
Synthesis of (S)- and (R)-β-Tyrosine by Redesigned Phenylalanine Aminomutase


Abstract
1. Introduction
2. Results and Discussion
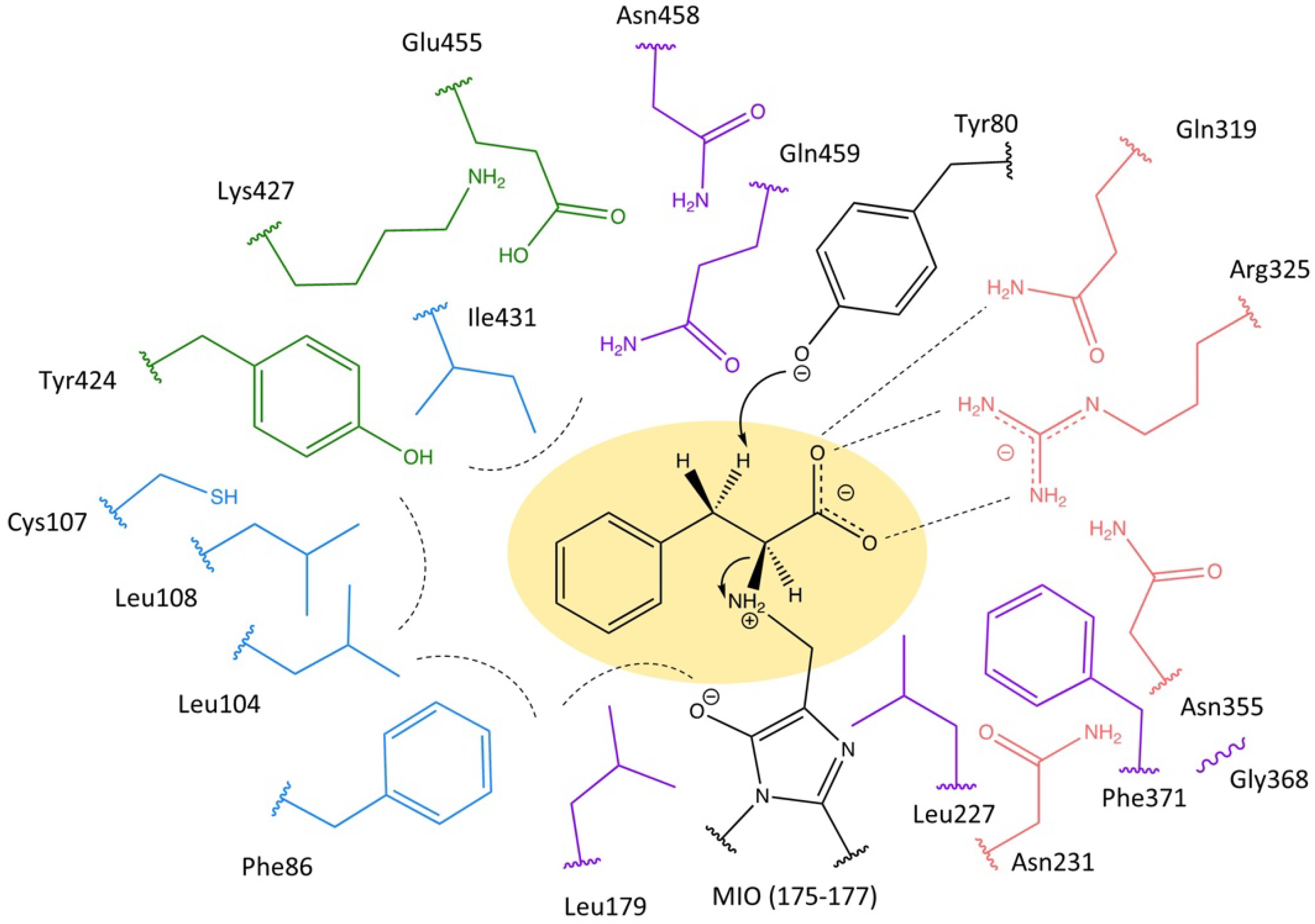
2.1. Highly Conserved Residues
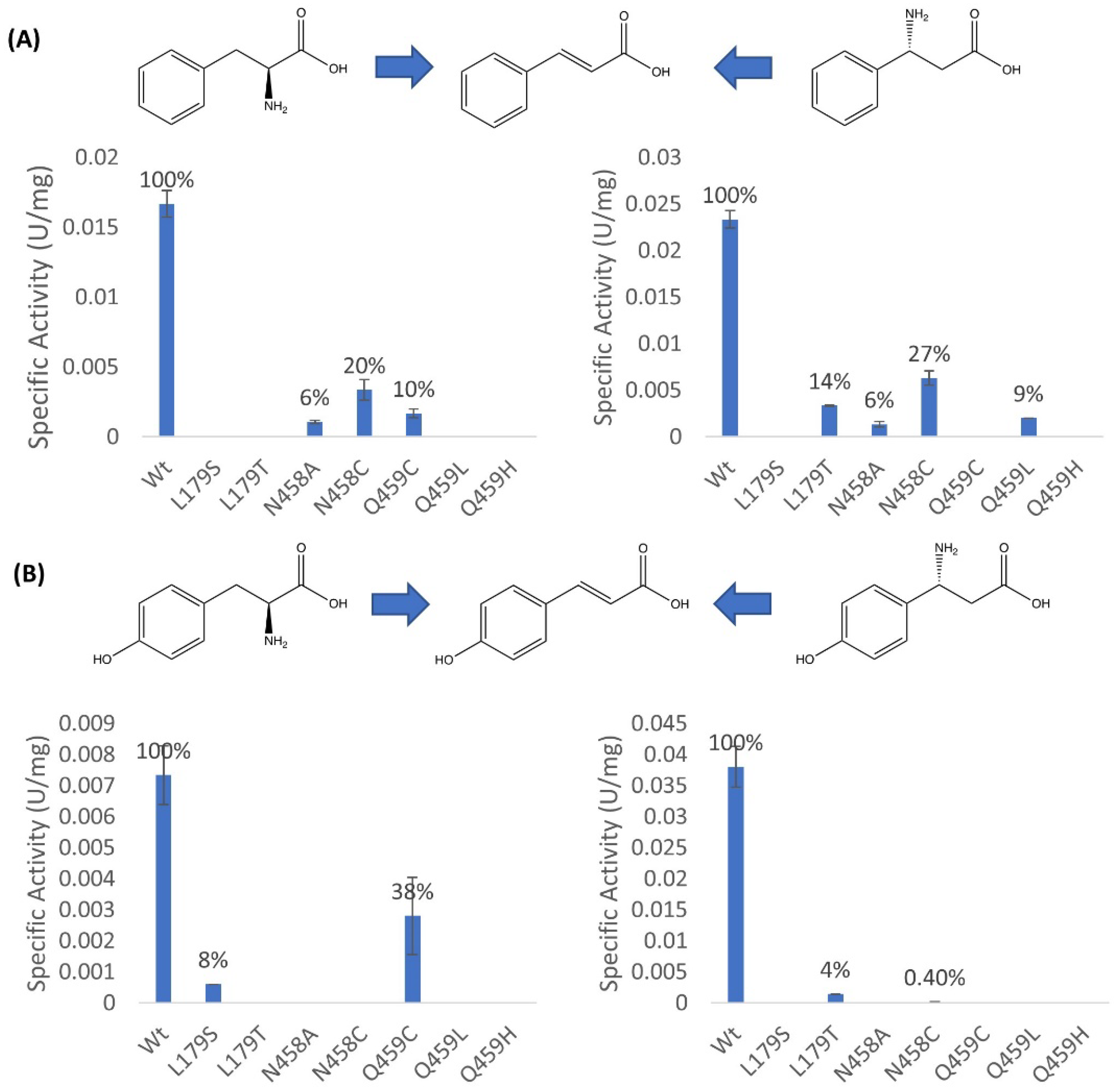
2.2. Mutagenesis and Kinetic Analyses of the Residues Surrounding the Aryl Ring on Substrates
2.3. Investigations into the Enantioselectivity of the Ammonia Addition Reactions
3. Conclusions
4. Materials and Methods
4.1. Computational Enzyme Design and Conservation Analysis
4.2. Construction of the TchPAM Mutants
4.3. Expression, Purification, and Concentration of TchPAM
4.4. Kinetic Analysis of the Ammonia Elimination Reaction
4.5. HPLC Analysis of the Ammonia Addition Reaction
Author Contributions
Funding
Conflicts of Interest
References
- Juaristi, E.; Soloshonok, V.A. Enantioselective Synthesis of Beta-Amino Acids; John Wiley & Sons: London, UK, 2005. [Google Scholar]
- Cabrele, C.; Martinek, T.A.; Reiser, O.; Berlicki, Ł. Peptides containing β-amino acid patterns: Challenges and successes in medicinal chemistry. J. Med. Chem. 2014, 57, 9718–9739. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.-P.; Cao, C.-H.; Zheng, Y.-G. Enzymatic asymmetric synthesis of chiral amino acids. Chem. Soc. Rev. 2018, 47, 1516–1561. [Google Scholar] [CrossRef] [PubMed]
- Slichenmyer, W.J.; Von Hoff, D.D. Taxol: A new and effective anti-cancer drug. Anticancer Drugs 1991, 2, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Cusidó, R.M.; Mirjalili, M.H.; Moyano, E.; Palazón, J.; Bonfill, M. Production of the anticancer drug taxol in Taxus baccata suspension cultures: A review. Process Biochem. 2011, 46, 23–34. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Thornburg, C.K.; Walter, T.; Walker, K.D. Biocatalysis of a paclitaxel analogue: Conversion of baccatin III to N-debenzoyl-N-(2-furoyl) paclitaxel and characterization of an amino phenylpropanoyl CoA transferase. Biochemistry 2017, 56, 5920–5930. [Google Scholar] [CrossRef]
- Zhang, G.; Liang, Y.; Qin, T.; Xiong, T.; Liu, S.; Guan, W.; Zhang, Q. Copper-Catalyzed Asymmetric Hydroamination: A Unified Strategy for the Synthesis of Chiral $β$-Amino Acid and Its Derivatives. CCS Chem. 2021, 3, 1737–1745. [Google Scholar] [CrossRef]
- Turner, N.J. Ammonia lyases and aminomutases as biocatalysts for the synthesis of α-amino and β-amino acids. Curr. Opin. Chem. Biol. 2011, 15, 234–240. [Google Scholar] [CrossRef]
- Schwede, T.F.; Rétey, J.; Schulz, G.E. Crystal structure of histidine ammonia-lyase revealing a novel polypeptide modification as the catalytic electrophile. Biochemistry 1999, 38, 5355–5361. [Google Scholar] [CrossRef]
- Baedeker, M.; Schulz, G.E. Autocatalytic peptide cyclization during chain folding of histidine ammonia-lyase. Structure 2002, 10, 61–67. [Google Scholar] [CrossRef][Green Version]
- Reid, B.G.; Flynn, G.C. Chromophore formation in green fluorescent protein. Biochemistry 1997, 36, 6786–6791. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Murcia, P.A.; Bueren-Calabuig, J.A.; Camacho-Artacho, M.; Cortés-Cabrera, Á.; Gago, F. Stepwise Simulation of 3,5-Dihydro-5-methylidene-4H-imidazol-4-one (MIO) Biogenesis in Histidine Ammonia-lyase. Biochemistry 2016, 55, 5854–5864. [Google Scholar] [CrossRef] [PubMed]
- Punekar, N.S. Enzymes: Catalysis, Kinetics and Mechanisms; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Chesters, C.; Wilding, M.; Goodall, M.; Micklefield, J. Thermal bifunctionality of bacterial phenylalanine aminomutase and ammonia lyase enzymes. Angew. Chem. Int. Ed. 2012, 51, 4344–4348. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, S.; Wybenga, G.G.; Jansen, M.; Heberling, M.M.; Wu, B.; Dijkstra, B.W.; Janssen, D.B. Redesign of a phenylalanine aminomutase into a phenylalanine ammonia lyase. ChemCatChem 2013, 5, 1797–1802. [Google Scholar] [CrossRef]
- Feng, L.; Wanninayake, U.; Strom, S.; Geiger, J.; Walker, K.D. Mechanistic, mutational, and structural evaluation of a taxus phenylalanine aminomutase. Biochemistry 2011, 50, 2919–2930. [Google Scholar] [CrossRef]
- Ratnayake, N.D.; Wanninayake, U.; Geiger, J.H.; Walker, K.D. Stereochemistry and mechanism of a microbial phenylalanine aminomutase. J. Am. Chem. Soc. 2011, 133, 8531–8533. [Google Scholar] [CrossRef]
- Wang, K.; Hou, Q.; Liu, Y. Insight into the mechanism of aminomutase reaction: A case study of phenylalanine aminomutase by computational approach. J. Mol. Graph. Model. 2013, 46, 65–73. [Google Scholar] [CrossRef]
- Wu, B.; Szymański, W.; Wybenga, G.G.; Heberling, M.M.; Bartsch, S.; Dewildeman, S.; Poelarends, G.J.; Feringa, B.L.; Dijkstra, B.W.; Janssen, D.B. Mechanism-inspired engineering of phenylalanine aminomutase for enhanced β-regioselective asymmetric amination of cinnamates. Angew. Chem. Int. Ed. 2012, 51, 482–486. [Google Scholar] [CrossRef]
- Wybenga, G.G.; Szymanski, W.; Wu, B.; Feringa, B.L.; Janssen, D.B.; Dijkstra, B.W. Structural investigations into the stereochemistry and activity of a phenylalanine-2,3-aminomutase from Taxus chinensis. Biochemistry 2014, 53, 3187–3198. [Google Scholar] [CrossRef]
- Peng, F.; Aliyu, H.; Delavault, A.; Engel, U.; Rudat, J. Computational-Designed Enzyme for β-Tyrosine Production in Lignin Valorization. Catalysts 2021, 11, 1310. [Google Scholar] [CrossRef]
- Ribeiro, A.J.M.; Tyzack, J.D.; Borkakoti, N.; Holliday, G.L.; Thornton, J.M. A global analysis of function and conservation of catalytic residues in enzymes. J. Biol. Chem. 2020, 295, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Janiak, C. A critical account on π-π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalt. Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- McGaughey, G.B.; Gagné, M.; Rappé, A.K. π-Stacking interactions. Alive and well in proteins. J. Biol. Chem. 1998, 273, 15458–15463. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Szymanski, W.; Wietzes, P.; de Wildeman, S.; Poelarends, G.J.; Feringa, B.L.; Janssen, D.B. Enzymatic synthesis of enantiopure α- and β-amino acids by phenylalanine aminomutase-catalysed amination of cinnamic acid derivatives. ChemBioChem 2009, 10, 338–344. [Google Scholar] [CrossRef]
- Zhu, L.; Ge, F.; Li, W.; Song, P.; Tang, H.; Tao, Y.; Liu, Y.; Du, G. One step synthesis of unnatural β-arylalanines using mutant phenylalanine aminomutase from Taxus chinensis with high β-regioselectivity. Enzym. Microb. Technol. 2018, 114, 22–28. [Google Scholar] [CrossRef]
- Nagy, E.Z.A.; Tork, S.D.; Lang, P.A.; Filip, A.; Irimie, F.D.; Poppe, L.; Toşa, M.I.; Schofield, C.J.; Brem, J.; Paizs, C.; et al. Mapping the Hydrophobic Substrate Binding Site of Phenylalanine Ammonia-Lyase from Petroselinum crispum. ACS Catal. 2019, 9, 8825–8834. [Google Scholar] [CrossRef]
- Strom, S.; Wanninayake, U.; Ratnayake, N.D.; Walker, K.D.; Geiger, J.H. Insights into the mechanistic pathway of the Pantoea agglomerans phenylalanine aminomutase. Angew. Chem. Int. Ed. 2012, 51, 2898–2902. [Google Scholar] [CrossRef]
- Pilbák, S.; Tomin, A.; Rétey, J.; Poppe, L. The essential tyrosine-containing loop conformation and the role of the C-terminal multi-helix region in eukaryotic phenylalanine ammonia-lyases. FEBS J. 2006, 273, 1004–1019. [Google Scholar] [CrossRef]
- Röther, D.; Poppe, L.; Morlock, G.; Viergutz, S.; Rétey, J. An active site homology model of phenylalanine ammonia-lyase from Petroselinum crispum. Eur. J. Biochem. 2002, 269, 3065–3075. [Google Scholar] [CrossRef]
- Heberling, M.M.; Masman, M.F.; Bartsch, S.; Wybenga, G.G.; Dijkstra, B.W.; Marrink, S.J.; Janssen, D.B. Ironing out their differences: Dissecting the structural determinants of a phenylalanine aminomutase and ammonia lyase. ACS Chem. Biol. 2015, 10, 989–997. [Google Scholar] [CrossRef]
- Chaudhury, S.; Lyskov, S.; Gray, J.J. PyRosetta: A script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 2010, 26, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Boyle, N.M.O.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision A.02; Gaussian Inc.: Oxford, UK, 2016. [Google Scholar]
- Kothiwale, S.; Mendenhall, J.L.; Meiler, J. BCL::Conf: Small molecule conformational sampling using a knowledge based rotamer library. J. Cheminform. 2015, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. The {PyMOL} Molecular Graphics System; Version 1.8; Schrodinger Inc.: New York, NY, USA, 2015. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Brückner, H.; Wittner, R.; Godel, H. Automated enantioseparation of amino acids by derivatization with o-phthaldialdehyde and n-acylated cysteines. J. Chromatogr. 1989, 476, 73–82. [Google Scholar] [CrossRef]
- Gord Noshahri, N.; Fooladi, J.; Syldatk, C.; Engel, U.; Heravi, M.M.; Zare Mehrjerdi, M.; Rudat, J. Screening and comparative characterization of microorganisms from Iranian soil samples showing ω-transaminase activity toward a plethora of substrates. Catalysts 2019, 9, 874. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (S)-α-Phenylalanine | |||
|---|---|---|---|
| TchPAM | KM (mM) | kcat (s−1) | kcat/KM (mM−1 s−1) |
| wt | 0.032 ± 0.001 | 0.02 ± 0.001 | 0.625 ± 0.02 |
| L104S | 0.856 ± 0.019 | 0.133 ± 0.001 | 0.156 ± 0.003 |
| L108S | 0.02 ± 0.003 | 0.006 [a] | 0.278 ± 0.004 |
| I431V | 0.028 ± 0.001 | 0.019 [a] | 0.681 ± 0.026 |
| (R)-β-Phenylalanine | |||
| TchPAM | KM (mM) | kcat (s−1) | kcat/KM (mM−1 s−1) |
| wt | 0.062 ± 0.012 | 0.026 ± 0.002 | 0.427 ± 0.078 |
| L104S | 1.311 ± 0.308 | 0.098 ± 0.019 | 0.077 ± 0.019 |
| L108S | 0.039 ± 0.003 | 0.013 [a] | 0.337 ± 0.035 |
| I431V | 0.076 ± 0.013 | 0.027 [a] | 0.357 ± 0.059 |
| (S)-α-Tyrosine | |||
| TchPAM | KM (mM) | kcat (s−1) | kcat/KM (mM−1 s−1) |
| wt | 2.435 ± 0.160 | 0.029 ± 0.005 | 0.011 ± 0.002 |
| L104S | - | - | - |
| L108S | - | - | - |
| I431V | 0.506 ± 0.063 | 0.013 [a] | 0.026 ± 0.002 |
| (R)-β-Tyrosine | |||
| TchPAM | KM (mM) | kcat (s−1) | kcat/KM (mM−1 s−1) |
| wt | 0.465 ± 0.008 | 0.06 ± 0.001 | 0.130 ± 0.01 |
| L104S | 2.715 ± 0.014 | 0.08 ± 0.016 | 0.030 ± 0.006 |
| L108S | 0.472 ± 0.054 | 0.021 ± 0.002 | 0.045 ± 0.001 |
| I431V | 0.618 ± 0.031 | 0.084 ± 0.003 | 0.137 ± 0.003 |
| Group Number | Containing the Residues | Function | Reference |
|---|---|---|---|
| 1 | Tyr80 | Activation | [21,31] |
| 2 | Ala175, Ser176, Gly177, Asp178, Tyr322 | MIO moiety Formation | [11,13,21] |
| 3 | Asn231, Gln319, Arg325, Asn458 | Carboxylate Binding | [20], this work |
| 4 | Phe86, Leu104, Leu108, Leu179, Ile431 | Aromatic group Binding | [20], this work |
| 5 | Cys107, Tyr424, Lys427, Glu455, Gln459 | Influence on the neighboring Residues | [22], this work |
| 6 | Leu227, Gly368, Phe371 | unknown | |
| 7 | Ala77, Ile79, Cys89, Leu97 | Flexibility of inner loop, selectivity of mutase/lyase activity | [32] |
| Mutant | Direction | Primer-Sequence |
|---|---|---|
| L104S | fw | CTG CAG GAA AGT AGC ATT CGC TGT CT |
| rw | ACT TTC CTG CAG TTC GCT CAG G | |
| L108S | fw | CTG ATT CGC TGT AGC CTG GCA G |
| rw | ACA GCG AAT CAG ACT TTC CTG CA | |
| L179S | fw | GCA AGC GGC GAT AGC ATT CCG |
| rw | ATC GCC GCT TGC GCT AAC A | |
| L179T | fw | GCA AGC GGC GAT ACC ATT CCG |
| rw | ATC GCC GCT TGC GCT AAC A | |
| I431V | fw | AAA GGT CTG GAT GTT GCA ATG GCC |
| rw | ATC CAG ACC TTT CAG GCC ATA ATC | |
| N458A | fw | GCA GAA CAG CAT GCA CAG GAT ATT AAT A |
| rw | ATG CTG TTC TGC ACT ATG CAC A | |
| N458C | fw | GCA GAA CAG CAT TGC CAG GAT ATT AAT A |
| rw | ATG CTG TTC TGC ACT ATG CAC A | |
| Q459C | fw | AGA ACA GCA TAA TTG CGA TAT TAA TAG TCT G |
| rw | ATT ATG CTG TTC TGC ACT ATG CAC | |
| Q459L | fw | AGA ACA GCA TAA TCT GGA TAT TAA TAG T |
| rw | ATT ATG CTG TTC TGC ACT ATG CAC | |
| Q459H | fw | AGA ACA GCA TAA TCA CGA TAT TAA TAG T |
| rw | ATT ATG CTG TTC TGC ACT ATG CAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, F.; Aliyu, H.; Delavault, A.; Engel, U.; Rudat, J. Synthesis of (S)- and (R)-β-Tyrosine by Redesigned Phenylalanine Aminomutase. Catalysts 2022, 12, 397. https://doi.org/10.3390/catal12040397
Peng F, Aliyu H, Delavault A, Engel U, Rudat J. Synthesis of (S)- and (R)-β-Tyrosine by Redesigned Phenylalanine Aminomutase. Catalysts. 2022; 12(4):397. https://doi.org/10.3390/catal12040397
Chicago/Turabian StylePeng, Fei, Habibu Aliyu, André Delavault, Ulrike Engel, and Jens Rudat. 2022. "Synthesis of (S)- and (R)-β-Tyrosine by Redesigned Phenylalanine Aminomutase" Catalysts 12, no. 4: 397. https://doi.org/10.3390/catal12040397
APA StylePeng, F., Aliyu, H., Delavault, A., Engel, U., & Rudat, J. (2022). Synthesis of (S)- and (R)-β-Tyrosine by Redesigned Phenylalanine Aminomutase. Catalysts, 12(4), 397. https://doi.org/10.3390/catal12040397