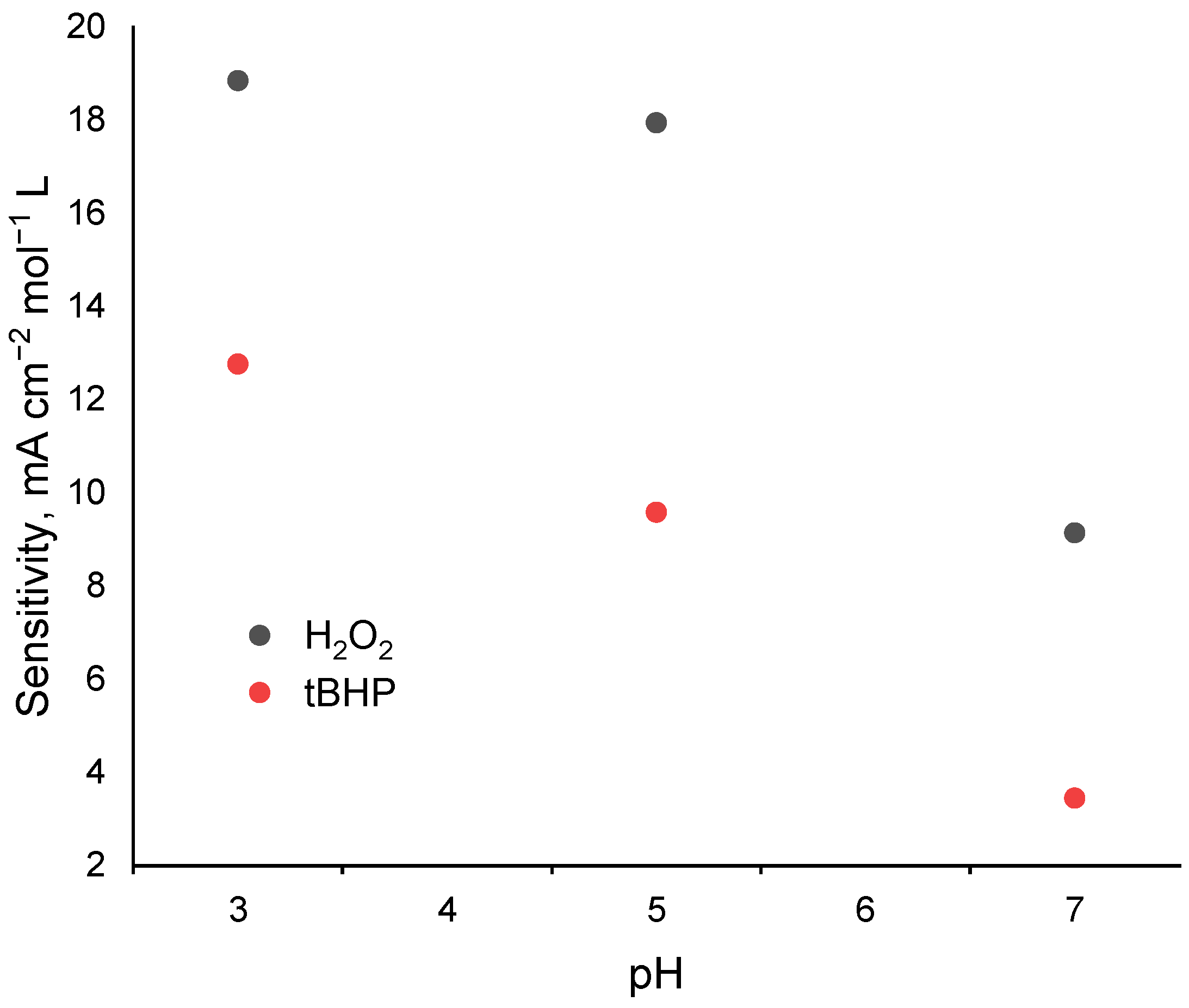
2.3. Electrocatalytic Reduction of Hydroperoxides by Co-g-C3N4/Nafion Modified Electrode
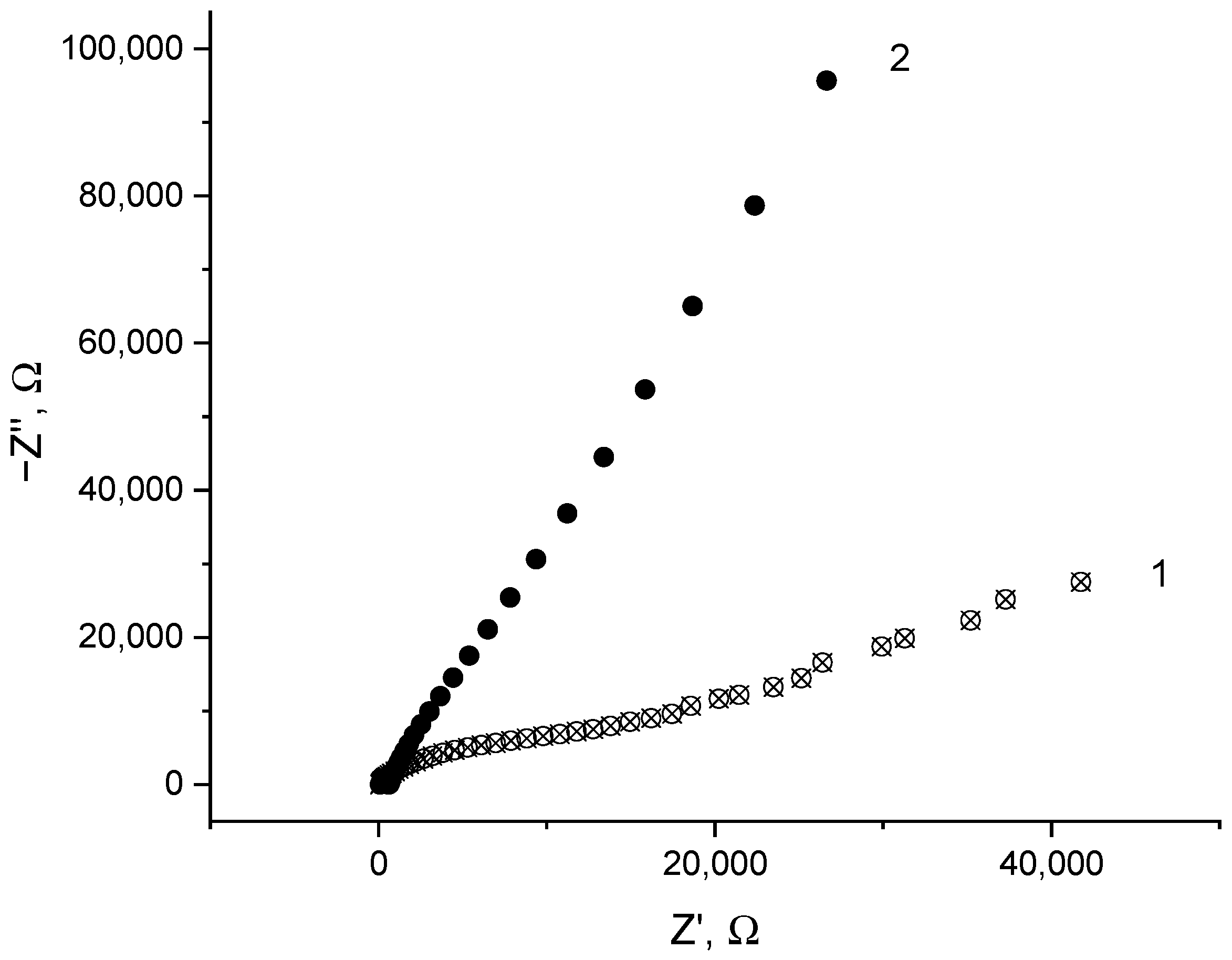
2.4. Stability of the Co-Doped C3N4/Nafion Modified Electrode
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Apparatus and Measurements
4.3. Synthesis of Graphitic Carbon Nitride g-C3N4 and Co-Doped g-C3N4
4.4. Modification of Working Electrode
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Talbot, G. The Stability and Shelf Life of Fats and Oils. In The Stability and Shelf Life of Food, 2nd ed.; Subramaniam, P., Ed.; Woodhead Publishing: Sawston, UK, 2016; pp. 461–503. [Google Scholar]
- Barsukova, M.E.; Veselova, I.A.; Shekhovtsova, T.N. Main Methods and Approaches to the Determination of Markers of Oxidative Stress—Organic Peroxide Compounds and Hydrogen Peroxide. J. Anal. Chem. 2019, 74, 425–436. [Google Scholar] [CrossRef]
- Ames, B.N. Dietary Carcinogens and Anti-Carcinogens. J. Toxicol. Clin. Toxicol. 1984, 22, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Miyata, R.; Tanuma, N. Oxidative stress in developmental brain disorders. Adv. Exp. Med. Biol. 2012, 724, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Singh, R.P. Advances in instrumental methods to determine food quality deterioration. In Food and Beverage Stability and Shelf Life; Kilcast, D., Subramaniam, P., Eds.; Woodhead Publishing: Sawston, UK, 2011; pp. 381–404. [Google Scholar]
- Beutner, G.L.; Ayers, S.; Chen, T.; Leung, S.W.; Tai, H.C.; Wang, Q. General Method for Quantitation of Organic Hydroperoxides. Org. Process Res. Dev. 2020, 24, 1321–1327. [Google Scholar] [CrossRef]
- Sun, J.; Ning, Y.; Chen, X.; Zhang, X.; Ren, Y.; Li, B. Comparison of Fluorescent Techniques Using Two Enzymes Catalysed for Measurement of Atmospheric Peroxides. Atmosphere 2022, 13, 659. [Google Scholar] [CrossRef]
- Ahmed, A.; Hayat, A.; Nawaz, M.H.; Chaudhry, A.A.; John, P.; Nasir, M. Fluorescence quenching mediated detection of hydrogen peroxide using tungsten incorporated graphitic carbon nitride nanoflakes. RSC Adv. 2021, 11, 7479–7491. [Google Scholar] [CrossRef]
- Luo, M.; Zhang, Y.; Zhao, S. Non-Enzymatic Hydrogen Peroxide Sensor Based on Fe3O4@Polydopamine-Ag Nanocomposite Modified Magnetic Glassy Carbon Electrode. J. Electrochem. Soc. 2021, 168, 067511. [Google Scholar] [CrossRef]
- Liu, W.; Zhan, W.; Jia, X.; Liu, Q.; Chen, R.; Li, D.; Huang, Y.; Zhang, G.; Ni, H. Rapid synthesis of vertically-aligned zinc oxide nanorods on stainless steel for non-enzymatic glucose and H2O2 photoelectrochemical sensor. Appl. Surf. Sci. 2019, 480, 341–348. [Google Scholar] [CrossRef]
- Liu, W.; Hiekel, K.; Hübner, R.; Sun, H.; Ferancova, A.; Sillanpää, M. Pt and Au bimetallic and monometallic nanostructured amperometric sensors for direct detection of hydrogen peroxide: Influences of bimetallic effect and silica support. Sens. Actuators B Chem. 2018, 255, 1325–1334. [Google Scholar] [CrossRef]
- Moozarm Nia, P.; Woi, P.M.; Alias, Y. Facile one-step electrochemical deposition of copper nanoparticles and reduced graphene oxide as nonenzymatic hydrogen peroxide sensor. Appl. Surf. Sci. 2017, 413, 56–65. [Google Scholar] [CrossRef]
- Baj, S. Quantitative determination of organic peroxides. Fresenius’ J. Anal. Chem. 1994, 350, 159–161. [Google Scholar] [CrossRef]
- German, N.; Ramanavicius, A.; Ramanaviciene, A. Amperometric Glucose Biosensor Based on Electrochemically Deposited Gold Nanoparticles Covered by Polypyrrole. Electroanalysis 2017, 29, 1267–1277. [Google Scholar] [CrossRef]
- Stasyuk, N.; Gayda, G.; Zakalskiy, A.; Zakalska, O.; Serkiz, R.; Gonchar, M. Amperometric biosensors based on oxidases and PtRu nanoparticles as artificial peroxidase. Food Chem. 2019, 285, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Dimcheva, N. Nanostructures of noble metals as functional materials in biosensors. Curr. Opin. Electrochem. 2020, 19, 35–41. [Google Scholar] [CrossRef]
- Bollella, P.; Gorton, L. Enzyme based amperometric biosensors. Curr. Opin. Electrochem. 2018, 10, 157–173. [Google Scholar] [CrossRef]
- Achari, D.S.; Santhosh, C.; Deivasegamani, R.; Nivetha, R.; Bhatnagar, A.; Jeong, S.K.; Grace, A.N. A non-enzymatic sensor for hydrogen peroxide based on the use of α-Fe2O3 nanoparticles deposited on the surface of NiO nanosheets. Microchim. Acta 2017, 184, 3223–3229. [Google Scholar] [CrossRef]
- Bai, H.; Zhang, L.; Shen, H.; Liu, L. Facile Synthesis of Cuprous Oxide/Gold Nanocomposites for Nonenzymatic Amperometric Sensing of Hydrogen Peroxide. Electroanalysis 2017, 29, 2773–2779. [Google Scholar] [CrossRef]
- Li, S.J.; Xing, Y.; Yang, H.Y.; Huang, J.Y.; Wang, W.T.; Liu, R.T. Electrochemical synthesis of a binary Mn-Co oxides decorated graphene nanocomposites for application in nonenzymatic H2O2 sensing. Int. J. Electrochem. Sci. 2017, 12, 6566–6576. [Google Scholar] [CrossRef]
- Gu, H.; Yang, Y.; Tian, J.; Shi, G. Photochemical synthesis of noble metal (Ag, Pd, Au, Pt) on graphene/ZnO multihybrid nanoarchitectures as electrocatalysis for H2O2 reduction. ACS Appl. Mater. Interfaces 2013, 5, 6762–6768. [Google Scholar] [CrossRef]
- Alves, I.C.B.; Santos, J.R.N.; Viégas, D.S.S.; Marques, E.P.; Lacerda, C.A.; Zhang, L.; Zhang, J.; Marques, A.L.B. Nanoparticles of Fe2O3 and Co3O4 as efficient electrocatalysts for oxygen reduction reaction in acid medium. J. Braz. Chem. Soc. 2019, 30, 2681–2690. [Google Scholar] [CrossRef]
- Hu, T.; Mei, X.; Wang, Y.; Weng, X.; Liang, R.; Wei, M. Two-dimensional nanomaterials: Fascinating materials in biomedical field. Sci. Bull. 2019, 64, 1707–1727. [Google Scholar] [CrossRef] [Green Version]
- Mamba, G.; Mishra, A.K. Graphitic carbon nitride (g-C3N4) nanocomposites: A new and exciting generation of visible light driven photocatalysts for environmental pollution remediation. Appl. Catal. B Environ. 2016, 198, 347–377. [Google Scholar] [CrossRef]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer to Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef] [PubMed]
- Baranowska, D.; Kędzierski, T.; Aleksandrzak, M.; Mijowska, E.; Zielińska, B. Influence of hydrogenation on morphology, chemical structure and photocatalytic efficiency of graphitic carbon nitride. Int. J. Mol. Sci. 2021, 22, 13096. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef]
- Yew, Y.T.; Lim, C.S.; Eng, A.Y.S.; Oh, J.; Park, S.; Pumera, M. Electrochemistry of Layered Graphitic Carbon Nitride Synthesised from Various Precursors: Searching for Catalytic Effects. ChemPhysChem 2016, 17, 481–488. [Google Scholar] [CrossRef]
- Li, J.; Fang, J.; Ye, P.; Wu, D.; Wang, M.; Li, X.; Xu, A. Peroxymonosulfate activation by iron oxide modified g-C3N4 under visible light for pollutants degradation. J. Photochem. Photobiol. A Chem. 2017, 342, 85–93. [Google Scholar] [CrossRef]
- Ivanova-Kolcheva, V.V.; Stoyanova, M.K. Catalytic activation of PMS by Co3O4 modified g-C3N4 for oxidative degradation of the azo dye Acid Orange 7 in aqueous solutions. Bulg. Chem. Commun. 2019, 51, 158–164. [Google Scholar]
- Ivanova-Kolcheva, V.; Sygellou, L.; Stoyanova, M. Enhanced Catalytic Degradation of Acid Orange 7 Dye by Peroxymonosulfate on Co3O4 Promoted by Bi2O3. Acta Chim. Slov. 2020, 67, 609–621. [Google Scholar] [CrossRef]
- Ivanova-Kolcheva, V.V.; Stoyanova, M.K. Enhancing the PMS activation ability of Co3O4 by doping with Bi and Mg. Bulg. Chem. Commun. 2018, 50, 144–150. [Google Scholar]
- Cappadonia, M.; Erning, J.W.; Niaki, S.M.S.; Stimming, U. Conductance of Nafion 117 membranes as a function of temperature and water content. Solid State Ion. 1995, 77, 65–69. [Google Scholar] [CrossRef]
- Huffstutler, J.D.; Wasala, M.; Richie, J.; Barron, J.; Winchester, A.; Ghosh, S.; Yang, C.; Xu, W.; Song, L.; Kar, S.; et al. High performance graphene-based electrochemical double layer capacitors using 1-butyl-1-methylpyrrolidinium tris (pentafluoroethyl) trifluorophosphate Ionic Liquid as an Electrolyte. Electronics 2018, 7, 229. [Google Scholar] [CrossRef] [Green Version]
- Horozova, E.; Dodevska, T.; Dimcheva, N.; Mussarlieva, R. Electrocatalytic Reduction of Hydrogen Peroxide on Palladium-Gold Codeposits on Glassy Carbon: Applications to the Design of Interference-Free Glucose Biosensor. Int. J. Electrochem. 2011, 2011, 697698. [Google Scholar] [CrossRef] [Green Version]
- Napoli, L.; Franco, J.; Fasoli, H.; Sanguinetti, A. Conductivity of Nafion® 117 membrane used in polymer electrolyte fuel cells. Int. J. Hydrogen Energy 2014, 39, 8656–8660. [Google Scholar] [CrossRef]
- Hu, S.; Chen, X.; Li, Q.; Li, F.; Fan, Z.; Wang, H.; Wang, Y.; Zheng, B.; Wu, G. Fe3+ doping promoted N2 photofixation ability of honeycombed graphitic carbon nitride: The experimental and density functional theory simulation analysis. Appl. Catal. B Environ. 2017, 201, 58–69. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pimpilova, M.; Ivanova-Kolcheva, V.; Stoyanova, M.; Dimcheva, N. 2D Nanomaterial—Based Electrocatalyst for Water Soluble Hydroperoxide Reduction. Catalysts 2022, 12, 807. https://doi.org/10.3390/catal12080807
Pimpilova M, Ivanova-Kolcheva V, Stoyanova M, Dimcheva N. 2D Nanomaterial—Based Electrocatalyst for Water Soluble Hydroperoxide Reduction. Catalysts. 2022; 12(8):807. https://doi.org/10.3390/catal12080807
Chicago/Turabian StylePimpilova, Mariya, Vanina Ivanova-Kolcheva, Maria Stoyanova, and Nina Dimcheva. 2022. "2D Nanomaterial—Based Electrocatalyst for Water Soluble Hydroperoxide Reduction" Catalysts 12, no. 8: 807. https://doi.org/10.3390/catal12080807
APA StylePimpilova, M., Ivanova-Kolcheva, V., Stoyanova, M., & Dimcheva, N. (2022). 2D Nanomaterial—Based Electrocatalyst for Water Soluble Hydroperoxide Reduction. Catalysts, 12(8), 807. https://doi.org/10.3390/catal12080807